

Abstract
During an ischemic stroke normal brain endothelial function is perturbed, resulting in blood brain barrier (BBB) breakdown with subsequent infiltration of activated inflammatory blood cells, ultimately leading to neuronal cell death. Kruppel-like factor 2 (KLF2) is regulated by flow, is highly expressed in vascular endothelial cells (ECs), and serves as a key molecular switch regulating endothelial function and promoting vascular health. In this study we sought to determine the role of KLF2 in cerebrovascular function and the pathogenesis of ischemic stroke. Transient middle cerebral artery occlusion was performed in KLF2-deficient (KLF2−/−), KLF2 overexpressing (KLF2tg), and control mice, and stroke volume was analyzed. BBB function was assessed in vivo by real-time neuroimaging using positron emission tomography and Evan's blue dye assay. KLF2−/− mice exhibited significantly larger strokes and impairment in BBB function. In contrast, KLF2tg mice were protected against ischemic stroke and demonstrated preserved BBB function. In concordance, gain- and loss-of-function studies in primary brain microvascular ECs using transwell assays revealed KLF2 to be BBB protective. Mechanistically, KLF2 was demonstrated, both in vitro and in vivo, to regulate the critical BBB tight junction factor occludin. These data are first to identify endothelial KLF2 as a key regulator of the BBB and a novel neuroprotective factor in ischemic stroke.
Keywords: Kruppel-like factor 2, stroke, blood brain barrier, endothelial cells, cerebrovascular disease
the brain endothelium plays a critical role in maintaining cerebrovascular homeostasis. Brain endothelial cells (ECs) interact with astrocytes, pericytes, and neurons to form the neurovascular unit (2, 13, 15) and serve as a dynamic interface between the peripheral circulation and the central nervous system (CNS). One of the key functions of the brain endothelium is the formation of the blood brain barrier (BBB) (1, 12, 15, 40), which is characterized by tight interendothelial junctions, formed by factors such as occludin (15). Oxygen and nutrient deprivation during ischemic stroke activates proteases, resulting in degradation of EC tight junction proteins and increased permeability of the BBB. This breakdown allows activated inflammatory cells from the blood stream to migrate across the brain endothelium and infiltrate into the brain parenchyma, initiating an inflammatory cascade that ultimately leads to neuronal damage and cell death (22, 32, 40). Although many of the factors involved in establishing the BBB have been identified, the upstream molecular control of BBB formation remains poorly understood. More insights into the molecular mechanisms of brain EC function are needed to develop new therapies to prevent and treat ischemic stroke.
Kruppel-like factors are members of the zinc finger family of transcription factors that have been shown to play key roles in the regulation of cellular growth and differentiation (26). KLF2 is highly expressed in ECs, and its expression is induced by laminar shear stress and inhibited by proinflammatory cytokines (35). Compelling in vitro observations over the past decade identify KLF2 as a “molecular switch” regulating key aspects of vascular function and disease (4). More recently, in vivo studies, have confirmed the important role of KLF2 in several biological processes including atherosclerosis (5) and systemic vascular permeability (20). However, the in vivo role of endothelial KLF2 in cerebrovascular function remains unknown.
In this study, we investigated the role of KLF2 in the pathogenesis of ischemic stroke. Herein, we have identified KLF2 as a novel stroke-protective factor in the cerebrovasculature through its regulation of BBB function and the tight junction factor occludin.
MATERIALS AND METHODS
Animals.
KLF2-green fluorescent protein (GFP) fusion mice were created in S. C. Jameson's laboratory at the University of Minnesota as previously described (39). CAG-CreERT2 mice were obtained from Jackson Laboratory. CAG-CreERT2 mice were crossed with KLF2 floxed mice [described previously (23)] or KLF2 floxed stop mice (generated as described in text and Fig. 3A), and 4-wk-old mice were intraperitoneally injected with tamoxifen (1 mg/day for 10 days). All mice colonies were maintained in a clean animal facility. All animal experimentation protocols were submitted to and approved by the Case Western Reserve University Institutional Animal Care and Use Committee.
Fig. 3.

KLF2-deficient mice have increased stroke volume and blood brain barrier (BBB) permeability. A: 2,3,5-Triphenyltetrazolium (TTC) staining of KLF2-deficient (K2−/−) and control brains after 1 h transient middle cerebral artery occlusion (tMCAO) and 48 h reperfusion. Representative results shown. B: quantitative analysis of stroke volume (n = 10–20/group). *P = 0.001 vs. control. C: quantitative positron emission tomography (PET) assessment of radiotracer uptake in K2−/− and control brains at baseline and after stereotactic injection of TNF-α into the cerebral cortex (n = 2–4/group). D: quantification of Evan's blue dye (EBD) permeability in K2−/− and control brains after sham or stereotactic injection of TNF-α (n = 3/group). *P = 0.0015; #P = 0.002.
India ink analysis.
Mice were deeply anesthetized using isoflurane and transcardially perfused using 50 ml of room temperature phosphate-buffered saline (pH 7.4), followed by 3.0 ml of undiluted India ink. Brains were then harvested, and the cerebrovasculature anatomy was assessed.
Mouse physiology analysis.
For measuring physiological variables in mice, catheterization of the carotid artery was performed in each mouse. Anesthesia was induced with isoflurane (2.5% isoflurane in 10% oxygen balanced with medical air) and maintained with 1.5% isoflurane through a nasal cone for the duration of the experiment. A polyethylene tubing (PE-10; 0.28 mm i.d., 0.6 mm o.d.) was placed in the left carotid artery to monitor arterial blood pressure and heart rate (Blood Pressure analyzer; Digi-Med) and obtain blood samples for measuring arterial blood gases (Blood Gas Analyzer; Nova Biomedical).
Mouse model of focal cerebral ischemia.
Temporary focal cerebral ischemia was induced by intraluminal occlusion of the middle cerebral artery (MCAO) as previously described (41). The success of MCAO was confirmed by examination of neurological dysfunction in all mice once they fully recovered from anesthesia (see below) and the formation of infarct as assessed by TTC staining (see below). Mice were evaluated for neurological dysfunction by assessing for forelimb flexion deficit on the contralateral side, or decreased resistance to lateral push and torso turning to the ipsilateral side when held by the tail, or very significant circling to the affected side and reduced capacity to bear weight on the affected side, or rare spontaneous movement. Mice that showed no neurological deficit or died were excluded from the study. For the experimental cohort including KLF2−/− mice, male 6- to 8-wk-old (25–30 g) KLF2−/− and littermate controls (tamoxifen treated KLF2 floxed and CAG-CreERT2 mice) underwent left MCAO for 60 min followed by reperfusion. Due to difficulty of the production of measurable strokes in KLF2tg mice, a slightly modified procedure was used for this experimental cohort. After 60 min of tMCAO both KLF2tg mice and littermate controls (tamoxifen-treated CAG-CreERT2 mice) were maintained in a warmed (30°C) postanesthesia recovery unit for 60 min after reperfusion, resulting in measurable strokes in this cohort. Body temperature of all mice was monitored during the surgical procedure using a rectal temperature probe and maintained at 37 ± 2°C with a heating pad. At 48 h after MCA occlusion, brains were removed and sliced into seven coronal sections, each 1 mm thick. Sections were stained with a 2% solution of 2,3,5-triphenyltetrazolium (TTC). Infarct areas were measured blinded using the National Institutes of Health (NIH) ImageJ software and summed to determine stroke volumes.
Stereotactic intracerebral injection of TNF-α.
For the experimental cohort including KLF2−/− mice, male 15- to 20-wk-old KLF2−/− and littermate controls (tamoxifen-treated CAG-CreERT2 mice) were stereotactically injected with TNF-α (1 μg/kg in 4 μl 1% BSA/PBS over 20–30 min) into the striatum using the following coordinates from bregma: anteroposterior, 0.0 mm; lateral, 1.5 mm; ventral, 2.1 mm. To assess the BBB protective effects in the experimental cohort including KLF2tg mice the same procedure was followed except 25 μg/kg TNF-α was used.
In vivo real-time microPET imaging with C-11 labeled N-methylserotonin.
At baseline or 2 h after stereotactic injection mice were placed onto the micro-PET/CT (Inveon; Siemens) scanner. C-11 labeled N-methylserotonin ([11C]Me-5-HT) (ca. 2 mCi/kg) was then administered through tail vein injection via a catheter. Dynamic microPET acquisition was performed over 90 min in a list-mode immediately after injection. Body temperature of the mice was monitored by using a rectal temperature probe and maintained at 37 ± 2°C with a heating pad. After PET data acquisition, 10 min of transmission data were acquired using Co-57 as a source.
Quantitative image analysis.
After microPET study, we conducted quantitative image analysis to evaluate BBB permeability in vivo. List-mode PET data were reconstructed into a time sequence of volumetric images: 6 frames of 10-s duration followed by 4 frames at 60-s duration, and the remainder of the data in 300-s duration sections. Each frame was reconstructed based on transmission data with correction for photon attenuation, scatter, and random events. The value of each image voxel in the unit of body-mass standard uptake value (SUV) was calculated by comparing reconstructed count values to a calibration image of phantom of known activity. The total activity was corrected for residual after injection. We defined the whole brain as the region of interest. The radioactivity concentration in the brain region is expressed in terms of standardized uptake values (SUV) [(μCi/cc)/(μCi/g)] as a function of time.
Evans blue dye assays.
Immediately after stereotactic injection 2% Evans blue dye (EBD; 4 ml/kg) was injected into the tail vein. For comparison, mice with burr hole only (sham) were used. After 2 h mice were euthanized and saline perfused, and brains were rapidly removed, weighed, and homogenized in 50% TCA. After extraction, the concentration of EBD (ng EBD/mg brain) was determined by fluorescent intensity (excitation 620, emission 680) as calculated from an EBD standard curve.
Cell culture, reagents, and immunoblotting.
Primary human brain microvascular ECs were acquired from Cell Systems and cultured in EBM-2 media with supplemental growth factors according to manufacturer's instructions. 293T cells were cultured in DMEM with 5% FBS. Mouse anti-occludin antibody was from Invitrogen, and mouse anti-β actin antibody was from Santa Cruz. All adenoviral constructs and small interfering (si)RNA oligos were generated, obtained, and used as previously described (20, 35). Protein isolation and Western analysis using the indicated antibodies was performed as previously described (20).
Transwell assays and oxygen glucose deprivation.
All transwell assays were conducted as described earlier (20). For oxygen glucose deprivation, cells were exposed to deoxygenated PBS in a modular incubator chamber flushed with 1% O2, 5% CO2, and 94% N2; sealed; and placed at 37°C for 30 min followed by 2 h of exposure to normal culture media under normoxic conditions (21% O2, 5% CO2) at 37°C in a tissue culture incubator. FITC dextran was added to the transwell insert immediately upon reexposure to normal media and normoxia.
Real-time quantitative RT-PCR.
Total RNA was extracted from primary human brain microvascular ECs and whole brain tissue with TRIzol Reagent (Invitrogen) according to the manufacturers instructions. Total RNA (2 μg) was reverse transcribed with M-MuLV reverse transcriptase (New England BioLabs) and oligo-dT primers. Real-time PCR was performed with Universal SYBR Green PCR Master Mix on Applied Biosystems Step One Real-Time PCR system with gene specific primers.
Transfection analysis.
Transient transfections were performed in 293T cells with a 480 bp occludin promoter-reporter construct containing the majority of the promoter activity (24) and increasing concentrations of KLF2 expression plasmid as indicated, using Lipofectamine 2000 (Invitrogen) according to manufacturer's protocol. β-Galactosidase was used as an internal control, and total DNA was kept constant at 4 μg. The cells were harvested after 48 h, and luciferase activity was measured using the Luciferase Reporter System (Promega), according to the manufacturer's recommendations. Luciferase activity was normalized to whole cell protein concentration.
Immunofluorescence microscopy.
Brain cryosections (8 μm) were fixed in 4% paraformaldehyde for 10 min, washed, and blocked for 30 min with 5% BSA in PBS-T (0.1 M PBS containing 0.2% Tween 20) and incubated overnight at 4°C with mouse anti-occludin and rabbit anti-GFP antibodies (Invitrogen), mouse anti-HA (Sigma), and rat anti-mouse CD31 (BD Biosciences) as indicted and followed by Alexa-488 or 594 conjugated secondary antibodies (Invitrogen). All imaging was performed using a Leica video imaging system.
Statistics.
Data are expressed as means ± SE. Differences between statistical groups were evaluated for statistical significance using the Student's t-test for unpaired data. P < 0.05 was considered statistically significant.
RESULTS
KLF2 deficiency augments ischemic stroke and BBB dysfunction.
KLF2 has been previously demonstrated to be highly expressed in systemic vascular ECs (4, 35). As a first step we assessed cellular KLF2 expression in brain tissues using a recently described KLF2-GFP fusion mouse (39). Mouse brain sections from KLF2-GFP fusion mice were stained with GFP and the EC specific cell surface marker CD31. KLF2 immunoreactivity is predominantly localized to the nuclei of ECs of both the macrovasculature (Fig. 1A) and the microvasculature (Fig. 1B) of the cerebral cortex.
Fig. 1.
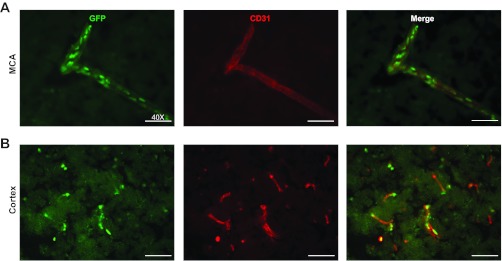
Kruppel-like factor 2 (KLF2) is expressed in brain macrovascular and microvascular endothelial cells (ECs). Green fluorescent protein (GFP) and CD31 immunofluorescence staining of brain cerebral cortex sections from KLF2 GFP fusion mice are shown. A: cerebral macrovascular middle cerebral artery (MCA). B: cerebral microvasculature (n = 6–9/group). Representative results shown.
Recognizing the critical function of KLF2 in endothelial biology and understanding the important role of brain ECs in cerebrovascular ischemia, we sought to assess KLF2 deficiency in a mouse model of stroke. Deletion of KLF2 during development results in embryonic lethality (17, 38). Thus to study KLF2 deficiency in adult mice we generated a conditional global KLF2 knockout mouse (KLF2−/−) by crossing the floxed KLF2 mouse (39) to the CAG CreERT2 mouse (a tamoxifen inducible Cre recombinase driven by the chicken β-actin promoter). Adult mice were treated with intraperitoneal tamoxifen, and >95% KLF2 deletion was confirmed by RT-PCR in multiple tissues (data not shown) including the brain (0.02 ± 0.003 fold vs. control; P = <0.0005) (Fig. 7A). Remarkably, adult KLF2−/− mice are viable and exhibit no gross abnormalities. India ink analysis reveals no anatomical differences in the cerebrovasculature between KLF2−/− mice and controls (Fig. 2A). In addition, there is no significant difference in basic physiologic parameters including blood pressure, heart rate, and arterial blood gases (Fig. 2B).
Fig. 7.

KLF2 regulates occludin expression in mouse brain. A: quantitative RT-PCR analysis of KLF2 and occludin in whole brain tissue from control and K2−/− mice (n = 4–7/group). *P < 0.0001, #P = 0.01. B: quantitative RT-PCR analysis of KLF2 and occludin in whole brain tissue from control and K2tg mice (n = 3 to 4/group). *P = 0.001, #P < 0.05. C: CD31 and occludin immunofluorescence staining of brain cerebral cortex sections from K2−/−, K2tg, and control mice. Representative results shown (n = 3 to 4/group).
Fig. 2.
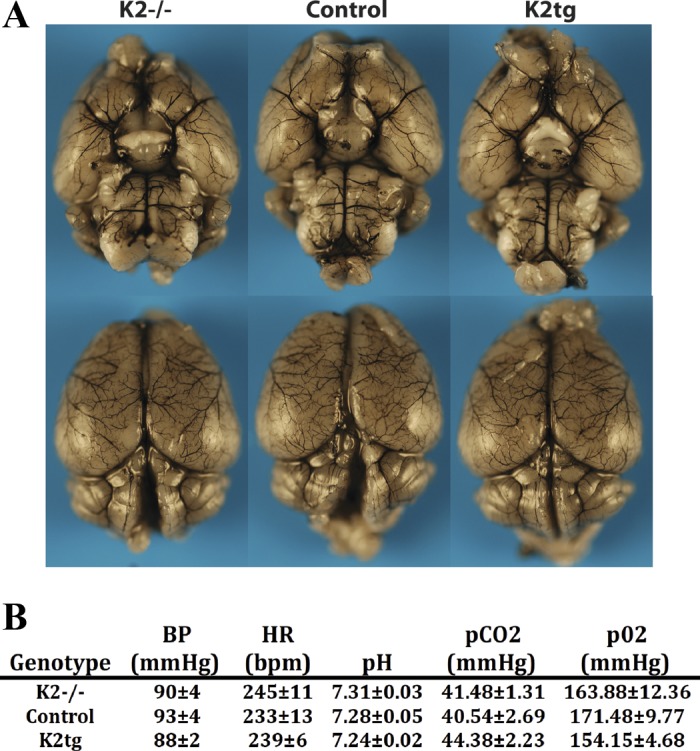
Cerebrovascular anatomy and basic physiology is unchanged in KLF2-deficient and KLF2 overexpressing mice. A: India ink staining of brains from KLF2-deficient (K2−/−), control, and KLF2 overexpressing (K2tg) mice. Representative results shown (n = 4 to 5/group). B: assessment of mean arterial blood pressure (BP), heart rate (HR), pH, pCO2, and pO2 in K2−/−, K2tg, and control mice (n = 4 to 5/group). P = nonsignificant for statistical comparisons between all groups. Bpm, beats/min.
To determine whether KLF2 regulates brain EC function in vivo, we subjected mice rendered postnatally deficient in KLF2 and control mice to temporary focal cerebral ischemia. Mice underwent 1 h of tMCAO followed by 48 h of reperfusion and brains were harvested and analyzed for stroke volume by TTC staining. KLF2−/− mice exhibited significantly larger stroke volumes compared with controls (68.6 ± 9.3 mm3 vs. 35.1 ± 4.6 mm3; P = 0.001) (Fig. 3, A and B), implicating KLF2 as an important factor in the biological response to stroke.
We next assessed the role of KLF2 in BBB function in vivo. TNF-α has been extensively studied in ischemic stroke and has been demonstrated to be induced in rodent models as well as in human disease (18, 37). Several studies implicate TNF-α as playing a pathologic role in stroke (18), and TNF-α has been shown to produce BBB disruption (25). KLF2−/− and control mice were stereotactically injected into the striatum with the inflammatory cytokine TNF-α. BBB function was then assessed using real-time in vivo PET neuroimaging analysis using C-11 labeled N-methylserotonin as radiotracer, which is impermeable across the intact BBB. In response to intracerebral TNF-α injection, KLF2−/− mice exhibited significantly greater BBB permeability (Fig. 3C). These findings were further confirmed by EBD assays (2.66 ± 0.10 ng/mg vs. 1.43 ± 0.13 ng/mg; P = 0.002) (Fig. 3D), thereby identifying KLF2 as playing an important role in BBB function.
Generation of KLF2 overexpressing mice.
To assess the effect of KLF2 overexpression in the pathogenesis of stroke we generated an inducible KLF2 transgenic mouse (KLF2tg). The full-length KLF2 cDNA was cloned downstream of a loxP flanked stop codon and targeted to the ROSA26 locus (Fig. 4A). Mice were then bred to the CAGCreERT2 mice and treated with tamoxifen. In the presence of an activated Cre recombinase the stop codon is excised and a hemagglutinin (HA) tagged KLF2 protein is expressed. As with KLF2−/− mice, KLF2tg mice are viable and exhibit no gross abnormalities. Specifically, we assessed cerebrovascular anatomy and basic physiologic parameters and observed no significant differences (Fig. 2, A and B). RT-PCR analysis of whole brain tissue from these mice reveals greater than sixfold KLF2 overexpression (6.30 ± 0.66 fold vs. control; P = 0.001) (Fig. 7B). Immunostaining of brain sections confirms KLF2 overexpression within the microvascular endothelium (Fig. 4B).
Fig. 4.
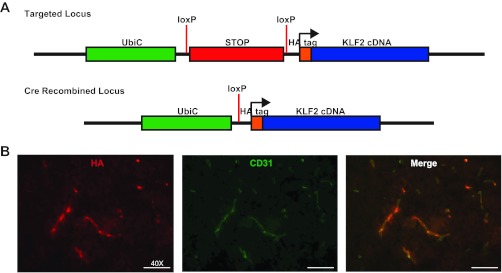
Generation of KLF2 overexpressing mouse. A: schematic diagram of the targeted (ROSA26) locus and Cre recombined locus for the generation of the KLF2tg mouse. UbiC, Ubiquitin promoter; HA, hemagglutinin. B: HA and CD31 immunofluorescence staining of brain cerebral cortex sections from KLF2tg mice (n = 6). Representative results shown.
KLF2 overexpression protects against ischemic stroke and BBB dysfunction.
As with the KLF2−/− mice, KLF2tg mice were subjected to ischemic strokes and BBB functional analysis. After tMCAO, KLF2tg mice had significantly smaller stroke volumes compared with controls (37.4 ± 8.2 mm3 vs. 73.0 ± 6.9 mm3; P = 0.004) (Fig. 5, A and B). In addition, KLF2tg mice were protected against TNF-α-mediated BBB dysfunction as assessed by neuroimaging analysis (Fig. 5C) and EBD assays (0.95 ± 0.04 ng/mg vs. 1.36 ± 0.05 ng/mg; P = 0.003) (Fig. 5D). Taken together, these in vivo gain of function studies strongly implicate KLF2 as a stroke and BBB protective factor within the brain vascular endothelium.
Fig. 5.

KLF2 overexpressing mice have decreased stroke volume and BBB permeability. A: TTC staining of KLF2 overexpressing (K2tg) and control brains after 1 h tMCAO and 48 h reperfusion. Representative results shown. B: quantitative analysis of stroke volume (n = 8 to 9/group). *P = 0.004 vs. control. C: quantitative PET assessment of radiotracer uptake in K2tg and control brains at baseline and after stereotactic injection of TNF-α into the cerebral cortex (n = 3/group). D: quantification of EBD permeability in K2tg and control brains after sham or stereotactic injection of TNF-α (n = 3/group). *P = 0.03; #P = 0.003.
KLF2 regulates key BBB factors in brain microvascular ECs.
To more specifically evaluate the role of KLF2 in mediating BBB function in brain ECs, in vitro transwell assays were performed using primary human brain microvascular ECs. Adenoviral overexpression of KLF2 protects against oxygen glucose deprivation (OGD) mediated BBB permeability (2.27 ± 0.13 units vs. 3.32 ± 0.29 units; P = 0.005) (Fig. 6A). In contrast, siRNA-mediated knockdown of KLF2 (>60% knockdown, data not shown) results in a marked increase in BBB permeability at baseline (2.07 ± 0.05 units vs. control; P < 0.0005) (Fig. 6B).
Fig. 6.

KLF2 regulates BBB permeability and occludin expression in brain microvascular ECs. A: transwell assays quantifying permeability of FITC dextran across primary human brain microvascular ECs infected with KLF2 (Ad-KLF2) or control (Ad-GFP) adenovirus at baseline and after 30 min oxygen glucose deprivation (OGD) followed by 2 h of normal media (n = 8/group). *P = 0.005. B: Transwell assays as in A using cells transfected with small interfering (si)-RNA (si-Control or si-KLF2) assessed at baseline (n = 8/group). *P < 0.0005 vs. si-Control. C: quantitative RT-PCR analysis of tight junction factors in human brain microvascular ECs infected with control (Ad-GFP) and KLF2 (Ad-KLF2) adenovirus (n = 3/group). *P < 0.0001, #P < 0.005, **P < 0.01, ##P < 0.05. D: quantitative RT-PCR analysis of tight junction factors in human brain microvascular ECs infected with si-RNA (si-Control or siKLF2) (n = 3 to 4/group). *P < 0.0001, #P = 0.01, **P = 0.01, ##P < 0.0001. E: Western blot analysis of occludin in human brain microvascular ECs infected with Ad-GFP or Ad-KLF2. Quantification of Western blot is shown. *P < 0.0001. F: transient transfection of 293T cells with occludin promoter luciferase reporter and increasing concentrations of KLF2 plasmid (n = 3/group). *P < 0.005, #P < 0.0005 vs. 0 μg. Representative results of 3 independent experiments shown.
To investigate the molecular mechanisms of the observed functions of KLF2 in the brain microvasculature, we assessed the ability of KLF2 to regulate several key brain EC factors known to play important roles in BBB tight junction formation. KLF2 overexpression in primary human brain ECs resulted in a marked induction of occludin (7.0 ± 0.3-fold; P < 0.0001) and a more modest induction of claudin-12 (3.1 ± 0.3-fold; P = 0.002), junction adhesion molecule-1 (JAM-1) (2.0 ± 0.1-fold; P = 0.008), and AF6 (2.6 ± 0.4-fold; P = 0.03) (Fig. 6C). In correlation with KLF2 overexpression, KLF2 knockdown results in decreased expression of occludin (0.51 ± 0.01 fold; P = 0.01), claudin-12 (0.66 ± 0.07-fold; P = 0.01), and JAM-1 (0.61 ± 0.03-fold; P < 0.0005) (Fig. 6D). The KLF2-dependent regulation of occludin was confirmed by Western blot analysis (3.02 ± 0.07-fold; P < 0.0005) (Fig. 6E). Transient transfections with an occludin promoter luciferase reporter plasmid were performed in 293T cells and demonstrate the ability of KLF2 to regulate the occludin promoter in a dose-dependent manner (Fig. 6F).
To confirm the ability of KLF2 to regulate occludin expression in vivo RT-PCR analysis of brain whole cell extract from KLF2−/−, KLF2tg, and control mice was performed. CD31 staining demonstrates that EC density is unchanged between the three groups of mice (Fig. 7C). However, brains from KLF2−/− mice demonstrated a significant decrease in occludin expression (0.56 ± 0.11-fold expression vs. control; P = 0.01) (Fig. 7A), and KLF2tg brains had a significant increase in occludin expression (2.5 ± 0.5-fold expression vs. control; P < 0.05) (Fig. 7B). Immunohistochemical staining of mouse brain sections provide corroborative findings. KLF2−/− brains exhibit decreased occludin staining, whereas KLF2tg brains display more intense staining compared with control mice (Fig. 7C).
DISCUSSION
Experimental, clinical, and pathologic studies demonstrate that brain microvascular ECs are an important cell type in the pathogenesis of stroke. Our findings are the first to implicate the KLF gene family in general, and KLF2 in particular, as an essential regulator of brain EC BBB function and the in vivo response to cerebral ischemia. We demonstrate that KLF2 is expressed within the cerebrovascular endothelium and serves as a neuroprotective factor in a mouse model of ischemic stroke. KLF2 regulates several key BBB tight junction factors, most notably occludin, thereby serving as a positive mediator of BBB function.
Brain ECs differ from the ECs of other vascular beds due to their high expression of tight junction proteins. This unique property contributes to the ability of the brain endothelium to selectively control permeability between blood and the CNS, thereby forming the BBB. The primary tight junction proteins expressed in brain ECs are occludin, claudins, and JAMs. Various accessory factors such as ZO-1, AF6, and cingulin are also expressed in brain ECs and serve as scaffolding proteins helping tether the tight junctions to the actin cytoskeleton of cells (15, 32, 40). Within the brain, occludin is highly expressed in a continuous fashion along the brain EC cell margins (14, 21), whereas in ECs of other vascular beds occludin expression is significantly less with a more sparse distribution pattern (16, 36), implicating occludin as being actively involved in BBB function. Although studies using knockdown of occludin suggest that occludin is not essential for tight junction formation (31), decreased occludin expression has been associated with BBB dysfunction of a number of disease states (6, 7, 29). One current explanation for these apparently disparate findings is that occludin may mediate tight junction responses to acute inflammation, thereby serving as a sort of “shock absorber” (32). Our findings support this hypothesis. EBD assays in KLF2−/− mice demonstrate no apparent difference in BBB permeability from control mice at baseline, despite a significant decrease in occludin expression. Yet, when stimulated with the inflammatory stimulus, TNF-α, there is a marked impairment of BBB function. Furthermore, the stereotactic injection of 1 μg/kg of TNF-α resulted in a nonsignificant increase in EBD permeability in control mice, whereas a near twofold increase in EBD permeability was observed in the KLF2−/− mice with the same TNF-α dose. In contrast, 25 μg/kg of TNF-α was needed to induce BBB disruption in control mice, whereas KLF2tg mice, with a significant increase in occludin expression, were protected from BBB dysfunction. These findings implicate occludin as playing a role in mediating the response of BBB to inflammatory stimuli. Although KLF2 more strongly regulates occludin, we show that KLF2 also regulates other tight junction factors in brain ECs, and this regulation likely plays some additional role in the observed effects of KLF2 on BBB function and response to ischemic stroke.
Interestingly, KLF2−/− mice have no apparent BBB dysfunction in the absence of an inflammatory stimulus, but when KLF2 is knocked-down in vitro in an isolated culture of primary brain ECs we observe significant BBB permeability at baseline (i.e., without OGD). One potential explanation for these findings is the role of the neurovascular unit (NVU) in cerebrovascular homeostasis and regulation of BBB function. Recent studies have demonstrated that there is significant interaction between all cells of the NVU (endothelial cells, astrocytes, pericytes, and neurons), resulting in a dynamic BBB (27). Emerging evidence indicates that this crosstalk is critical for the regulation of BBB function. Studies are just beginning to elucidate the complex mechanisms of NVU-mediated BBB function, and this area of research has recently been identified by the NIH National Institute of Neurological Disorders and Stroke as a significant area of interest for future investigation. Another potential explanation is the possibility that in vivo in KLF2−/− mice there may be compensatory mechanisms at baseline, such as upregulation of other tight junction proteins. Interestingly, KLF4, another KLF that is highly expressed in ECs, has the ability to regulate many of the same EC factors as KLF2. Using KLF2 hemizygous mice in a model of atherosclerosis, we have previously demonstrated a compensatory increase in endothelial KLF4 expression, thereby limiting the observed EC phenotype (5). The role of KLF4 in the cerebrovasculature and BBB function is unknown and is an interesting area for future investigation.
In addition to the BBB, brain ECs also regulate cerebral blood flow and vasoreactivity (33), as well as cerebrovascular inflammation and thrombosis (9). Although the current work underscores the importance of KLF2 in BBB regulation, we recognize that additional mechanisms may be operative in the observed stroke phenotype. For example, previous studies have indicated that KLF2 inhibits expression of endothelial pro-inflammatory and prothrombotic targets (4, 19, 35). Furthermore, KLF2 regulates endothelial nitric oxide synthase (eNOS)-derived nitric oxide (35), an important determinate of regional cerebral blood flow (rCBF) during ischemic stroke (11). Consequently, it will be of interest to examine rCBF in KLF2 mutant mice under basal conditions and at various time points during the tMCAO stroke model. In addition to rCBF, complementary studies assessing cerebrovascular vasoreactivity, brain microvascular EC eNOS regulation, and NO bioavailability are also likely to be informative.
One limitation of our studies is the use of global knockout and overexpressing mice. KLF2 has been demonstrated to be expressed and exert functions in other cell types besides endothelial cells. KLF2 is expressed in blood immune cells, including monocytes and macrophages (8, 23). Although we recognize that the dominant expression of KLF2 in the brain is localized to the ECs, once the BBB is breached KLF2 function in other cell types may impact the stroke phenotype. Future studies in immune cells will be required to gain a comprehensive understanding of the biological role of KLF2 in stroke.
Previous studies have demonstrated that endothelial KLF2 expression is induced by HMG CoA-reductase inhibitors (i.e., statins) and that some of the pleiotropic effects of statins in endothelial cells are KLF2 dependent (28, 34). As KLF2 has already been identified as a target for therapeutic manipulation, it may serve as a novel target for the treatment of ischemic stroke. Additionally, recent studies have implicated statins as playing a role in cerebrovascular disease and stroke prevention. Clinical studies show about a 20% stroke reduction in patients treated with statins (3, 30), and they have been demonstrated to be neuroprotective in mouse models of stroke (10). Studies investigating the role of KLF2 in mediating the stroke protective effects of statins will be revealing and are currently being conducted in our laboratory. Finally, brain ECs and the BBB have been implicated in several other pathologic processes within the brain (27). Therefore, the findings of this study may bear implications for other disease processes including Alzheimer's disease and multiple sclerosis.
GRANTS
This work was supported by NIH Grants HL-088740 (to G. B. Atkins); HL-076754, HL-086548, HL-097593 (to M. K. Jain); NS-056118, NS-045048, NS-036736 (to J. Chen); HL-087595 (to Z. Lin); NS-061837 (to Y. Wang); Alcoholic Beverage Medical Research Foundation grant (to Z. Lin); Department of Defense Grant W81XWH-10-1-0842 (to Y. Wang); and National Multiple Sclerosis Society Grant RG4339-A-2 (to Y. Wang).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: H.S., B.S., F.Z., C.W., R.Z., J.Z., K.X., Y.K., and G.B.A. performed experiments; H.S., B.S., F.Z., C.W., and G.B.A. analyzed data; H.S., B.S., C.W., and G.B.A. prepared figures; H.S., B.S., F.Z., C.W., R.Z., J.Z., K.X., Y.K., S.C.J., Z.L., Y.W., J.C., M.K.J., and G.B.A. edited and revised manuscript; H.S., B.S., F.Z., C.W., R.Z., J.Z., K.X., Y.K., S.C.J., Z.L., Y.W., J.C., M.K.J., and G.B.A. approved final version of manuscript; S.C.J., Z.L., Y.W., J.C., M.K.J., and G.B.A. conception and design of research; S.C.J., Z.L., Y.W., J.C., M.K.J., and G.B.A. interpreted results of experiments; G.B.A. drafted manuscript.
REFERENCES
- 1. Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiology of disease 37: 13–25, 2010 [DOI] [PubMed] [Google Scholar]
- 2. Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci 7: 41–53, 2006 [DOI] [PubMed] [Google Scholar]
- 3. Amarenco P, Labreuche J. Lipid management in the prevention of stroke: review and updated meta-analysis of statins for stroke prevention. Lancet Neurol 8: 453–463, 2009 [DOI] [PubMed] [Google Scholar]
- 4. Atkins GB, Jain MK. Role of Kruppel-like transcription factors in endothelial biology. Circ Res 100: 1686–1695, 2007 [DOI] [PubMed] [Google Scholar]
- 5. Atkins GB, Wang Y, Mahabeleshwar GH, Shi H, Gao H, Kawanami D, Natesan V, Lin Z, Simon DI, Jain MK. Hemizygous deficiency of Kruppel-like factor 2 augments experimental atherosclerosis. Circ Res 103: 690–693, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bolton SJ, Anthony DC, Perry VH. Loss of the tight junction proteins occludin and zonula occludens-1 from cerebral vascular endothelium during neutrophil-induced blood-brain barrier breakdown in vivo. Neuroscience 86: 1245–1257, 1998 [DOI] [PubMed] [Google Scholar]
- 7. Brown RC, Davis TP. Hypoxia/aglycemia alters expression of occludin and actin in brain endothelial cells. Biochem Biophys Res Commun 327: 1114–1123, 2005 [DOI] [PubMed] [Google Scholar]
- 8. Das H, Kumar A, Lin Z, Patino WD, Hwang PM, Feinberg MW, Majumder PK, Jain MK. Kruppel-like factor 2 (KLF2) regulates proinflammatory activation of monocytes. Proc Natl Acad Sci USA 103: 6653–6658, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. del Zoppo GJ. Virchow's triad: the vascular basis of cerebral injury. Rev Neurol Dis 5, Suppl 1: S12–S21, 2008 [PMC free article] [PubMed] [Google Scholar]
- 10. Endres M. Statins and stroke. J Cereb Blood Flow Metab 25: 1093–1110, 2005 [DOI] [PubMed] [Google Scholar]
- 11. Endres M, Laufs U, Liao JK, Moskowitz MA. Targeting eNOS for stroke protection. Trends Neurosci 27: 283–289, 2004 [DOI] [PubMed] [Google Scholar]
- 12. Engelhardt B. Development of the blood-brain barrier. Cell Tissue Res 314: 119–129, 2003 [DOI] [PubMed] [Google Scholar]
- 13. Fisher M. Pericyte signaling in the neurovascular unit. Stroke 40: S13–S15, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hawkins BT, Abbruscato TJ, Egleton RD, Brown RC, Huber JD, Campos CR, Davis TP. Nicotine increases in vivo blood-brain barrier permeability and alters cerebral microvascular tight junction protein distribution. Brain Res 1027: 48–58, 2004 [DOI] [PubMed] [Google Scholar]
- 15. Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev 57: 173–185, 2005 [DOI] [PubMed] [Google Scholar]
- 16. Hirase T, Staddon JM, Saitou M, Ando-Akatsuka Y, Itoh M, Furuse M, Fujimoto K, Tsukita S, Rubin LL. Occludin as a possible determinant of tight junction permeability in endothelial cells. J Cell Sci 110: 1603–1613, 1997 [DOI] [PubMed] [Google Scholar]
- 17. Kuo CT, Veselits ML, Barton KP, Lu MM, Clendenin C, Leiden JM. The LKLF transcription factor is required for normal tunica media formation and blood vessel stabilization during murine embryogenesis. Genes Dev 11: 2996–3006, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lambertsen KL, Biber K, Finsen B. Inflammatory cytokines in experimental and human stroke. J Cereb Blood Flow Metab 32: 1677–1698, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lin Z, Kumar A, SenBanerjee S, Staniszewski K, Parmar K, Vaughan DE, Gimbrone MA, Jr, Balasubramanian V, Garcia-Cardena G, Jain MK. Kruppel-like factor 2 (KLF2) regulates endothelial thrombotic function. Circ Res 96: e48–e57, 2005 [DOI] [PubMed] [Google Scholar]
- 20. Lin Z, Natesan V, Shi H, Dong F, Kawanami D, Mahabeleshwar GH, Atkins GB, Nayak L, Cui Y, Finigan JH, Jain MK. Kruppel-like factor 2 regulates endothelial barrier function. Arterioscler Thromb Vasc Biol 30: 1952–1959, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lippoldt A, Kniesel U, Liebner S, Kalbacher H, Kirsch T, Wolburg H, Haller H. Structural alterations of tight junctions are associated with loss of polarity in stroke-prone spontaneously hypertensive rat blood-brain barrier endothelial cells. Brain Res 885: 251–261, 2000 [DOI] [PubMed] [Google Scholar]
- 22. Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci 4: 399–415, 2003 [DOI] [PubMed] [Google Scholar]
- 23. Mahabeleshwar GH, Kawanami D, Sharma N, Takami Y, Zhou G, Shi H, Nayak L, Jeyaraj D, Grealy R, White M, McManus R, Ryan T, Leahy P, Lin Z, Haldar SM, Atkins GB, Wong HR, Lingrel JB, Jain MK. The myeloid transcription factor KLF2 regulates the host response to polymicrobial infection and endotoxic shock. Immunity 34: 715–728, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mankertz J, Tavalali S, Schmitz H, Mankertz A, Riecken EO, Fromm M, Schulzke JD. Expression from the human occludin promoter is affected by tumor necrosis factor alpha and interferon gamma. J Cell Sci 113: 2085–2090, 2000 [DOI] [PubMed] [Google Scholar]
- 25. Mayhan WG. Cellular mechanisms by which tumor necrosis factor-alpha produces disruption of the blood-brain barrier. Brain Res 927: 144–152, 2002 [DOI] [PubMed] [Google Scholar]
- 26. McConnell BB, Yang VW. Mammalian Kruppel-like factors in health and diseases. Physiol Rev 90: 1337–1381, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Neuwelt EA, Bauer B, Fahlke C, Fricker G, Iadecola C, Janigro D, Leybaert L, Molnar Z, O′Donnell ME, Povlishock JT, Saunders NR, Sharp F, Stanimirovic D, Watts RJ, Drewes LR. Engaging neuroscience to advance translational research in brain barrier biology. Nat Rev Neurosci 12: 169–182, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Parmar KM, Nambudiri V, Dai G, Larman HB, Gimbrone MA, Jr, Garcia-Cardena G. Statins exert endothelial atheroprotective effects via the KLF2 transcription factor. J Biol Chem 280: 26714–26719, 2005 [DOI] [PubMed] [Google Scholar]
- 29. Persidsky Y, Heilman D, Haorah J, Zelivyanskaya M, Persidsky R, Weber GA, Shimokawa H, Kaibuchi K, Ikezu T. Rho-mediated regulation of tight junctions during monocyte migration across the blood-brain barrier in HIV-1 encephalitis (HIVE). Blood 107: 4770–4780, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Prinz V, Endres M. Statins and stroke: prevention and beyond. Curr Opin Neurol 24: 75–80, 2011 [DOI] [PubMed] [Google Scholar]
- 31. Saitou M, Furuse M, Sasaki H, Schulzke JD, Fromm M, Takano H, Noda T, Tsukita S. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell 11: 4131–4142, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sandoval KE, Witt KA. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis 32: 200–219, 2008 [DOI] [PubMed] [Google Scholar]
- 33. Sawada N, Liao JK. Targeting eNOS and beyond: emerging heterogeneity of the role of endothelial Rho proteins in stroke protection. Expert Rev Neurother 9: 1171–1186, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sen-Banerjee S, Mir S, Lin Z, Hamik A, Atkins GB, Das H, Banerjee P, Kumar A, Jain MK. Kruppel-like factor 2 as a novel mediator of statin effects in endothelial cells. Circulation 112: 720–726, 2005 [DOI] [PubMed] [Google Scholar]
- 35. SenBanerjee S, Lin Z, Atkins GB, Greif DM, Rao RM, Kumar A, Feinberg MW, Chen Z, Simon DI, Luscinskas FW, Michel TM, Gimbrone MA, Jr, Garcia-Cardena G, Jain MK. KLF2 Is a novel transcriptional regulator of endothelial proinflammatory activation. J Exp Med 199: 1305–1315, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vorbrodt AW, Dobrogowska DH. Molecular anatomy of intercellular junctions in brain endothelial and epithelial barriers: electron microscopist's view. Brain Res Brain Res Rev 42: 221–242, 2003 [DOI] [PubMed] [Google Scholar]
- 37. Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. J Neuroimmunol 184: 53–68, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wani MA, Means RT, Jr, Lingrel JB. Loss of LKLF function results in embryonic lethality in mice. Transgenic Res 7: 229–238, 1998 [DOI] [PubMed] [Google Scholar]
- 39. Weinreich MA, Takada K, Skon C, Reiner SL, Jameson SC, Hogquist KA. KLF2 transcription-factor deficiency in T cells results in unrestrained cytokine production and upregulation of bystander chemokine receptors. Immunity 31: 122–130, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Weiss N, Miller F, Cazaubon S, Couraud PO. The blood-brain barrier in brain homeostasis and neurological diseases. Biochim Biophys Acta 1788: 842–857, 2009 [DOI] [PubMed] [Google Scholar]
- 41. Zhang F, Wang S, Signore AP, Chen J. Neuroprotective effects of leptin against ischemic injury induced by oxygen-glucose deprivation and transient cerebral ischemia. Stroke 38: 2329–2336, 2007 [DOI] [PubMed] [Google Scholar]