

Abstract
In response to cellular and environmental stresses, mitochondria undergo morphology transitions regulated by dynamic processes of membrane fusion and fission. These events of mitochondrial dynamics are central regulators of cellular activity, but the mechanisms linking mitochondrial shape to cell function remain unclear. One possibility evaluated in this review is that mitochondrial morphological transitions (from elongated to fragmented, and vice-versa) directly modify canonical aspects of the organelle's function, including susceptibility to mitochondrial permeability transition, respiratory properties of the electron transport chain, and reactive oxygen species production. Because outputs derived from mitochondrial metabolism are linked to defined cellular signaling pathways, fusion/fission morphology transitions could regulate mitochondrial function and retrograde signaling. This is hypothesized to provide a dynamic interface between the cell, its genome, and the fluctuating metabolic environment.
Keywords: mitochondrial dynamics, morphology, fusion and fission, bioenergetics, retrograde signaling
mitochondria are crucial organelles involved in cellular energy production and in the regulation of numerous aspects of cellular activity, including, but not limited to, apoptosis, Ca2+ signaling, and redox homeostasis (41). Over the last decade, mitochondria have moved to the forefront of cell biology research for their ability to undergo regulated events of membrane fusion and fission, which remodel the architecture of the mitochondrial network within the cytoplasm (43, 36, 146). Processes of mitochondrial fusion and fission are collectively termed mitochondrial dynamics. Importantly, abnormal mitochondrial dynamics cause bioenergetic defects (26), induce embryonic lethality in transgenic animal models (27), and underly a heterogenous group of human diseases (164), attesting to the pivotal role of these processes in physiopathology (25, 50, 91). In addition, experimental work has shown that mitochondrial dynamics are involved in regulating cellular activity and that mitochondria participate in important cellular signaling pathways (136, 158).
Mitochondrial retrograde signaling is defined as the transfer of information from mitochondria to the cytoplasm and nucleus. Retrograde signaling pathways have been established to occur in response to metabolic perturbations, with the aim to trigger adaptive cellular responses orchestrated by coordinated changes in gene expression (reviewed in Refs. 21 and 93). Among the retrograde signals produced by mitochondria that are known to regulate different aspects of cellular activity, the most studied include reactive oxygen species (ROS), pro-apoptotic molecules, ATP/ADP/AMP, metabolic intermediates (NAD+/NADH), and Ca2+ (20, 39, 58). From recent discoveries revealing that mitochondrial dynamics regulate cellular activity in response to various stressors, it is hypothesized that changes in mitochondrial morphology regulate, either directly or indirectly, various aspects of mitochondrial function involved in retrograde signaling.
The purpose of this review is to integrate current knowledge on the mitochondrial morphology-function relationship to gain insights into the potential role of mitochondrial dynamics in shaping cellular function. In so doing, we note that most of the available evidence on the functional consequences of mitochondrial fusion/fission, and hence reviewed here, is derived from molecular studies in which chronic and widespread perturbations were induced either by knocking down or overexpressing fusion/fission proteins. Such chronic perturbations of mitochondrial dynamics may also compromise autophagic quality control system (147), lead to morphological abnormalities (10), impair interactions between mitochondria and endoplasmic reticulum (34), and cause accumulation of mitochondrial DNA (mtDNA) damage (mutations and deletions) (29). It should therefore be kept in mind that these aftereffects inherent to long-term experimental perturbations of mitochondrial dynamics could alter mitochondrial functions independently from morphology itself. In comparison, it is less clear what the immediate functional effects of stochastic, localized, and temporary fusion and fission events are on mitochondrial function in vivo.
Here, we review experimental evidence linking mitochondrial morphology and sensitivity to permeability transition (PT) and apoptotic signaling, mitochondrial respiratory chain function, and ROS production. We then outline putative mitochondrial retrograde signals relevant to these functions and outline their known roles in regulating cellular activity. While minimal direct evidence currently exists that mitochondrial morphology transitions result into altered retrograde signaling, modulation of mitochondrial signals by fusion and fission appears as a candidate mechanism to promote normal cellular responses to metabolic stresses, and therefore, as a possible contributor to the etiology of metabolic diseases.
Mitochondrial Structure-Function Relationship
Shaping the mitochondrion: molecular machinery.
The notion that mitochondrial dynamics bears functional significance impacting on cellular life and death is relatively recent. This has largely been established through the identification and molecular genetic manipulation of the major fusion and fission proteins interacting with the double membrane system of mitochondria (reviewed in Refs. 136 and 158). Briefly, the fusion machinery consists of the dynamin-like proteins mitofusins 1 and 2 (Mfn1 and Mfn2) and optic atrophy 1 (OPA1), which, respectively, have a role in the sequential fusion of the outer (OMM) and inner mitochondrial membranes (IMM) (87, 135). Mitofusins and OPA1 form an antiparallel intermolecular coiled coil, and GTP hydrolysis by the GTPase domains of Mfn1, Mfn2, and OPA1 induces a conformational change generating the mechanical force necessary for fusion of adjacent organelles (70, 81), effectively resulting in elongation and formation of mitochondrial tubules in the cytoplasm.
On the other hand, mitochondrial fission, the splitting of one mitochondrion into two or more smaller mitochondria, mainly results from the constriction of mitochondria by dynamin-related protein 1 (DRP1, also DLP1) (134). The cytoplasmic dynamin-related protein DRP1 is presumably recruited at the OMM by Fis1 (human fission protein 1) to assemble into oligomeric rings around the OMM to fragment the mitochondrion (36, 97). Some other structural membrane-bending proteins are also suggested to assist in mitochondrial tubulation by insertion of β-barrels, hydrophobic and scaffolding proteins (reviewed in Refs. 130 and 158), but comparatively little is known about those accessory proteins. We therefore focus our discussion below on the major pro-fusion and pro-fission proteins that ultimately control gross mitochondrial morphology. Importantly, maintenance of mitochondrial shape depends on a constant equilibrium between fusion and fission events, and interfering with either of these processes results in the extension of the mitochondrial network or its fragmentation. As detailed below, altering mitochondrial fusion and fission has several potential functional consequences (Fig. 1).
Fig. 1.

Hypothesized relationship linking mitochondrial morphology and function. Dynamic regulation of mitochondrial morphology involves regulated processes of fusion and fission, which modify mitochondrial function. Pro-fusion [mitofusin 1 and 2 (Mfn1, Mfn2), optic atrophy 1 (OPA1)] and pro-fission [dynamin-related protein (DRP1), fission 1 (Fis1)] core proteins modulate mitochondrial morphology and physiology. Altering the balance of fusion/fission dynamics results in mitochondrial elongation and branching (pro-fusion) or mitochondrial fragmentation (pro-fission). For detailed review of these processes, see Ref. 158. Listed on both sides of the figure are the putative functional consequences generally associated with mitochondrial fusion and fission. Insets: fluorescence microscopy images of mitochondria from cultured human myoblasts labeled with Mitotracker Green (45 min, 125 nM) depicting different states of mitochondrial morphology.
Morphology, mitochondrial permeability transition, and apoptosis.
Among the consequences of changes in mitochondrial morphology on mitochondrial function, the sensitivity of mitochondria to permeability transition and apoptotic signaling are the most extensively documented (39) (Table 1). The mitochondrial permeability transition pore (mPTP) is presumed to consist in a multiprotein complex located in the IMM, which is modulated by interactions with OMM proteins (32, 144). The composition and regulation of the mitochondrial PTP (mPTP) is still a matter of debate (89), but putative mPTP components include cyclophilin D (CypD) and the adenine nucleotide translocase (ANT), and postulated regulatory proteins include hexokinase, creatine kinase, and peripheral benzodiazepine receptor (PBR) (89, 131, 132). Although the full details of these events are still only partially characterized and are in need of further clarification (13, 62), it is established that irreversible opening of the mPTP in a high conductance mode occurs in response to diverse cellular stressors such as matrix Ca2+, loss of membrane potential, and ROS (14, 17, 57), leading to the release of proapoptotic factors inducing cell death (131, 155). Furthermore, because mitochondrial respiration becomes uncoupled when the mPTP is open, prolonged permeability transition can lead to ATP depletion and bioenergetic crisis contributing to cell death.
Table 1.
Effect of mitochondrial morphology regulation on sensitivity to mitochondrial PT and apoptotic cell death
| Manipulation/Condition | Cell Type | Effect on Susceptibility to Permeability Transition and Apoptotic Cell Death | Reference |
|---|---|---|---|
| Pro-fusion (elongation) | |||
| DRP1 RNAi | HeLa | Delayed but does not inhibit apoptosis (STS, Act D) | 46 |
| DRP1 RNAi | HeLa | Reduced apoptosis (Eto) 40–50% at 24 h | 141 |
| DRP1K38A overexpression | COS-7 | Prevented release of cyt. c, decreased number of apoptotic cells with STS. | 48 |
| DRP1 shRNA | HeLa | Decreased apoptosis in response to pharmacological inducers (STS, Act D, α-Fas); less potent than DRP1 | 86 |
| DRP1K38A overexpression | HL-1 (cardiac) | Decreased cell death 60–80% 2 h after ischemia-reperfusion, decreased time to PT 50–60%* | 106 |
| DRP1K38A overexpression | H9c2 myoblasts | Prevented 1–7 fold induction of apoptosis (annexin V, caspase activation) during hyperglycemia | 162 |
| DRP1 inhibition (mdivi-1) | HL-1 (cardiac) | Decreased cell death 50–60% 40 mins after ischemia-reperfusion; prevented loss of membrane potential in permeabilized cardiomyocytes | 106 |
| αDRP1 antibody microinjection | HeLa | Decreased nuclear fragmentation 20 h after STS | 48 |
| Fis1 shRNA | HeLa | Decreased apoptosis in response to pharmacological inducers (STS, Act D, Eto, α-Fas) | 86 |
| MNF2 overexpression | Primary CGNs | Decreased cyt. c release and cell death after H2O2 treatment and DNA damage | 71 |
| MFN2 overexpression | HL-1 (cardiac) | Decreased cell death 50–70% 2 h after ischemia-reperfusion, decreased time to PT by 50–60%* | 106 |
| MFN1 overexpression | HL-1 (cardiac) | Decreased cell death 60–80% 2 h after ischemia-reperfusion, decreased time to PT by 50–60%* | 106 |
| OPA1 overexpression | Primary CGNs | Protected against cell death from excitotoxicity (Ca2+ stress) | 72 |
| Pro-fission (fragmentation) | |||
| OPA1 RNAi | HeLa | Increased sensitivity to apoptosis and increased spontaneous death | 86 |
| Fis1 overexpression | HL-1 (cardiac) | Increased cell death 40–60% 2 h after ischemia-reperfusion, no effect on time to PT | 106 |
| MFN1/2 RNAi | Hela | Increased apoptosis (Eto) 40–50% at 24 h | 141 |
| MFN2 KO | CGN (in vivo) | Neurodegeneration and increased abundance of TUNEL-positive CGN. | 28 |
| Mechanical isolation of mitochondria | Myofibers | Reduced time to Ca2+-induced PT opening by 98%, reduced calcium retention capacity by 50% | 115 |
PT, permeability transition; STS, sautosporine; Act D, actinomycin D; Eto, etoposide; α-Fas, anti-Fas receptor agonist; CGN, cerebellar granular neurons; DRP1K38A, dominant negative version of DRP1.
Effect similar in magnitude to that of cyclosporin A. MFN, mitofusin; OPA1, optic atrophy 1; shRNA, short hairpin RNA; RNAi, interfering RNA.
In addition to its role in apoptosis, transient opening of the mPTP in a low-conductance mode that allows diffusion of ions but not higher molecular weight solutes serves a physiological function (113). Indeed, such mPTP flickering is suggested to 1) serve as a Ca2+ release valve to regulate mitochondrial Ca2+ levels and likely cellular Ca2+ transients (45); 2) provide a means to rapidly and reversibly depolarize mitochondria, possibly controlling their autophagic removal (44); 3) modulate superoxide production since transient mPTP opening yields superoxide flashes in myocytes (156); and 4) allow the release from the mitochondrial matrix into the cytoplasm of other molecules including the nicotinamide adenine nucleotides (NAD+ and NADH) (42) and possibly metabolic intermediates (see first 5 headings under Mitochondrial Retrograde Signaling).
Despite the fact that overt (or unbalanced) mitochondrial fission and network fragmentation are well known to be associated with cell death, it is only recently that some of the molecular mechanisms underlying this phenomenon have been described (157). Mitochondrial fission alone does not necessarily induce apoptosis, but it sensitizes cells to death stimuli (86). In cultured cardiomyocytes, enhancing fission by overexpressing Fis1 induced mitochondrial fragmentation, decreased the time required for the opening of the mPTP in response to oxidative stress, and promoted cell death following ischemia-reperfusion (106). Mitochondrial network fragmentation and concomitant sensitization to apoptotic death was also obtained in various cell types upon inhibition of mitochondrial fusion by RNA interference (RNAi) against the fusion proteins Mfn1, Mfn2, and OPA1 (71, 86, 141). Furthermore, in an in vivo model, normal expression of Mfn2 conferred protection against apoptotic cell death in mice Purkinje neurons and its ablation increased cell death (28). Collectively, evidence indicates that cells containing extensively fragmented mitochondria resulting from chronic disruption of mitochondrial dynamics are more susceptible to apoptotic cell death through mechanisms possibly involving the mPTP.
Conversely, overexpression of the fusion proteins, which promote the formation of elongated and branched mitochondria, protects against mitochondria-induced apoptosis. For instance, induction of apoptosis with oxidative stress in different cell types was reduced by overexpressing the fusion proteins Mfn1 (106), Mfn2 (71, 106), or OPA1 (72). Inhibition of mitochondrial fission by downregulating Fis1 and DRP1 yielded a similar hyperfused morphology, preventing fragmentation and conferring resistance to mitochondrial depolarization and cell death in response to a variety of exogenous apoptotic inducers (48, 86, 141, 162). Furthermore, overexpression of a dominant-negative DRP1K38A, which inhibits fission-induced mitochondrial fragmentation, also prevented the loss of the mitochondrial membrane potential, the release of cytochrome c, and reduced apoptosis in cardiomyocytes and COS-7 cells (48, 106). However, careful examination of these processes revealed that the majority of cases, inhibition of DRP1-mediated fission does not completely prevent apoptosis but delays it (46), raising the possibility that studies assessing apoptosis at only one time point may confuse inhibition and retardation of cell death. Nevertheless, these findings demonstrate a role of fusion and fission proteins in regulating mitochondrial permeability transition and apoptotic signaling (63).
In addition to apoptotic signaling, mPTP dynamics influences mitochondrial Ca2+ signaling and physiological regulation of mitochondrial function (120, 154). Likewise, mitochondrial shape also influences cytoplasmic Ca2+ handling (reviewed in Ref. 143). In high-resolution microscopy experiments, histamine-induced elevations in endothelial cell cytoplasmic [Ca2+], small fragmented mitochondria experienced significantly greater rises in matrix [Ca2+] (and greater increase in superoxide production) than adjacent elongated mitochondria (109). In line with this, compared with reticular mitochondria within permeabilized muscle fibers, fragmentation of skeletal muscle mitochondria during mechanical isolation reduced the ability of mitochondria to buffer exogenous Ca2+ and dramatically increased mPTP sensitivity in response to an exogenous Ca2+ challenge (115). Because high mitochondrial [Ca2+] is a trigger of mPTP opening, mitochondrial morphology may impact the response of mitochondria to given intracellular Ca2+ levels, whereby elongated branched mitochondria may be more resistant to Ca2+-induced mPTP opening than smaller fragmented mitochondria. The molecular mechanisms accounting for differential sensitivity in permeability transition between elongated branched mitochondria and fragmented mitochondria remain unclear but may be caused by alterations of the IMM structure upon fission/fragmentation (111, 128) differences in the distribution volume of Ca2+ within the mitochondrial matrix (15) or possibly by other nonmorphological factors (see Altering mitochondrial dynamics: effects on quality control and beyond).
Morphology and respiration.
Oxygen consumption and ATP synthesis by the oxidative phosphorylation (OXPHOS) system are also affected by mitochondrial dynamics (12) (Table 2). Mitochondrial fragmentation induced by downregulation of OPA1, or gene invalidation of Mfn1 and Mfn2, strongly decreased mitochondrial respiratory capacity in permeabilized cells when fueled with substrate for electron transport chain (ETC) complexes I, II, and IV (26). Other experiments have demonstrated similar decreases in cellular respiration or glucose oxidation in response to Mfn2 downregulation (6, 117). In line with this, fibroblasts from patients with mutations in either Mfn2 (24, 94) or OPA1 (105) showed decreased ATP synthesis and/or impaired respiratory coupling efficiency (the ratio of ATP generated per oxygen molecule consumed). DRP-1-mediated mitochondrial fragmentation also increased oxygen consumption in response to high-glucose exposure, possibly suggesting that fragmentation of mitochondrial tubules favors respiratory uncoupling (162).
Table 2.
Effect of mitochondrial morphology regulation on mitochondrial respiratory properties
| Manipulation/Condition | Cell Type | Effect on Mitochondrial Respiration and ETC Function | Reference |
|---|---|---|---|
| Pro-fusion (elongation) | |||
| DRP1 RNAi | HeLa | Decreased respiration 60–70%, decreased ATP synthesis (abnormal budding of perinuclear mitochondria, experiments performed 96 h postinduction) | 10 |
| DRP1K38A overexpression | Clone 9 hepatocytes | Prevented a 40–50% increase in respiration during hyperglycemia | 162 |
| Mfn2 overexpression | H6E9 myotubes | Increased membrane potential and glucose oxidation rate (in the absence of network remodeling) | 117 |
| Pro-fission (fragmentation)* | |||
| Mfn1/Mfn2 double mutant | MEFs | Decreased respiration 30–70% under different energized states | 26 |
| Mfn2 antisense cDNA | 10T1/2 fibroblasts | Decreased respiration 20–40%, decreased membrane potential (in L6E9 myotubes) | 6 |
| Mfn2 mutations | Human fibroblasts | Decreased coupling efficiency 20–30% (ATP/O ratio) | 94 |
| Mfn2 mutations | Human fibroblasts | Decreased coupling efficiency 50–70% (ATP/O ratio), increased respiration with complex II-linked substrate | 24 |
| OPA1 mutation | Human fibroblasts | Decreased coupling efficiency 50% (ATP/O ratio) | 105 |
| OPA1 RNAi | MEFs | Decreased respiration 50–80% under different energized states | 26 |
| OPA1 mutations | Human fibroblasts | Decreased complex I-driven ATP synthesis | 166 |
| OPA1 KO | MEFs | Decreased ATP levels with starvation, impaired dimerization of ATP synthase | 54 |
MEF, mouse embryonic fibroblasts;
Loss of function of MFN2 and OPA1 has been linked to mitochondrial DNA instability (see text for details), which may impair respiratory chain function via mtDNA damage over periods exceeding several days, rather than via direct of mitochondrial morphology.
On the contrary, overexpression of Mfn2 had the opposite effect and doubled maximal glucose oxidation in one study (117), but this may have been caused by upregulation of respiratory chain genes, which appeared to be independent from changes in mitochondrial morphology. Although mitochondrial fusion may preserve and/or enhance respiration, excessive fusion in the absence of fission is deleterious. In HeLa cells, 4 days postinduction with RNAi targeting DRP1 caused excessive budding and branching of poorly ramified mitochondria along with decreased mitochondrial respiration (10). Thus altering the expression of mitochondrial fusion and fission proteins, which leads to abnormal mitochondrial morphology, impairs mitochondrial respiration and possibly coupling efficiency.
Studies in patients with OPA1 mutations indicate that OPA1 promotes mitochondrial fusion in tandem with complex I-driven oxidative phosphorylation (166). In mouse embryonic fibroblasts (MEFs), ATP synthase activity was enhanced in fused elongated mitochondria induced by starvation, a phenomenon that was absent in OPA-1-deficient MEFs (54). Recent work also suggested that OPA1 is involved in remodeling IMM cristae morphology, and that this may impact respiratory properties of mitochondria through action on the assembly of respiratory supercomplexes (complexes I1, III2, IV1) (54), which are believed to optimize downstream electron transport from complex I during normal mitochondrial respiration (126, 149).
At the transcriptional level, Mfn2 overexpression in myotubes increased the expression of oxidative phosphorylation genes, mitochondrial membrane potential, and maximal glucose oxidation rates (117). In obese individuals, increasing body mass index was associated with lower levels of Mfn2 transcripts in skeletal muscle, which was in turn strongly correlated with lower glucose oxidation rates (99). In addition, exercise training, which enhances mitochondrial biogenesis and muscles' oxidative capacity (66), also increased Mfn1 and Mfn2 gene expression (23), suggesting a genetic synergy and a possible evolutionary overlap between the signaling pathways regulating mitochondrial morphology and respiration (167, 168). Increases in Mfn2 expression with exercise could be expected to link muscle activity and mitochondrial fusion, but because fusion and fission processes are heavily regulated by posttranslational mechanisms (106, 136), whether physical activity acutely or chronically translates into more fused mitochondria is uncertain. Moreover, Mfn2 levels are more abundant in skeletal muscle than in other tissues and are even more abundant in cardiac muscle (where mitochondrial content is the highest) (64), suggesting that Mfn2 levels may simply mirror mitochondrial density and that the activity of fusion proteins may be mostly regulated by posttranslational modifications in response to diverse cellular cues, such as local redox state and [Ca2+] (64, 74, 133).
Collectively, available evidence point to a bidirectional relationship between mitochondrial morphology and oxidative metabolism (12, 18), whereby fused mitochondrial tubules have generally been regarded as having greater respiratory capacity than fragmented globular units. However, Mfn2 may directly increase expression of oxidative phosphorylation (OXPHOS) genes (117), and long-term disruption of mitochondrial dynamics may have deleterious effects on bioenergetics through other processes (see Altering mitochondrial dynamics: effects on quality control and beyond). Therefore, whether manipulating fusion and fission proteins influences mitochondrial respiration directly through effect on mitochondrial morphology, or indirectly via other effects, remains unclear. Notably, a study where mitochondria were fragmented via mechanical isolation showed no difference in maximal coupled respiration between reticular and fragmented mitochondria (115). Additional properly controlled studies investigating the effects of acute (within minutes to hours) changes in mitochondrial morphology on mitochondrial respiration are required to clarify this point.
Morphology and reactive oxygen species.
Mitochondria produce ROS as a function of electron transport and redox status within the IMM (7). A bidirectional relationship between ROS and mitochondrial fragmentation has been documented in certain conditions (Table 3). In response to a hyperglycemic challenge, mitochondrial fission was a prerequisite for increasing mitochondrial ROS production in cultured cells (162). In congruence with this, abrogating mitochondrial fission effectively prevented hyperglycemia-induced ROS production. Inhibition of mitochondrial fission by expression of a dominant negative construct of DRP1 prevented mitochondrial fragmentation and subsequent ROS production (162, 163), thus suggesting that mitochondrial fragmentation allows increases in ROS production under these conditions. Given that diabetes is thought to involve oxidative stress and fragmented mitochondria (161), and that diabetic insulin resistance is associated with skeletal muscle mitochondrial dysfunction, it has been suggested that mitochondrial dynamics mediates the deleterious effects of high blood glucose on skeletal muscle (161, 167). This possibility has recently received strong empirical support from in vitro (129) and in vivo (76) models of diabetes where mitochondrial fragmentation was required for hyperglycemia- and hyperlipidemia-induced increases in mitochondrial ROS production in endothelial cells and skeletal muscle, respectively. In line with this, mitochondria isolated using homogenization, which induces fragmentation of the mitochondrial network, released 5- to 10-fold more H2O2 compared mitochondria within permeabilized fibers, which retain a more intact network morphology (115), substantiating molecular genetic experiments where mitochondrial fragmentation might directly enable increased ROS production.
Table 3.
Effect of mitochondrial morphology regulation on mitochondrial ROS production
| Manipulation/Condition | Cell Type | Effect on Mitochondrial ROS Production | Reference |
|---|---|---|---|
| Pro-fusion (elongation) | |||
| DRP1K38A overexpression | Clone 9 hepatocytes | Prevented 1- to 1-fold increase in ROS [DHE] during hyperglycemia | 162 |
| MFN2 overexpression | H9c2 myoblasts | Prevented 1.5-fold increase in ROS [DHE] during hyperglycemia | 162 |
| MFN2 overexpression | H9c2 myoblasts, endothelial cells, smooth muscle cells | Prevented 50% increase in ROS [DHE] during hyperglycemia | 163 |
| DRP1 RNAi | Endothelial cells | Prevented 3-fold increase in ROS [MitoSOX] and further reduced ROS by 60–70% during hyperglycemia | 129 |
| Fis1 RNAi | Endothelial cells | Prevented 3-fold increase in ROS [MitoSOX] during hyperglycemia | 129 |
| DRP1 inhibition (Mdivi-1) | C2C12 myoblasts | Prevented 15–20% increase in ROS [H2DCFDA] with palmitate | 76 |
| Pro-fission (fragmentation) | |||
| Hyperglycemia | Endothelial cells, myoblasts, hepatocytes | Increases ROS 1- to 3-fold, which can be prevented by blocking fission (see above) | |
| Mechanical isolation of mitochondria | Myofibers | Increased total ROS 5- to 10-fold [Amplex Red], and free radical leak 9- to 23-fold (per unit O2 consumed) | 115 |
ROS, reactive oxygen species; DHE, dihydroethidium is a superoxide probe; H2DCFDA, carboxy-dichlorodihydrofluorescein is a broad-scope ROS probe; MitoSox is a mitochondria-targeted superoxide probe; Amplex Red is a H2O2 probe.
Although mitochondrial morphology might influence ROS production, experimental support also exists for the reverse scenario whereby ROS induces alterations in mitochondrial morphology. Fibroblasts from patients with genetic mtDNA defects demonstrated that severe complex I deficiency yield increased ROS production was associated with mitochondrial fragmentation (79). Similarly, exposure of cultured cells to sublethal doses of exogenous ROS (H2O2) induced mitochondrial fragmentation in mouse embryonic fibroblasts (MEFs) (31, 73, 92), neuronal cells (71), and in C2C12 myocytes (47). However, observations contrary to these have also been made showing that ROS production secondary to inhibition of complex I with rotenone, induced mitochondrial fusion (78). Furthermore, in a cell-free isolated mitochodria fusion assay, oxidized glutathione (GSSG) induced fusion, whereas antioxidants inhibited mitochondrial fusion (133), suggesting that the oxidized cytosol favors mitochondrial fusion. Discrepancies in intact cells could have been confounded by the different timelines (e.g., initial fusion and subsequent fragmentation) and the fact that rotenone may alter the cytoskeleton (microtubule assembly) that interacts with mitochondria (19). It must also be noted that experiments conducted with inhibitors of the ETC may be confounded by the critical role of the ETC in maintaining mitochondrial membrane potential, as mitochondrial depolarization leads to mitochondrial network fragmentation and autophagy (56).
Globally, available data linking mitochondrial dynamics and ROS production suggest a causal relationship, at least in the context of hyperglycemic stress, where fragmented mitochondrial release significantly more ROS than fused mitochondrial tubules. A bidirectional relationship, whereby mitochondrial fission increases ROS emission, and ROS causes mitochondrial network fragmentation appears to pervade (161). Properly controlled studies are needed to clarify the exact sequence of events linking mitochondrial morphology and ROS production in different physiological conditions.
Altering mitochondrial dynamics: effects on quality control and beyond.
In interpreting the above-reviewed literature, one must consider that studies performed in stable cell lines lacking fusion proteins may be confounded by deleterious side effects of halted mitochondrial dynamics on other cellular functions, including the cellular quality control process. The removal of damaged/dysfunctional mitochondria requires cycles of mitochondrial fusion and fission enabling the selective removal of poorly functioning organelles via mitochondrial autophagy—mitophagy (145). Mitophagy is in turn important to eliminate damaged (mutated) copies of mtDNA (140). The mtDNA partially encodes the enzymatic protein complexes that perform mitochondrial respiration (150). Mutations and deletions in mtDNA therefore impair mitochondrial function and are as a result a cause of human diseases (55, 80).
Disruption of mitochondrial fusion (and likely fission) results in the rapid accumulation of mtDNA damage and prevents normal functional complementation of mtDNA gene products (29, 102, 107). Preventing mitochondrial fission has also been shown to deplete mtDNA (110). In skeletal muscle, lack of both Mfn1 and Mfn2 chronically disrupted mitochondrial fusion in vivo and resulted in accumulation of mtDNA damage and mutations, mtDNA depletion, and impairment of maximal respiratory capacity (29). In addition to the role of mitochondrial dynamics for mitophagy, an adequate level of mitochondrial fusion appears essential for functional complementation of mtDNA and gene products (e.g., mRNA, proteins) within the mitochondrial network (25, 29, 86, 102, 107). Eliminating Mfn2 from mouse embryonic fibroblasts impaired the distribution of mtDNA nucleoids (28) and respiratory chain proteins (29). In humans, it was recently demonstrated that loss-of-function mutations in both OPA1 (2, 165) and Mfn2 (123) genes also impair mtDNA stability, leading to multisystemic disease.
Furthermore, mtDNA damage and respiratory chain deficiencies do not only impair ETC function but also increases apoptosis (5) and ROS production (69). Chronic impairments of mitochondrial dynamics could thus impair all three aspects of mitochondrial function discussed above. Globally, long-term impairments in mitochondrial dynamics, such as those induced by chronic/widespread abrogation of dynamics proteins in cell lines, directly impact the integrity of the mitochondrial genome, which can translate into altered mitochondrial function through mechanisms other than primary changes mitochondrial morphology.
Further to the inhibitory effect on mitophagy and damage to mtDNA, altering the function of the fusion (Mfn1, Mfn2, OPA1) and fission (Fis1 and DRP1) proteins may impact other cellular processes that could confound the effects of their experimental knockdown/overexpression on mitochondrial bioenergetics and retrograde signaling. As mentioned above, Mfn2 does not only allow fusion of mitochondria but also regulates the expression of oxidative phosphorylation genes independently from its role in fusion (117); and OPA1 can remodel cristae independent of organelle fusion or permeation of the OMM (51), suggesting that fusion proteins also influence mitochondrial function without necessarily involving overt changes in mitochondrial network morphology and OMM permeability (4).
Moreover, Mfn2 tethers mitochondria to the endo/sarcoplasmic reticulum and is thus important for Ca2+ homeostasis (34), Mfn1 may regulate the activation of the proapoptotic Bax on the mitochondrial surface (125), OPA1 is found in lipid droplets where it acts to regulate lipolysis (118), and both Fis1 and DRP1 localize to peroxisomes where they mediate peroxisomal fission (77, 90). Mitochondrial fusion proteins also participate in mitochondrial transport and localization; Mfn2 interacts with the Miro/Milton complex on the OMM (100) and contributes to axonal mitochondrial positioning, for example (101), and OPA1 could indirectly modulate mitochondrial motility by regulating mitochondrial Ca2+ uptake (52). Thus it is clear that mitochondrial dynamics proteins execute multiple functional roles, involving other cellular organelles, and it cannot therefore be assumed than their experimental manipulation selectively affects mitochondrial morphology (Fig. 2).
Fig. 2.

Potential consequences of altering the mitochondrial fusion and fission proteins in experimental systems. Mitochondrial fusion and fission proteins directly influence mitochondrial morphology and the architecture of the mitochondrial network but also influence other cellular processes that impact bioenergetics. These include the mitochondrial autophagic quality control processes (mitophagy), interactions with other intracellular structures (endo-/sarcoplasmic reticulum, cytoskeleton, lipid droplets), membrane interactions between mitochondria, transcriptional effects (transcriptional regulation of OXPHOS genes by Mfn2), and possibly other processes not depicted here. Experimental manipulation of mitochondrial dynamics proteins, especially if of long duration, may impact mitochondrial bioenergetics and downstream retrograde signals through mechanisms independent from mitochondrial morphology transitions. LD, lipid droplet; ER, endoplasmic reticulum.
Gaining insight into the functional effects of stochastic, localized, and temporary mitochondrial fusion/fission events will require experimental conditions where mitochondrial morphology can be acutely disrupted, thus eliminating the chronic “side effects” of altered mitophagy and/or accumulation of mtDNA damage. Ideally, experimental paradigms should be developed where involvement of other potential influence on mitochondrial bioenergetics can be ruled out. This will allow to more accurately investigate the specific effects of morphology transitions on defined mitochondrial functions and resulting retrograde signals.
Mitochondrial Retrograde Signaling
Key aspects of cellular function are influenced by signaling molecules produced and released by mitochondria as part of their normal metabolism (reviewed in Refs. 11 and 124). Below, we briefly review fundamental principles of mitochondrial biology and their link to five groups of molecules that act as retrograde signals (Fig. 3). We then discuss direct evidence that mitochondrial morphology transitions are involved in retrograde signaling pathways, which may contribute to regulate cellular activity and adaptive responses under certain physiological states.
Fig. 3.

Substrates of mitochondrial retrograde signaling. Shown are selected second messengers derived from mitochondrial function, which directly yield cellular effects (see first 5 headings under Mitochondrial Retrograde Signaling for discussion). Second messengers can in turn influence molecular targets that induce specific cellular effects. mPTP, mitochondrial permeability transition pore.
Calcium ion.
Upon its entry into the cytoplasm from extracellular or intracellular stores, Ca2+ is removed from the cytoplasm by active mitochondrial and sarcoplasmic reticulum (SR) import (8, 35). Ca2+ is also released back into the cytoplasm by the mitochondrial Na+/Ca2+ exchanger and by opening of the mPTP (68, 154), making the mitochondrion a central component of intracellular Ca2+ signaling (142). In many cell types, Ca2+ and other proapoptotic factors released by mitochondria can activate Ca2+-dependent pathways of cell death (both apoptosis and necrosis) (120). In the presynaptic terminals of axons, where mitochondria are far removed from the SR and must buffer Ca2+ locally in a confined space (i.e., small volume, acting over short time scales), mitochondrial Ca2+ uptake and release might have profound effects on synaptic transmission. In other excitable cells like skeletal muscle fibers, mitochondrial Ca2+ dynamics act in tandem with their close neighbor the SR to define cytoplasmic Ca2+ levels (49), which regulate Ca2+-sensitive kinases/phosphatases, proteases, and contractile activity (21, 33, 121). The loss of mitochondrial membrane potential can increase cytoplasmic [Ca2+] three- to eightfold, activating Ca2+-sensitive proteins such as calcineurin, protein kinase C (PKC), calmodulin kinase IV (CamK IV), and MAPK (mitogen-activated protein kinase)/JNK (21). Furthermore, alteration of mPTP dynamics in CypD knockout mice altered Ca2+ dynamics and resulted in abnormal skeletal muscle metabolic profiles and death upon physiological stress (e.g., exercise) (45). Thus Ca2+ signals derived from mitochondria interact closely with intracellular signaling pathways that play roles in regulating cellular activity.
Reactive oxygen species.
ROS are potent oxidizing molecules that influence the cellular redox state. The redox state is reflected in the balance of reduced glutathione/glutathione disulfide (GSH/GSSG) and the cysteine/cystine (Cys/CySS) couples, which regulate the activity of several enzymes and cellular processes (40). Redox-sensitive targets and processes include the binding of transcription factors to DNA, activation of the ubiquitin/proteasome and autophagic quality control pathways (30), and the activity of several metabolic enzymes (reviewed in Refs. 53 and 58). As further evidence that mitochondrial ROS are signaling molecules, mitochondria-targeted antioxidant molecules have been shown to abolish retrograde signaling and osteoclast differentiation in vitro (138). Likewise, pharmacological closure of the open mPTP during embryonic development reduced mitochondrial ROS production (and allowed fusion and elongation of mitochondria), and induced progenitor cell differentiation into cardiomyocytes as a result (65). In vivo, administration of a mitochondria-targeted antioxidant molecule (SS-31) reduced mitochondrial ROS-emitting potential by 50% in rat muscle cells (3) and consequently prevented dietary glucose-induced cellular oxidation of GSH into GSSG and inhibited the development of insulin resistance in a model of high-fat diet (3). Likewise, abolishing mitochondrial ROS release with SS-31 also prevented the activation of the muscle ubiquitin-proteasome atrophy pathway in a rat model of diaphragm disuse (119); similar results were obtained by overexpression of the endogenous mitochondrial antioxidant enzyme peroxiredoxin 3 (PRX3) in mice (114). Furthermore, hypertriglyceridemia-induced mitochondrial ROS production in the hypothalamus was found to alter cellular oxidative stress and signal satiety, such that blocking this mitochondrial signal with antioxidants prevented satiety in rats and increased calorie intake (9). Collectively, significant evidence demonstrate a role of mitochondria-derived ROS in cellular signaling and function (88). Because cellular ROS are mainly produced by mitochondria under normal conditions (84), cytoplasmic redox regulation by mitochondria is considered an important signal of cellular adaptation (58).
ATP/ADP/AMP.
Mitochondria are the major source of ATP in the cell. The mitochondrial membrane potential (ΔΨ) generated by the transport of electrons through proton pumps within the OXPHOS system is harnessed to resynthesize ATP from ADP and Pi. In turn, ATP is the primary substrate for protein phosphorylation, which is a highly conserved type of reversible posttranslational modification involved in signal transduction processes and the regulation of a large number of enzymes (1, 127). In addition, ATP is the substrate for the synthesis by adenylyl cyclases of the second messenger cAMP (cAMP) involved in signal transduction pathways (59). Importantly, mitochondrial OXPHOS dysfunction that impairs ATP synthesis activates AMP-activated protein kinase (AMPK) (148). Failure to resynthesize ATP increases both cytoplasmic [AMP] and [ADP], which directly activates AMPK through allosteric effects (61). AMPK is a broadly acting protein kinase that regulates whole body and cellular energy metabolism (98, 139), and its activation constitutes an important retrograde signal that controls cellular functions such as mitochondrial biogenesis, autophagy, cell cycle progression, and proliferation (60, 108). Thus the ability of mitochondria to actively synthesize ATP can impact overall cell function via defined signaling mechanisms such as AMPK.
Acetyl coenzyme A.
Acetyl coenzyme A (Ac-CoA) is derived from the metabolism of pyruvate (the catabolic intermediate of glucose) by pyruvate dehydrogenase within the mitochondrial matrix and can be exported to the cytoplasm (159). In the cytosol, Ac-CoA acts as substrate for lysine acetylation by protein acetyltransferases (137). Some evidence suggests that mitochondrial acetylcarnitine, a derivative of Ac-CoA, is exported to the cytoplasm by carnitine/acylcarnitine translocase where it can enter the nucleus and be converted back to AcCoA to act as source of acetyl group for histone acetylation (95). Like protein phosphorylation, acetylation and deacetylation (by NAD+-dependent deacetylases) are function-defining posttranslational modifications regulating the function of numerous proteins, including membrane-associated receptors, transcriptional coactivators and transcription factors, and histones involved in chromatin remodeling (137, 160). Acetylation/deacetylation reactions thus modulate cell signaling pathways and gene expression (137, 160). To the authors' knowledge, besides one study (95), data demonstrating direct mitochondrial signaling by Ac-CoA is lacking. However, changes in the production and export of Ac-CoA by mitochondria could theoretically impact cellular function.
NAD+/NADH.
The nicotinamide adenine dinucleotide couple NAD+/NADH is at the heart of mitochondrial bioenergetics and regulates key cellular elements (reviewed in Ref. 67). The metabolism of energetic substrates by the Krebs cycle enzymes results in the reduction of NAD+ to NADH; and NADH is then reoxidized to NAD+ by NADH oxidoreductase (Complex I of the ETC) in respiring mitochondria (7). In muscle cells (both cardiac and skeletal), mitochondria contain the majority of total intracellular NAD+ (38). The mitochondrial and cytoplasmic NAD(H) redox states are directly connected via the activity of the malate/aspartate NAD(H) redox and a-glycerophosphate shuttle (67) and mitochondrial permeability transition rapidly releases NAD+ and NADH into the cytoplasm (37, 38, 42). In the cytoplasm, NAD+ acts as substrate for various enzymes, including poly(ADP-ribose) polymerases (PARPs) and sirtuins, which include the NAD+-dependent class III deacetylase sirtuins (e.g., SIRT1) (22, 67) that play important roles in DNA repair, signal transduction events, chromatin remodeling, and regulation of gene expression partly through interaction with the circadian clock components CLOCK:BMAL1 (103). Thus the ratio of NAD+/NADH within mitochondria and its propagation within the cytoplasm can fundamentally impact cellular activity.
Linking morphology function and signaling.
A number of mitochondrial “outputs” fulfill the criteria of second messengers. As shown in Fig. 3, mitochondria release specific signals which affect molecular targets that bring about adaptive responses within the cell. The most influential hypothesized outcomes of mitochondrial second messengers are posttranslational modifications of proteins that regulate cellular metabolism and gene expression. This may occur as a result of both transient posttranslational modifications of cytoplasmic proteins and posttranslational modifications of nuclear histone tails that stably affect chromatin remodeling and gene “expressibility” through cooperative epigenetic mechanisms (153). Strikingly, all the major known posttranslational modifications of proteins (phosphorylation, acetylation and deacetylation, succinylation, malonylation, methylation) use as substrates key byproducts produced and released by mitochondria (153). This likely reflects the simultaneous evolution of the eukaryotic protein-control machinery and mitochondrial bioenergetics to optimize physiological function (151).
In addition to well-defined retrograde signals listed above, other mitochondrial signaling mechanisms may contribute to intracellular signaling events. For example, mitochondria play a permissive role in cellular signaling among T lymphocytes and are thus involved in the regulation of normal immune responses through mechanisms suggested to involve mitochondrial membrane potential (82). In addition, it was recently discovered that mitochondria secrete vesicles mitochondria-derived vesicles (MDVs) (104). Although of unknown significance, it is interesting to consider that mitochondria's ancestors, the bacteria, use vesicular release as a mode of signaling (83, 96). Aside from these speculative signaling roles, evidence reviewed here and elsewhere by Benard et al. (11) and by Soubannier and McBride (136) demonstrate that these organelles are positioned, both topologically and functionally, to alter cellular function through retrograde signaling pathways.
Mitochondria: hub of intracellular signaling?
This brings us to our central postulate, namely, that changes in mitochondrial morphology, because they are intimately linked to changes in mitochondrial function, represent an important cellular signaling mechanism (Fig. 4). The notion that mitochondrial morphology transitions directly control cellular signals is reinforced by the fact that mitochondrial fission and fusion constitute early events having downstream consequences on cell function and survival (112). For example, inhibiting mitochondrial fragmentation in vitro and in vivo with the DRP1 inhibitor Mdivi-1 conferred protection against the deleterious effects of hyperglycemia/hyperlipidemia on skeletal muscle (insulin resistance) (76), suggesting that mitochondrial fragmentation-induced ROS production plays a causal role in the development of insulin resistance with metabolic oversupply in vivo (i.e., hyperglycemia) (3, 76). In endothelial cells subjected to hyperglycemia, increased ROS production secondary to mitochondrial fragmentation blunted the normal activation of nitric oxide synthase and production of cGMP, which were restored by blocking Fis1 or DRP1-mediated mitochondrial fragmentation (129). Also with hyperglycemia, mitochondrial fission is a required early event that mediates cell death, such that blocking mitochondrial fission in cultured cells prevented hyperglycemia-induced ROS production and apoptotic cell death (163).
Fig. 4.
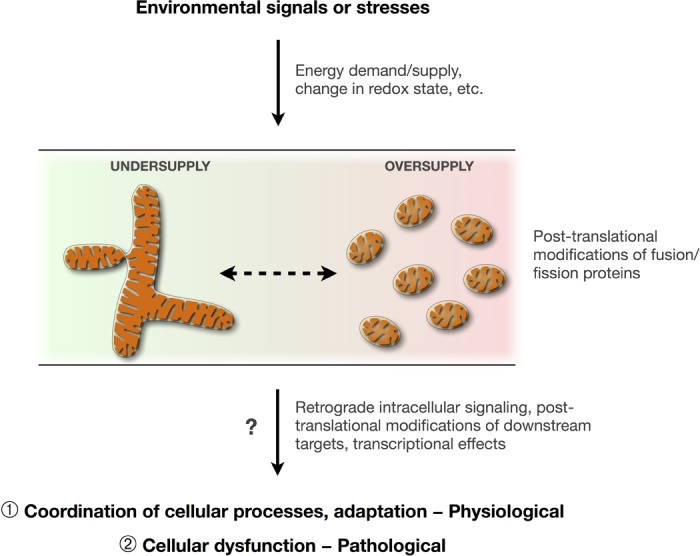
Potential role of mitochondrial morphology transitions as an integrating mechanism influencing intracellular signaling. Specific environmental stimuli and stresses impact cellular function via signaling mechanisms that may involve mitochondrial morphology transitions. Mitochondrial dynamics (fusion and fission) are linked to certain aspects of mitochondrial function. Mitochondrial dynamics could participate in both normal physiological adaptation (see circled 1) [e.g., promoting cell survival in response to nutrient deprivation (54, 112)], and to pathological responses (see circled 2) [e.g., hyperglycemia-induced cell death death (163), diabetic insulin resistance in skeletal muscle (76)]. Metabolic states of “undersupply” (negative energy balance) and “oversupply” (positive energy balance) tend to induce fusion and fragmentation of the mitochondrial network, respectively, possibly accounting for the health effects of physical activity/inactivity and energy intake (116).
Additional mechanistic studies have shown that mitochondrial fragmentation in mouse skeletal muscle was a required upstream event from AMPK activation and induced transcriptional activation of the atrophy program to permit skeletal muscle atrophy (122). Together, these reports demonstrate that inhibiting the mitochondrial fission machinery prevents mitochondrial fragmentation and blocks specific retrograde signals (e.g., ROS) and downstream cellular processes ranging from insulin resistance, apoptosis, and muscle atrophy. On the other hand, mitochondrial fusion is required for normal cell survival in periods of starvation, possibly due to an increased efficiency of the ATP synthase machinery (16). One report showed that abrogating OPA1 or Mfn1/2-mediated mitochondrial fusion during starvation in mice prevented mitochondrial elongation in skeletal muscle, decreased cellular ATP levels, and reduced survival upon starvation in vitro (54).
Currently, only limited data directly demonstrate that functional changes accompanying mitochondrial fusion and fission provide decisive intracellular signals. Most of the available evidence is derived from experiments investigating the effects of alterations in the metabolic state, such as metabolic oversupply (hyperglycemia, hyperlipidemia; i.e., positive metabolic balance) or undersupply (low glucose, fasting; i.e., negative metabolic balance), which are both inducers of mitochondrial fragmentation and fusion, respectively (116). This limited number of mechanistic studies performed under conditions of fluctuating energy supply demonstrates that mitochondrial morphology transitions, via their effect on intracellular signaling pathways, can significantly impact cellular function and survival.
Conclusions
The findings reviewed here suggest the existence of a bidirectional relationship linking mitochondrial morphology, mPTP sensitivity, respiration, and ROS production. Because functional outputs from mitochondria are second messengers that may operate as retrograde signals regulating key aspects of cellular function, it has been postulated that mitochondrial morphology transitions, through their effects on mitochondrial function, regulate intracellular signaling. However, only a limited number of studies yet provide direct support for this presumption. More research is thus needed to determine the exact mechanisms by which mitochondrial dynamics contribute to cellular signaling cascades under both normal and pathological conditions. In particular, it will be important to differentiate, conceptually and experimentally, the functional consequences of chronic/widespread alterations of mitochondrial dynamics in current experimental models, from the stochastic, localized and temporary events of mitochondrial fusion and fission occurring in vivo. In addition, approaches enabling to rule out nonmorphological (and nonmitochondrial) effects resulting from altering the fusion and fission proteins are required to establish that morphology and function are directly linked.
The exact role that mitochondria fulfill in the pathogenesis of specific chronic diseases such as cancer, diabetes, neurodegeneration, and in aging itself is still unclear. Most existing studies have sought to detect overt mitochondrial biochemical defects that result in molecular damage or cell death as only valid evidence for a role of mitochondrial dysfunction in disease. However, more subtle and/or transient in vivo changes in mitochondrial morphology and function; e.g., in response to altered metabolic state, hyperglycemia, hyperlipidemia, could possibly lead to chronically aberrant intracellular signaling sufficient to cause cellular and organ dysfunction. Given that the eukaryotic cell evolved its complexity through a process that depended heavily on mitochondrial metabolism (85, 152), it is logical that mammalian cells have developed astute sensitivity to numerous functional outputs from these organelles. New discoveries about mitochondrial dynamics will undoubtedly continue to evolve our perception of mitochondria: from a static peanut-shaped organelle primarily invested in ATP synthesis to networks of dynamic organelles actively contributing to cellular adaptation in health and disease.
GRANTS
The work of the authors is supported by a grant from the National Science and Engineering Research Council (NSERC) of Canada to Y. Burelle and from the Canadian Institute of Health Research (CIHR) Grant 86725 to B. J. Gentil. M. Picard was supported by CIHR Fellowships in Systems Biology and in Psychosocial Oncology, a PhD Scholarship from the NSERC of Canada, and holds a CIHR Postdoctoral Fellowship from the Institute of Neuroscience as part of the Canadian Epigenetics, Environment and Health Research Consortium. Y. Burelle is a Junior 2 Investigator from the Fonds de Recherche en Santé du Québec (FRSQ).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: M.P. conception and design of research; M.P. prepared figures; M.P. and Y.B. drafted manuscript; M.P., O.S.S., B.J.G., and Y.B. edited and revised manuscript; M.P., O.S.S., B.J.G., and Y.B. approved final version of manuscript.
ACKNOWLEDGMENTS
We are grateful to Russell Hepple and Paolo Bernardi for providing comments on parts of this work and to the authors whose diligent research inspired this review.
REFERENCES
- 1. Acin-Perez R, Gatti DL, Bai Y, Manfredi G. Protein phosphorylation and prevention of cytochrome oxidase inhibition by ATP: coupled mechanisms of energy metabolism regulation. Cell Metab 13: 712–719, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Amati-Bonneau P, Valentino ML, Reynier P, Gallardo ME, Bornstein B, Boissiere A, Campos Y, Rivera H, de la Aleja JG, Carroccia R, Iommarini L, Labauge P, Figarella-Branger D, Marcorelles P, Furby A, Beauvais K, Letournel F, Liguori R, La Morgia C, Montagna P, Liguori M, Zanna C, Rugolo M, Cossarizza A, Wissinger B, Verny C, Schwarzenbacher R, Martin MA, Arenas J, Ayuso C, Garesse R, Lenaers G, Bonneau D, Carelli V. OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain 131: 338–351, 2008 [DOI] [PubMed] [Google Scholar]
- 3. Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, Price JW, 3rd, Kang L, Rabinovitch PS, Szeto HH, Houmard JA, Cortright RN, Wasserman DH, Neufer PD. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest 119: 573–581, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Arnoult D. Mitochondrial fragmentation in apoptosis. Trends Cell Biol 17: 6–12, 2007 [DOI] [PubMed] [Google Scholar]
- 5. Aure K, Fayet G, Leroy JP, Lacene E, Romero NB, Lombes A. Apoptosis in mitochondrial myopathies is linked to mitochondrial proliferation. Brain 129: 1249–1259, 2006 [DOI] [PubMed] [Google Scholar]
- 6. Bach D, Pich S, Soriano FX, Vega N, Baumgartner B, Oriola J, Daugaard JR, Lloberas J, Camps M, Zierath JR, Rabasa-Lhoret R, Wallberg-Henriksson H, Laville M, Palacin M, Vidal H, Rivera F, Brand M, Zorzano A. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J Biol Chem 278: 17190–17197, 2003 [DOI] [PubMed] [Google Scholar]
- 7. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, aging. Cell 120: 483–495, 2005 [DOI] [PubMed] [Google Scholar]
- 8. Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476: 341–345, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Benani A, Troy S, Carmona MC, Fioramonti X, Lorsignol A, Leloup C, Casteilla L, Penicaud L. Role for mitochondrial reactive oxygen species in brain lipid sensing: redox regulation of food intake. Diabetes 56: 152–160, 2007 [DOI] [PubMed] [Google Scholar]
- 10. Benard G, Bellance N, James D, Parrone P, Fernandez H, Letellier T, Rossignol R. Mitochondrial bioenergetics and structural network organization. J Cell Sci 120: 838–848, 2007 [DOI] [PubMed] [Google Scholar]
- 11. Benard G, Bellance N, Jose C, Melser S, Nouette-Gaulain K, Rossignol R. Multi-site control and regulation of mitochondrial energy production. Biochim Biophys Acta 1797: 698–709, 2010 [DOI] [PubMed] [Google Scholar]
- 12. Benard G, Rossignol R. Ultrastructure of the mitochondrion and its bearing on function and bioenergetics. Antioxid Redox Signal 10: 1313–1342, 2008 [DOI] [PubMed] [Google Scholar]
- 13. Bernardi P. Pathophysiology of mitochondrial permeability transition. Society of General Physiologists Conference. Proceedings: Mitochondrial Physiology in Medicine (Abstract). J Gen Physiol 138: 131, 2011. 21788608 [Google Scholar]
- 14. Bernardi P, Veronese P, Petronilli V. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore. I. Evidence for two separate Me2+ binding sites with opposing effects on the pore open probability. J Biol Chem 268: 1005–1010, 1993 [PubMed] [Google Scholar]
- 15. Bianchi K, Vandecasteele G, Carli C, Romagnoli A, Szabadkai G, Rizzuto R. Regulation of Ca2+ signalling and Ca2+-mediated cell death by the transcriptional coactivator PGC-1alpha. Cell Death Differ 13: 586–596, 2006 [DOI] [PubMed] [Google Scholar]
- 16. Blackstone C, Chang CR. Mitochondria unite to survive. Nat Cell Biol 13: 521–522, 2011 [DOI] [PubMed] [Google Scholar]
- 17. Blattner JR, He L, Lemasters JJ. Screening assays for the mitochondrial permeability transition using a fluorescence multiwell plate reader. Anal Biochem 295: 220–226, 2001 [DOI] [PubMed] [Google Scholar]
- 18. Bo H, Zhang Y, Ji LL. Redefining the role of mitochondria in exercise: a dynamic remodeling. Ann NY Acad Sci 1201: 121–128, 2010 [DOI] [PubMed] [Google Scholar]
- 19. Brinkley BR, Barham SS, Barranco SC, Fuller GM. Rotenone inhibition of spindle microtubule assembly in mammalian cells. Exp Cell Res 85: 41–46, 1974 [DOI] [PubMed] [Google Scholar]
- 20. Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol 287: C817–C833, 2004 [DOI] [PubMed] [Google Scholar]
- 21. Butow RA, Avadhani NG. Mitochondrial signaling: the retrograde response. Mol Cell 14: 1–15, 2004 [DOI] [PubMed] [Google Scholar]
- 22. Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458: 1056–1060, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cartoni R, Leger B, Hock MB, Praz M, Crettenand A, Pich S, Ziltener JL, Luthi F, Deriaz O, Zorzano A, Gobelet C, Kralli A, Russell AP. Mitofusins 1/2 and ERRalpha expression are increased in human skeletal muscle after physical exercise. J Physiol 567: 349–358, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Casasnovas C, Banchs I, Cassereau J, Gueguen N, Chevrollier A, Martinez-Matos JA, Bonneau D, Volpini V. Phenotypic spectrum of MFN2 mutations in the Spanish population. J Med Genet 47: 249–256, 2010 [DOI] [PubMed] [Google Scholar]
- 25. Chen H, Chan DC. Physiological functions of mitochondrial fusion. Ann NY Acad Sci 1201: 21–25, 2010 [DOI] [PubMed] [Google Scholar]
- 26. Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem 280: 26185–26192, 2005 [DOI] [PubMed] [Google Scholar]
- 27. Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160: 189–200, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 130: 548–562, 2007 [DOI] [PubMed] [Google Scholar]
- 29. Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141: 280–289, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen Y, Azad MB, Gibson SB. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ 16: 1040–1052, 2009 [DOI] [PubMed] [Google Scholar]
- 31. Cole NB, Daniels MP, Levine RL, Kim G. Oxidative stress causes reversible changes in mitochondrial permeability and structure. Exp Gerontol 45: 596–602, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Crompton M, Virji S, Doyle V, Johnson N, Ward JM. The mitochondrial permeability transition pore. Biochem Soc Symp 66: 167–179, 1999 [DOI] [PubMed] [Google Scholar]
- 33. Csordas G, Hajnoczky G. SR/ER-mitochondrial local communication: calcium and ROS. Biochim Biophys Acta 1787: 1352–1362, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456: 605–610, 2008 [DOI] [PubMed] [Google Scholar]
- 35. De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476: 336–340, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Detmer SA, Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol 8: 870–879, 2007 [DOI] [PubMed] [Google Scholar]
- 37. Di Lisa F, Menabo R, Canton M, Barile M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem 276: 2571–2575, 2001 [DOI] [PubMed] [Google Scholar]
- 38. Di Lisa F, Ziegler M. Pathophysiological relevance of mitochondria in NAD(+) metabolism. FEBS Lett 492: 4–8, 2001 [DOI] [PubMed] [Google Scholar]
- 39. Dimmer KS, Scorrano L. (De)constructing mitochondria: what for? Physiology (Bethesda) 21: 233–241, 2006 [DOI] [PubMed] [Google Scholar]
- 40. Droge W. Free radicals in the physiological control of cell function. Physiol Rev 82: 47–95, 2002 [DOI] [PubMed] [Google Scholar]
- 41. Duchen MR, Szabadkai G. Roles of mitochondria in human disease. Essays Biochem 47: 115–137, 2010 [DOI] [PubMed] [Google Scholar]
- 42. Dumas JF, Argaud L, Cottet-Rousselle C, Vial G, Gonzalez C, Detaille D, Leverve X, Fontaine E. Effect of transient and permanent permeability transition pore opening on NAD(P)H localization in intact cells. J Biol Chem 284: 15117–15125, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Eisner V, Hajnoczky G. Mitochondrial Fusion Dynamics in Skeletal Muscle. Society of General Physiologists Conference Proceedings: Mitochondrial Physiology in Medicine. J Gen Physiol 138: 23, 2011 [Google Scholar]
- 44. Elmore SP, Qian T, Grissom SF, Lemasters JJ. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J 15: 2286–2287, 2001 [DOI] [PubMed] [Google Scholar]
- 45. Elrod JW, Wong R, Mishra S, Vagnozzi RJ, Sakthievel B, Goonasekera SA, Karch J, Gabel S, Farber J, Force T, Brown JH, Murphy E, Molkentin JD. Cyclophilin D controls mitochondrial pore-dependent Ca(2+) exchange, metabolic flexibility, and propensity for heart failure in mice. J Clin Invest 120: 3680–3687, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Estaquier J, Arnoult D. Inhibiting Drp1-mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ 14: 1086–1094, 2007 [DOI] [PubMed] [Google Scholar]
- 47. Fan X, Hussien R, Brooks GA. H2O2-induced mitochondrial fragmentation in C2C12 myocytes. Free Radic Biol Med 49: 1646–1654, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell 1: 515–525, 2001 [DOI] [PubMed] [Google Scholar]
- 49. Franzini-Armstrong C. ER-mitochondria communication. How privileged? Physiology (Bethesda) 22: 261–268, 2007 [DOI] [PubMed] [Google Scholar]
- 50. Frazier AE, Kiu C, Stojanovski D, Hoogenraad NJ, Ryan MT. Mitochondrial morphology and distribution in mammalian cells. Biol Chem 387: 1551–1558, 2006 [DOI] [PubMed] [Google Scholar]
- 51. Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, Scorrano L. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126: 177–189, 2006 [DOI] [PubMed] [Google Scholar]
- 52. Fulop L, Szanda G, Enyedi B, Varnai P, Spat A. The effect of OPA1 on mitochondrial Ca(2)(+) signaling. PLos One 6: e25199, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Go YM, Jones DP. Redox control systems in the nucleus: mechanisms and functions. Antioxid Redox Signal 13: 489–509, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 13: 589–598, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Greaves LC, Reeve AK, Taylor RW, Turnbull DM. Mitochondrial DNA and disease. J Pathol 226: 274–286, 2012 [DOI] [PubMed] [Google Scholar]
- 56. Guillery O, Malka F, Frachon P, Milea D, Rojo M, Lombes A. Modulation of mitochondrial morphology by bioenergetics defects in primary human fibroblasts. Neuromuscul Disord 18: 319–330, 2008 [DOI] [PubMed] [Google Scholar]
- 57. Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol Cell Physiol 258: C755–C786, 1990 [DOI] [PubMed] [Google Scholar]
- 58. Handy DE, Loscalzo J. Redox regulation of mitochondrial function. Antioxid Redox Signal 16: 1323–1367, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hanoune J, Defer N. Regulation and role of adenylyl cyclase isoforms. Annu Rev Pharmacol Toxicol 41: 145–174, 2001 [DOI] [PubMed] [Google Scholar]
- 60. Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev 25: 1895–1908, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hardie DG, Carling D, Gamblin SJ. AMP-activated protein kinase: also regulated by ADP? Trends Biochem Sci 36: 470–477, 2011 [DOI] [PubMed] [Google Scholar]
- 62. He L, Lemasters JJ. Regulated and unregulated mitochondrial permeability transition pores: a new paradigm of pore structure and function? FEBS Lett 512: 1–7, 2002 [DOI] [PubMed] [Google Scholar]
- 63. Heath-Engel HM, Shore GC. Mitochondrial membrane dynamics, cristae remodelling and apoptosis. Biochim Biophys Acta 1763: 549–560, 2006 [DOI] [PubMed] [Google Scholar]
- 64. Hom J, Sheu SS. Morphological dynamics of mitochondria–a special emphasis on cardiac muscle cells. J Mol Cell Cardiol 46: 811–820, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hom JR, Quintanilla RA, Hoffman DL, de Mesy Bentley KL, Molkentin JD, Sheu SS, Porter GA., Jr The permeability transition pore controls cardiac mitochondrial maturation and myocyte differentiation. Dev Cell 21: 469–478, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hood DA. Mechanisms of exercise-induced mitochondrial biogenesis in skeletal muscle. Appl Physiol Nutr Metab 34: 465–472, 2009 [DOI] [PubMed] [Google Scholar]
- 67. Houtkooper RH, Canto C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev 31: 194–223, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ichas F, Jouaville LS, Mazat JP. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell 89: 1145–1153, 1997 [DOI] [PubMed] [Google Scholar]
- 69. Indo HP, Davidson M, Yen HC, Suenaga S, Tomita K, Nishii T, Higuchi M, Koga Y, Ozawa T, Majima HJ. Evidence of ROS generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage. Mitochondrion 7: 106–118, 2007 [DOI] [PubMed] [Google Scholar]
- 70. Ishihara N, Eura Y, Mihara K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J Cell Sci 117: 6535–6546, 2004 [DOI] [PubMed] [Google Scholar]
- 71. Jahani-Asl A, Cheung EC, Neuspiel M, MacLaurin JG, Fortin A, Park DS, McBride HM, Slack RS. Mitofusin 2 protects cerebellar granule neurons against injury-induced cell death. J Biol Chem 282: 23788–23798, 2007 [DOI] [PubMed] [Google Scholar]
- 72. Jahani-Asl A, Pilon-Larose K, Xu W, Maclaurin JG, Park DS, McBride HM, Slack RS. The mitochondrial inner membrane GTPase, Optic Atrophy 1 (Opa1), restores mitochondrial morphology and promotes neuronal survival following excitotoxicity. J Biol Chem, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jendrach M, Mai S, Pohl S, Voth M, Bereiter-Hahn J. Short- and long-term alterations of mitochondrial morphology, dynamics and mtDNA after transient oxidative stress. Mitochondrion 8: 293–304, 2008 [DOI] [PubMed] [Google Scholar]
- 74. Jeyaraju DV, Cisbani G, Pellegrini L. Calcium regulation of mitochondria motility and morphology. Biochim Biophys Acta 1787: 1363–1373, 2009 [DOI] [PubMed] [Google Scholar]
- 76. Jheng HF, Tsai PJ, Guo SM, Kuo LH, Chang CS, Su IJ, Chang CR, Tsai YS. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol Cell Biol 32: 309–319, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kikuchi M, Hatano N, Yokota S, Shimozawa N, Imanaka T, Taniguchi H. Proteomic analysis of rat liver peroxisome: presence of peroxisome-specific isozyme of Lon protease. J Biol Chem 279: 421–428, 2004 [DOI] [PubMed] [Google Scholar]
- 78. Koopman WJ, Verkaart S, Visch HJ, van der Westhuizen FH, Murphy MP, van den Heuvel LW, Smeitink JA, Willems PH. Inhibition of complex I of the electron transport chain causes O2−-mediated mitochondrial outgrowth. Am J Physiol Cell Physiol 288: C1440–C1450, 2005 [DOI] [PubMed] [Google Scholar]
- 79. Koopman WJ, Visch HJ, Verkaart S, van den Heuvel LW, Smeitink JA, Willems PH. Mitochondrial network complexity and pathological decrease in complex I activity are tightly correlated in isolated human complex I deficiency. Am J Physiol Cell Physiol 289: C881–C890, 2005 [DOI] [PubMed] [Google Scholar]
- 80. Koopman WJ, Willems PH, Smeitink JA. Monogenic mitochondrial disorders. N Engl J Med 366: 1132–1141, 2012 [DOI] [PubMed] [Google Scholar]
- 81. Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science 305: 858–862, 2004 [DOI] [PubMed] [Google Scholar]
- 82. Koshiba T, Yasukawa K, Yanagi Y, Kawabata S. Mitochondrial membrane potential is required for MAVS-mediated antiviral signaling. Sci Signal 4: ra7, 2011 [DOI] [PubMed] [Google Scholar]
- 83. Kulp A, Kuehn MJ. Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Annu Rev Microbiol 64: 163–184, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lambert AJ, Brand MD. Reactive oxygen species production by mitochondria. Methods Mol Biol 554: 165–181, 2009 [DOI] [PubMed] [Google Scholar]
- 85. Lane N, Martin W. The energetics of genome complexity. Nature 467: 929–934, 2010 [DOI] [PubMed] [Google Scholar]
- 86. Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell 15: 5001–5011, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Legros F, Lombes A, Frachon P, Rojo M. Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol Biol Cell 13: 4343–4354, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Leloup C, Casteilla L, Carriere A, Galinier A, Benani A, Carneiro L, Penicaud L. Balancing mitochondrial redox signaling: a key point in metabolic regulation. Antioxid Redox Signal 14: 519–530, 2011 [DOI] [PubMed] [Google Scholar]
- 89. Lemasters JJ. Mitochondrial permeability transition. In: Mitochondrial Biogenesis: Processes, Regulation, Functions and Disease, edited by Hood DA. London: (online at http://hstalks.com/bio): The Biomedical & Life Sciences Collection, Henry Stewart Talks, Ltd., 2007 [Google Scholar]
- 90. Li X, Gould SJ. The dynamin-like GTPase DLP1 is essential for peroxisome division and is recruited to peroxisomes in part by PEX11. J Biol Chem 278: 17012–17020, 2003 [DOI] [PubMed] [Google Scholar]
- 91. Liesa M, Palacin M, Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol Rev 89: 799–845, 2009 [DOI] [PubMed] [Google Scholar]
- 92. Liot G, Bossy B, Lubitz S, Kushnareva Y, Sejbuk N, Bossy-Wetzel E. Complex II inhibition by 3-NP causes mitochondrial fragmentation and neuronal cell death via an NMDA- and ROS-dependent pathway. Cell Death Differ 16: 899–909, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Liu Z, Butow RA. Mitochondrial retrograde signaling. Annu Rev Genet 40: 159–185, 2006 [DOI] [PubMed] [Google Scholar]
- 94. Loiseau D, Chevrollier A, Verny C, Guillet V, Gueguen N, Pou de Crescenzo MA, Ferre M, Malinge MC, Guichet A, Nicolas G, Amati-Bonneau P, Malthiery Y, Bonneau D, Reynier P. Mitochondrial coupling defect in Charcot-Marie-Tooth type 2A disease. Ann Neurol 61: 315–323, 2007 [DOI] [PubMed] [Google Scholar]
- 95. Madiraju P, Pande SV, Prentki M, Madiraju SR. Mitochondrial acetylcarnitine provides acetyl groups for nuclear histone acetylation. Epigenetics 4: 399–403, 2009 [DOI] [PubMed] [Google Scholar]
- 96. Mashburn LM, Whiteley M. Membrane vesicles traffic signals and facilitate group activities in a prokaryote. Nature 437: 422–425, 2005 [DOI] [PubMed] [Google Scholar]
- 97. McBride H, Soubannier V. Mitochondrial function: OMA1 and OPA1, the grandmasters of mitochondrial health. Curr Biol 20: R274–R276, 2010 [DOI] [PubMed] [Google Scholar]
- 98. McGee SL, Hargreaves M. AMPK-mediated regulation of transcription in skeletal muscle. Clin Sci (Lond) 118: 507–518, 2010 [DOI] [PubMed] [Google Scholar]
- 99. Mingrone G, Manco M, Calvani M, Castagneto M, Naon D, Zorzano A. Could the low level of expression of the gene encoding skeletal muscle mitofusin-2 account for the metabolic inflexibility of obesity? Diabetologia 48: 2108–2114, 2005 [DOI] [PubMed] [Google Scholar]
- 100. Misko A, Jiang S, Wegorzewska I, Milbrandt J, Baloh RH. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci 30: 4232–4240, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Misko AL, Sasaki Y, Tuck E, Milbrandt J, Baloh RH. Mitofusin 2 mutations disrupt axonal mitochondrial positioning and promote axon degeneration. J Neurosci 32: 4145–4155, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Nakada K, Inoue K, Ono T, Isobe K, Ogura A, Goto YI, Nonaka I, Hayashi JI. Inter-mitochondrial complementation: mitochondria-specific system preventing mice from expression of disease phenotypes by mutant mtDNA. Nat Med 7: 934–940, 2001 [DOI] [PubMed] [Google Scholar]
- 103. Nakahata Y, Kaluzova M, Grimaldi B, Sahar S, Hirayama J, Chen D, Guarente LP, Sassone-Corsi P. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell 134: 329–340, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Neuspiel M, Schauss AC, Braschi E, Zunino R, Rippstein P, Rachubinski RA, Andrade-Navarro MA, McBride HM. Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr Biol 18: 102–108, 2008 [DOI] [PubMed] [Google Scholar]
- 105. Nochez Y, Arsene S, Gueguen N, Chevrollier A, Ferre M, Guillet V, Desquiret V, Toutain A, Bonneau D, Procaccio V, Amati-Bonneau P, Pisella PJ, Reynier P. Acute and late-onset optic atrophy due to a novel OPA1 mutation leading to a mitochondrial coupling defect. Mol Vis 15: 598–608, 2009 [PMC free article] [PubMed] [Google Scholar]
- 106. Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 121: 2012–2022, 2010 [DOI] [PubMed] [Google Scholar]
- 107. Ono T, Isobe K, Nakada K, Hayashi JI. Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat Genet 28: 272–275, 2001 [DOI] [PubMed] [Google Scholar]
- 108. Owusu-Ansah E, Yavari A, Mandal S, Banerjee U. Distinct mitochondrial retrograde signals control the G1-S cell cycle checkpoint. Nat Genet 40: 356–361, 2008 [DOI] [PubMed] [Google Scholar]
- 109. Paltauf-Doburzynska J, Malli R, Graier WF. Hyperglycemic conditions affect shape and Ca2+ homeostasis of mitochondria in endothelial cells. J Cardiovasc Pharmacol 44: 423–436, 2004 [DOI] [PubMed] [Google Scholar]
- 110. Parone PA, Da Cruz S, Tondera D, Mattenberger Y, James DI, Maechler P, Barja F, Martinou JC. Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLos One 3: e3257, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Perkins G, Bossy-Wetzel E, Ellisman MH. New insights into mitochondrial structure during cell death. Exp Neurol 218: 183–192, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Picard M, Burelle Y. Mitochondria: Starving to reach quorum? Bioessays 34: 272–274, 2012 [DOI] [PubMed] [Google Scholar]
- 113. Picard M, Hepple RT, Burelle Y. Mitochondrial functional specialization in glycolytic and oxidative muscle fibers: tailoring the organelle for optimal function. Am J Physiol Cell Physiol 302: C629–C641, 2012 [DOI] [PubMed] [Google Scholar]
- 114. Picard M, Jung B, Liang F, Azuelos I, Hussain SN, Goldberg AL, Godin R, Danialou G, Chaturvedi R, Rygiel K, Matecki S, Jaber S, Des Rosiers C, Karpati G, Ferri L, Burelle Y, Turnbull DM, Taivassalo T, Petrof BJ. Mitochondrial dysfunction and lipid accumulation in the human diaphragm during mechanical ventilation. Am J Respir Crit Care Med 186: 1140–1149, 2012 [DOI] [PubMed] [Google Scholar]
- 115. Picard M, Taivassalo T, Ritchie D, Wright KJ, Thomas MT, Romestaing C, Hepple RT. Mitochondrial Structure and Function are Disrupted by Standard Isolation Methods. PLos One 6: e18317, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Picard M, Turnbull DM. Linking the metabolic state and mitochondrial DNA in chronic disease, health and aging. Diabetes 62: 672–678, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Pich S, Bach D, Briones P, Liesa M, Camps M, Testar X, Palacin M, Zorzano A. The Charcot-Marie-Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum Mol Genet 14: 1405–1415, 2005 [DOI] [PubMed] [Google Scholar]
- 118. Pidoux G, Witczak O, Jarnaess E, Myrvold L, Urlaub H, Stokka AJ, Kuntziger T, Tasken K. Optic atrophy 1 is an A-kinase anchoring protein on lipid droplets that mediates adrenergic control of lipolysis. EMBO J 30: 4371–4386, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Powers SK, Hudson MB, Nelson WB, Talbert EE, Min K, Szeto HH, Kavazis AN, Smuder AJ. Mitochondria-targeted antioxidants protect against mechanical-ventilation-induced diaphragm weakness. Crit Care Med 39: 1749–1759, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Rasola A, Bernardi P. Mitochondrial permeability transition in Ca(2+)-dependent apoptosis and necrosis. Cell Calcium 50: 222–233, 2011 [DOI] [PubMed] [Google Scholar]
- 121. Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280: 1763–1766, 1998 [DOI] [PubMed] [Google Scholar]
- 122. Romanello V, Guadagnin E, Gomes L, Roder I, Sandri C, Petersen Y, Milan G, Masiero E, Del Piccolo P, Foretz M, Scorrano L, Rudolf R, Sandri M. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J 29: 1774–1785, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Rouzier C, Bannwarth S, Chaussenot A, Chevrollier A, Verschueren A, Bonello-Palot N, Fragaki K, Cano A, Pouget J, Pellissier JF, Procaccio V, Chabrol B, Paquis-Flucklinger V. The MFN2 gene is responsible for mitochondrial DNA instability and optic atrophy “plus” phenotype. Brain 135: 23–34, 2012 [DOI] [PubMed] [Google Scholar]
- 124. Ryan MT, Hoogenraad NJ. Mitochondrial-nuclear communications. Annu Rev Biochem 76: 701–722, 2007 [DOI] [PubMed] [Google Scholar]
- 125. Ryu SW, Choi K, Park JH, Park YM, Kim S, Choi C. Mitofusin 1 inhibits an apoptosis-associated amino-terminal conformational change in Bax, but not its mitochondrial translocation, in a GTPase-dependent manner. Cancer Lett 323: 62–68, 2012 [DOI] [PubMed] [Google Scholar]
- 126. Schafer E, Seelert H, Reifschneider NH, Krause F, Dencher NA, Vonck J. Architecture of active mammalian respiratory chain supercomplexes. J Biol Chem 281: 15370–15375, 2006 [DOI] [PubMed] [Google Scholar]
- 127. Schreiber TB, Mausbacher N, Breitkopf SB, Grundner-Culemann K, Daub H. Quantitative phosphoproteomics–an emerging key technology in signal-transduction research. Proteomics 8: 4416–4432, 2008 [DOI] [PubMed] [Google Scholar]
- 128. Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA, Korsmeyer SJ. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell 2: 55–67, 2002 [DOI] [PubMed] [Google Scholar]
- 129. Shenouda SM, Widlansky ME, Chen K, Xu G, Holbrook M, Tabit CE, Hamburg NM, Frame AA, Caiano TL, Kluge MA, Duess MA, Levit A, Kim B, Hartman ML, Joseph L, Shirihai OS, Vita JA. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation 124: 444–453, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Shibata Y, Hu J, Kozlov MM, Rapoport TA. Mechanisms shaping the membranes of cellular organelles. Annu Rev Cell Dev Biol 25: 329–354, 2009 [DOI] [PubMed] [Google Scholar]
- 131. Shoshan-Barmatz V, De Pinto V, Zweckstetter M, Raviv Z, Keinan N, Arbel N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol Aspects Med 31: 227–285, 2010 [DOI] [PubMed] [Google Scholar]
- 132. Shoshan-Barmatz V, Gincel D. The voltage-dependent anion channel: characterization, modulation, and role in mitochondrial function in cell life and death. Cell Biochem Biophys 39: 279–292, 2003 [DOI] [PubMed] [Google Scholar]
- 133. Shutt T, Geoffrion M, Milne R, McBride HM. The intracellular redox state is a core determinant of mitochondrial fusion. EMBO Rep 13: 909–915, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell 12: 2245–2256, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Song Z, Ghochani M, McCaffery JM, Frey TG, Chan DC. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol Biol Cell 20: 3525–3532, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Soubannier V, McBride HM. Positioning mitochondrial plasticity within cellular signaling cascades. Biochim Biophys Acta 1793: 154–170, 2009 [DOI] [PubMed] [Google Scholar]
- 137. Spange S, Wagner T, Heinzel T, Kramer OH. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol 41: 185–198, 2009 [DOI] [PubMed] [Google Scholar]
- 138. Srinivasan S, Koenigstein A, Joseph J, Sun L, Kalyanaraman B, Zaidi M, Avadhani NG. Role of mitochondrial reactive oxygen species in osteoclast differentiation. Ann NY Acad Sci 1192: 245–252, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev 89: 1025–1078, 2009 [DOI] [PubMed] [Google Scholar]
- 140. Suen DF, Narendra DP, Tanaka A, Manfredi G, Youle RJ. Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc Natl Acad Sci USA 107: 11835–11840, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Sugioka R, Shimizu S, Tsujimoto Y. Fzo1, a protein involved in mitochondrial fusion, inhibits apoptosis. J Biol Chem 279: 52726–52734, 2004 [DOI] [PubMed] [Google Scholar]
- 142. Szabadkai G, Duchen MR. Mitochondria: the hub of cellular Ca2+ signaling. Physiology (Bethesda) 23: 84–94, 2008 [DOI] [PubMed] [Google Scholar]
- 143. Szabadkai G, Simoni AM, Bianchi K, De Stefani D, Leo S, Wieckowski MR, Rizzuto R. Mitochondrial dynamics and Ca2+ signaling. Biochim Biophys Acta 1763: 442–449, 2006 [DOI] [PubMed] [Google Scholar]
- 144. Tsujimoto Y, Nakagawa T, Shimizu S. Mitochondrial membrane permeability transition and cell death. Biochim Biophys Acta 1757: 1297–1300, 2006 [DOI] [PubMed] [Google Scholar]
- 145. Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 27: 433–446, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Twig G, Liu X, Liesa M, Wikstrom JD, Molina AJ, Las G, Yaniv G, Hajnoczky G, Shirihai OS. Biophysical properties of mitochondrial fusion events in pancreatic beta-cells and cardiac cells unravel potential control mechanisms of its selectivity. Am J Physiol Cell Physiol 299: C477–C487, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Twig G, Shirihai OS. The interplay between mitochondrial dynamics and mitophagy. Antioxid Redox Signal 14: 1939–1951, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Viscomi C, Bottani E, Civiletto G, Cerutti R, Moggio M, Fagiolari G, Schon EA, Lamperti C, Zeviani M. In vivo correction of COX deficiency by activation of the AMPK/PGC-1alpha axis. Cell Metab 14: 80–90, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Vonck J, Schafer E. Supramolecular organization of protein complexes in the mitochondrial inner membrane. Biochim Biophys Acta 1793: 117–124, 2009 [DOI] [PubMed] [Google Scholar]
- 150. Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet 39: 359–407, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Wallace DC. Bioenergetics and the epigenome: interface between the environment and genes in common diseases. Dev Disabil Res Rev 16: 114–119, 2010 [DOI] [PubMed] [Google Scholar]
- 152. Wallace DC. Bioenergetics, the origins of complexity, and the ascent of man. Proc Natl Acad Sci USA 107, Suppl 2: 8947–8953, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Wallace DC, Fan W. Energetics epigenetics, mitochondrial genetics. Mitochondrion 10: 12–31, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Walsh C, Barrow S, Voronina S, Chvanov M, Petersen OH, Tepikin A. Modulation of calcium signalling by mitochondria. Biochim Biophys Acta 1787: 1374–1382, 2009 [DOI] [PubMed] [Google Scholar]
- 155. Wang C, Youle RJ. The role of mitochondria in apoptosis. Annu Rev Genet 43: 95–118, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, Wang X, Li K, Han P, Zheng M, Yin J, Mattson MP, Kao JP, Lakatta EG, Sheu SS, Ouyang K, Chen J, Dirksen RT, Cheng H. Superoxide flashes in single mitochondria. Cell 134: 279–290, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Wasilewski M, Scorrano L. The changing shape of mitochondrial apoptosis. Trends Endocrinol Metab 20: 287–294, 2009 [DOI] [PubMed] [Google Scholar]
- 158. Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 11: 872–884, 2010 [DOI] [PubMed] [Google Scholar]
- 159. Wieland OH. The mammalian pyruvate dehydrogenase complex: structure and regulation. Rev Physiol Biochem Pharmacol 96: 123–170, 1983 [DOI] [PubMed] [Google Scholar]
- 160. Yang XJ, Seto E. Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol Cell 31: 449–461, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Yoon Y, Galloway CA, Jhun BS, Yu T. Mitochondrial Dynamics in Diabetes. Antioxid Redox Signal. doi:10.1089/ars.2010.3286, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci USA 103: 2653–2658, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Yu T, Sheu SS, Robotham JL, Yoon Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc Res 79: 341–351, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Yu-Wai-Man P, Chinnery PF. Dysfunctional mitochondrial maintenance: what breaks the circle of life? Brain 135: 9–11, 2012 [DOI] [PubMed] [Google Scholar]
- 165. Yu-Wai-Man P, Sitarz KS, Samuels DC, Griffiths PG, Reeve AK, Bindoff LA, Horvath R, Chinnery PF, OPA1 mutations cause cytochrome c oxidase deficiency due to loss of wild-type mtDNA molecules. Hum Mol Genet 19: 3043–3052, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Zanna C, Ghelli A, Porcelli AM, Karbowski M, Youle RJ, Schimpf S, Wissinger B, Pinti M, Cossarizza A, Vidoni S, Valentino ML, Rugolo M, Carelli V. OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain 131: 352–367, 2008 [DOI] [PubMed] [Google Scholar]
- 167. Zorzano A, Liesa M, Palacin M. Mitochondrial dynamics as a bridge between mitochondrial dysfunction and insulin resistance. Arch Physiol Biochem 115: 1–12, 2009 [DOI] [PubMed] [Google Scholar]
- 168. Zorzano A, Liesa M, Sebastian D, Segales J, Palacin M. Mitochondrial fusion proteins: dual regulators of morphology and metabolism. Semin Cell Dev Biol 21: 566–574, 2010 [DOI] [PubMed] [Google Scholar]