

Abstract
The SLC37 family consists of four sugar-phosphate exchangers, A1, A2, A3, and A4, which are anchored in the endoplasmic reticulum (ER) membrane. The best characterized family member is SLC37A4, better known as the glucose-6-phosphate (G6P) transporter (G6PT). SLC37A1, SLC37A2, and G6PT function as phosphate (Pi)-linked G6P antiporters catalyzing G6P:Pi and Pi:Pi exchanges. The activity of SLC37A3 is unknown. G6PT translocates G6P from the cytoplasm into the lumen of the ER where it couples with either glucose-6-phosphatase-α (G6Pase-α) or G6Pase-β to hydrolyze intraluminal G6P to glucose and Pi. The functional coupling of G6PT with G6Pase-α maintains interprandial glucose homeostasis and the functional coupling of G6PT with G6Pase-β maintains neutrophil energy homeostasis and functionality. A deficiency in G6PT causes glycogen storage disease type Ib, an autosomal recessive disorder characterized by impaired glucose homeostasis, neutropenia, and neutrophil dysfunction. Neither SLC37A1 nor SLC37A2 can functionally couple with G6Pase-α or G6Pase-β, and there are no known disease associations for them or SLC37A3. Since only G6PT matches the characteristics of the physiological ER G6P transporter involved in blood glucose homeostasis and neutrophil energy metabolism, the biological roles for the other SLC37 proteins remain to be determined.
Keywords: SLC37, sugar phosphate exchanger, glucose-6-phosphate transporter, endoplasmic reticulum, glycogen storage disease type Ib, glucose-6-phosphatase
1. Introduction
The solute carrier (SLC) gene series consists of over 50 gene families that are predicted to encode membrane-bound transporters (He et al., 2009). The families are organized largely based on predicted protein sequence homology and predicted substrate specificity. The SLC37 family consists of 4 characterized sugar-phosphate exchange (SPX) proteins. SLC37A1 is also known as SPX1 (Bartoloni, et al., 2000; Bartoloni and Antonarakis, 2004; Iacopetta et al., 2010; Pan et al., 2011); SLC37A2 is SPX2 (Bartoloni and Antonarakis, 2004; Kim et al., 2007; Pan et al., 2011; Takahashi et al., 2000); SLC37A3 is SPX3 (Bartoloni and Antonarakis, 2004; Pan et al., 2011); and SLC37A4 is SPX4 (Annabi et al., 1998; Bartoloni and Antonarakis, 2004; Chen et al., 2008; Gerin et al., 1997; Hiraiwa et al., 1999; Pan et al., 2011). Each of these proteins is localized in the endoplasmic reticulum (ER) membrane (Pan et al., 2011). The archetype of the family is SLC37A4, which is better known as the G6P transporter (G6PT). Topological analysis shows that G6PT (SLC37A4) is anchored in the ER by 10 transmembrane helices (Fig.1) (Pan et al., 1999; Pan et al., 2009) and the other members are predicted to similarly contain between 10 and 12 transmembrane domains. The family members were originally grouped into the SLC37 family based on sequence homology to the bacterial organo-phosphate:phosphate (Pi) exchangers (Pao et al., 1998). In the Transport Classification Database (TCDB) the SLC37 transporters are members of family 2.A.1.4 (http://www.tcdb.org/). Within the SLC37 family, the gene localization, structure, and amino acid compositions vary significantly suggesting the proteins have evolved independently and do not arise through gene duplication (Table 1). SLC37A1 and SLC37A2 are the most closely related at the protein level, while the G6PT protein is the most distant member of the family. Experimentally, SLC37A1, SLC37A2, and SLC37A4/G6PT have been shown to function as Pi-linked glucose-6-phosphate (G6P) antiporters while SLC37A3 lacks the activity (Chen et al., 2008; Pan et al., 2011). The primary in vivo function of G6PT is well characterized. In the liver, kidney, and intestine, G6PT couples functionally with glucose-6-phosphatase-α (G6Pase-α or G6PC) to maintain interprandial blood glucose homeostasis (Chou et al., 2010a; Chou et al., 2010b), while in neutrophils (Chou et al., 2010a; Chou et al., 2010b; Jun et al., 2010) it couples functionally with G6Pase-β (or G6PC3) to maintain neutrophil energy homeostasis and functionality (Fig. 1). Deficiencies in G6PT cause glycogen storage disease type Ib (GSD-Ib, OMIM232220) (Chou et al., 2010a; Chou et al., 2010b). No diseases have yet been linked to the other members of the family and their physiological function remains unknown. The characteristics of the SLC37 family members are summarized in Table 1.
Fig.1.

The topology of G6PT and its functional coupling with G6Pase within the ER. The diagram shows a cross-section of the ER within two different cell types. The cell cytoplasm lies outside the ER membrane that encircles the ER lumen. In the gluconeogenic tissues of the liver, kidney and intestine, G6PT couples with G6Pase-α, while in neutrophils, G6PT couples with G6Pase-β. The proteins are shown as spatially separated for clarity, but evidence suggests that they reside in physical contact with each other as a G6PT/G6Pase complex. The G6PT/G6Pase-α complex expressed in gluconeogenic tissues maintains interprandial blood glucose homeostasis and disruption of either protein leads to the phenotype associated with impaired glucose homeostasis listed. The G6PT/G6Pase-β complex, which is ubiquitously expressed, is critical for maintaining neutrophil energy homeostasis and functionality, and disruption of either protein leads to neutropenia and the neutrophil dysfunctions listed.
Table 1.
The SLC37 family of sugar-phosphate/phosphate exchangers
| Human gene name | SLC37A1 | SLC37A2 | SLC37A3 | SLC37A4 |
|---|---|---|---|---|
| Protein name(s) | SLC37A1, SPX1 | SLC37A2, SPX2 | SLC37A3, SPX3 | SLC37A4, G6PT, SPX4 |
| Known substrate | Glucose-6-phosphate | Glucose-6-phosphate | Unknown | Glucose-6-phosphate |
|
Transport type and
coupling ion |
Exchanger Inorganic phosphate |
Exchanger Inorganic phosphate |
Exchanger Inorganic phosphate |
|
| Tissue expression | Ubiquitous. High levels in prostate, pancreas, thymus, bone marrow, spleen |
Ubiquitous. High levels in spleen, thymus, bone marrow, prostate, lung, pancreas, liver, kidney, macrophages |
Ubiquitous. High levels in spleen, thymus, bone marrow, lung, brain, prostate, pancreas |
Ubiquitous. High levels in liver, kidney, prostate, pancreas, thymus, bone marrow, spleen |
| Link to disease | None reported | None reported | None reported | GSD-Ib/GSD-Ic (OMIM232220/232240) |
| Human gene locus | 21q22.3 | 11q24.2 | 7q34 | 11q23.3 |
| Sequence accession ID | NM_018964 | NM_198277 | NM_207113 | NM_001467 |
|
Mouse model status
(IMSR)a |
Targeted mutation and gene trap – phenotype not reported |
Targeted mutation – phenotype not reported |
Targeted mutation and gene trap – phenotype not reported |
Targeted mutation – GSD-Ib phenotype |
|
Major protein isoform/
Number of isoformsb,c |
533-amino-acid 1 |
505-amino-acid 4 |
494-amino-acid 3 |
429-amino-acid 2 |
|
Amino acid identity (%)
(NCBI)b |
||||
| SLC37A1 | 100 | 59 | 35 | 22 |
| SLC37A2 | 59 | 100 | 36 | 23 |
| SLC37A3 | 35 | 36 | 100 | 22 |
| SLC37A4 | 22 | 23 | 22 | 100 |
The International Mouse Search Resource (IMSR): http://www.findmice.org/
NCBI blast for amino acid identity and isoform prediction: http://blast.ncbi.nlm.nih.gov/Blast.cgi
Nextprot for isoform prediction: http://www.nextprot.org/
2. SLC37A1 (SPX1)
The human SLC37A1 gene, which consists of 19 coding exons on chromosome 21q22.3, was originally isolated by exon trapping in a study seeking to identify genes on chromosome 21 involved in Down syndrome (Bartoloni, et al., 2000). Subsequent studies excluded SLC37A1 as a contributing factor to Down syndrome.
SLC37A1 is a 533 amino-acid protein with a calculated molecular weight of 58 kDa. The protein shares 57% homology to SLC37A2 (Table 1) and 30% sequence identity to glycerol-3-phosphate transporter, suggesting that SLC37A1 could be a glycerol-3-phosphate transporter. However analyses of the SLC37A1 gene in seven patients with glyceroluria, lacking mutations in the glycerol kinase gene, revealed only non-pathogenetic sequence variants, excluding SLC37A1 as the causative gene in these patients (Bartoloni, et al., 2000). Despite mapping to the critical region of the autosomal recessive deafness locus, DFNB10 on chromosome 21q22.3, mutational analyses have also excluded it as the DFNB10 gene (Bartoloni, et al., 2000). There are some data suggesting SLC37A1 expression may correlate with breast cancer. In estrogen receptor negative SkBr3 breast cancer cells, expression of the SLC37A1 transcript is up-regulated by epidermal growth factor (EGF) via the EGF receptor/mitogen-activated protein kinase/Fos transduction pathway (Iacopetta et al., 2010). Interestingly, similar up-regulation is reported in estrogen receptor positive endometrial cancer cells. One hypothesis is that SLC37A1 is involved in phospholipid biosynthesis (Iacopetta et al., 2010), which has a role in the proliferation of tumor cells. However, to date, there is no direct evidence for a role in breast cancer, or any other disease.
Experimentally, SLC37A1 has been shown to be a Pi -linked G6P antiporter capable of G6P:Pi and Pi:Pi exchanges (Fig. 2A) (Pan et al., 2011). However, the transport activity of SLC37A1 is not sensitive to inhibition by chlorogenic acid (Fig. 2B) and SLC37A1 cannot couple functionally with G6Pase-α or G6Pase-β to mediate microsomal G6P uptake (Fig. 3A). Each of these findings excludes SLC37A1 as the primary physiological G6P transporter involved in blood and neutrophil glucose homeostasis (Pan et al., 2011).
Fig. 2.

The antiporter activities of the SLC37 members. The antiporter activity was determined in 50 mM Pi -loaded proteoliposomes expressing SLC37A1, SLC37A2, SLC37A3, or G6PT. (A) G6P or Pi uptake activity. (B) Effects of chlorogenic acid. Data are presented as the mean ± SEM.
Fig. 3.

Microsomal G6P transport activity of the SLC37 members. (A) Effects of G6Pase-α and G6Pase-β. G6P uptake activity was determined in microsomal membranes expressing SLC37A1, SLC37A2, or G6PT in the absence or presence of G6Pase-α or G6Pase-β. (B) G6P uptake activity in hepatic microsomes isolated from wild type (+/+), GSD-Ia (-/-) or Ad-G6Pase-α-treated GSD-Ia (Ad-G6Pase-α) mice. Data are presented as the mean ± SEM.
The SLC37A1 gene is widely expressed, with the highest transcript levels reported in adult kidney, bone marrow, intestine, spleen, and liver as well as in fetal liver, spleen and brain (Bartoloni, et al., 2000). When compared to the expression of other members of the SLC37 family, in the liver and kidney, the primary gluconeogenic organs, the SLC37A1 transcripts constitute less than 2% of the levels seen for SLC37A4 (Fig. 4) (Pan et al., 2011). In the intestine, pancreas, and macrophage, the SLC37A1 transcripts constitute 60%, 69%, and 43%, respectively, of the levels seen for SLC37A4 (Fig. 4). In neutrophils, the SLC37A1 transcripts are significantly higher being 2.8-fold over that of SLC37A4. These observations suggest that a search for the biological role of SLC37A1, if related to G6P metabolism, might best focus on neutrophils.
Fig. 4.
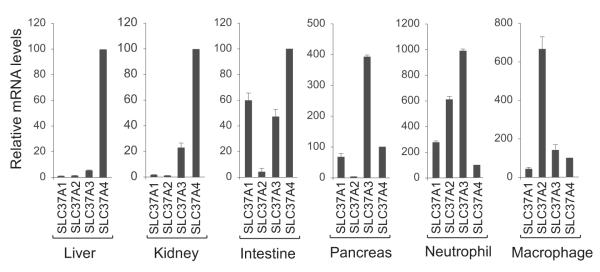
The mRNA levels of the SLC37 members, relative to Slc37a4, in mouse liver, kidney, intestine, pancreas, neutrophils, and macrophages. The expression levels of the Slc37a1, Slc37a2, Slc37a3, and Slc37a4 transcripts were normalized to β-actin RNA and then scaled, for each tissue, relative to the Slc37a4 transcript which was arbitrarily assigned as 100%. Results are expressed as mean ± SEM.
3. SLC37A2 (SPX2)
Slc37a2 was first identified in a murine study to identify cAMP-inducible genes in RAW264 macrophages (Takahashi et al., 2000). The murine Slc37a2 gene was mapped to chromosome 9q and shown to be highly expressed in white adipose tissue of obese mice following significant macrophage infiltration (Kim et al., 2007). Two murine Slc37a2 transcripts of 3.1 kb and 2.8 kb, generated by the alternative usage of polyadenylation sites, were identified, both encoding a 510 amino-acid polypeptide with a calculated molecular mass of 55 kDa (Takahashi et al., 2000). The murine protein contains 3 potential N-linked glycosylation sites and migrates as a heterogeneous species of 55-75 kDa (Kim et al., 2007). The expression of the Slc37a2 transcript in mice is restricted to spleen, thymus, and macrophages (Kim et al., 2007). The transcript is also expressed in murine liver, kidney, intestine, and pancreas but the expression levels are less than 5% of those of murine Slc37a4 (Fig. 4) (Pan et al., 2011). Compared to Slc37a4, the Slc37a2 transcript is highly expressed in neutrophils and macrophages (Fig. 4).
The human SLC37A2 gene maps to chromosome 11q24.2 and encodes 4 transcripts generated by alternative splicing of 18 coding exons. The longest SLC37A2 isoform of 505-amino acids has been characterized (Pan et al., 2011). Like the murine protein it is an ER-associated transmembrane protein that is post-translational modified by N-linked glycosylation. The expression of SLC37A2 increases markedly during differentiation of THP-1 human monocytes to macrophages (Kim et al., 2007) but no physiological role in the immune system has yet been identified. Experimentally, SLC37A2 protein functions as a Pi-linked G6P antiporter catalyzing G6P:Pi and Pi:Pi exchanges (Fig. 2A) (Pan et al., 2011). As with the SLC37A1 activity, the antiport activity of SLC37A2 is insensitive to chlorogenic acid inhibition (Fig. 2B), and SLC37A2 does not couple functionally with either G6Pase-α or G6Pase-β to mediate microsomal G6P uptake (Fig. 3A). Together, the data suggest that SLC37A2 is also unlikely to be a physiological G6P transporter involved in blood and neutrophil glucose homeostasis (Pan et al., 2011). The tissue-specific expression of SLC37A2 in macrophages and neutrophils may point to a biological transport role in these cells.
4. SLC37A3 (SPX3)
The least is known about SLC37A3. The human SLC37A3 gene maps to chromosome 7q34 and consists of 17 coding exons that are alternatively spliced to generate three different transcripts (Bartoloni and Antonarakis, 2004). The 494 amino acid isoform has been shown to be a transmembrane protein localized in the ER (Pan et al., 2011). Unlike the other SLC37 members, SLC37A3 lacks G6P antiporter activity (Fig. 2A) (Pan et al., 2011) and there are no other reported activities for the protein. The SLC37A3 transcripts are expressed in liver, kidney, intestine, pancreas, neutrophils and macrophages (Fig. 4) (Pan et al., 2011). Interestingly, the levels of the SLC37A3 transcript in the pancreas and neutrophils are significantly higher than the levels of other SLC37 members (Fig. 4), suggesting a yet undiscovered role for this protein in the pancreas and neutrophils.
5. SLC37A4 (G6PT, SPX4)
SLC37A4 is more commonly known as the G6PT. It was the first SLC37 member to be functionally characterized (Chen et al., 2000; Chen et al., 2002; Chen et al., 2008; Hiraiwa et al., 1999) and is the best characterized protein in this family. Human SLC37A4 is a single copy gene consisting of 9 coding exons on chromosome 11q23 (Annabi et al., 1998; Haraiwa et al., 1999). Transcription gives rise to two alternately spliced transcripts, SLC37A4 and variant SLC37A4, which encode polypeptides of 429 and 451 amino acids respectively, differing in the absence or presence of exon-7 sequence (Lin et al., 2000). SLC37A4 is ubiquitously expressed (Lin et al., 1998) while the variant SLC37A4 is expressed primarily in the brain, heart, and skeletal muscle (Lin et al., 2000). To date, there is no apparent difference in the activity and function of the two G6PT proteins and so further discussion will refer to both species as G6PT, although most experiments use G6PT, not the variant. Human G6PT is a hydrophobic protein anchored in the ER via 10 transmembrane helices with both N- and C-termini on the cytoplasmic side of the ER membrane (Fig. 1) (Pan et al., 1999; Pan et al., 2009), where it acts as a Pi-linked G6P antiporter catalyzing G6P:Pi and Pi:Pi exchanges between the cytoplasm and the ER lumen (Fig. 2A) (Chen et al., 2008). The in vivo function of G6PT depends upon its ability to couple functionally with either G6Pase-α (Lei et al., 1996; Chou et al., 2010a; Chou et al., 2010b) or G6Pase-β (Shieh et al., 2003; Chou et al., 2010a; Chou et al., 2010b). The resulting G6PT/G6Pase complex is essential for catalyzing intracellular glucose production (Fig. 1). G6Pase-α and G6Pase-β are also ER transmembrane proteins, but their active sites are localized inside the ER lumen (Ghosh et al., 2002; Ghosh et al., 2004). Therefore they depend on the antiporter activity of G6PT to transport cytoplasmic G6P into the ER lumen (Fig. 1).
The tissue expression profiles of G6Pase-α or G6Pase-β determine the different phenotypes for the G6PT/G6Pase complexes. The G6PT/G6Pase- complex, expressed primarily in the gluconeogenic organs, liver, kidney, and intestines, maintains interprandial glucose homeostasis while the G6PT/G6Pase-β complex in neutrophils maintains energy homeostasis and functionality (Fig.1) (Chou et al., 2010a; Chou et al., 2010b; Jun et al., 2010). Mutations of G6Pase-α underlie the metabolic disease, GSD-Ia which is characterized by impaired blood glucose homeostasis (Chou et al., 2010a; Chou et al., 2010b). Mutations in G6Pase-β underlie severe congenital neutropenia syndrome type 4 which is characterized by neutropenia and neutrophil dysfunction (Chou et al., 2010a; Chou et al., 2010b). Mutations of G6PT underlie GSD-Ib, which shares the abnormal metabolic phenotype of GSD-Ia, but with the additional complications of neutropenia and neutrophil dysfunction that mimic G6Pase-β deficiency (Chou et al., 2010a; Chou et al., 2010b). Although G6PT is the dominant SLC37 member in the gluconeogenic organs (Fig. 4) (Pan et al., 2011), surprisingly in neutrophils, despite its vital role, SLC37A4 is the least abundant of the SLC37 transcripts (Fig. 4).
5.1. The function of the G6PT protein
The primary in vivo function of G6PT is to translocate G6P from the cytoplasm into the lumen of the ER. The protein is capable of both heterologous G6P:Pi and homologous Pi:Pi exchanges (Chen et al., 2008). Unlike SLC37A1 or SLC37A2, the antiporter activity of G6PT is sensitive to inhibition by chlorogenic acid (Chen et al., 2008) (Fig. 2B). Microsomal G6P uptake activity in the liver is sensitive to inhibition by chlorogenic acid (Hemmerle et al., 1997) and in peripheral blood neutrophils is sensitive to inhibition by S3483, a chlorogenic acid derivative (Leuzzi et al., 2003). Therefore, chlorogenic acid and its derivative are specific inhibitors for the G6PT component of the G6PT/G6Pase-α and the G6PT/G6Pase-β complexes. The G6PT-mediated microsomal G6P uptake activity requires an active G6Pase (Fig. 3) (Chen et al., 2008; Lei et al., 1996; Pan et al., 2011; Shieh et al., 2003). Hepatic microsomes prepared from G6Pase-α-deficient (GSD-Ia) mice, with an intact G6PT, exhibit markedly lower G6P uptake activity compared to wild type hepatic microsomes (Fig. 3B) (Lei et al., 1996). However, this can be reversed if G6Pase-α activity is restored via gene transfer (Fig. 3B) (Zingone et al., 2000). Based on these characteristics, functional assays to detect physiologically important G6PT activity have been developed (Chen et al., 2000; Chen et al., 2002; Chen et al., 2008; Hiraiwa et al., 1999; Pan et al., 2011) and excluded SLC37A1, and SLC37A2 as a partner for G6Pase in intracellular glucose production (Pan et al., 2011). In addition to explaining why a deficiency in G6PT results in impaired glucose homeostasis and myeloid dysfunction, it also suggests that the G6Pase/G6PT functionality depends on a specific physical association between the G6Pase isozyme and G6PT transporter, otherwise the presence of the other transporters should suffice.
5.2. Genetics of SLC37A4 (G6PT) Deficiency - GSD-Ib
GSD-Ib is the heritable disease due to SLC37A4 (G6PT) deficiency. Eighty-two separate SLC37A4 mutations have been identified in ~ 170 GSD-Ib patients studied to date (Chou et al., 2010a; http://www.hgmd.cf.ac.uk/ac/gene.php?gene=SLC37A4). These include 34 missense, 11 nonsense, 19 insertion/deletion, 17 splicing mutations, and one gross deletion upstream of exon-7, which are scattered throughout the coding and exon-intron junction regions. Of the ~170 patients studied to date, 7 patients carrying deleterious SLC37A4 mutations have not been reported to manifest either neutropenia or frequent infections (Angaroni et al., 2006; Kure et al., 2000; Melis et al., 2005; Martens et al., 2006). A genotype-phenotype study with a cohort of 22 GSD-Ib patients carrying 16 different SLC37A4 mutations, including 9 patients homozygous or compound heterozygous for nonsense mutations failed to show any correlation between individual mutations and the presence of neutropenia, bacterial infections or systemic complications (Melis et al., 2005). While these numbers are low, they do suggest there may be genetic modifiers of G6PT activity that can compensate for or stabilize low level expression in vivo.
For years GSD-I had been considered to contain a third subtype, GSD-Ic, postulated to have a deficiency in exporting phosphate from the ER following hydrolysis of G6P to glucose and Pi (Chen, 2001; Chou et al., 2002). However, genetic analysis of these patients revealed mutations in the SLC37A4 gene seen in GSD-Ib patients (Galli et al., 1999; Janecke et al., 2000; Veiga-da-Cunha et al., 1998; Veiga-da-Cunha et al., 1999), raising the question whether GSD-Ic was real or a misdiagnosis of GSD-Ib. The recognition of the Pi:Pi and G6P:Pi exchange activities of G6PT (Chen et al., 2008) has resolved this issue showing GSD-Ic patients may have defective Pi exchange but are indeed GSD-Ib patients harboring SLC37A4 mutations. While the results show that G6PT has a dual transport role, it raises the question whether GSD-Ib and Ic are distinct diseases. Using reconstituted proteoliposomes, the G6P and Pi uptake activities of 23 G6PT mutations, including 8 missense G6PT mutations identified in GSD-Ic/Id patients have been characterized (Pan et al., 2009). While most G6P mutations abolish G6P and Pi uptake activities to similar extents, the G6PT p.Q133P mutation identified in a GSD-Ic patient (Veiga-da-Cunha et al., 1999) exhibits differential G6P and Pi transport activities (Pan et al., 2009). Therefore GSD-Ib and Ic may manifest as different clinically although the evidence is that they are very similar, if not the same disease.
5.3. Clinical phenotype of GSD-Ib (SLC37A4 deficiency)
Most mutations in the SLC37A4 gene that decrease or completely abolish microsomal G6P uptake activity result in both an abnormal metabolic phenotype and an abnormal myeloid phenotype (Chou et al., 2010a; Chou et al., 2010b).
5.3.1. GSD-Ib metabolic phenotype
The liver, and to a lesser extent, the kidney and intestine, are the primary gluconeogenic organs involved in the regulation of blood glucose homeostasis between meals. As blood glucose levels fall between meals, G6P produced in the terminal step of gluconeogenesis and glycogenolysis in the liver, kidney, and intestine is transported by G6PT into the ER where G6P is hydrolyzed by G6Pase-α to glucose for release back into the blood (Chen, 2001; Chou et al., 2002) (Fig. 1). In G6PT deficiency, the defective G6PT/G6Pase-α complex in the liver, kidney and intestine results in impaired blood glucose homeostasis and the hallmark is hypoglycemia following a short fast. Associated with this is an elevation of G6P in the cytoplasm of the cells leading to an excessive accumulation of glycogen, which promotes progressive hepatomegaly and nephromegaly (Chen, 2001; Chou et al., 2002). Other major metabolic consequences of elevated liver and kidney cytoplasmic G6P are hypercholesterolemia, hypertriglyceridemia, hyperuricemia, and lactic acidemia that characterize the clinical pathophysiology of GSD-Ib. Longer-term presentations include hepatocellular adenomas with risk for malignancy, and renal disease (Chen, 2001; Chou et al., 2002).
5.3.2. GSD-Ib myeloid phenotype
G6PT deficiency also results in neutropenia and myeloid dysfunction (Chou et al., 2010a; Chou et al., 2010b). Neutrophils from GSD-Ib patients exhibit impairment in chemotaxis, calcium mobilization, respiratory burst, and phagocytic activities (Chou et al., 2010a; Kilpatrick et al., 1990) (Fig. 1). As a result, recurrent bacterial infections are commonly seen and up to 75% of patients manifesting neutropenia also develop inflammatory bowel disease or enterocolitis, indistinguishable from idiopathic Crohn disease (Dieckgraefe et al., 2002; Visser et al., 2000). All patients with enterocolitis also had neutropenia. Oral manifestations of GSD-Ib include dental caries, gingivitis, periodontal disease, delayed dental maturation and eruption, oral bleeding diathesis, and oral ulcers (Mortellaro et al., 2005).
The mechanism underlying neutropenia in G6PT deficiency (GSD-Ib) and G6Pase-β deficiency is due to a defective G6PT/G6Pase-β complex in neutrophils (Chou et al., 2010a; Chou et al., 2010b). This complex is dysfunctional if either G6PT or G6Pase-β loses activity, therefore a study of either GSD-Ib or G6Pase-β deficiency reveals information about the other. Kuijpers et al. (2003) showed that neutrophils of GSD-Ib patients exhibit enhanced apoptosis, suggesting a causal relationship between apoptosis and neutropenia, but the underlying cause for neutrophil dysfunction remained unclear. More recently, neutrophils from G6PT-deficient mice that are unable to translocate G6P from the cytoplasm into the lumen of the ER, were shown to exhibit enhanced ER stress and apoptosis (Kim et al., 2008). Likewise, G6Pase- -deficient neutrophils which are unable to hydrolyze endolumenal G6P to glucose also manifested enhanced ER stress and apoptosis (Boztug et al., 2009; Cheung et al., 2007; Jun et al., 2010). Neutrophil apoptosis in GSD-Ib mice was shown to result from increased oxidative stress and to be mediated, at least in part, by the intrinsic mitochondrial stress pathway (Kim et al., 2008).
The molecular mechanism underlying neutrophil dysfunction in GSD-Ib and G6Pase- deficiency is also beginning to be understood. It is well established that neutrophils require a constant supply of glucose to both function and survive, yet they are unable to produce glucose via gluconeogenesis. Therefore, the primary source of glucose is uptake from the blood. In neutrophils, there are three primary pathways that compete for intracellular glucose/G6P, namely: glycolysis; the hexose monophosphate shunt (HMS); and ER cycling of G6P/glucose (Jun et al., 2010). The latter pathway is mediated by the G6PT/G6Pase-β complex. G6Pase-β-deficient neutrophils exhibit reduced expression and translocation of glucose transporter-1 along with impaired neutrophil glucose uptake. In parallel, levels of G6P, lactate, and ATP are markedly lower in G6Pase-β-deficient neutrophils, compared with the controls (Jun et al., 2010). Moreover, the expression and activation of NADPH oxidase are down-regulated in G6Pase-β-deficient neutrophils. Consequently, G6Pase-β inactivation disrupts neutrophil energy homeostasis, leading to impaired neutrophil respiratory burst, chemotaxis, calcium flux, and phagocytosis. Taken together, the underlying cause of neutrophil dysfunction in G6Pase-β deficiency is a disturbance in ER energy homeostasis caused by a loss of glucose/G6P recycling in neutrophils (Jun et al., 2010).
The cycling of G6P and glucose between the cytoplasm and ER via the coupled action of G6PT/G6Pase- , that regulates cytoplasmic pathways of glycolysis and HMS, suggests that GSD-Ib neutrophils should exhibit similar defects. Indeed, neutrophils from GSD-Ib patients have a decreased rate of glucose transport (Bashan et al., 1987) and their intracellular G6P levels are markedly lower, compared to neutrophils in control subjects (Verhoeven et al., 1999). In conclusion, the G6PT/G6Pase-β complex plays a key role in neutrophil energy homeostasis and functionality.
5.4. Treatment and prognosis of GSD-Ib (SLC37A4 deficiency)
The metabolic abnormalities of G6PT deficiency can be adequately controlled with dietary therapies augmented by drug therapy. Patients 3 years or older are prescribed uncooked cornstarch, a slow release carbohydrate, that prolongs the length of euglycemia between meals (Chen et al., 1984). Younger patients typically receive nocturnal nasogastric infusion of glucose (Greene et al., 1976) because their pancreatic amylase activity, required to hydrolyze raw starch, is insufficient (Chen et al., 1984). These dietary therapies maintain normoglycemia and enable patients to attain near normal growth and pubertal development, with fewer complications as they age. However, dietary therapies fail to improve myeloid functions. Moreover, the underlying pathological process remains uncorrected and long-term complications still persist in GSD-Ib patients. For patients exhibiting persistent, therapy-refractory hypoglycemic episodes, orthotopic liver transplantation (Adachi et al., 2004; Bhattacharya et al., 2004; Martin et al., 2006; Martinez-Olmos et al., 2001; Matern et al., 1999) and hepatocyte transplantation have been performed in a few GSD-Ib patients (Lee et al., 2007). Both transplantations improved the metabolic abnormalities for the patients and neutrophil function also appeared to improve.
GSD-Ib patients manifesting neutropenia, and/or neutrophil dysfunction that predispose to frequent infections and enterocolitis are treated by granulocyte colony-stimulating factor (G-CSF) therapy which improves neutropenia and decreases the number and severity of infections (Calderwood et al., 2001; Visser et al., 2002). G-CSF therapy shows no significant short-term toxicity, although all patients receiving chronic G-CSF therapy do develop splenomegaly (Calderwood et al., 2001). One major complication of patients with severe chronic neutropenia receiving G-CSF therapy is myelodysplasia/acute myeloid leukemia (AML) (Donadieu et al., 2005). Although no cases of AML have been found in GSD-Ib patients receiving short term G-CSF therapy (Calderwood et al., 2001), three separate studies have reported the development of AML in GSD-Ib patients (Pinsk et al., 2002; Schroeder et al., 2008; Simmons et al., 1984). This suggests that GSD-Ib patients receiving long-term G-CSF therapy should receive regular bone marrow examinations.
5.4.1. Animal model of GSD-Ib
The G6PT-deficient GSD-Ib mice, generated by gene targeting (Chen et al., 2003), manifest all the metabolic and myeloid defects of human GSD Ib, namely hypoglycemia, growth retardation, hepatomegaly, nephromegaly, hyperlipidemia, hyperuricemia, mild lactic acidemia, neutropenia, neutrophil dysfunction, and elevated serum G-CSF levels. Studies showed that wild type mice harboring bone marrow transplants from G6PT-deficient mice manifest neutropenia and neutrophil dysfunction (Kim et al., 2006). These findings suggest that abnormal G6PT expression in bone marrow and resident neutrophils leads to abnormal myeloid function and that bone marrow transplantation may be considered for treating neutrophil complications in GSD-Ib. Indeed, bone marrow transplantation into a GSD-Ib patient manifesting severe enterocolitis and recurrent infections did improve neutrophil function and reduced enterocolitis severity although a mild neutropenia persisted (Pierre et al., 2008). While an isolated case, this promising outcome may support further exploration of this approach in addressing severe myeloid complications in GSD-Ib.
5.4.2. Gene therapy for GSD-Ib
Somatic gene therapy is a promising therapeutic approach for the hydrophobic, transmembrane protein G6PT. Gene therapy for GSD-Ib has been evaluated in GSD-Ib mice using both adenovirus (Ad) (Yiu et al., 2007) and adeno-associated virus (AAV) (Yiu et al., 2009) vectors carrying human G6PT. The Ad vector, injected systemically via the temporal vein, transduces the liver and transiently corrects metabolic and myeloid abnormalities in GSD-Ib mice. While the longer term efficacy of this therapy is unknown, there are significant vector safety concerns that require extensive evaluation. An AAV8 vector expressing G6PT directed by the chicken β-actin promoter/cytomegalovirus enhancer (AAV8-G6PT) has also been evaluated following systemic administration in neonatal GSD-Ib mice. The AAV8-G6PT successfully delivered the G6PT transgene to the liver and bone marrow. In the treated mice, hepatic G6PT activity dropped rapidly from 50% of wild-type levels at age 2 weeks post-infusion to 3% by age 6 to 72 weeks. However, long term metabolic correction was achieved and the infused mice maintained normalized serum glucose and metabolite profiles over the entire 72 weeks of the study. Moreover a transient 6-week myeloid correction was also achieved. Despite these successes, metabolic normalization is not a sufficient endpoint for therapy. All five treated GSD-Ib mice that lived over age 50 weeks exhibited excessive accumulation of glycogen in their hepatocytes, marked hepatic steatosis, neutrophil infiltration, fibrosis, and hepatocellular injury reminiscent of non-alcoholic steatohepatitis (Yiu et al., 2009). Two mice developed multiple hepatocellular adenomas with one undergoing malignant transformation. Clearly, while a promising start, challenges remain. A next step forward would be to use the native SLC37A4 promoter/enhancer to improve gene expression. There is also evidence that infusing the virus at a later age in the mice, when liver cell replication has slowed, might stabilize gene expression at a higher level (Yiu et al., 2010).
Correction of neutropenia and neutrophil dysfunction are also critical aspects for GSD-Ib gene therapy. Neutrophils are terminally differentiated, short-lived, leukocytes that are replenished from the multi-potent hematopoietic stem cells (HSCs) in the bone marrow. Modification of neutrophil responses will depend on efficient gene transfer into the bone marrow HSC. Recent studies have demonstrated that a sustained transgene expression in HSCs can be obtained using self-complementary AAV vectors in conjunction with an HSC-specific promoter/enhancer (Han et al., 2008; Srivastava 2008). These modifications are worthy of further consideration for GSD-Ib gene therapy. The question remains, however, whether a single vector will be able to balance the needs of hepatic and renal correction alongside myeloid correction or whether a multi-pronged approach, one for metabolism, one for myeloid dysfunction, may be necessary.
6. Future perspectives
The biological role of G6PT is now well established and gene therapy holds promise for metabolic and myeloid correction in GSD-Ib. However, very little is known of the other SLC37 proteins beyond the P:P and G6P:P exchange activities of SLC37A1 and SLC37A2. Despite expressing similar or higher levels than G6PT in key tissues, they do not appear to have roles in regulating neutrophil energy metabolism or blood glucose homeostasis. Moreover, there are few clues to their biological roles as no diseased phenotype has been associated with a deficiency in SLC37A1, A2, or A3. Mouse models of the other SLC37 family members do exist but their phenotypes remain to be characterized. Study of these models may give valuable insights into the biological and functional roles of the other SLC37 family members. Differential expression in tissues may also hold clues to their roles. SLC37A1 is expressed at significant high levels in the intestine, pancreas, and neutrophils and SLC37A2 in macrophages. Given the gene similarity, future study could also examine if SLC37A1 is a glycerol-3-phosphate transporter involved in phospholipid biosynthesis. Of this family, SLC37A3 remains the least studied despite being the predominant gene in the SLC37 family expressed in pancreas and neutrophils. There is still much to learn about the other ER transmembrane proteins of the SLC37 family.
Acknowledgements
This research was supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health
Biography
Janice Y. Chou Janice Y. Chou, Ph.D. is a Section Chief on Cellular Differentiation at Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health. Her research interests focus on the molecular genetics and pathogenesis of heritable human metabolic disorders, with specific emphasis on type I glycogen storage diseases and glucose/energy homeostasis.
Hyun Sik Jun Hyun Sik Jun, Ph.D. is a Staff Scientist in J.Y. Chou’ laboratory. He completed his doctoral program at University of Guelph, Canada. His current research projects in J.Y. Chou’ laboratory focus on elucidation of the molecular mechanisms that underlie myeloid dysfunction in glycogen storage disease type Ib and G6PC3 deficiency.
Brian C. Mansfield Brian C. Mansfield, Ph.D. is Deputy Chief Research Officer of the Foundation Fighting Blindness. He is also a long-standing adjunct scientist in the Section on Cellular Differentiation at Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health. His current research interests are in the genetics of, and metabolic defects in, the inherited orphan retinal degenerative diseases and type I glycogen storage diseases.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no conflict of interest.
References
- Adachi M, Shinkai M, Ohhama Y, Tachibana K, Kuratsuji T, Saji H, Maruya E. Improved neutrophil function in a glycogen storage disease type 1b patient after liver transplantation. Eur. J. Pediatr. 2004;163(4-5):202–206. doi: 10.1007/s00431-004-1405-1. [DOI] [PubMed] [Google Scholar]
- Angaroni CJ, Labrune P, Petit F, Sastre D, Capra AE, Dodelson de Kremer R, Argaraña CE. Glycogen storage disease type Ib without neutropenia generated by a novel splice-site mutation in the glucose-6-phosphate translocase gene. Mol. Genet. Metab. 2006;88(1):96–99. doi: 10.1016/j.ymgme.2005.12.011. [DOI] [PubMed] [Google Scholar]
- Annabi B, Hiraiwa H, Mansfield BC, Lei K-J, Ubagai T, Polymeropoulos MH, Moses SW, Parvari R, Hershkovitz E, Mandel H, Frydman M, Chou JY. The gene for glycogen storage disease type 1b maps to chromosome 11q23. Am. J. Hum. Genet. 1998;62(2):400–405. doi: 10.1086/301727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartoloni L, Antonarakis SE. The human sugar-phosphate/phosphate exchanger family SLC37. Pflugers Arch. 2004;447(5):780–783. doi: 10.1007/s00424-003-1105-0. [DOI] [PubMed] [Google Scholar]
- Bartoloni L, Wattenhofer M, Kudoh J, Berry A, Shibuya K, Kawasaki K, Wang J, Asakawa S, Talior I, Bonne-Tamir B, Rossier C, Michaud J, McCabe ER, Minoshima S, Shimizu N, Scott HS, Antonarakis SE. Cloning and characterization of a putative human glycerol 3-phosphate permease gene (SLC37A1 or G3PP) on 21q22.3: mutation analysis in two candidate phenotypes, DFNB10 and a glycerol kinase deficiency. Genomics. 2000;70(2):190–200. doi: 10.1006/geno.2000.6395. [DOI] [PubMed] [Google Scholar]
- Bashan N, Potashnik R, Hagay Y, Moses SW. Impaired glucose transport in polymorphonuclear leukocytes in glycogen storage disease Ib. J. Inherit. Metab. Dis. 1987;10(3):234–241. doi: 10.1007/BF01800068. [DOI] [PubMed] [Google Scholar]
- Bhattacharya N, Heaton N, Rela M, Walter JH, Lee PJ. The benefits of liver transplantation in glycogenosis type Ib. J. Inherit. Metab. Dis. 2004;27(4):539–540. doi: 10.1023/b:boli.0000037400.49488.20. [DOI] [PubMed] [Google Scholar]
- Boztug K, Appaswamy G, Ashikov A, Schäffer AA, Salzer U, Diestelhorst J, Germeshausen M, Brandes G, Lee-Gossler J, Noyan F, Gatzke AK, Minkov M, Greil J, Kratz C, Petropoulou T, Pellier I, Bellanné-Chantelot C, Rezaei N, Mönkemöller K, Irani-Hakimeh N, Bakker H, Gerardy-Schahn R, Zeidler C, Grimbacher B, Welte K, Klein C. A syndrome with congenital neutropenia and mutations in G6PC3. N. Engl. J. Med. 2009;360(1):32–43. doi: 10.1056/NEJMoa0805051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderwood S, Kilpatrick L, Douglas SD, Freedman M, Smith-Whitley K, Rolland M, Kurtzberg J. Recombinant human granulocyte colony-stimulating factor therapy for patients with neutropenia and/or neutrophil dysfunction secondary to glycogen storage disease type 1b. Blood. 2001;97(2):376–382. doi: 10.1182/blood.v97.2.376. [DOI] [PubMed] [Google Scholar]
- Chen L-Y, Shieh J-J, Lin B, Pan C-J, Gao J-L, Murphy PM, Roe TF, Moses S, Ward JM, Westphal H, Lee EJ, Mansfield BC, Chou JY. Impaired glucose homeostasis, neutrophil trafficking and function in mice lacking the glucose-6-phosphate transporter. Hum. Mol. Genet. 2003;12(19):2547–2558. doi: 10.1093/hmg/ddg263. [DOI] [PubMed] [Google Scholar]
- Chen L-Y, Lin B, Pan C-J, Hiraiwa H, Chou JY. Structural requirements for the stability and microsomal transport activity of the human glucose-6-phosphate transporter. J. Biol. Chem. 2000;275(44):34280–34286. doi: 10.1074/jbc.M006439200. [DOI] [PubMed] [Google Scholar]
- Chen L-Y, Pan C-J, Shieh J-J, Chou JY. Structure-function analysis of the glucose-6-phosphate transporter deficient in glycogen storage disease type Ib. Hum. Mol. Genet. 2002;11(25):3199–3207. doi: 10.1093/hmg/11.25.3199. [DOI] [PubMed] [Google Scholar]
- Chen SY, Pan CJ, Nandigama K, Mansfield BC, Ambudkar SV, Chou JY. The Glucose-6-phosphate transporter is a phosphate-linked antiporter deficient in glycogen storage disease type Ib and Ic. FASEB J. 2008;22(7):2206–2213. doi: 10.1096/fj.07-104851. [DOI] [PubMed] [Google Scholar]
- Chen YT. Glycogen storage diseases. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th edn McGraw-Hill; New York: 2001. pp. 1521–1551. [Google Scholar]
- Chen YT, Cornblath M, Sidbury JB. Cornstarch therapy in type I glycogen storage disease. N. Engl. J. Med. 1984;310(3):171–175. doi: 10.1056/NEJM198401193100306. [DOI] [PubMed] [Google Scholar]
- Cheung YY, Kim SY, Yiu WH, Pan CJ, Jun HS, Ruef RA, Lee EJ, Westphal H, Mansfield BC, Chou JY. Impaired neutrophil activity and increased susceptibility to bacterial infection in mice lacking glucose-6-phosphatase-beta. J. Clin. Invest. 2007;117(3):784–793. doi: 10.1172/JCI30443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou JY, Jun HS, Mansfield BC. Neutropenia in type Ib glycogen storage disease. Curr. Opin. Hematol. 2010a;17(1):36–42. doi: 10.1097/MOH.0b013e328331df85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou JY, Jun HS, Mansfield BC. Glycogen storage disease type I and G6Pase-β deficiency: etiology and therapy. Nat. Rev. Endocrinol. 2010b;6(12):676–688. doi: 10.1038/nrendo.2010.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou JY, Matern D, Mansfield BC, Chen Y-T. Type I glycogen storage diseases: disorders of the glucose-6-phosphatase complex. Curr. Mol. Med. 2002;2(12):121–143. doi: 10.2174/1566524024605798. [DOI] [PubMed] [Google Scholar]
- Dieckgraefe BK, Korzenik JR, Husain A, Dieruf L. Association of glycogen storage disease 1b and Crohn disease: results of a North American survey. Eur. J. Pediatr. 2002;161(Suppl. 1):S88–S92. doi: 10.1007/s00431-002-1011-z. [DOI] [PubMed] [Google Scholar]
- Donadieu J, Barkaoui M, Fenneteau O, Bertrand Y, Maier-Redelsperger M, Micheau M, Stephan JL, Phillipe N, Bordigoni P, Babin-Boilletot A, Bensaid P, Manel AM, Vilmer E, Thuret I, Blanche S, Gluckman E, Fischer A, Mechinaud F, Joly B, Lamy T, Hermine O, Cassinat B, Bellanné-Chantelot C, Chomienne C, French Severe Chronic Neutropenia Study Group Analysis of risk factors for myelodysplasias, leukemias and death from infection among patients with congenital neutropenia. Experience of the French Severe Chronic Neutropenia Study Group. Haematologica. 2005;90(1):45–53. [PubMed] [Google Scholar]
- Galli L, Orrico A, Marcolongo P, Fulceri R, Burchell A, Melis D, Parini R, Gatti R, Lam C, Benedetti A, Sorrentino V. Mutations in the glucose-6-phosphate transporter (G6PT) gene in patients with glycogen storage diseases type 1b and 1c. FEBS Lett. 1999;459(2):255–258. doi: 10.1016/s0014-5793(99)01248-x. [DOI] [PubMed] [Google Scholar]
- Gerin I, Veiga-da-Cunha M, Achouri Y, Collet JF, Van Schaftingen E. Sequence of a putative glucose-6-phosphate translocase, mutated in glycogen storage disease type 1b. FEBS Lett. 1997;419(2-3):235–238. doi: 10.1016/s0014-5793(97)01463-4. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Shieh J-J, Pan C-J, Chou JY. Histidine-167 is the phosphate acceptor in glucose-6-phosphatase- forming a phosphohistidine-enzyme intermediate during catalysis. J Biol. Chem. 2004;279(13):12479–12483. doi: 10.1074/jbc.M313271200. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Shieh J-J, Pan C-J, Sun M-S, Chou JY. The catalytic center of glucose-6-phosphatase: His176 is the nucleophile forming the phosphohistidine-enzyme intermediate during catalysis. J. Biol. Chem. 2002;277(36):32837–32842. doi: 10.1074/jbc.M201853200. [DOI] [PubMed] [Google Scholar]
- Greene HL, Slonim AE, O’Neill JA, Jr., Burr IM. Continuous nocturnal intragastric feeding for management of type 1 glycogen-storage disease. N. Engl. J. Med. 1976;294(8):423–425. doi: 10.1056/NEJM197602192940805. [DOI] [PubMed] [Google Scholar]
- Han Z, Zhong L, Maina N, Hu Z, Li X, Chouthai NS, Bischof D, Weigel-Van Aken KA, Slayton WB, Yoder MC, Srivastava A. Stable integration of recombinant adeno-associated virus vector genomes after transduction of murine hematopoietic stem cells. Hum. Gene. Ther. 2008;19(3):267–78. doi: 10.1089/hum.2007.161. [DOI] [PubMed] [Google Scholar]
- He L, Vasiliou K, Nebert DW. Analysis and update of the human solute carrier (SLC) gene superfamily. Hum. Genomics. 2009;3(2):195–206. doi: 10.1186/1479-7364-3-2-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmerle H, Burger HJ, Below P, Schubert G, Rippel R, Schindler PW, Paulus E, Herling AW. Chlorogenic acid and synthetic chlorogenic acid derivatives: novel inhibitors of hepatic glucose-6-phosphate translocase. J. Med. Chem. 1997;40(2):137–145. doi: 10.1021/jm9607360. [DOI] [PubMed] [Google Scholar]
- Hiraiwa H, Pan C-J, Lin B, Moses SW, Chou JY. Inactivation of the glucose-6-phosphate transporter causes glycogen storage disease type 1b. J. Biol. Chem. 1999;274(9):5532–5536. doi: 10.1074/jbc.274.9.5532. [DOI] [PubMed] [Google Scholar]
- Iacopetta D, Lappano R, Cappello AR, Madeo M, De Francesco EM, Santoro A, Curcio R, Capobianco L, Pezzi V, Maggiolini M, Dolce V. SLC37A1 gene expression is up-regulated by epidermal growth factor in breast cancer cells. Breast Cancer Res. Treat. 2010;122(3):755–764. doi: 10.1007/s10549-009-0620-x. [DOI] [PubMed] [Google Scholar]
- Janecke AR, Linder M, Erdel M, Mayatepek E, Moslinger D, Podskarbi T, Fresser F, Stockler-Ipsirogly S, Hoffmann GF, Utermann G. Mutation analysis in glycogen storage disease type 1 non-a. Hum. Genet. 2000;107(3):285–289. doi: 10.1007/s004390000371. [DOI] [PubMed] [Google Scholar]
- Jun HS, Lee YM, McDermott DH, DeRavin SS, Murphy PM, Mansfield BC, Chou JY. Lack of glucose recycling between endoplasmic reticulum and cytoplasm underlies cellular dysfunction in glucose-6-phosphatase-beta-deficient neutrophils in a congenital neutropenia syndrome. Blood. 2010;116(15):2783–2792. doi: 10.1182/blood-2009-12-258491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpatrick L, Garty BZ, Lundquist KF, Hunter K, Stanley CA, Baker L, Douglas SD, Korchak HM. Impaired metabolic function and signaling defects in phagocytic cells in glycogen storage disease type 1b. J. Clin. Invest. 1990;86(1):196–202. doi: 10.1172/JCI114684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Tillison K, Zhou S, Wu Y, Smas CM. The major facilitator superfamily member Slc37a2 is a novel macrophage- specific gene selectively expressed in obese white adipose tissue. Am. J. Physiol. Endocrinol. Metab. 2007;293(1):E110–E120. doi: 10.1152/ajpendo.00404.2006. [DOI] [PubMed] [Google Scholar]
- Kim SY, Jun HS, Mead PA, Mansfield BC, Chou JY. Neutrophil stress and apoptosis underlie myeloid dysfunction in glycogen storage disease type Ib. Blood. 2008;111(12):5704–5711. doi: 10.1182/blood-2007-12-129114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Nguyen ATD, Gao J-L, Murphy PM, Mansfield BC, Chou JY. Bone-marrow derived cells require a functional glucose-6-phosphate transporter for normal myeloid functions. J. Biol. Chem. 2006;281(39):28794–28801. doi: 10.1074/jbc.M604964200. [DOI] [PubMed] [Google Scholar]
- Kuijpers TW, Maianski NA, Tool AT, Smit GP, Rake JP, Roos D, Visser G. Apoptotic neutrophils in the circulation of patients with glycogen storage disease type 1b (GSD1b) Blood. 2003;101(12):5021–5024. doi: 10.1182/blood-2002-10-3128. [DOI] [PubMed] [Google Scholar]
- Kure S, Hou DC, Suzuki Y, Yamagishi A, Hiratsuka M, Fukuda T, Sugie H, Kondo N, Matsubara Y, Narisawa K. Glycogen storage disease type Ib without neutropenia. J. Pediatr. 2000;137(2):253–256. doi: 10.1067/mpd.2000.107472. [DOI] [PubMed] [Google Scholar]
- Lee KW, Lee JH, Shin SW, Kim SJ, Joh JW, Lee DH, Kim JW, Park HY, Lee SY, Lee HH, Park JW, Kim SY, Yoon HH, Jung DH, Choe YH, Lee SK. Hepatocyte transplantation for glycogen storage disease type Ib. Cell Transplant. 2007;16(6):629–637. doi: 10.3727/000000007783465019. [DOI] [PubMed] [Google Scholar]
- Lei K-J, Chen H, Pan C-J, Ward JM, Mosinger B, Lee EJ, Westphal H, Chou JY. Glucose-6-phosphatase dependent substrate transport in the glycogen storage disease type 1a mouse. Nat. Genet. 1996;13(2):203–209. doi: 10.1038/ng0696-203. [DOI] [PubMed] [Google Scholar]
- Leuzzi R, Bánhegyi G, Kardon T, Marcolongo P, Capecchi PL, Burger HJ, Benedetti A, Fulceri R. Inhibition of microsomal glucose-6-phosphate transport in human neutrophils results in apoptosis: a potential explanation for neutrophil dysfunction in glycogen storage disease type 1b. Blood. 2003;101(6):2381–2387. doi: 10.1182/blood-2002-08-2576. [DOI] [PubMed] [Google Scholar]
- Lin B, Annabi B, Hiraiwa H, Pan C-J, Chou JY. Cloning and characterization of cDNAs encoding a candidate glycogen storage disease type 1b protein in rodents. J. Biol. Chem. 1998;273(48):31656–31670. doi: 10.1074/jbc.273.48.31656. [DOI] [PubMed] [Google Scholar]
- Lin B, Pan C-J, Chou JY. Human variant glucose-6-phosphate transporter is active in microsomal transport. Hum. Genet. 2000;107(5):526–52. doi: 10.1007/s004390000404. [DOI] [PubMed] [Google Scholar]
- Martens DH, Kuijpers TW, Maianski NA, Rake JP, Smit GP, Visser G. A patient with common glycogen storage disease type Ib mutations without neutropenia or neutrophil dysfunction. J. Inherit. Metab. Dis. 2006;29(1):224–225. doi: 10.1007/s10545-006-0146-x. [DOI] [PubMed] [Google Scholar]
- Martin AP, Bartels M, Schreiber S, Buehrdel P, Hauss J, Fangmann J. Successful staged kidney and liver transplantation for glycogen storage disease type Ib: A case report. Transplant. Proc. 2006;38(10):3615–3619. doi: 10.1016/j.transproceed.2006.10.160. [DOI] [PubMed] [Google Scholar]
- Martinez-Olmos MA, López-Sanromán Sanromán A, Martín-Vaquero P, Molina-Pérez E, Bárcena R, Vicente E, Candela A, Pallardo-Sánchez LF. Liver transplantation for type Ib glycogenosis with reversal of cyclic neutropenia. Clin. Nutr. 2001;20(4):375–377. doi: 10.1054/clnu.2001.0432. [DOI] [PubMed] [Google Scholar]
- Matern D, Starzl TE, Arnaout W, Barnard J, Bynon JS, Dhawan A, Emond J, Haagsma EB, Hug G, Lachaux A, Smit GP, Chen YT. Liver transplantation for glycogen storage disease types I, III, and IV. Eur. J. Pediatr. 1999;158(Suppl. 2):S43–S48. doi: 10.1007/pl00014320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melis D, Fulceri R, Parenti G, Marcolongo P, Gatti R, Parini R, Riva E, Della Casa R, Zammarchi E, Andria G, Benedetti A. Genotype/phenotype correlation in glycogen storage disease type 1b: a multicentre study and review of the literature. Eur. J. Pediatr. 2005;164(8):501–508. doi: 10.1007/s00431-005-1657-4. [DOI] [PubMed] [Google Scholar]
- Mortellaro C, Garagiola U, Carbone V, Cerutti F, Marc i V, Bonda PL. Unusual oral manifestations and evolution in glycogen storage disease type Ib. J. Craniofac. Surg. 2005;16(1):45–52. doi: 10.1097/00001665-200501000-00010. [DOI] [PubMed] [Google Scholar]
- Pan C-J, Lin B, Chou JY. Transmembrane topology of human Glucose-6-phosphate transporter. J. Biol. Chem. 1999;274(20):13865–13869. doi: 10.1074/jbc.274.20.13865. [DOI] [PubMed] [Google Scholar]
- Pan CJ, Chen SY, Jun HS, Lin SR, Mansfield BC, Chou JY. SLC37A1 and SLC37A2 are phosphate-linked, glucose-6-phosphate antiporters. PloS ONE. 2011;6(9):e23157. doi: 10.1371/journal.pone.0023157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan CJ, Chen SY, Lee S, Chou JY. Structure-function study of glucose-6-phosphate transporter, an eukaryotic antiporter deficient in glycogen storage disease type Ib. Mol. Genet. Metab. 2009;96(1):32–37. doi: 10.1016/j.ymgme.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao SS, Paulsen IT, Saier MH., Jr. Major facilitator superfamily. Microbiol. Mol. Biol. Rev. 1998;62(1):1–34. doi: 10.1128/mmbr.62.1.1-34.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierre G, Chakupurakal G, McKiernan P, Hendriksz C, Lawson S, Chakrapani A. Bone marrow transplantation in glycogen storage disease type 1b. J. Pediatr. 2008;152(2):286–288. doi: 10.1016/j.jpeds.2007.09.031. [DOI] [PubMed] [Google Scholar]
- Pinsk M, Burzynski J, Yhap M, Fraser RB, Cummings B, Ste-Marie M. Acute myelogenous leukemia and glycogen storage disease 1b. J. Pediatr. Hematol. Oncol. 2002;24(9):756–758. doi: 10.1097/00043426-200212000-00015. [DOI] [PubMed] [Google Scholar]
- Schroeder T, Hildebrandt B, Mayatepek E, Germing U, Haas R. A patient with glycogen storage disease type Ib presenting with acute myeloid leukemia (AML) bearing monosomy 7 and translocation t(3;8)(q26;q24) after 14 years of treatment with granulocyte colony-stimulating factor (G-CSF): A case report. J. Med. Case Reports. 2008;2:319. doi: 10.1186/1752-1947-2-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh J-J, Pan C-J, Mansfield BC, Chou JY. Glucose-6-phosphate hydrolase, widely expressed outside the liver, can explain age-dependent resolution of hypoglycemia in glycogen storage disease type Ia. J. Biol. Chem. 2003;278(47):47098–47103. doi: 10.1074/jbc.M309472200. [DOI] [PubMed] [Google Scholar]
- Simmons PS, Smithson WA, Gronert GA, Haymond MW. Acute myelogenous leukemia and malignant hyperthermia in a patient with type 1b glycogen storage disease. J. Pediatr. 1984;105(3):428–431. doi: 10.1016/s0022-3476(84)80020-7. [DOI] [PubMed] [Google Scholar]
- Srivastava A. Adeno-associated virus-mediated gene transfer. J. Cell. Biochem. 2008;105(1):17–24. doi: 10.1002/jcb.21819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Miyata M, Zheng P, Imazato T, Horwitz A, Smith JD. Identification of cAMP analogue inducible genes in RAW264 macrophages. Biochim. Biophys. Acta. 2000;1492(2-3):385–394. doi: 10.1016/s0167-4781(00)00133-0. [DOI] [PubMed] [Google Scholar]
- Veiga-da-Cunha M, Gerin I, Chen Y-T, de Barsy T, de Lonlay P, Dionisi-Vici C, Fenske CD, Lee PJ, Leonard JV, Maire I, McConkie-Rosell A, Schweitzer S, Vikkula M, Van Schaftingen E. A gene on chromosome 11q23 coding for a putative glucose- 6-phosphate translocase is mutated in glycogen-storage disease types Ib and Ic. Am. J. Hum. Genet. 1998;63(4):976–983. doi: 10.1086/302068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga-da-Cunha M, Gerin I, Chen Y-T, Lee PJ, Leonard JV, Maire I, Wendel U, Vikkula M, Van Schaftingen E. The putative glucose 6-phosphate translocase gene is mutated in essentially all cases of glycogen storage disease type I non-a. Eur. J. Hum. Genet. 1999;7(16):717–723. doi: 10.1038/sj.ejhg.5200366. [DOI] [PubMed] [Google Scholar]
- Verhoeven AJ, Visser G, van Zwieten R, Gruszczynska B, Tien Poll-The DW, Smit GP. A convenient diagnostic function test of peripheral blood neutrophils in glycogen storage disease type I. Pediatr. Res. 1999;45(6):881–885. doi: 10.1203/00006450-199906000-00018. [DOI] [PubMed] [Google Scholar]
- Visser G, Rake JP, Fernandes J, Labrune P, Leonard JV, Moses S, Ullrich K, Smit GP. Neutropenia, neutrophil dysfunction, and inflammatory bowel disease in glycogen storage disease type Ib: results of the European Study on Glycogen Storage Disease type I. J. Pediatr. 2000;137(2):187–191. doi: 10.1067/mpd.2000.105232. [DOI] [PubMed] [Google Scholar]
- Visser G, Rake JP, Labrune P, Leonard JV, Moses S, Ullrich K, Wendel U, Groenier KH, Smit GP. Granulocyte colony-stimulating factor in glycogen storage disease type 1b. Results of the European Study on Glycogen Storage Disease Type 1. Eur. J. Pediatr. 2002;161(Suppl. 1):S83–S87. doi: 10.1007/s00431-002-1010-0. [DOI] [PubMed] [Google Scholar]
- Yiu WH, Pan CC--J, Allamarvdasht A, Kim SY, Chou JY. Glucose-6-phosphate transporter gene therapy corrects metabolic and myeloid abnormalities in glycogen storage disease type Ib mice. Gene Ther. 2007;14(3):219–226. doi: 10.1038/sj.gt.3302869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiu WH, Pan C-J, Mead PA, Starost MF, Mansfield BC, Chou JY. Normoglycemia alone is insufficient to prevent long term complications of hepatocellular adenoma in glycogen storage disease type Ib mice. J. Hepatol. 2009;51(5):909–917. doi: 10.1016/j.jhep.2008.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiu WH, Lee YM, Peng WT, Pan C-J, Mead PA, Mansfield BC, Chou JY. Complete normalization of hepatic G6PC deficiency in murine glycogen storage disease type Ia using gene therapy. Mol. Ther. 2010;18(6):1076–1084. doi: 10.1038/mt.2010.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingone A, Hiraiwa H, Pan C-J, Lin B, Chen H, Ward JM, Chou JY. Correction of glycogen storage disease type 1a in a mouse model by gene therapy. J. Biol. Chem. 2000;275(2):828–832. doi: 10.1074/jbc.275.2.828. [DOI] [PubMed] [Google Scholar]