

Abstract
Objective
To describe relationships of risk alleles in complement factor H (CFH, rs1061170) and Age-Related Maculopathy susceptibility 2 (ARMS2, rs10490924) to the incidence and progression of age-related macular degeneration (AMD) over a 20-year period.
Methods
There were 4282 persons aged 43–86 years at the baseline examination in 1988–1990 enrolled in a population-based cohort study who participated in at least 1 pair of examinations spaced 5 years apart over a 20-year period and had gradable fundus photographs for AMD and genotype information on CFH and ARMS2. Low, intermediate, and high genetic risk for AMD was defined by the presence of 0–1, 2, or 3–4 risk alleles for CFH and ARMS2, respectively. Multi-state models (MSMs) were used to estimate progression of AMD over the entire age range.
Results
There were 2820 (66%), 1129 (26%), and 333 persons (8%) with low, intermediate, and high genetic risk for AMD, respectively. The 5-year incidences of early and late AMD were 9.1% and 1.6%, respectively, and increased with age but did not differ by sex. Using the MSM, of persons aged 45 years with no AMD in the low, intermediate, and high AMD genetic risk groups, 33.0%, 39.9%, and 46.5%, respectively were estimated to develop early AMD, and 1.4%, 5.2%, and 15.3%, respectively were estimated to develop late AMD by age 80 years.
Conclusions
These population-based data provide estimates of the long-term risk of the incidence and progression of AMD and its lesions by age and genetic risk alleles for CFH and ARMS2.
Keywords: age-related macular degeneration, ARMS2, CFH, epidemiology
Following the observations of the associations of specific single nucleotide polymorphisms (SNPs) in the Complement Factor H region (CFH, rs1061170) and in the Age-Related Maculopathy Susceptibility 2 region (ARMS2, rs10490924) with late age-related macular degeneration (AMD), there have been a growing number of studies examining the relationships of these and other AMD candidate genes and their interactions with environmental and host risk factors.1–21 Most of these studies have been either clinical case series or case-control studies, and have focused largely on late AMD. Few long-term studies have examined the relationships of these genetic risk factors along the continuum of the disease, from its earliest to its most advanced stages.2,22 The purpose of this report is to describe the relationships of age and CFH and ARMS2 risk alleles to the incidence of AMD over a 20-year period, building upon previous reports in the Beaver Dam Eye Study (BDES) cohort.23–26
METHODS
Population
Methods used to identify the population and descriptions of the population have appeared in previous reports.27–31 A private census of the population of Beaver Dam, Wisconsin was performed from fall 1987 to spring 1988.27 There were 5924 eligible individuals, of whom 4926 participated in the examination phase between March 1, 1988 and September 15, 1990, 3721 participated in the 5-year follow-up examination phase between March 1, 1993 and June 15, 1995, 2962 participated in the 10-year follow-up examination phase between March 16, 1998 and June 9, 2000, 2375 participated in the 15-year follow-up examination phase between March 31, 2003 and June 1, 2005, and 1913 participated in the 20-year follow-up examination phase between November 5, 2008 and November 16, 2010. Ninety-nine percent of the population was white.
Approval for this study was granted by the Institutional Review Board at the University of Wisconsin. Informed consent was obtained from each participant before every examination. The tenets of the Declaration of Helsinki were observed.
Comparisons between participants and nonparticipants at each examination have appeared elsewhere.28–31 In general, those who participated at the 20-year follow-up were more likely to be younger than nonparticipants who were alive or those who died before follow-up, and while adjusting for age, were less likely to have AMD. The mean and median times between the baseline and 20-year follow-up examinations were 20.4 years (standard deviation = 0.6) and 20.3 years, respectively.
Procedures
Similar procedures were used at baseline and follow-up examinations.23–26,32–37 A standardized interview and examination were administered at each visit. Information on demographic characteristics, were obtained from the questionnaire. Stereoscopic 30° color fundus photographs centered on the disc (Diabetic Retinopathy Study standard field 1) and macula (Diabetic Retinopathy Study standard field 2) and a nonstereoscopic color fundus photograph temporal to but including the fovea of each eye.
Details of the grading procedure have been described previously.23,36,37 In brief, a circular grid was placed on 1 photographic slide of the stereoscopic pair, which divided the macular area into 9 subfields, consisting of a central circle (a single subfield), inner ring (comprised of the 4 inner subfields), and outer ring (comprised of 4 outer subfields). Some lesions were graded in each subfield, other lesions only in Diabetic Retinopathy Study field 2 as a whole, and still others in additional fields. For the purpose of this report, measurements made only within the 9 subfields defined by the grid are presented. Circles of defined size (63, 125, 175, 250, 325, 350, and 650 μm in diameter) printed on clear acetate were used to estimate drusen size and areas involved by drusen, increased retinal pigment, and retinal pigment epithelial (RPE) depigmentation.
Two gradings were performed on photographs of each eye at each examination with graders masked to any information about the fellow eye and the participant.23–26,36,37 First, 1 of 2 senior graders performed a preliminary grading. Next, a detailed grading was performed by 1 of 3 other experienced graders. The assessment consisted of a subfield-by-subfield, lesion-by-lesion evaluation of each photograph set using the Wisconsin Age-Related Maculopathy Grading System.36,37 Next, a series of edits and reviews was performed. The presence and severity of specific lesions of AMD (eg, maximum drusen size/type/area, pigmentary abnormalities) at the fifth examination as determined by detail grading were compared to that of the preliminary grading. Standardized edit rules were used to adjudicate disagreements.23–26 Finally, the detail graders were asked to make side-by-side comparisons between 15- and 20-year follow-up photographs randomly ordered so that photography dates were masked for eyes that showed change for AMD lesions between these 2 examinations; in cases where there were no photos at the 15-year examination, the photos from the next most recent examination were used for comparison. After this masked longitudinal review of 15- and 20-year photographs was complete, the senior grader (SMM) and principal investigator (RK) performed a final unmasked review of all 5 visits for progression and regression. All eyes newly classified with late AMD were also confirmed at this time. Additional information on gradability at previous examinations can be found elsewhere.23–26
Genetic Measurements
DNA was extracted from buffy coat specimens collected at the baseline exam. The 2 most common AMD-associated SNPs, Y402H in CFH and A69S in ARMS2, were used in this study. The A69S variant was genotyped in 5188 individuals using two different platforms, Taqman (Applied Biosystems, Foster City, California, USA) and Illumina (San Diego, California, USA). Assays were performed at two separate times in 2248 and 2940 samples, respectively. Five hundred and eighty-eight samples were genotyped with both platforms, with a genotype concordance rate of 99.7%. The genotype calls from each assay were combined to create a single dataset for analysis. The Y402H variant was directly genotyped using a Taqman assay in 3015 samples in the BDES, which is described elsewhere.38,39 To increase sample size for the Y402H variant, we used data imputation techniques (MACH program version 1.032) on 2940 samples genotyped for 70 markers in the CFH region using a custom Illumina array. Using the surrounding linkage disequilibrium structure at CFH, we inferred genotypes at Y402H in both typed and untyped samples, keeping only genotypes that could be imputed with a high probability (r2 ≥ 0.9). A concordance rate of 99.8% was observed among 1476 samples for which both genotyped and imputed data was available.40
Definitions
Age was defined at the time of each participant visit and treated categorically in the following age groups: 43–54, 55–64, 65–74, 75–84, and ≥85 years.
Three genetic risk groups were defined based on distributions of late AMD by risk allele status: low genetic risk included persons with 0–1 risk alleles (no risk alleles for either CFH or ARMS2, or 1 risk allele for either CFH or ARMS2); intermediate genetic risk included persons with 2 risk alleles (2 risk alleles for either CFH or ARMS2 but none for the other or 1 risk allele for each); high genetic risk included persons with 3–4 risk alleles (2 risk alleles for either CFH or ARMS2 and at least 1 risk allele for the other, or 2 risk alleles for both).
The severity of AMD was determined using the 5-step BDES AMD Severity Scale (eFigure 1). The definitions of each level are as follows.
10 (No AMD)
Hard drusen or small soft drusen (<125 μm in diameter) only, regardless of area of involvement and no pigmentary abnormalities (defined as increased retinal pigment or RPE depigmentation present); or no definite drusen with any pigmentary abnormality.
20 (Minimally severe early AMD)
Hard drusen or small soft drusen (<125 μm in diameter), regardless of area of involvement, with any pigmentary abnormality; or soft drusen (≥125 μm in diameter) with drusen area <331,820 μm2 (equivalent to O2, a circle with a diameter of 650 μm) and no pigmentary abnormalities.
30 (Moderately severe early AMD)
Soft drusen (≥125 μm in diameter) with drusen area <331,820 μm2 (equivalent to O2) and with any pigmentary abnormality; or soft drusen (≥125 μm in diameter) with drusen area ≥331,820 μm2 (equivalent to O2) with or without increased retinal pigment but no RPE depigmentation.
40 (Severe early AMD)
Soft drusen (≥125 μm in diameter) with drusen area ≥331,820 μm2 (equivalent to O2) and RPE depigmentation present, with or without increased retinal pigment.
50 (Late AMD)
Pure geographic atrophy (GA) in the absence of exudative macular degeneration; or exudative macular degeneration with or without GA present.
As noted above, there were additional severity classifications based on scales for retinal drusen size, drusen type, and pigmentary abnormalities. Details of these scales appear in eFigures 3–5.
Persons at risk for developing early AMD were those without any lesion defining early or late AMD at baseline or the beginning of a 5-year examination interval. Incidence of early AMD in the worse eye was defined by developing Level 20, 30, or 40 in at least 1 eye when both eyes were Level 10 at the previous examination. When 1 eye was ungradable, it was assumed to have the same AMD level as the fellow eye. Incidence was determined for presence of signs of early AMD, eg, large drusen (size ≥125 μm in diameter), drusen type (soft indistinct/reticular), and pigmentary abnormalities. The incidence of a specific lesion was defined by its presence at follow-up when it was not present at the previous examination in any of the subfields. Similarly, persons at risk for developing late AMD (Level 50) were those without late AMD at the beginning of a 5-year examination interval who developed late AMD in 1 or both eyes at follow-up.
The progression or regression of AMD over the 20-year period was analyzed in the worse eye and evaluated using a multi-state model (MSM). Progression of AMD was defined for an individual as either eye transitioning to a more severe AMD level, and regression of AMD was defined as either eye transitioning to a less severe AMD level. In the model, individuals were not allowed to regress from Level 50 to less severe levels. Progression and regression along the pigment, drusen type, and drusen size scales were defined similarly (eFigures 3–5) using data from the right eye only. For most analyses, age and other characteristics were defined at the beginning of an examination interval.
Statistical Methods
Incidence analyses were conducted using chi-square tables with SAS version 9.2 (SAS Institute, Cary, North Carolina, USA). Incidence of AMD was calculated for each 5-year period and accumulated over the 20 years of the study. The values of the incident outcomes were updated for each consecutive 5-year period. Once a person developed an incident outcome, they no longer contributed to later 5-year periods. Relationships were further stratified by age group and genetic risk. Incidence was evaluated in the worse eye in all analyses.
Next, MSM analyses were conducted in R41 using the msm package.42 Covariate effects on transition intensities were summarized as hazard ratios. Using matrix exponentiation, we obtained annual transition matrices for each initial state, age, sex, and genetic risk group as described above. From these transition matrices, we calculated estimated transition probabilities to each drusen, pigment or AMD state (and death) after 5 years and the cumulative incidence of each AMD state by genetic risk group. Cumulative incidence calculations were based on model estimates, not individual data. Estimated cumulative incidence calculations used annual assessments of AMD status; subjects were assigned to the most severe AMD state observed at or before their current age. Confidence intervals (CIs) for these non-linear functions of the transition intensity parameters were obtained from a parametric bootstrap.43 Transitions along the drusen size and type and pigment scales were analyzed in right eyes only, and transitions along the AMD severity scale were analyzed in the worse eye.
Population attributable risk was defined as the portion of the incidence of a disease in the population that is due to exposure and was calculated by subtracting the incidence in the unexposed group (no risk alleles present) from the incidence in the total population. Change in area under the receiver operating characteristic curve (AUC) was used to measure improvement in prediction when traditional AMD risk factors and CFH and ARMS2 were added to the model based on AMD severity. AUC was calculated for each set of predictors using the ROC statement in SAS Proc Logistic and was plotted using the ODS Graphics statement.
RESULTS
The cohort
There were 4362 individuals who participated in at least 1 BDES examination and had genotype data for both CFH and ARMS2, of whom 4282 had at least 1 eye gradable for AMD lesions during at least 1 pair of BDES exams. Person-specific analyses were based on the worse eye at each interval. Reliable AMD data for at least 1 eye were available for 13721 person-visits, 4232 at baseline, 3217 at 5-year follow-up, 2565 at 10-year follow-up, 2068 at 15-year follow-up and 1639 at the 20-year follow-up. eFigure 2 shows the number of individuals who were included in analyses at each pair of exams. There were 4270, 4262, and 4266 unique individuals in which the right eye was gradable for transitions along scales of retinal pigment (eFigure 3), drusen size (eFigure 4), and drusen type (eFigure 5), respectively and 4282 unique individuals in which the worse eye was gradable for transitions along the 5-step AMD scale (eFigure 1).
Incidence and rate of progression relationships
The overall 5-year incidence of early AMD over the 20-year period was 9.1% and for late AMD it was 1.6%. There were 2820 (66%), 1129 (26%), and 333 persons (8%) with low, intermediate, and high genetic risk for AMD, respectively. The genotype distribution for CFH was 39.4% TT, 46.8% TC, and 13.8% CC and for ARMS2 it was 60.3% GG, 35.0% GT, and 4.7% TT. The incidence of early and late AMD increased with increasing number of risk alleles for CFH and ARMS2 (Table 1).
Table 1.
Five-Year Incidence of Early and Late Age-related Macular Degeneration by Complement Factor H and Age-Related Maculopathy Susceptibility 2 Genotype in the Beaver Dam Eye Study, 1988–2010.
| ARMS2 Genotype | Early AMD | Late AMD | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| CFH Genotype | CFH Genotype | |||||
|
| ||||||
| T/T | C/T | C/C | T/T | C/T | C/C | |
|
|
||||||
| G/G | 1784 (7.3%) | 1873 (9.4%) | 558 (11.1%) | 2184 (0.5%) | 2432 (1.4%) | 718 (1.1%) |
| G/T | 886 (8.5%) | 1168 (8.9%) | 341 (11.4%) | 1145 (0.7%) | 1520 (2.4%) | 441 (3.4%) |
| T/T | 122 (11.5%) | 80 (18.8%) | 28 (32.1%) | 160 (3.6%) | 48 (12.1%) | 55 (10.9%) |
AMD, age-related macular degeneration; ARMS2, Age-Related Maculopathy Susceptibility 2; CFH, Complement Factor H.
Reported as N at risk (% events).
The 5-year incidences of both early and late AMD and specific AMD lesions increased with age over the 20-year period (Table 2). The 5-year incidence of early AMD over the 20-year period was 8.4%, 9.7%, and 14.0% and for late AMD it was 0.9%, 2.1%, and 5.9% for low, intermediate, and high genetic risk groups, respectively. While adjusting for age, there were associations of increasing genetic risk with all incident AMD outcomes.
Table 2.
Incidence of Age-related Macular Degeneration Outcomes by Age and Genetic Risk in the Beaver Dam Eye Study, 1988–2010.
| Outcome* and Genetic Risk† | Overall | Age (years)
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 43–54 | 55–64 | 65–74 | 75–84 | ≥ 85 | ||||||||
|
| ||||||||||||
| N at risk | N (%)events | N at risk | N (%)events | N at risk | N (%)events | N at risk | N (%)events | N at risk | N (%)events | N at risk | N (%)events | |
|
|
||||||||||||
| Early AMD | ||||||||||||
| Overall | 6840 | 624 (9.1) | 1567 | 62 (4.0) | 2503 | 147 (5.9) | 2018 | 251 (12.4) | 700 | 143 (20.4) | 52 | 21 (40.4) |
| Low | 4543 | 381 (8.4) | 998 | 40 (4.0) | 1621 | 76 (4.7) | 1383 | 155 (11.2) | 502 | 96 (19.1) | 39 | 14 (35.9) |
| Intermediate | 1848 | 180 (9.7) | 458 | 19 (4.2) | 704 | 45 (6.4) | 509 | 74 (14.5) | 166 | 37 (22.3) | 11 | 5 (45.5) |
| High | 449 | 63 (14.0) | 111 | 3 (2.7) | 178 | 26 (14.6) | 126 | 22 (17.5) | 32 | 10 (31.3) | 2 | 2 (100.0) |
| Drusen size >125 μm | ||||||||||||
| Overall | 7396 | 646 (8.7) | 1648 | 56 (3.4) | 2680 | 153 (5.7) | 2187 | 261 (11.9) | 813 | 153 (18.8) | 68 | 23 (33.8) |
| Low | 4887 | 386 (7.9) | 1047 | 39 (3.7) | 1717 | 74 (4.3) | 1483 | 152 (10.3) | 588 | 105 (17.9) | 52 | 16 (30.8) |
| Intermediate | 2011 | 189 (9.4) | 485 | 14 (2.9) | 765 | 52 (6.8) | 562 | 83 (14.8) | 185 | 35 (18.9) | 14 | 5 (35.7) |
| High | 498 | 71 (14.3) | 116 | 3 (2.6) | 198 | 27 (13.6) | 142 | 26 (18.3) | 40 | 13 (32.5) | 2 | 2 (100.0) |
| Soft indistinct drusen | ||||||||||||
| Overall | 7627 | 473 (6.2) | 1664 | 33 (2.0) | 2768 | 98 (3.5) | 2275 | 194 (8.5) | 845 | 129 (15.3) | 75 | 19 (25.3) |
| Low | 5017 | 269 (5.4) | 1058 | 21 (2.0) | 1766 | 46 (2.6) | 1535 | 109 (7.1) | 602 | 80 (13.3) | 56 | 13 (23.2) |
| Intermediate | 2091 | 149 (7.1) | 487 | 10 (2.1) | 798 | 32 (4.0) | 591 | 68 (11.5) | 198 | 35 (17.7) | 17 | 4 (23.5) |
| High | 519 | 55 (10.6) | 119 | 2 (1.7) | 204 | 20 (9.8) | 149 | 17 (11.4) | 45 | 14 (31.1) | 2 | 2 (100.0) |
| Any pigmentary abnormality | ||||||||||||
| Overall | 7893 | 548 (6.9) | 1638 | 33 (2.0) | 2755 | 105 (3.8) | 2436 | 206 (8.5) | 966 | 176 (18.2) | 98 | 28 (28.6) |
| Low | 5207 | 315 (6.1) | 1046 | 18 (1.7) | 1776 | 55 (3.1) | 1638 | 113 (6.9) | 678 | 113 (16.7) | 69 | 16 (23.2) |
| Intermediate | 2132 | 164 (7.7) | 475 | 11 (2.3) | 775 | 31 (4.0) | 627 | 71 (11.3) | 232 | 43 (18.5) | 23 | 8 (34.8) |
| High | 554 | 69 (12.5) | 117 | 4 (3.4) | 204 | 19 (9.3) | 171 | 22 (12.9) | 56 | 20 (35.7) | 6 | 4 (66.7) |
| Late AMD | ||||||||||||
| Overall | 8814 | 140 (1.6) | 1741 | 2 (0.1) | 2992 | 9 (0.3) | 2745 | 47 (1.7) | 1204 | 66 (5.5) | 132 | 16 (12.1) |
| Low | 5772 | 52 (0.9) | 1109 | 1 (0.1) | 1908 | 0 (0.0) | 1832 | 12 (0.7) | 832 | 30 (3.6) | 91 | 9 (9.9) |
| Intermediate | 2398 | 50 (2.1) | 509 | 0 (0.0) | 851 | 5 (0.6) | 710 | 21 (3.0) | 296 | 20 (6.8) | 32 | 4 (12.5) |
| High | 644 | 38 (5.9) | 123 | 1 (0.8) | 233 | 4 (1.7) | 203 | 14 (6.9) | 76 | 16 (21.1) | 9 | 3 (33.3) |
| Pure geographic atrophy | ||||||||||||
| Overall | 8775 | 49 (0.6) | 1743 | 0 (0.0) | 2992 | 3 (0.1) | 2730 | 15 (0.6) | 1179 | 22 (1.9) | 131 | 9 (6.9) |
| Low | 5770 | 21 (0.4) | 1109 | 0 (0.0) | 1910 | 0 (0.0) | 1832 | 4 (0.2) | 826 | 11 (1.3) | 93 | 6 (6.5) |
| Intermediate | 2383 | 17 (0.7) | 512 | 0 (0.0) | 851 | 1 (0.1) | 704 | 9 (1.3) | 286 | 6 (2.1) | 30 | 1 (3.3) |
| High | 622 | 11 (1.8) | 122 | 0 (0.0) | 231 | 2 (0.9) | 194 | 2 (1.0) | 67 | 5 (7.5) | 8 | 2 (25.0) |
| Exudative AMD | ||||||||||||
| Overall | 8867 | 91 (1.0) | 1742 | 2 (0.1) | 2998 | 6 (0.2) | 2753 | 32 (1.2) | 1234 | 44 (3.6) | 140 | 7 (5.0) |
| Low | 5798 | 31 (0.5) | 1109 | 1 (0.1) | 1911 | 0 (0.0) | 1835 | 8 (0.4) | 846 | 19 (2.3) | 97 | 3 (3.1) |
| Intermediate | 2416 | 33 (1.4) | 510 | 0 (0.0) | 854 | 4 (0.5) | 713 | 12 (1.7) | 306 | 14 (4.6) | 33 | 3 (9.1) |
| High | 653 | 27 (4.1) | 123 | 1 (0.8) | 233 | 2 (0.9) | 205 | 12 (5.9) | 82 | 11 (13.4) | 10 | 1 (10.0) |
AMD, age-related macular degeneration.
In worse eye.
Genetic risk groups: Low=0–1 risk alleles; intermediate=2 risk alleles; high=3–4 risk alleles.
Cumulative incidence of AMD for individuals with no AMD at age 45 and 65 years by increasing genetic risk
Using an MSM, Figure 1 shows estimated cumulative incidence of increasing severity of early AMD (Level 20 to Level 40) and late AMD (Level 50) through age 100 years based on assessments of AMD severity state for persons free of AMD at age 45 and 65 years for the 3 genetic risk groups. For individuals in the low genetic risk group who were free of AMD at age 45 years (Figure 1, Panel A), the cumulative incidence of late AMD (Level 50) was estimated to increase to 9.9%, the cumulative incidence of Level 40 or higher to 15.9%, the cumulative incidence of Level 30 or higher to 32.2%, and the cumulative incidence of Level 20 or higher to 51.5% by age 100 years. The estimated cumulative incidences were increasingly higher at a given age for early and late AMD in the intermediate and high risk groups (Figure 1, Panels B and C compared to Panel A). At age 80 years, estimated cumulative incidence of late AMD in individuals with no AMD at age 45 was 1.4% (Figure 1, Panel A), 5.2% (Panel B), and 15.3% (Panel C) in the low, intermediate, and high risk groups, respectively.
Figure 1.

Estimated cumulative incidence of age related macular degeneration (AMD) by genetic risk and age (assuming no AMD at the starting age) in the Beaver Dam Eye Study, 1988–2010. Rows are ordered by increasing starting age from top to bottom (age 45 and 65 years) and columns are ordered by increasing genetic risk from left to right (low, intermediate, high). For example, Panel A shows the cumulative incidence of AMD in individuals assuming no AMD at age 45 and low genetic risk and Panel E shows the cumulative incidence of AMD in individuals assuming no AMD at age 65 and intermediate genetic risk. Numbers to right of lines indicate AMD severity level on the Beaver Dam AMD Severity Scale.
The estimated cumulative incidence of early and late AMD was smaller in individuals who survived to age 65 without any signs of AMD than in individuals who had no signs of AMD at age 45 (Figure 1, Panel D compared to Panel A, Panel E compared to Panel B and Panel F compared to Panel C). For example, in persons with no evidence of early AMD at age 45 and 65 years, the estimated cumulative incidence of late AMD by age 80 years in those with high genetic risk was 15.3% (Panel C) and 7.9% (Panel F), respectively, while for those aged 45 and 65 years with no evidence of AMD and low genetic risk, the estimates for developing late AMD were lower (1.7% [Panel D] and 0.6% [Panel A], respectively).
Cumulative incidence of AMD for individuals with increasing severity of AMD by genetic risk at age 45 years
Figure 2 shows estimated cumulative incidence of more severe stages of AMD in persons who, at age 45 years, had AMD levels 10, 20, 30, and 40. Individuals with low genetic risk who already had early AMD at age 45 years were estimated to have a higher cumulative incidence of late AMD by age 80 years compared to individuals without AMD at age 45 years (Figure 2 Panels D, G, and J compared to Panel A). This was also true for the intermediate (Panels E, H, and K compared to Panel B) and high (Panels F, I, and L compared to Panel C) risk groups. In those in the low genetic risk group at age 45 years, 1.4%, 7.0%, 15.0%, and 33.4% of individuals with AMD levels 10 (Panel A), 20 (Panel D), 30 (Panel G), and 40 (Panel J), respectively, were estimated to have developed late AMD by age 80 years.
Figure 2.

Estimated cumulative incidence of more severe age related macular degeneration (AMD) in persons at age 45 years by increasing beginning AMD level (rows top [Level 10] to bottom [Level 40]) and increasing genetic risk (columns left [low] to right [high]) in the Beaver Dam Eye Study, 1988–2010. For example, Panel A shows the cumulative incidence of AMD levels 20–50 for an individual whose AMD level is 10 at age 45 and low genetic risk and Panel I shows the cumulative incidence of AMD levels 40 and 50 for an individual who had AMD level 30 age age 45 and high genetic risk. Numbers to right of lines indicate level on the Beaver Dam AMD Severity Scale.
Cumulative incidence of AMD for individuals with increasing severity of drusen size, type, and pigmentary abnormalities by increasing genetic risk at age 45 years
Figure 3 shows the MSM estimates of cumulative incidence of increasing size and severity of retinal drusen and increasing severity of pigmentary abnormalities in individuals with low, intermediate, and high genetic risk who were free from those lesions at age 45 years. For example, Figure 3A shows that the estimated cumulative incidence of intermediate size drusen (≥63 to <125 μm diameter) was 23.8%, 24.4%, and 23.3% and large size drusen (≥125 μm in diameter) was 11.0%, 15.8%, and 18.6% at age 80 years in individuals with low (Panel A1), intermediate (Panel A2), and high (Panel A3) genetic risk, respectively who had no or small drusen at age 45 years. Figure 3B shows that the estimated cumulative incidence of hard distinct drusen was 68.3%, 57.6%, and 43.6%, for soft distinct drusen it was 13.6%, 15.4%, and 17.0%, at age 80 and for soft indistinct/reticular drusen it was 7.3%, 13.4%, and 18.8% at age 80 years in individuals with low (Panel B1), intermediate (Panel B2), and high (Panel B3) genetic risk, respectively, who had no or hard indistinct drusen at age 45 years. Because individuals with higher genetic risk are more likely to develop a more severe drusen type, there is an inverse association of genetic risk groups with the cumulative incidence of hard distinct drusen at age 80 years (Figure 3B). Figure 3C shows the estimated cumulative incidence of increased retinal pigment was 13.9%, 21.4%, and 28.9% at age 80, for RPE depigmentation it was 1.8%, 3.5%, and 8.1% at age 80 years, and for geographic atrophy it was 0.1%, 0.4%, and 1.8%, at age 80 years in individuals with low (Panel C1), intermediate (Panel C2), and high (Panel C3) genetic risk, respectively who had no pigmentary abnormalities at age 45 years.
Figure 3.
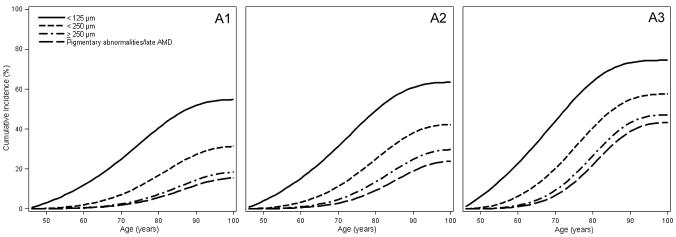
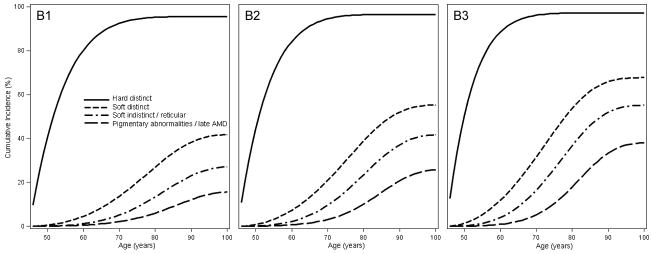
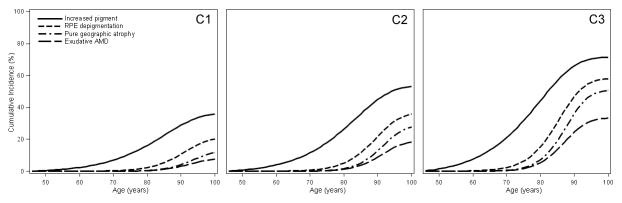
Estimated cumulative incidence by genetic risk level (increasing left [low] to right [high]) of A. intermediate and large size drusen in individuals with no or small drusen at age 45 years; B. hard distinct, soft distinct, and soft indistinct/reticular drusen in individuals with no or hard indistinct drusen at age 45 years; and C. increased retinal pigment, retinal pigment epithelial depigmentation, and pure geographic atrophy in individuals with no pigmentary abnormalities at age 45 years.
Population attributable risk
The estimated population attributable risk fraction for early and late AMD was 9.6% and 53.2%, respectively when at least 1 CFH risk allele was present and 5.0% and 43.0%, respectively when at least 1 ARMS2 risk allele was present.
Prognostic assessment of risk
The AUC for progression from no or early AMD to late AMD for a model that included AMD severity level only was 0.9316 (Figure 4). Adding traditional risk factors (eg, age, sex, history of smoking, hypertension, history of physical activity, and history of multivitamin use) showed an incremental gain of 0.0280, and a further incremental gain of 0.0066 with the addition of CFH and ARMS2 to the model. ARMS2 and CFH added a small incremental gain after AMD severity level and traditional risk factors had been added to a model with GA (0.0019) or exudative AMD (0.0099) as the endpoint.
Figure 4.

Area under the receiver operating characteristic curves (AUCs) for various models of progression from no or early age-related macular degeneration (AMD) to late AMD over a 5-year interval in the Beaver Dam Eye Study, 1988–2010. ARMS2, age-related maculopathy susceptibility 2; CFH, complement factor H; TR, traditional risk.
COMMENT
We examined the relationships of genetic risk defined by the number of allelic variants of SNPs of 2 AMD candidate genes, CFH (rs1061170) and ARMS2 (rs10490924), to the estimated cumulative incidence and progression of AMD in the population-based BDES cohort over a 20-year period. Using MSMs, the estimated cumulative incidences of early AMD at age 80 years in persons without AMD at age 45 years in the genetically low, intermediate, and high risk groups were 33.0%, 39.9%, and 46.5%, respectively and for late AMD they were 1.4%, 5.2%, and 15.3%, respectively.
Genetic risk group status was directly associated with the incidence of more severe drusen type, large size drusen, and pigmentary abnormalities. The association of genetic risk with late AMD (OR per increasing genetic risk group=2.93) was stronger than for early AMD (OR=1.38). The population attributable risk for late AMD was 53% when at least 1 CFH risk allele was present and 43% when at least 1 ARMS2 risk allele was present.
Using 20 years of BDES data and MSMs, we estimated the cumulative incidence for developing late AMD in persons aged 45 years without AMD who survive to age 80 years, the current estimated life expectancy for a 45-year-old in the US, to vary from 1.4% in persons with low genetic risk to 15.3% in those with high genetic risk. These findings are consistent with virtually all earlier studies showing a strong association of increasing age and genetic risk with high long term incidence of AMD.1–26,44–48 The relatively high estimated overall cumulative incidence in persons with high genetic risk and the availability of preventive approaches have led others to develop risk assessment models based on genetic and environmental exposures.2,22,49,50 The rationale for such screening is that earlier detection of those at high risk of developing late AMD may lead to changes in behaviors such as cessation of smoking, changes in diet (eg, eating more foods containing omega-3 fatty acids and more leafy green vegetables), and increasing physical activity levels that might, in part, prevent or reduce the incidence and progression of AMD in these individuals. For example, data from a pilot study showed that smokers reported they would be more likely to quit smoking if told they were at high genetic risk of developing late AMD.51 However, in the BDES, smoking status has not proven to be a risk factor for the cumulative incidence of late AMD,52 and to date, it has not been demonstrated that interventions to alter behaviors (eg, change in diet, smoking cessation) affect the incidence of late AMD.
While we have demonstrated the effects of the two most common, and arguably strongest, genetic correlates of AMD in this study, about 80% of those at high genetic risk for late AMD are estimated to be free of this disease if they live until age 80 years. For every 1000 persons in the cohort screened for genetic risk, only 78 persons aged 45 years would be expected to have the high risk genotype, and of those, only 12 are estimated to develop late AMD if they live to age 80 while 66 will not. Thus, data from our study and others suggest that such screening using genotyping in young persons without early AMD is not indicated.53 Once early AMD is present, defining genetic risk using CFH and ARMS2 status, while statistically significantly adding to predicting the risk of late AMD, the addition is small and contributes little beyond that of knowing the phenotypic stage of the disease (Figure 4). It has also been shown by Klein and colleagues22 using data from the Age-Related Eye Disease Study that genotyping adds little to the AUC analyses beyond that of knowing the phenotype once signs of early AMD are present.
Genetic risk of the two candidate SNPs were shown for both early and late AMD lesions. Candidate SNPs from both CFH and ARMS2 have consistently shown stronger associations with both exudative AMD and GA than with signs of early AMD.1–5,10–16,18–22,49–51,53–67 Our results are consistent with cross-sectional findings from the Blue Mountains Eye Study, the Rotterdam Study and the ALIENOR study where persons who were homozygous for the CFH variant had increased odds ranging from 1.2 to 2.4 for early AMD compared to no AMD.54,55,64 In addition, in the Rotterdam Study, the association became stronger with increasing severity of early AMD. Our results are also consistent with findings of Yu and colleagues,2 who used a similar MSM in data from 2560 patients without late AMD in the Age-Related Eye Disease Study and found that CFH and ARMS2 were related to the development of large size drusen but not intermediate sized drusen (≥63 to <125 μm in diameter). They did not examine the relation of these genes to pigmentary abnormalities.
The reason for the weaker relation of genetic risk to early than to late AMD is not understood. Delcourt and colleagues54 speculated that the weaker odds of approximately 2 found between those homozygous for the C allele of the CFH Y402H polymorphism and early AMD compared to odds of 16–23 for late AMD may be due to lesions considered to be specific for early AMD really being a heterogeneous group of abnormalities, some of which actually bear a very low risk of developing late AMD. Another possibility is that environmental factors may have more impact on the lesions now considered to be characteristic of early AMD compared to late AMD.
The biological mechanisms of the CFH gene in the pathogenesis of AMD have been well described, and include changes in CFH protein levels that lead to alterations in the regulation of complement activation in response to inflammation. CFH also affects the metabolism of lipids such as malondialdehyde that accumulate in response to oxidative stress and are thought to contribute to RPE cell death.68 The role of ARMS2 is also uncertain. It may work through different mechanisms, eg, stabilization of the extracellular matrix in Bruch’s membrane and in protecting against oxidative stress.69–71 Smailhodzic and colleagues72 reported complement deregulation to be associated not only with CFH high-risk alleles but also with ARMS2 high-risk alleles, suggesting that ARMS2 may also be involved in the activation of the complement system. We did not find an interaction between CFH and ARMS2 and incident early AMD (Klein R, unpublished data), and we were underpowered to examine such an interaction for late AMD.
In the analyses presented in this paper we used the MSM model, which permits staged modeling of AMD progression incorporating all facets of the natural history of AMD as well as death into a single, biologically plausible model rather than modeling aspects of the disease process in isolation. It uses a biological meaningful time scale (participant age) rather than an artificial time scale (time of study) and incorporates time-varying covariates by updating covariate values at observation times. The MSM accounts for the correlation between past outcomes and future outcomes by conditioning on the current state (the Markov assumption). That is, a subject’s AMD state at the next scheduled visit given the current AMD state is assumed to be independent of the past history of AMD. This assumption is fairly standard in survival analysis applications (it is a key assumption for the Cox regression model). A key advantage of the MSM model over the Cox model is the ability to more fully use the available information on AMD progression over time. The primary disadvantage of the MSM model is computational complexity; the assumptions underlying the MSM model are either the same or less restrictive than the alternatives.
Caution should be observed in interpreting our data. First, we only used SNPs from 2 AMD candidate genes; it is possible that inclusion of all the identified possible loci, many of which are likely to be unknown, might have influenced our findings. Second, because the study population is racially/ethnically homogeneous (99.6% white), this limits our inferences to whites. Third, power to examine relationships for some infrequent endpoints (eg, reticular drusen, geographic atrophy, exudative AMD) was limited, as well as our ability to assess the possibility of gene×gene and gene×age interactions. Mortality may limit the interpretation of associations due to selective survival. On the other hand, the BDES has many strengths, including repeated examinations over a 20-year period using standardized detailed procedures for obtaining stereoscopic color fundus photographs of the macula, and an objective system for grading those photographs for AMD phenotypes.
This report provides long-term population-based observations regarding the relationships of genetic risk as defined by the number of CFH and ARMS2 risk alleles and age to the natural history of AMD from its earliest to its latest stages. The high incidence of AMD at older ages and increased survival suggest a growing burden of this disease in the future. The value of risk assessment will be determined as the pathogenesis of the disease becomes better understood and new evidence emerges to support cost-effective interventions prior to onset or at the earliest stages of the disease. At present, knowing the phenotype once early AMD is present contributes more to risk assessment than knowing the genetic risk based on the 2 AMD candidate genes with the largest attributable risk.
Supplementary Material
Acknowledgments
The National Institutes of Health grant EY06594 (Dr R. Klein, Dr B. E. K. Klein) provided funding for entire study, including collection and analyses of data; further support for data analyses was provided by Research to Prevent Blindness (Dr R. Klein and Dr B. E. K. Klein, Senior Scientific Investigator Awards), New York, NY. Dr R. Klein had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. The authors acknowledge the members of the Beaver Dam Eye Study Data Monitoring and Oversight Committee for their helpful feedback.
ROLE OF SPONSORS
The organizations that provided funding were in no way involved in the design and conduct of the study, in the collection, analysis, and interpretation of the data, or in the preparation, review, or approval of the manuscript.
Footnotes
DISCLAIMER
The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Eye Institute or the National Institutes of Health.
DISCLOSURE
None reported.
References
- 1.Seddon JM, Reynolds R, Maller J, Fagerness JA, Daly MJ, Rosner B. Prediction model for prevalence and incidence of advanced age-related macular degeneration based on genetic, demographic, and environmental variables. Invest Ophthalmol Vis Sci. 2009;50(5):2044–2053. doi: 10.1167/iovs.08-3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yu Y, Reynolds R, Rosner B, Daly MJ, Seddon JM. Prospective assessment of genetic effects on progression to different stages of age-related macular degeneration using multi-state Markov models. Invest Ophthalmol Vis Sci. 2012;53(3):1548–1556. doi: 10.1167/iovs.11-8657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Edwards AO, Ritter R, III, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308(5720):421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 4.Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308(5720):419–421. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 5.Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308(5720):385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fagerness JA, Maller JB, Neale BM, Reynolds RC, Daly MJ, Seddon JM. Variation near complement factor I is associated with risk of advanced AMD. Eur J Hum Genet. 2009;17(1):100–104. doi: 10.1038/ejhg.2008.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maller JB, Fagerness JA, Reynolds RC, Neale BM, Daly MJ, Seddon JM. Variation in complement factor 3 is associated with risk of age-related macular degeneration. Nat Genet. 2007;39(10):1200–1201. doi: 10.1038/ng2131. [DOI] [PubMed] [Google Scholar]
- 8.Gold B, Merriam JE, Zernant J, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38(4):458–462. doi: 10.1038/ng1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nozaki M, Raisler BJ, Sakurai E, et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci U S A. 2006;103(7):2328–2333. doi: 10.1073/pnas.0408835103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jakobsdottir J, Conley YP, Weeks DE, Mah TS, Ferrell RE, Gorin MB. Susceptibility genes for age-related maculopathy on chromosome 10q26. Am J Hum Genet. 2005;77(3):389–407. doi: 10.1086/444437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fisher SA, Abecasis GR, Yashar BM, et al. Meta-analysis of genome scans of age-related macular degeneration. Hum Mol Genet. 2005;14(15):2257–2264. doi: 10.1093/hmg/ddi230. [DOI] [PubMed] [Google Scholar]
- 12.Weeks DE, Conley YP, Tsai HJ, et al. Age-related maculopathy: a genomewide scan with continued evidence of susceptibility loci within the 1q31, 10q26, and 17q25 regions. Am J Hum Genet. 2004;75(2):174–189. doi: 10.1086/422476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iyengar SK, Song D, Klein BE, et al. Dissection of genomewide-scan data in extended families reveals a major locus and oligogenic susceptibility for age-related macular degeneration. Am J Hum Genet. 2004;74(1):20–39. doi: 10.1086/380912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seddon JM, Santangelo SL, Book K, Chong S, Cote J. A genomewide scan for age-related macular degeneration provides evidence for linkage to several chromosomal regions. Am J Hum Genet. 2003;73(4):780–790. doi: 10.1086/378505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Majewski J, Schultz DW, Weleber RG, et al. Age-related macular degeneration--a genome scan in extended families. Am J Hum Genet. 2003;73(3):540–550. doi: 10.1086/377701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu Y, Reynolds R, Fagerness J, Rosner B, Daly MJ, Seddon JM. Association of variants in the LIPC and ABCA1 genes with intermediate and large drusen and advanced age-related macular degeneration. Invest Ophthalmol Vis Sci. 2011;52(7):4663–4670. doi: 10.1167/iovs.10-7070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seddon JM, Reynolds R, Rosner B. Associations of smoking, body mass index, dietary lutein, and the LIPC gene variant rs10468017 with advanced age-related macular degeneration. Mol Vis. 2010;16:2412–2424. [PMC free article] [PubMed] [Google Scholar]
- 18.Seitsonen SP, Onkamo P, Peng G, et al. Multifactor effects and evidence of potential interaction between complement factor H Y402H and LOC387715 A69S in age-related macular degeneration. PLoS One. 2008;3(12):e3833. doi: 10.1371/journal.pone.0003833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schaumberg DA, Hankinson SE, Guo Q, Rimm E, Hunter DJ. A prospective study of 2 major age-related macular degeneration susceptibility alleles and interactions with modifiable risk factors. Arch Ophthalmol. 2007;125(1):55–62. doi: 10.1001/archopht.125.1.55. [DOI] [PubMed] [Google Scholar]
- 20.Schmidt S, Hauser MA, Scott WK, et al. Cigarette smoking strongly modifies the association of LOC387715 and age-related macular degeneration. Am J Hum Genet. 2006;78(5):852–864. doi: 10.1086/503822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rivera A, Fisher SA, Fritsche LG, et al. Hypothetical LOC387715 is a second major susceptibility gene for age-related macular degeneration, contributing independently of complement factor H to disease risk. Hum Mol Genet. 2005;14(21):3227–3236. doi: 10.1093/hmg/ddi353. [DOI] [PubMed] [Google Scholar]
- 22.Klein ML, Francis PJ, Ferris FL, III, Hamon SC, Clemons TE. Risk assessment model for development of advanced age-related macular degeneration. Arch Ophthalmol. 2011;129(12):1543–1550. doi: 10.1001/archophthalmol.2011.216. [DOI] [PubMed] [Google Scholar]
- 23.Klein R, Klein BE, Linton KL. Prevalence of age-related maculopathy. The Beaver Dam Eye Study. Ophthalmology. 1992;99(6):933–943. doi: 10.1016/s0161-6420(92)31871-8. [DOI] [PubMed] [Google Scholar]
- 24.Klein R, Klein BE, Jensen SC, Meuer SM. The five-year incidence and progression of age-related maculopathy: The Beaver Dam Eye Study. Ophthalmology. 1997;104(1):7–21. doi: 10.1016/s0161-6420(97)30368-6. [DOI] [PubMed] [Google Scholar]
- 25.Klein R, Klein BE, Tomany SC, Meuer SM, Huang GH. Ten-year incidence and progression of age-related maculopathy: The Beaver Dam Eye Study. Ophthalmology. 2002;109(10):1767–1779. doi: 10.1016/s0161-6420(02)01146-6. [DOI] [PubMed] [Google Scholar]
- 26.Klein R, Klein BE, Knudtson MD, Meuer SM, Swift M, Gangnon RE. Fifteen-year cumulative incidence of age-related macular degeneration: the Beaver Dam Eye Study. Ophthalmology. 2007;114(2):253–262. doi: 10.1016/j.ophtha.2006.10.040. [DOI] [PubMed] [Google Scholar]
- 27.Linton KL, Klein BE, Klein R. The validity of self-reported and surrogate-reported cataract and age-related macular degeneration in the Beaver Dam Eye Study. Am J Epidemiol. 1991;134(12):1438–1446. doi: 10.1093/oxfordjournals.aje.a116049. [DOI] [PubMed] [Google Scholar]
- 28.Klein R, Klein BE, Linton KL, De Mets DL. The Beaver Dam Eye Study: visual acuity. Ophthalmology. 1991;98(8):1310–1315. doi: 10.1016/s0161-6420(91)32137-7. [DOI] [PubMed] [Google Scholar]
- 29.Klein R, Klein BE, Lee KE. Changes in visual acuity in a population. The Beaver Dam Eye Study. Ophthalmology. 1996;103(8):1169–1178. doi: 10.1016/s0161-6420(96)30526-5. [DOI] [PubMed] [Google Scholar]
- 30.Klein R, Klein BE, Lee KE, Cruickshanks KJ, Chappell RJ. Changes in visual acuity in a population over a 10-year period: The Beaver Dam Eye Study. Ophthalmology. 2001;108(10):1757–1766. doi: 10.1016/s0161-6420(01)00769-2. [DOI] [PubMed] [Google Scholar]
- 31.Klein R, Klein BE, Lee KE, Cruickshanks KJ, Gangnon RE. Changes in visual acuity in a population over a 15-year period: the Beaver Dam Eye Study. Am J Ophthalmol. 2006;142(4):539–549. doi: 10.1016/j.ajo.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 32.Klein R, Klein BE. The Beaver Dam Eye Study. Manual of Operations: Revised. Springfield, VA: National Technical Information Service; 1991. NTIS Publication PB91–149823. [Google Scholar]
- 33.Klein R, Klein BE. The Beaver Dam Eye Study II. Manual of Operations. Springfield, VA: National Technical Information Service; 1995. NTIS Publication PB95–273827. [Google Scholar]
- 34.Klein R, Klein BE. The Beaver Dam Eye Study III. Manual of Operations. Springfield, VA: National Technical Information Service; 1999. NTIS Publication PB99–137861. [Google Scholar]
- 35.Klein BE, Klein R. The Beaver Dam Eye Study V. Manual of Operations. Springfield, VA: National Technical Information Service; 2010. NTIS Publication PB2010–114194. [Google Scholar]
- 36.Klein R, Davis MD, Magli YL, Segal P, Klein BE, Hubbard L. The Wisconsin Age-Related Maculopathy Grading System. Springfield, VA: National Technical Information Service; 1991. NTIS Publication PB91–184267. [DOI] [PubMed] [Google Scholar]
- 37.Klein R, Davis MD, Magli YL, Segal P, Klein BE, Hubbard L. The Wisconsin Age-Related Maculopathy Grading System. Ophthalmology. 1991;98(7):1128–1134. doi: 10.1016/s0161-6420(91)32186-9. [DOI] [PubMed] [Google Scholar]
- 38.Sivakumaran TA, Igo RP, Jr, Kidd JM, et al. A 32 kb critical region excluding Y402H in CFH mediates risk for age-related macular degeneration. PLoS One. 2011;6(10):e25598. doi: 10.1371/journal.pone.0025598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thompson CL, Klein BE, Klein R, et al. Complement factor H and hemicentin-1 in age-related macular degeneration and renal phenotypes. Hum Mol Genet. 2007;16(17):2135–2148. doi: 10.1093/hmg/ddm164. [DOI] [PubMed] [Google Scholar]
- 40.Huang L, Li Y, Singleton AB, et al. Genotype-imputation accuracy across worldwide human populations. Am J Hum Genet. 2009;84(2):235–250. doi: 10.1016/j.ajhg.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.R Development Core Team. R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2009. [Google Scholar]
- 42.Jackson CH. Multi-state models for panel data: the msm package for R. J Stat Softw. 2011;38(8):1–28. [Google Scholar]
- 43.Efron B, Tibshirani RJ. An Introduction to the Bootstrap. Boca Raton: Chapman and Hall/CRC; 1994. [Google Scholar]
- 44.Bressler NM, Munoz B, Maguire MG, et al. Five-year incidence and disappearance of drusen and retinal pigment epithelial abnormalities: Waterman Study. Arch Ophthalmol. 1995;113(3):301–308. doi: 10.1001/archopht.1995.01100030055022. [DOI] [PubMed] [Google Scholar]
- 45.Klaver CC, Assink JJ, van Leeuwen R, et al. Incidence and progression rates of age-related maculopathy: the Rotterdam Study. Invest Ophthalmol Vis Sci. 2001;42(10):2237–2241. [PubMed] [Google Scholar]
- 46.Mitchell P, Wang JJ, Foran S, Smith W. Five-year incidence of age-related maculopathy lesions: the Blue Mountains Eye Study. Ophthalmology. 2002;109(6):1092–1097. doi: 10.1016/s0161-6420(02)01055-2. [DOI] [PubMed] [Google Scholar]
- 47.van Leeuwen R, Klaver CC, Vingerling JR, Hofman A, de Jong PT. The risk and natural course of age-related maculopathy: follow-up at 6 1/2 years in the Rotterdam Study. Arch Ophthalmol. 2003;121(4):519–526. doi: 10.1001/archopht.121.4.519. [DOI] [PubMed] [Google Scholar]
- 48.Wang JJ, Foran S, Smith W, Mitchell P. Risk of age-related macular degeneration in eyes with macular drusen or hyperpigmentation: the Blue Mountains Eye Study cohort. Arch Ophthalmol. 2003;121(5):658–663. doi: 10.1001/archopht.121.5.658. [DOI] [PubMed] [Google Scholar]
- 49.Ying GS, Maguire MG. Development of a risk score for geographic atrophy in complications of the age-related macular degeneration prevention trial. Ophthalmology. 2011;118(2):332–338. doi: 10.1016/j.ophtha.2010.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zanke B, Hawken S, Carter R, Chow D. A genetic approach to stratification of risk for age-related macular degeneration. Can J Ophthalmol. 2010;45(1):22–27. doi: 10.3129/i09-209. [DOI] [PubMed] [Google Scholar]
- 51.Rennie CA, Stinge A, King EA, Sothirachagan S, Osmond C, Lotery AJ. Can genetic risk information for age-related macular degeneration influence motivation to stop smoking? A pilot study. Eye (Lond) 2012;26(1):109–118. doi: 10.1038/eye.2011.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klein R, Klein BE, Moss SE. Relation of smoking to the incidence of age-related maculopathy: the Beaver Dam Eye Study. Am J Epidemiol. 1998;147(2):103–110. doi: 10.1093/oxfordjournals.aje.a009421. [DOI] [PubMed] [Google Scholar]
- 53.Klein R, Klein BE, Myers CE. Risk assessment models for late age-related macular degeneration. Arch Ophthalmol. 2011;129(12):1605–1606. doi: 10.1001/archophthalmol.2011.372. [DOI] [PubMed] [Google Scholar]
- 54.Delcourt C, Delyfer MN, Rougier MB, et al. Associations of complement factor H and smoking with early age-related macular degeneration: the ALIENOR study. Invest Ophthalmol Vis Sci. 2011;52(8):5955–5962. doi: 10.1167/iovs.10-6235. [DOI] [PubMed] [Google Scholar]
- 55.Despriet DD, Klaver CC, Witteman JC, et al. Complement factor H polymorphism, complement activators, and risk of age-related macular degeneration. JAMA. 2006;296(3):301–309. doi: 10.1001/jama.296.3.301. [DOI] [PubMed] [Google Scholar]
- 56.Ferris FL, Davis MD, Clemons TE, et al. A simplified severity scale for age-related macular degeneration: AREDS Report No. 18. Arch Ophthalmol. 2005;123(11):1570–1574. doi: 10.1001/archopht.123.11.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gangnon RE, Lee KE, Klein BE, Iyengar SK, Sivakumaran TA, Klein R. The Y402H variant in the complement factor H gene affects incidence and progression of age-related macular degeneration: results from multi-state models applied to the Beaver Dam Eye Study. Arch Ophthalmol. 2012 doi: 10.1001/archophthalmol.2012.693. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hughes AE, Orr N, Patterson C, et al. Neovascular age-related macular degeneration risk based on CFH, LOC387715/HTRA1, and smoking. PLoS Med. 2007;4(12):e355. doi: 10.1371/journal.pmed.0040355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Klein R, Cruickshanks KJ, Nash SD, et al. The prevalence of age-related macular degeneration and associated risk factors. Arch Ophthalmol. 2010;128(6):750–758. doi: 10.1001/archophthalmol.2010.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Magnusson KP, Duan S, Sigurdsson H, et al. CFH Y402H confers similar risk of soft drusen and both forms of advanced AMD. PLoS Med. 2006;3(1):e5. doi: 10.1371/journal.pmed.0030005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ryu E, Fridley BL, Tosakulwong N, Bailey KR, Edwards AO. Genome-wide association analyses of genetic, phenotypic, and environmental risks in the Age-Related Eye Disease Study. Mol Vis. 2010:162811–2821. [PMC free article] [PubMed] [Google Scholar]
- 62.Thompson CL, Klein BE, Klein R, et al. Complement factor H and hemicentin-1 in age-related macular degeneration and renal phenotypes. Hum Mol Genet. 2007;16(17):2135–2148. doi: 10.1093/hmg/ddm164. [DOI] [PubMed] [Google Scholar]
- 63.Thompson CL, Jun G, Klein BE, et al. Genetics of pigment changes and geographic atrophy. Invest Ophthalmol Vis Sci. 2007;48(7):3005–3013. doi: 10.1167/iovs.06-1325. [DOI] [PubMed] [Google Scholar]
- 64.Xing C, Sivakumaran TA, Wang JJ, et al. Complement factor H polymorphisms, renal phenotypes and age-related macular degeneration: the Blue Mountains Eye Study. Genes Immun. 2008;9(3):231–239. doi: 10.1038/gene.2008.10. [DOI] [PubMed] [Google Scholar]
- 65.Munch IC, Ek J, Kessel L, et al. Small, hard macular drusen and peripheral drusen: associations with AMD genotypes in the Inter99 Eye Study. Invest Ophthalmol Vis Sci. 2010;51(5):2317–2321. doi: 10.1167/iovs.09-4482. [DOI] [PubMed] [Google Scholar]
- 66.Klein R, Knudtson MD, Klein BE, et al. Inflammation, complement factor H, and age-related macular degeneration: the Multi-Ethnic Study of Atherosclerosis. Ophthalmology. 2008;115(10):1742–1749. doi: 10.1016/j.ophtha.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tedeschi-Blok N, Buckley J, Varma R, Triche TJ, Hinton DR. Population-based study of early age-related macular degeneration: role of the complement factor H Y402H polymorphism in bilateral but not unilateral disease. Ophthalmology. 2007;114(1):99–103. doi: 10.1016/j.ophtha.2006.07.043. [DOI] [PubMed] [Google Scholar]
- 68.Weismann D, Hartvigsen K, Lauer N, et al. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature. 2011;478(7367):76–81. doi: 10.1038/nature10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fritsche LG, Loenhardt T, Janssen A, et al. Age-related macular degeneration is associated with an unstable ARMS2 (LOC387715) mRNA. Nat Genet. 2008;40(7):892–896. doi: 10.1038/ng.170. [DOI] [PubMed] [Google Scholar]
- 70.Kortvely E, Hauck SM, Duetsch G, et al. ARMS2 is a constituent of the extracellular matrix providing a link between familial and sporadic age-related macular degenerations. Invest Ophthalmol Vis Sci. 2010;51(1):79–88. doi: 10.1167/iovs.09-3850. [DOI] [PubMed] [Google Scholar]
- 71.Kanda A, Chen W, Othman M, et al. A variant of mitochondrial protein LOC387715/ARMS2, not HTRA1, is strongly associated with age-related macular degeneration. Proc Natl Acad Sci U S A. 2007;104(41):16227–16232. doi: 10.1073/pnas.0703933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smailhodzic D, Klaver CC, Klevering BJ, et al. Risk alleles in CFH and ARMS2 are independently associated with systemic complement activation in age-related macular degeneration. Ophthalmology. 2012;119(2):339–346. doi: 10.1016/j.ophtha.2011.07.056. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.