

Abstract
Chronic global N-methyl-d-aspartate receptor (NMDAR) blockade leads to changes in glutamatergic transmission. The impact of more subunit-selective NMDAR inhibition on glutamatergic circuits remains incomplete. To this end, organotypic hippocampal slice cultures were treated for 17–21 days with the high-affinity competitive antagonist d-aminophosphonovaleric acid (d-APV), the allosteric GluN2B-selective antagonist Ro25-6981, or the newer competitive GluN2A-preferring antagonist NVP-AAM077. Electrophysiological recordings from dentate granule cells revealed that chronic d-APV treatment increased, whereas chronic Ro25-6981 reduced, epileptiform event-associated large-amplitude spontaneous excitatory postsynaptic currents (sEPSC) compared with all other treatment groups, consistent with opposite effects on glutamatergic networks. Presynaptically, chronic d-APV or Ro25-6981 increased small-amplitude sEPSCs and AMPA/kainate receptor-mediated miniature EPSCs (mEPSCAMPAR) frequency. Chronic d-APV or NVP-AAM077, but not Ro25-6981, increased putative vGlut1-positive glutamatergic synapses. Postsynaptically, chronic d-APV dramatically increased mEPSCAMPAR and profoundly decreased NMDAR-mediated mEPSC (mEPSCNMDAR) measures, suggesting increased AMPAR/NMDAR ratio. Ro25-6981 decreased mEPSCAMPAR charge transfer and modestly decreased mEPSCNMDAR frequency and decay, suggesting downward scaling of AMPAR and NMDAR function without dramatically altering AMPAR/NMDAR ratio. Extrasynaptically, threo-β-benzyloxyaspartate-enhanced “tonic” NMDAR current amplitude and activated channel number estimates were significantly increased only by chronic Ro25-6981. For intrinsic excitability, action potential threshold was slightly more negative following chronic d-APV or NVP-AAM077. The predominant pro-excitatory effects of chronic d-APV are consistent with increased glutamatergic transmission and network excitability. The minor effects of chronic NVP-AAM077 on action potential threshold and synapse number are consistent with minimal effects on circuit function. The chronic Ro25-6981-induced downward scaling of synaptic AMPAR and NMDAR function is consistent with decreased postsynaptic glutamate receptors and reduced network excitability.
Keywords: AMPA receptor, hippocampus, GluN2A, GluN2B, NR2A, NR2B, Ro25-6981
n-methyl-d-aspartate receptors (NMDAR) are heteromeric ligand-gated ion channels composed of GluN1 and GluN2 (A–D) subunits. Hippocampus and cortex predominantly express GluN2A and GluN2B (Monyer et al. 1994; Yamakura and Shimoji 1999). NMDAR activation is required for normal brain function including synaptic plasticity, learning and memory, and neuronal survival (Dingledine et al. 1999; Hardingham and Bading 2003; Mori and Mishina 1995; Morris et al. 1986). However, excessive NMDAR activation also can contribute to pathophysiology associated with epilepsy, brain trauma, stroke, depression, and neurodegenerative disorders (Dingledine et al. 1990; Lipton 2007).
Early high-affinity competitive and uncompetitive antagonists that dramatically reduce global NMDAR function caused cognitive and psychotomimetic dysfunction. Chronic use of these antagonists led to neuronal vacuolization, paradoxical seizure exacerbation, increased synaptic connectivity, and increased ionotropic glutamate receptor expression (Gould et al. 1994; Ikonomidou et al. 1999; Loscher 1998; Muir and Lees 1995; Olney et al. 1989; Parsons et al. 1999; Sveinbjornsdottir et al. 1993; Wang and Bausch 2004). A number of newer NMDAR antagonists appear to reduce pathological overactivation while better maintaining the physiological NMDAR function required for normal brain function. For example, the newer allosteric GluN2B-selective NMDAR antagonists remain in clinical trials but are well-tolerated in broad patient populations (Kemp and McKernan 2002; Muir and Lees 1995; Palmer 2001). However, despite the obvious clinical relevance, the physiological consequences of chronic treatment with these newer NMDAR antagonists at the cellular and network levels are just beginning to be explored.
The purpose of this study was to determine the consequences of chronic treatment with the allosteric GluN2B-selective NMDAR antagonist Ro25-6981 and the newer competitive GluN2A-preferring NMDAR antagonist NVP-AAM077 on glutamatergic networks and to compare these effects to chronic treatment with d-aminophosphonovaleric acid (d-APV) in a model of deafferentation-induced pathophysiology. A 17- to 21-day chronic treatment period was chosen to mimic the treatment time necessary to ameliorate primary and secondary pathophysiological processes that occur due to deafferentation following stroke or traumatic brain injury. We focused on glutamatergic networks because of the documented effects of chronic global NMDAR blockade with d-APV on glutamatergic neurotransmission (Liao et al. 1999; Rao and Craig 1997). We hypothesized that unlike chronic treatment with d-APV, which increased excitatory glutamatergic transmission, chronic treatment with Ro25-6981 would decrease glutamatergic transmission. This hypothesis was based on our initial studies showing that seizure-like events (SLE) involving dentate granule cells were exacerbated following chronic treatment of organotypic hippocampal slice cultures with d-APV but dramatically reduced following chronic treatment with Ro25-6981 (Wang and Bausch 2004).
MATERIALS AND METHODS
Choice of antagonists.
Representative NMDAR antagonists were chosen on the basis of their pharmacological properties as described previously (He et al. 2012; Wang and Bausch 2004). Concentrations were selected to 1) elicit maximal antagonism while maintaining selectivity and 2) allow direct comparison with our previous studies (Bausch et al. 2006; Wang and Bausch 2004).
d-APV (50 μM) was chosen as the high-affinity competitive NMDAR antagonist because of its high affinity and selectivity for the NMDAR (Evans et al. 1982) and its frequent use in physiological experiments. d-APV (50 μM; IC100) completely blocks NMDAR-mediated responses and was included to facilitate comparison with previous studies documenting neuroplasticity following NMDAR blockade.
Ro25-6981 (1 μM) was selected as the GluN2B-selective antagonist because Ro25-6981 has high affinity and high specificity for GluN2B-containing NMDAR (Fischer et al. 1997). Ro25-6981 was chosen over ifenprodil because ifenprodil also interacts with β-adrenergic receptors, serotonin receptors, and calcium channels (Chenard et al. 1991; Church et al. 1994; McCool and Lovinger 1995). Ro25-6981 (1 μM) imparts maximal binding at GluN2B-containing NMDAR, but the degree of functional NMDAR inhibition is use dependent and thus is proportional to the level of NMDAR activation. The greatest NMDAR inhibition (∼80%) occurs at high glutamate concentrations (such as those seen during synaptic transmission), whereas a slight potentiation occurs at very low glutamate concentrations (such as those seen under normal physiological conditions at extrasynaptic sites) because ifenprodil derivatives such as Ro25-6981 increase glutamate affinity for NMDAR (Fischer et al. 1997; Kew et al. 1996).
NVP-AAM077 (50 nM) was chosen as the GluN2A-preferring antagonist because of its preference for GluN2A-containing NMDAR at this concentration (Auberson et al. 2002). NVP-AAM077 (50 nM) corresponds to an ∼IC75 at GluN1/GluN2A and ∼IC25 at GluN1/GluN2B when rodent NMDAR were transfected into Xenopus oocytes and glutamate concentrations used to activate the receptors were normalized to EC50 values for GluN1/GluN2A and GluN1/GluN2B (Neyton and Paoletti 2006). NVP-AAM077 also displays a twofold and sevenfold preference for GluN1/GluN2A over GluN1/GluN2C and GluN1/GluN2D, respectively (Feng et al. 2004). However, although low to moderate GluN2D is expressed in glutamatergic hippocampal principal neurons during early postnatal development (Monyer et al. 1994), after postnatal day 7 GluN2C and GluN2D are expressed only at very low levels in subsets of hippocampal formation interneurons (Monyer et al. 1994; Scherzer et al. 1998; Standaert et al. 1996). We showed previously that chronic Ro25-6981, but not NVP-AAM077, altered GABAergic transmission (He et al. 2012).
Organotypic hippocampal slice cultures.
Organotypic hippocampal slice cultures were used as a model of deafferentation-induced pathophysiology (for review see Bausch 2009). Slice cultures were prepared using the method of Stoppini et al. (1991) as described previously (Bausch et al. 2006; Bausch and McNamara 2000). All treatment of animals complied with National Institutes of Health, Department of Defense, and institutional guidelines and was approved by our Institutional Animal Care and Use Committee. Briefly, postnatal day 10–11 Sprague-Dawley rat pups (Taconic, Germantown, NY) were anesthetized with pentobarbital sodium and decapitated. Brains were removed. Hippocampi were cut into 400-μm transverse sections using a McIlwain tissue chopper and placed into Gey's balanced salt solution composed of (in mM) 137 NaCl, 5 KCl, 0.25 MgSO4, 1.5 CaCl2, 1.05 MgCl2, 0.84 Na2HPO4, 0.22 K2HPO4, 2.7 NaHCO3, and 41.6 glucose. The entorhinal cortex was removed, and the middle four to six slices of each hippocampus were placed onto tissue culture membrane inserts (Millipore, Bedford, MA) in a culture dish containing medium. Medium consisted of 50% minimum essential medium, 25% Hank's buffered salt solution, 25% heat-inactivated horse serum, 0.5% GlutaMax, 10 mM HEPES (all from Invitrogen, Carlsbad, CA), and 6.5 mg/ml glucose (pH 7.2). Slice cultures were maintained at 37°C under room air plus 5% CO2, and medium was changed three times per week. Slice cultures were treated with the following NMDAR antagonists from 0 days in vitro (DIV) through the end of the culture period (17–21 DIV): d-APV (50 μM; Tocris Cookson, Ellisville, MO), Ro25-6981 hydrochloride (1 μM; Sigma), or NVP-AAM077 (50 nM; kind gift from Dr. Yves Auberson, Novartis Institutes for Biomedical Research, Basel, Switzerland). For vehicle-treated slice cultures, drugs were omitted. Cultures treated with vehicle and NMDAR antagonists were always studied concurrently under identical experimental conditions. Only cultures that displayed bright, well-defined cell layers were used.
Electrophysiological recordings.
Whole cell recordings were conducted in dentate granule cells from the suprapyramidal blade of the granule cell layer following >20 min washout of treatment drugs as described previously (Bausch et al. 2006). Granule cells were chosen because our previous results showed differential effects of chronic treatment with different classes of NMDAR antagonists on seizures involving granule cells (Wang and Bausch 2004) and because the dentate granule cells are thought to act as a “gate” to invasion of pathological hyperexcitability into the hippocampus (Behr et al. 1996, 1998; Collins et al. 1983). Briefly, a single cultured slice attached to the tissue culture insert membrane was placed into a submerged recording chamber mounted to a Zeiss Axioskop microscope with infrared-differential interference contrast (IR-DIC) optics (Carl Zeiss, Thornwood, NY). Recordings were conducted at room temperature (RT) to minimize the likelihood of electrographic seizures during antagonist washout (Bausch et al. 2006; Bausch and McNamara 2000, 2004). Slice cultures were superfused (2–3 ml/min) with physiological recording buffer composed of (in mM) 124 NaCl, 4.9 KCl, 1.2 KH2PO4, 2.4 MgSO4, 2.5 CaCl2, 25.6 NaHCO3, and 10 glucose, equilibrated with 95% O2-5% CO2. In recordings of NMDAR-mediated currents, 2.4 mM MgSO4 was replaced with 2.4 mM Na2SO4. Tetrodotoxin (TTX, 1 μM; Sigma), d-APV (50 μM), bicuculline methiodide (10 μM; Tocris Cookson), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 10 μM; Tocris Cookson), 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide disodium salt (NBQX, 10 μM; Tocris Cookson), d,l-threo-β-benzyloxyaspartate (dl-TBOA, 50 μM; Tocris Cookson), Ro25-6981 (1 μM), and NVP-AAM077 (500 nM) were diluted immediately before use and acutely applied by bath superfusion. Recording pipettes were filled with (in mM) 125 K-gluconate, 13 KCl, 10 HEPES, 10 EGTA, and 2 MgATP (pH 7.2 with KOH) for all whole cell recordings except those measuring NMDAR-mediated currents. QX-314 bromide (5 mM; Tocris Cookson) was added to the pipette solution immediately before use in experiments recording spontaneous excitatory postsynaptic currents (sEPSCs) to block sodium channel-mediated action potentials in the recorded neuron. For miniature (mEPSCs) and tonic NMDAR-mediated currents, recording pipettes were filled with (in mM) 125 Cs-methanesulfonic acid, 13 CsCl, 10 HEPES, 10 EGTA, and 2 Na2ATP (pH 7.2 with CsOH). Recordings were excluded if the resting membrane potential (RMP) was more positive than −50 mV or series resistance varied more than 15%. Data were collected using a Multiclamp 700A amplifier (2-kHz analog filter) and Digidata 1320 analog-to-digital converter and were sampled at 10 kHz using pCLAMP software (Axon Instruments, Union City, CA).
Electrophysiological analyses.
MiniAnalysis software (Synaptosoft, Fort Lee, NJ) was used for analyses of all postsynaptic currents and generation of cumulative probability plots. Cumulative probability plots were generated from events pooled from all cells. Detection threshold for sEPSCs and mEPSCs was set at 8 pA, amplitude was measured at peak negativity, and rise and decay times were measured from 10 to 90% and 100 to 10%, respectively. Detection limits for NMDAR-mediated mEPSCs were similar, except thresholds were 10 pA for amplitude, 50 pA·ms for area, and >20 ms for decay. All events were then evaluated manually in a blinded manner for shape and decay.
“Tonic” NMDAR currents were high-pass filtered at 1 Hz (8-pole Bessel, 3-dB cutoff) and low-pass filtered at 1 kHz (8-pole Butterworth, 3-dB cutoff) using Clampfit software. Power spectra (0.61-Hz resolution) were generated from 1-min data segments immediately before application of 1) 0 mM Mg2+ recording buffer, 2) d,l-TBOA, 3) NVP-AAM077, 4) physiological recording buffer, and 5) d-APV, as well as 6) at the peak of the TBOA-induced tonic current. With the use of the Levenberg-Marquardt algorithm, the TBOA-induced spectra was fit with the sum of two Lorentzian functions: S(f) = S(0)1/[1 + (f/fc1)2] + S(0)2/[1 + (f/fc2)2], where S(f) is the power at a given frequency; S(0)1 and S(0)2 are the powers for component 1 and 2 at zero frequency, respectively; and fc1 and fc2 are the corner frequencies at which the spectral power is ½S(0)1 and ½S(0)2, respectively. Single-channel conductance was estimated using stationary mean-variance noise analysis. The mean noise and variance from the mean were calculated from a 200-ms data segment during the slow rise phase of the TBOA-induced tonic NMDAR-mediated current. Single-channel currents (I) were estimated as the slope of the linear portion of the mean-variance plot. Single-channel conductance (g) was calculated as g = I/V, where V is the −70-mV holding potential. Channel number was calculated as tonic current amplitude divided by single-channel current (Kohr et al. 1993; Tammaro and Ashcroft 2007).
Immunohistochemistry.
Slice cultures were fixed with 4% paraformaldehyde in 0.1 M phosphate buffer (PB; pH 7.4) containing 0.15 M NaCl (PBS) for 20 min, removed from the membrane, and processed for immunohistochemistry as described previously (Bausch et al. 2006). All steps were performed at RT unless stated otherwise. Briefly, free-floating slice cultures were pretreated sequentially with 70% ethanol, 100% methanol, 70% ethanol, and 7% streptavidin in PBS, followed by 7% biotin in PBS. Cultures were blocked for 1 h at 37°C with PBS containing 2% gelatin and 10% normal goat serum. Slice cultures were then processed for double immunofluorescence against microtubule-associated protein (MAP2) to label dendrites and vesicular glutamate transporter 1 (vGlut1) to label glutamatergic terminals. Slice cultures were incubated for 1 h at RT, followed by 36 h at 4°C with mouse monoclonal anti-MAP2 IgG (clone HM-2 ascites; Sigma) diluted 1:1,000 and guinea pig polyclonal anti-vGlut1 antibody [Chemicon (now Millipore, Billerica, MA)] diluted 1:1,000 in PBS containing 2% BSA, 10% normal goat serum, and 0.1% Triton X-100. Immunoreactivity was visualized with Alexa 488-conjugated goat anti-mouse IgG [Molecular Probes (now Life Technologies, Grand Island, NY)] diluted 1:1,000 and Alexa 555-conjugated goat anti-guinea pig IgG (Molecular Probes) diluted 1:1,000 in PBS containing 1% BSA and 0.1% Triton X-100 for 1 h. Slice cultures were then mounted onto subbed glass slides and coverslipped with Vectashield mounting medium (Vector, Burlingame, CA).
Images were collected using a Zeiss Pascal LSM5 confocal microscope, ×63 oil objective, and multitrack scanning with an argon laser and 405/488/543-nm excitation, 505- to 530-nm band-pass emission, and 560-nm long-pass emission filters. Acquisition parameters were adjusted to minimize photobleaching and background labeling and were identical across treatment groups. Z series were collected from 5–10 consecutive optical sections (2,048 × 2,048 pixels, 0.1 μm/pixel; 0.3-μm z-axis interval). Z-stack reconstructions were used to identify dendrites; granule cells were identified morphologically. Only dendrites clearly attributable to a single granule cell were used for further analyses. All analyses were performed on background-subtracted images. The number of vGlut1-immunoreactive (-IR) puncta apposed to a single granule cell dendrite was counted manually from individual optical sections. Puncta were counted only once, even if they appeared in more than one sequential image. The integrated intensity of vGlut1-IR puncta was measured following spectral separation to isolate vGlut1 immunoreactivity. Images were thresholded to define puncta area, and the intensity of vGlut1 immunoreactivity was measured within the thresholded region. Data from five consecutive serial sections for each granule cell dendrite were averaged. All quantitative analyses were performed with MetaMorph software (Molecular Devices, Downingtown, PA).
Statistics.
Investigators were blinded to experimental groupings for all data analyses. Parametric data are represented as means ± SE; nonparametric data are represented as medians. Most statistical analyses were performed with SigmaStat software (SPSS, Chicago, IL). Data fitting a parametric distribution were tested for significance using analysis of variance (ANOVA) with Holm-Sidak post hoc comparison (multiple groups) or t-test (two groups). Data fitting a nonparametric distribution were tested for significance using ANOVA on ranks. Significance was defined as P ≤ 0.05. Cumulative probability distributions were tested for significance with a two-tailed Kolmogorov-Smirnov (KS) test using MiniAnalysis software; significance was defined as P ≤ 0.025.
RESULTS
Large- and small-amplitude sEPSCs were differentially affected by chronic treatment with NMDAR antagonists.
To begin to assess changes in excitatory glutamatergic circuits following chronic treatment with NMDAR antagonists, we first recorded sEPSCs in individual dentate granule cells. As described previously (Bausch and McNamara 2004), sEPSCs in granule cells from vehicle-treated organotypic hippocampal slice cultures fell into two distinct amplitude distributions, <300 pA (sEPSCsmall) and >2 nA (sEPSClarge). Spontaneous EPSClarge were characterized by low frequency, long duration, and multiple large peaks (Fig. 1A, top; Fig. 1B, top; and Table 1) and mirrored temporally associated epileptiform bursts recorded simultaneously with field potential recordings (Fig. 1A, bottom). Spontaneous EPSClarge were abolished by TTX or by a combination of d-APV and CNQX but were only partially inhibited by either d-APV or CNQX alone (data not shown). These data suggest that sEPSClarge were mediated by action potential-dependent glutamatergic transmission in recurrent polysynaptic circuits containing both NMDAR and AMPA/kainate receptors (AMPAR/KAR).
Fig. 1.

Spontaneous excitatory postsynaptic currents (sEPSCs) in granule cells were increased in hippocampal slice cultures treated chronically with N-methyl-d-aspartate receptor (NMDAR) antagonists. Whole cell voltage-clamp recordings were conducted at a −70-mV holding potential in physiological recording buffer containing bicuculline methiodide (BMI; 10 μM) and pipette solution containing QX-314 (5 mM) to block action potentials. Cumulative probability plots were generated from events pooled from all cells. A: simultaneous whole cell voltage-clamp (top) and extracellular field potential recordings (bottom) show BMI-induced epileptiform burst activity (arrowheads) in a dentate granule cell and dentate gyrus, respectively, in a vehicle-treated hippocampal slice culture. B: expanded time scale shows a whole cell voltage-clamp recording of a large-amplitude, long-duration sEPSC (sEPSClarge; line) associated with an epileptiform burst. The boxed area after the burst is expanded in the inset showing individual small-amplitude sEPSCs (sEPSCsmall; *). Cumulative probability plots show increased sEPSC frequency (C) in granule cells from slice cultures treated with d-aminophosphonovaleric acid (d-APV) compared with all other treatment groups and increased sEPSC amplitude (D) and charge transfer (E) for d-APV and Ro25-6981 compared with vehicle. Insets in D and E are expansions of regions enclosed in boxes. Legend in C applies to C–E; the number of granule cells/slice cultures is indicated in parentheses. See Table 1 for means.
Table 1.
EPSCs in granule cells
| Treatment | n | Frequency, Hz | Amplitude, pA | Rise Time, ms | Decay Time, ms | Charge Transfer, pA·ms | Duration, ms | No. of Peaks | Interpeak Interval, ms |
|---|---|---|---|---|---|---|---|---|---|
| sEPSClarge | |||||||||
| Vehicle | 11 | 0.026 ± 0.003 | −5,110 ± 531 | ND | ND | ND | 1,874 ± 70 | 7.6 ± 0.4 | 123 ± 6 |
| d-APV | 11 | 0.030 ± 0.002 | −5,315 ± 244 | ND | ND | ND | 2,227 ± 119‡ | 9.7 ± 0.6†‡ | 107 ± 5 |
| Ro25-6981 | 11 | 0.013 ± 0.003*‡§ | −4,864 ± 411 | ND | ND | ND | 1,869 ± 84 | 6.2 ± 0.9 | 123 ± 9 |
| NVP-AAM077 | 11 | 0.026 ± 0.003 | −5,056 ± 403 | ND | ND | ND | 1,751 ± 63 | 6.9 ± 0.3 | 110 ± 4 |
| sEPSCsmall | |||||||||
| Vehicle | 11 | 1.12 ± 0.17 | −25.6 ± 1.1 | 1.9 ± 0.1 | 7.3 ± 0.3 | 126 ± 9 | ND | ND | ND |
| d-APV | 11 | 1.90 ± 0.23 | −31.7 ± 3.0 | 2.0 ± 0.1 | 7.8 ± 0.2 | 178 ± 16* | ND | ND | ND |
| Ro25-6981 | 11 | 1.10 ± 0.15 | −29.7 ± 0.8 | 2.0 ± 0.1 | 7.5 ± 0.3 | 159 ± 6 | ND | ND | ND |
| NVP-AAM077 | 11 | 1.27 ± 0.24 | −28.2 ± 1.9 | 1.9 ± 0.1 | 7.4 ± 0.2 | 145 ± 12 | ND | ND | ND |
| mEPSC | |||||||||
| Vehicle | 18 | 0.35 ± 0.08 | −16.9 ± 0.7 | 2.2 ± 0.1 | 7.2 ± 0.3 | 84 ± 6 | ND | ND | ND |
| d-APV | 14 | 0.45 ± 0.05 | −21.8 ± 0.9* | 2.1 ± 0.1 | 8.5 ± 0.3*†‡ | 124 ± 7*†‡ | ND | ND | ND |
| Ro25-6981 | 25 | 0.49 ± 0.06 | −18.1 ± 1.0 | 2.0 ± 0.1 | 6.8 ± 0.2§ | 87 ± 6 | ND | ND | ND |
| NVP-AAM077 | 23 | 0.30 ± 0.03 | −17.9 ± 1.0 | 2.2 ± 0.1 | 7.2 ± 0.2 | 89 ± 5 | ND | ND | ND |
| mEPSCAMPAR | |||||||||
| Vehicle | 18 | 0.25 ± 0.06 | −17.8 ± 1.0 | 2.4 ± 0.2 | 8.0 ± 0.4 | 98 ± 9 | ND | ND | ND |
| d-APV | 14 | 0.37 ± 0.05 | −20.1 ± 0.9‡ | 2.2 ± 0.1 | 8.0 ± 0.2 | 112 ± 8†‡ | ND | ND | ND |
| Ro25-6981 | 18 | 0.49 ± 0.06*‡ | −16.8 ± 0.7 | 1.9 ± 0.1* | 6.7 ± 0.2*§ | 77 ± 4 | ND | ND | ND |
| NVP-AAM077 | 20 | 0.26 ± 0.03 | −16.2 ± 0.7 | 2.2 ± 0.1 | 7.3 ± 0.2 | 80 ± 4 | ND | ND | ND |
| mEPSCNMDAR | |||||||||
| Vehicle | 24 | 0.035 ± 0.003 | −40 ± 1 | 17.1 ± 0.4 | 162 ± 10 | 2,663 | ND | ND | ND |
| d-APV | 23 | 0.007 ± 0.001*†‡ | −28 ± 1*†‡ | 15.8 ± 0.8 | 113 ± 8* | 1,340 | ND | ND | ND |
| Ro25-6981 | 21 | 0.019 ± 0.003*‡§ | −38 ± 1 | 14.9 ± 0.5*‡ | 98 ± 7* | 1,741 | ND | ND | ND |
| NVP-AAM077 | 26 | 0.034 ± 0.003 | −38 ± 1 | 17.4 ± 0.4 | 134 ± 5*† | 3,209 | ND | ND | ND |
Values are means ± SE except for the charge transfer of N-methyl-d-aspartate receptor (NMDAR)-mediated miniature excitatory postsynaptic currents (mEPSCNMDAR), which was measured from the averaged trace from all events; n = no. of granule cells/hippocampal slice cultures investigated. sEPSClarge and sEPSCsmall, large and small spontaneous excitatory postsynaptic current events, respectively; ND, not determined.
P < 0.05, different from vehicle.
P < 0.05, different from Ro25-6981;
Comparing the effects of chronic treatment with distinct classes of NMDAR antagonists, we found that chronic NMDAR blockade with d-APV significantly increased sEPSClarge duration and peak number compared with other NMDAR antagonists. Conversely, chronic inhibition of GluN2B-containing NMDAR with Ro25-6981 reduced sEPSClarge frequency by 50% compared with vehicle and all other treatment groups (Table 1). Spontaneous EPSClarge in cultures treated with NVP-AAM077 were similar to those in cultures treated with vehicle (Table 1). These findings are consistent with our hypothesis that chronic d-APV would increase, whereas chronic Ro25-6981 would decrease, glutamatergic transmission. Findings are also consistent with a previous report describing opposite effects on pathological hyperactivity in excitatory networks following chronic NMDAR blockade with d-APV and chronic inhibition of GluN2B-containing receptors with Ro25-6981 (Bausch et al. 2006; Wang and Bausch 2004).
In contrast to sEPSClarge, sEPSCsmall were characterized by higher frequency, shorter decay, and a single peak (Fig. 1B, bottom, and Table 1). Consistent with our hypothesis that chronic d-APV would increase glutamatergic transmission, chronic NMDAR blockade with d-APV increased sEPSCsmall frequency and modestly increased amplitude and charge transfer compared with vehicle (Table 1 and Fig. 1, C–E; P ≤ 0.0001, KS test). However, contrary to our hypothesis that chronic Ro25-6981 would decrease glutamatergic transmission, chronic inhibition of GluN2B-containing NMDAR with Ro25-6981 also modestly increased sEPSCsmall amplitude and charge transfer (Fig. 1, D and E; P ≤ 0.025 and P ≤ 0.005, respectively, KS test).
Action potential threshold was slightly more negative following chronic NMDAR blockade and inhibition of GluN2A-containing NMDAR.
Alterations in action potential and membrane properties could contribute to changes in sEPSCs and therefore were examined using whole cell current-clamp recordings. Comparison between treatment groups revealed no differences in granule cell 1) resting membrane potential, 2) input resistance, or 3) action potential number at threshold or following a 200-pA input (Table 2). Action potential threshold in Ro25-6981-treated cultures was similar to that in vehicle-treated cultures. However, action potential threshold was slightly but significantly more negative (−1.5 mV) in granule cells from cultures treated with d-APV or NVP-AAM077 compared with vehicle (Table 2). These findings implicate GluN2A as a modulator of sodium channel function. Although this small shift in action potential threshold is unlikely to elicit dramatic hyperexcitability by itself, it could act in concert with other pro-excitatory changes in d-APV- and NVP-AAM077-treated cultures. Hence, subsequent experiments utilized TTX to prevent action potential generation.
Table 2.
Granule cell membrane properties
| Treatment | n | RMP, mV | Rin, mΩ | AP Threshold, mV | No. of APs at Threshold | No. of APs at 200 pA |
|---|---|---|---|---|---|---|
| Vehicle | 50 | −58.5 ± 0.2 | 149 ± 7 | −36.4 ± 0.3 | 2.0 | 6.0 |
| d-APV | 41 | −59.3 ± 0.3 | 136 ± 7 | −37.9 ± 0.3* | 2.0 | 6.0 |
| Ro25-6981 | 53 | −59.0 ± 0.3 | 148 ± 6 | −36.9 ± 0.3 | 2.0 | 6.0 |
| NVP-AAM077 | 59 | −59.1 ± 0.3 | 136 ± 5 | −37.4 ± 0.2* | 2.0 | 6.0 |
Values are means ± SE for resting membrane potential (RMP), input resistance (Rin), and action potential (AP) threshold and medians for no. of APs elicited at threshold or following a 200-pA current; n = no. of granule cells/hippocampal slice cultures investigated.
P < 0.05, different from vehicle (ANOVA).
Miniature EPSCs were altered following chronic treatment with NMDAR antagonists.
We next recorded action potential-independent mEPSCs (Table 1) in dentate granule cells. AMPAR/KAR- and NMDAR-mediated mEPSCs (mEPSCAMPAR and mEPSCNMDAR, respectively) were recorded individually to more precisely define changes in the two major ionotropic components of glutamatergic transmission. Quantitative analyses of mEPSCAMPAR showed that chronic NMDAR blockade with d-APV increased mEPSCAMPAR frequency compared with vehicle as measured by cumulative probability distributions (Fig. 2B; P ≤ 0.0001, KS test), whereas chronic inhibition of GluN2B-containing NMDAR with Ro25-6981 increased mEPSCAMPAR frequency compared with vehicle as measured by mean values and cumulative probability distributions (Table 1 and Fig. 2B; P ≤ 0.0001, KS test). These data suggest increased excitatory synapse number and/or glutamate release probability. To differentiate between these possibilities, we used double-immunofluorescence labeling for vGlut1 to detect presynaptic glutamatergic terminals and MAP2 to identify dendrites. Quantitative analyses revealed that the intensity of vGlut1 immunoreactivity in individual puncta apposed to granule cell dendrites was increased in cultures treated with d-APV compared with vehicle (Fig. 3C), suggesting a possible upregulation in vGlut1 transporter expression at glutamatergic synapses. More importantly, vGlut1-positive contacts onto granule cell dendrites were increased in cultures treated with d-APV and, to a lesser extent, NVP-AAM077 compared with vehicle (Fig. 3, A and B), but the distribution of vGlut1-positive contacts along granule cell dendrites was not significantly altered (Fig. 3D). These findings are consistent with previous reports showing that chronic NMDAR blockade with d-APV promotes synaptic reorganization (Bausch et al. 2006; Lin and Constantine-Paton 1998; McKinney et al. 1999). In contrast, Ro25-6981 had no significant effect on the number of vGlut1-positive contacts onto granule cell dendrites (Fig. 3, A–D). These data suggest that increased mEPSCAMPAR frequency (d-APV, Fig. 2B; Ro25-6981, Table 1 and Fig. 2B) most likely arises from increased excitatory synapse number following chronic d-APV and from increased neurotransmitter release probability following chronic Ro25-6981. Activation of presynaptic GluN2B-containing NMDARs can facilitate glutamate release and increase mEPSC frequency (Dalby and Mody 2003; Jourdain et al. 2007; Sjostrom et al. 2003; Woodhall et al. 2001). Therefore, increased presynaptic NMDAR function and subsequent increased release probability could explain increased mEPSC frequency following chronic Ro25-6981.
Fig. 2.
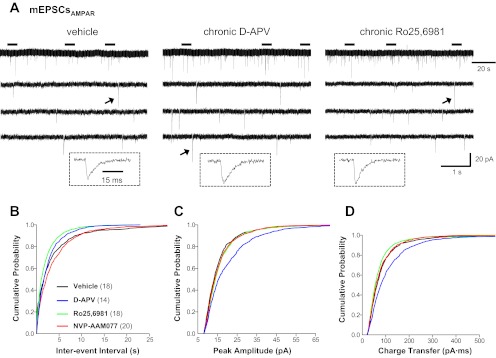
AMPA/kainate receptor (AMPAR/KAR)-mediated miniature EPSCs (mEPSCAMPAR) in granule cells were differentially altered in hippocampal slice cultures treated chronically with d-APV and Ro25-6982. Recordings were obtained at a −70-mV holding potential in physiological recording buffer containing tetrodotoxin (TTX; 1 μM), BMI (10 μM), and d-APV (50 μM). Cumulative probability plots were generated from events pooled from all cells. A: representative traces of mEPSCAMPAR recorded in granule cells from cultures treated chronically with vehicle (left), d-APV (middle), or Ro25-6981 (right). The 3 lower traces are expanded time scales of the periods marked by horizontal bars above the top trace. Traces in insets are an expanded time scale of the mEPSCAMPAR marked with an arrow. Cumulative probability plots show increased mEPSCAMPAR frequency (B) in granule cells from slice cultures treated with Ro25-6981 and d-APV. Miniature EPSCAMPAR peak amplitude (C) and charge transfer (D) were increased following chronic d-APV; charge transfer was decreased slightly following chronic Ro25-6981. Vertical scale bar in A, bottom right, applies to all traces in A. Horizontal scale bar in A, top right, applies to all top traces in A; that in A, bottom right, to all lower traces in A; and that in A, left inset, to all traces in insets. Legend in B applies to B–D; the number of granule cells/slice cultures is indicated in parentheses. See Table 1 for means.
Fig. 3.

The number and intensity of vesicular glutamate transporter 1 (vGlut1)-positive excitatory synapses onto granule cell dendrites were increased in hippocampal slice cultures treated chronically with d-APV or NVP-AAM077. Cultures were double-labeled for vGlut1 (red, for presynaptic glutamatergic terminals) and microtubule-associated protein (MAP2; green, for dendrites) using immunofluorescence. A: single optical sections from representative granule cells illustrate vGlut1-immunoreactive (vGlut1-IR) puncta (arrowheads) along a primary apical dendrite. Yellow indicates overlap of vGlut1 and MAP2 immunoreactivities. Surrounding elements were subtracted digitally for clarity. B: the number of vGlut1-IR puncta apposed to single granule cell dendrites was increased in cultures treated chronically with d-APV or NVP-AAM077. C: integrated intensity of vGlut1-IR puncta apposed to single granule cell dendrites was increased in cultures treated chronically with d-APV. D: plots of vGlut1-IR puncta distribution (10-μm bins) along apical granule cell dendrites substantiate compiled means. Inset: cumulative probability plots show no significant differences in the distribution of vGlut1-IR puncta along granule cell dendrites (P > 0.025, 2-tailed Kolmogorov-Smirnov test). Data were collected from 3–4 granule cell dendrites/slice culture in 5–7 individual slice cultures/independent experiments. *P < 0.05: **P < 0.01, different from vehicle. +P <0.05; ++P <0.001, different from d-APV (Kruskal Wallis ANOVA by ranks with Dunn's post hoc comparison).
Chronic d-APV also increased mEPSCAMPAR amplitude and charge transfer (Fig. 2, C and D; P ≤ 0.0001, KS test). In contrast, chronic Ro25-6981 slightly reduced mEPSCAMPAR charge transfer (Fig. 2D; P ≤ 0.005, KS test). These data suggest that, in line with our hypothesis that chronic d-APV would increase, whereas chronic Ro25-6981 would decrease, glutamatergic transmission, chronic NMDAR blockade with d-APV enhanced, whereas chronic inhibition of GluN2B-containing NMDAR with Ro25-6981 diminished, AMPAR/KAR function.
We next recorded mEPSCNMDAR at a holding potential of −45 mV, because the 0 mM Mg2+ recording buffer required to relieve Mg2+ block of NMDAR caused very large increases in baseline noise at −70 mV (see Fig. 6A), which made mEPSCNMDAR extremely difficult to detect and analyze. Miniature EPSCNMDAR were abolished by acute application of non-subunit-selective NMDAR antagonists (not shown). Quantitative analysis of mEPSCNMDAR revealed that mEPSCNMDAR frequency was decreased profoundly in granule cells from cultures treated chronically with d-APV (80%) or Ro25-6981 (46%) compared with vehicle (Table 1 and Fig. 4B; P = 0.0000 and P ≤ 0.005, respectively, KS test). In addition, mEPSCNMDAR amplitude was reduced following chronic NMDAR blockade with d-APV (Table 1 and Fig. 4C; P = 0.0000, KS test.). Insufficient washout of d-APV was unlikely to contribute to reduced mEPSCNMDAR because reduced mEPSCNMDAR frequency and amplitude persisted for up to 24 h after d-APV washout (Fig. 4, D and E). Since reduced mEPSCNMDAR frequency occurred in treatment groups displaying increased mEPSC frequency and in recording paradigms favoring NMDAR activation, these data suggest a dramatic reduction in the number of synapses containing functional NMDAR. Alternatively, the reduced mEPSCNMDAR frequency may be tied to diminished mEPSCNMDAR amplitude and subsequent failure to resolve the small synaptic events. Regardless of the reasons for reduced mEPSCNMDAR frequency, these findings were surprising because previous immunohistochemical studies reported increased postsynaptic NMDAR clusters following chronic NMDAR blockade with d-APV (O'Brien et al. 1998; Rao and Craig 1997). The most parsimonious explanations for this discrepancy include nonfunctional NMDAR, perisynaptically clustered NMDAR that were not activated by constitutive presynaptic glutamate release, nascent NMDAR-containing synapses without functional presynaptic elements (Liao et al. 1999), or reduced glutamate release probability at NMDAR only-containing synapses.
Fig. 6.

Tonic NMDAR-mediated currents and NMDAR channel number were increased in granule cells from Ro25-6981-treated hippocampal slice cultures. Recordings were conducted at a −70-mV holding potential in 0 mM Mg2+ recording buffer containing d,l-threo-β-benzyloxyaspartate (dl-TBOA; 50 μM), TTX (1 μM), BMI (10 μM), and NBQX (10 μM). Representative trace (A) and power spectra (B) of a tonic NMDAR-mediated current recorded in a granule cell from a vehicle-treated slice culture are shown. Note the change in holding current and increase in noise associated with application of 0 mM Mg2+ recording buffer that was accentuated by addition of the glutamate reuptake inhibitor TBOA and reversed by acute application of Ro25-6981 (1 μM) + NVP-AAM077 (500 nM) but not by Ro25-6981 (1 μM) alone. Acute application of 500 nM NVP-AAM077 corresponds to an approximate IC95 at GluN1/GluN2A and IC75 at GluN1/GluN2B at rodent NMDAR expressed in Xenopus oocytes (Neyton and Paoletti 2006) and thus is not subunit selective at this concentration. C: compiled data show significantly increased tonic NMDAR-mediated current amplitude (maximal change in holding current) in cultures treated with Ro25-6981. *P < 0.05, different from vehicle. #P < 0.05, different from d-APV. &P < 0.05, different from NVP-AAM077 (ANOVA with Holm-Sidak post hoc comparison). The number of granule cells/slice cultures is indicated in parentheses.
Fig. 4.

NMDAR-mediated mEPSCs (mEPSCNMDAR) in granule cells were dramatically reduced in d-APV- and Ro25-6981-treated hippocampal slice cultures. Recordings were conducted at a −45-mV holding potential in 0 mM Mg2+ recording buffer containing TTX (1 μM), BMI (10 μM), and 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide disodium salt (NBQX; 10 μM). A holding potential of −45 mV was chosen over −70 mV because 0 mM Mg2+ recording buffer caused very large increases in baseline noise at −70 mV (see Fig. 6A), which made mEPSCNMDAR extremely difficult to detect and analyze. Cumulative probability plots were generated from events pooled from all cells and included 1,456 mEPSCNMDAR for vehicle, 272 for chronic d-APV, 723 for chronic Ro25-6981, and 1,456 for chronic NVP-AAM077. A: representative mEPSCNMDAR (*) recorded in granule cells from cultures treated chronically with vehicle (left), d-APV (middle), or Ro25-6981 (right). The 3 lower traces are expanded time scales of the periods marked by horizontal bars above the top trace. Traces in insets are an expanded time scale of the mEPSCNMDAR marked with an arrow. Cumulative probability plots show that mEPSCNMDAR frequency (B) and amplitude (C) were dramatically reduced in granule cells from d-APV-treated cultures; smaller decreases in frequency are shown for Ro25-6981. When d-APV was removed from the culture medium 24 h before recordings, the chronic d-APV-induced decreases in mEPSCNMDAR frequency (D) and amplitude (E) persisted. Vertical scale bar in A, top right, applies to all traces in A. Horizontal scale bar in A, top right, applies to all top traces in A; that in A, bottom right, to all lower traces in A; and that in A, left inset, to all traces in insets. Legend in B applies to B and C, and that in D applies to D and E; the number of granule cells/slice cultures is indicated in parentheses. See Table 1 for means.
Further analyses of mEPSCNMDAR revealed that mEPSCNMDAR decay was decreased in all chronic NMDAR antagonist treatment groups but was most pronounced following treatment with d-APV or Ro25-6981 (Table 1). These data suggest decreased synaptic localization of GluN2B subunits, which convey longer decay times than GluN2A to NMDAR-mediated currents (Flint et al. 1997; Stocca and Vicini 1998). To investigate this possibility, the GluN2B antagonist Ro25-6981 was applied acutely following mEPSCNMDAR recordings. Acute application of Ro25-6981 reduced mEPSCNMDAR frequency in cultures treated chronically with vehicle (27%) or NVP-AAM077 (43%) but had no significant effect in cultures treated chronically with d-APV or Ro25-6981 (Fig. 5A). In contrast, acute application of Ro25-6981 reduced mEPSCNMDAR amplitude in chronic vehicle, d-APV, and Ro25-6981 treatment groups (Fig. 5B). Surprisingly, acute application of Ro25-6981 had no significant effect on mEPSCNMDAR decay in any chronic treatment group (Fig. 5C). However, acute Ro25-6981 effects on mEPSCNMDAR may underestimate synaptic GluN2B levels. When GluN2B is associated with other GluN2 subunits in triheteromeric NMDAR (GluN1/GluN2x/GluN2y), Ro25-6986 and ifenprodil still bind with high affinity (Chazot et al. 2002; Hatton and Paoletti 2005), but the degree of inhibition is much reduced (Hatton and Paoletti 2005). Thus, despite the lack of acute Ro25-6981 effect on mEPSCNMDAR decay, these findings suggest that chronic treatment with d-APV or Ro25-6981 reduced the number of individual synapses containing functional GluN2B-containing NMDAR. Taken together, these roughly similar alterations following chronic d-APV and chronic Ro25-6981 suggest that altered synaptic NMDAR alone do not contribute significantly to the opposite effects of chronic d-APV and chronic Ro25-6981 on pathological circuit hyperexcitability.
Fig. 5.

Miniature EPSCNMDAR in granule cells from hippocampal slice cultures were mediated predominantly by GluN2A-containing NMDAR. Recordings were conducted at a −45-mV holding potential in 0 mM Mg2+ recording buffer containing TTX (1 μM), BMI (10 μM), and NBQX (10 μM). A holding potential of −45 mV was chosen over −70 mV for reasons stated in Fig. 4 legend. Bar graphs were generated from 1,289 (decay)–1,703 (frequency, amplitude) mEPSCNMDAR for vehicle, 271 for chronic d-APV, 581–705 for chronic Ro25-6981, and 1,129–2,082 for chronic NVP-AAM077. Bar graphs show significant decreases in mEPSCNMDAR frequency (A) and amplitude (B) following acute application of Ro25-6981, but no significant change in decay (C). *P < 0.05; **P ≤ 0.01, different from value before acute Ro25-6981 application (paired t-test). Legend in A is for A–C; the number of granule cells/slice cultures is indicated in parentheses.
Tonic NMDAR currents were enhanced following chronic treatment with Ro25-6981.
Given the apparent reduction in functional postsynaptic GluN2B-containing NMDAR following chronic NMDAR antagonist treatment, we next focused on tonic NMDAR-mediated currents. Tonic NMDAR currents are mediated primarily by extrasynaptic NMDAR (Le Meur et al. 2007), which historically were thought to comprise GluN2B-containing NMDARs (Sheng et al. 1994; Tovar and Westbrook 1999). Tonic currents were enhanced with dl-TBOA (50 μM) to eliminate confounds associated with potential alterations in transporter expression/function following chronic NMDAR antagonist treatment (for example see Fig. 3C, d-APV). TBOA is a potent, highly selective competitive blocker of excitatory amino acid transporters. TBOA is not a neuronal transporter substrate and thus does not elicit transport currents or chloride fluxes (Shimamoto et al. 1998). TBOA-enhanced tonic currents were associated with a shift in holding current and increased recording noise (Fig. 6, A and B), both of which were blocked by acute application of d-APV (50 μM; data not shown), Mg2+-containing recording buffer (2.5 mM; data not shown), or a high concentration of NVP-AAM007 [500 nM, not subunit selective (Fig. 6, A and B)]. Tonic currents were unaffected by acute application of GluN2B-selective antagonist Ro25-6981 (Fig. 6, A and B) or specific blockade of neuronal vesicular glutamate release with tetanus toxin (Fellin et al. 2004; Schiavo et al. 1992) (data not shown). These findings suggest that the shift in holding current and increased noise required nonneuronal glutamate release and were mediated by non-GluN2B-containing NMDAR or triheteromeric NMDAR containing GluN2B (see results for mEPSCNMDAR). Comparison between treatment groups revealed a significant 78% increase in tonic NMDAR current amplitude following chronic inhibition of GluN2B-containing NMDAR with Ro25-6981 compared with all other treatment groups (Fig. 6C). Noise analysis estimates suggested that this change in amplitude was associated with a dramatic (154%) increase in open NMDAR channel number compared with vehicle (not shown; P < 0.05, ANOVA with Holm-Sidak post hoc comparison). No significant changes in tonic NMDAR current were apparent following chronic treatment with d-APV or NVP-AAM007. These findings show an association between increased NMDAR-mediated tonic currents and decreased pathological glutamatergic circuit activity.
DISCUSSION
To our knowledge, this is the first report demonstrating changes in glutamatergic circuits following chronic partial inhibition of GluN2B-containing NMDAR with Ro25-6981 or GluN2A-containing NMDAR with NVP-AAM077 and comparison of their effects with complete NMDAR blockade with d-APV. Our major findings can be summarized as follows. Chronic NMDAR blockade with d-APV significantly increased sEPSClarge duration and superimposed peak number, whereas chronic inhibition of GluN2B-containing NMDAR with Ro25-6981 reduced sEPSClarge frequency compared with vehicle and all other treatment groups. These findings are consistent with our hypothesis that chronic d-APV would increase, whereas chronic Ro25-6981 would decrease, glutamatergic transmission. Findings also are consistent with our previous reports describing opposite effects on pathological hyperactivity in excitatory networks following chronic complete global NMDAR blockade with d-APV and chronic partial inhibition of GluN2B-containing receptors with Ro25-6981 (Bausch et al. 2006; Wang and Bausch 2004). Potential mechanisms underlying these opposite effects on circuit behavior can be divided into presynaptic, postsynaptic, extrasynaptic, and intrinsic excitability (see Fig. 7 for schematic summary). In terms of presynaptic changes, chronic treatment with either d-APV or Ro25-6981 increased sEPSCsmall and mEPSCAMPAR frequency. Immunohistochemical labeling of putative glutamatergic synapses suggested that increased frequency likely arose from increased excitatory synapse number for chronic d-APV and increased neurotransmitter release probability for chronic Ro25-6981. The latter is consistent with the possibility of increased presynaptic GluN2B-containing NMDAR. On the postsynaptic side, chronic d-APV dramatically increased mEPSCAMPAR amplitude and charge transfer and profoundly decreased mEPSCNMDAR frequency, amplitude, and decay, suggesting a very large increase in the ratio of postsynaptic AMPAR/NMDAR function. In contrast, Ro25-6981 decreased mEPSCAMPAR charge transfer and modestly decreased mEPSCNMDAR frequency and decay, suggesting a downward scaling of synaptic AMPAR and NMDAR function without a dramatic change in the ratio of postsynaptic AMPAR/NMDAR function. For extrasynaptic alterations, TBOA-enhanced tonic NMDAR current amplitude was significantly enhanced only by chronic Ro25-6981. For intrinsic excitability, action potential threshold was slightly more negative following either chronic d-APV or chronic NVP-AAM077. Taken together, the predominant pro-excitatory effects of chronic d-APV are consistent with our hypothesis that chronic d-APV would increase glutamatergic transmission and likely contribute to the increased network excitability and SLEs reported previously following chronic NMDAR blockade (Bausch et al. 2006; McKinney et al. 1999; O'Brien et al. 1998; Wang and Bausch 2004). The small effects of chronic NVP-AAM077 on action potential threshold and synapse number are consistent with its minimal effects on circuit function. The chronic Ro25-6981-induced downward scaling of synaptic AMPAR and NMDAR function is consistent with our hypothesis that chronic Ro25-6981 would decrease glutamatergic function, but changes in presynaptic and extrasynaptic components were opposite to our expected results.
Fig. 7.

Schematic of major findings. Presynaptic NMDAR are depicted to show NMDAR-mediated changes in presynaptic function and do not validate presynaptic NMDAR localization. Increased numbers of extrasynaptic NMDAR are depicted to illustrate increased numbers of activated receptors and do not validate increased expression/localization of NMDAR at extrasynaptic sites.
The role of extrasynaptic NMDAR in neuronal excitability and network function.
Changes in tonic NMDAR-mediated currents are difficult to place into the context of network function without a better understanding of the physiological role of extrasynaptic NMDAR, which mediate tonic NMDAR-mediated currents. Extrasynaptic NMDAR are activated following high-frequency stimulation and subsequent synaptic glutamate spillover and/or nonneuronal glutamate release. Previous reports suggest that tonic NMDAR-mediated currents activated by ambient endogenous glutamate levels elicit plateau potentials and enhanced neuronal excitability via increased excitatory input-output gain (Sah et al. 1989; Suzuki et al. 2008). However, other studies reported no significant effect of small-amplitude tonic NMDAR-mediated currents on neuronal excitability (Cavelier and Attwell 2005; Le Meur et al. 2007). Theoretically, extrasynaptic NMDAR activation also may lead to a shunting effect, analogous to the shunting inhibition seen following extrasynaptic GABAA receptor activation, which reduces temporal integration of excitatory synaptic inputs and subsequent action potential generation (Brizzi et al. 2004; Cope et al. 2005; Farrant and Nusser 2005; Semyanov et al. 2004). These seemingly disparate mechanisms are not mutually exclusive. Increased input-output gain may predominate at normal resting membrane potentials, whereas shunting could prevail when dendrites are depolarized to near the NMDAR reversal potential during periods of high network activity and seizures. Further studies that selectively modulate extrasynaptic NMDAR without affecting synaptic or presynaptic NMDAR are clearly necessary to definitively document the functional consequences of extrasynaptic NMDAR activation under diverse physiological and pathological conditions.
Our finding showing that tetanus toxin blockade of neuronal vesicular glutamate release had no affect on TBOA-enhanced tonic NMDAR-mediated currents suggests the possibility of astrocytic glutamate release. This raises the possibility that chronic Ro25-6981-induced increases in tonic currents could have arisen from altered astrocytic function and astrocytic glutamate release rather than increased extrasynaptic NMDAR expression/localization. NMDAR are expressed in astrocytes (Conti et al. 1996; Lalo et al. 2006; Van Bockstaele and Colago 1996), and GluN2B-containing NMDAR are upregulated in astrocytic processes following central nervous system insults (Krebs et al. 2003). However, the functional role of astrocytic GluN2B-containing NMDAR and astrocytic glutamate release were not specifically investigated in this study, and their impact on circuit excitability remains elusive.
Chronic Ro25-6981-induced alterations in the context of other circuit alterations.
At first pass, chronic Ro25-6981-mediated increases in sEPSC measures and mEPSCAMPAR frequency would suggest increased excitability in glutamatergic circuits rather than the reduced pathological glutamatergic circuit activity observed following chronic Ro25-6981 treatment. However, shifts in sEPSCs and mEPSCs arise from a plethora of synaptic inputs onto individual neurons, and increased sEPSC and mEPSC measures may or may not be associated with circuits mediating pathological hyperexcitability. Furthermore, chronic Ro25-6981-mediated effects on circuit excitability may depend on the mechanisms underlying changes in sEPSC and mEPSCAMPAR frequency (i.e., increased synapse number vs. potential increased release probability). Finally, excitatory glutamatergic transmission is subject to GABAergic modulation, and we showed previously (He et al. 2012) that action potential-dependent GABA release was altered following chronic Ro25-6981, but not d-APV or NVP-AAM077, treatment. Any or all of these possibilities may contribute to the discrepancy between EPSC frequency measures and circuit excitability.
Summary.
Chronic NMDAR inhibition altered glutamatergic circuitry, individual glutamatergic synapses, ionotropic glutamate receptor function, and action potential threshold in a model of deafferentation-induced pathophysiology. These changes may contribute to the therapeutic efficacy of NMDAR antagonists. Conversely, they may represent homeostatic changes that have limited effects on disease processes. Further studies are needed to differentiate between these possibilities.
GRANTS
This work was supported by the Defense Brain and Spinal Cord Injury Program, Congressionally Directed Medical Research Programs Award W81XWH-04-1-0065/PR030035 and National Institute of Neurological Disorders and Stroke Grant NS045964 (to S. B. Bausch) and a Uniformed Services University of the Health Sciences (USUHS) Intramural Student Research Award (to S. He).
DISCLAIMER
The opinions or assertions contained herein are the private ones of the authors and are not to be construed as official or reflecting the views of the Department of Defense or USUHS.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.H., Y.W., and S.B.B. conception and design of research; S.H., L.-R.S., and Y.W. performed experiments; S.H., L.-R.S., and Y.W. analyzed data; S.H., L.-R.S., Y.W., and S.B.B. interpreted results of experiments; S.H., L.-R.S., and Y.W. prepared figures; S.H. and S.B.B. drafted manuscript; S.H., L.-R.S., and S.B.B. edited and revised manuscript; S.H., L.-R.S., Y.W., and S.B.B. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Yves Auberson for the kind gift of NVP-AAM077, Drs. Boris Gafurov and YingBing Liu for helpful discussions, and W. Bradley Rittase for assistance in preparing figures.
Preliminary portions of this work were presented previously as a short Workshop on the Neurobiology of Epilepsy (WONEOP) report (Bausch et al. 2010).
Current address of Y. Wang: Epilepsy and Headache Group, Department of Neurology, First Hospital of Anhui Medical University, Jixi Road 218, Hefei 230022, China.
REFERENCES
- Auberson YP, Allgeier H, Bischoff S, Lingenhoehl K, Moretti R, Schmutz M. 5-Phosphonomethylquinoxalinediones as competitive NMDA receptor antagonists with a preference for the human 1A/2A, rather than 1A/2B receptor composition. Bioorg Med Chem Lett 12: 1099–1102, 2002 [DOI] [PubMed] [Google Scholar]
- Bausch SB, He S, Dong Y. Inverse relationship between seizure expression and extrasynaptic NMDAR function following chronic NMDAR inhibition. Epilepsia 51, Suppl 3: 102–105, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bausch SB. Organotypic hippocampal slice cultures as a model of limbic epileptogenesis. In: Neuromethods. Innovations in Epilepsy Research, edited by Baraban SC. New York: Humana, 2009, vol. 40, p. 183–201 [Google Scholar]
- Bausch SB, He S, Petrova Y, Wang XM, McNamara JO. Plasticity of both excitatory and inhibitory synapses is associated with seizures induced by removal of chronic blockade of activity in cultured hippocampus. J Neurophysiol 96: 2151–2167, 2006 [DOI] [PubMed] [Google Scholar]
- Bausch SB, McNamara JO. Contributions of mossy fiber and CA1 pyramidal cell sprouting to dentate granule cell hyperexcitability in kainic acid-treated hippocampal slice cultures. J Neurophysiol 92: 3582–3595, 2004 [DOI] [PubMed] [Google Scholar]
- Bausch SB, McNamara JO. Synaptic connections from multiple subfields contribute to granule cell hyperexcitability in hippocampal slice cultures. J Neurophysiol 84: 2918–2932, 2000 [DOI] [PubMed] [Google Scholar]
- Behr J, Gloveli T, Gutierrez R, Heinemann U. Spread of low Mg2+ induced epileptiform activity from the rat entorhinal cortex to the hippocampus after kindling studied in vitro. Neurosci Lett 216: 41–44, 1996 [DOI] [PubMed] [Google Scholar]
- Behr J, Lyson KJ, Mody I. Enhanced propagation of epileptiform activity through the kindled dentate gyrus. J Neurophysiol 79: 1726–1732, 1998 [DOI] [PubMed] [Google Scholar]
- Brizzi L, Meunier C, Zytnicki D, Donnet M, Hansel D, D'Incamps BL, Van Vreeswijk C. How shunting inhibition affects the discharge of lumbar motoneurones: a dynamic clamp study in anaesthetized cats. J Physiol 558: 671–683, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavelier P, Attwell D. Tonic release of glutamate by a DIDS-sensitive mechanism in rat hippocampal slices. J Physiol 564: 397–410, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chazot PL, Lawrence S, Thompson CL. Studies on the subtype selectivity of CP-101,606: evidence for two classes of NR2B-selective NMDA receptor antagonists. Neuropharmacology 42: 319–324, 2002 [DOI] [PubMed] [Google Scholar]
- Chenard BL, Shalaby IA, Koe BK, Ronau RT, Butler TW, Prochniak MA, Schmidt AW, Fox CB. Separation of alpha 1 adrenergic and N-methyl-d-aspartate antagonist activity in a series of ifenprodil compounds. J Med Chem 34: 3085–3090, 1991 [DOI] [PubMed] [Google Scholar]
- Church J, Fletcher EJ, Baxter K, MacDonald JF. Blockade by ifenprodil of high voltage-activated Ca2+ channels in rat and mouse cultured hippocampal pyramidal neurones: comparison with N-methyl-d-aspartate receptor antagonist actions. Br J Pharmacol 113: 499–507, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins RC, Tearse RG, Lothman EW. Functional anatomy of limbic seizures: focal discharges from medial entorhinal cortex in rat. Brain Res 280: 25–40, 1983 [DOI] [PubMed] [Google Scholar]
- Conti F, DeBiasi S, Minelli A, Melone M. Expression of NR1 and NR2A/B subunits of the NMDA receptor in cortical astrocytes. Glia 17: 254–258, 1996 [DOI] [PubMed] [Google Scholar]
- Cope DW, Hughes SW, Crunelli V. GABAA receptor-mediated tonic inhibition in thalamic neurons. J Neurosci 25: 11553–11563, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalby NO, Mody I. Activation of NMDA receptors in rat dentate gyrus granule cells by spontaneous and evoked transmitter release. J Neurophysiol 90: 786–797, 2003 [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev 51: 7–61, 1999 [PubMed] [Google Scholar]
- Dingledine R, McBain CJ, McNamara JO. Excitatory amino acid receptors in epilepsy. Trends Pharmacol Sci 11: 334–338, 1990 [DOI] [PubMed] [Google Scholar]
- Evans RH, Francis AA, Jones AW, Smith DA, Watkins JC. The effects of a series of omega-phosphonic alpha-carboxylic amino acids on electrically evoked and excitant amino acid-induced responses in isolated spinal cord preparations. Br J Pharmacol 75: 65–75, 1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABAA receptors. Nat Rev Neurosci 6: 215–229, 2005 [DOI] [PubMed] [Google Scholar]
- Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron 43: 729–743, 2004 [DOI] [PubMed] [Google Scholar]
- Feng B, Tse HW, Skifter DA, Morley R, Jane DE, Monaghan DT. Structure-activity analysis of a novel NR2C/NR2D-preferring NMDA receptor antagonist: 1-(phenanthrene-2-carbonyl) piperazine-2,3-dicarboxylic acid. Br J Pharmacol 141: 508–516, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer G, Mutel V, Trube G, Malherbe P, Kew JN, Mohacsi E, Heitz MP, Kemp JA. Ro 25–6981, a highly potent and selective blocker of N-methyl-d-aspartate receptors containing the NR2B subunit. Characterization in vitro. J Pharmacol Exp Ther 283: 1285–1292, 1997 [PubMed] [Google Scholar]
- Flint AC, Maisch US, Weishaupt JH, Kriegstein AR, Monyer H. NR2A subunit expression shortens NMDA receptor synaptic currents in developing neocortex. J Neurosci 17: 2469–2476, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould E, Cameron HA, McEwen BS. Blockade of NMDA receptors increases cell death and birth in the developing rat dentate gyrus. J Comp Neurol 340: 551–565, 1994 [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. The Yin and Yang of NMDA receptor signalling. Trends Neurosci 26: 81–89, 2003 [DOI] [PubMed] [Google Scholar]
- Hatton CJ, Paoletti P. Modulation of triheteromeric NMDA receptors by N-terminal domain ligands. Neuron 46: 261–274, 2005 [DOI] [PubMed] [Google Scholar]
- He S, Shao LR, Rittase WB, Bausch SB. Increased Kv1 channel expression may contribute to decreased sIPSC frequency following chronic inhibition of NR2B-containing NMDAR. Neuropsychopharmacology 37: 1338–1356, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vockler J, Dikranian K, Tenkova TI, Stefovska V, Turski L, Olney JW. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 283: 70–74, 1999 [DOI] [PubMed] [Google Scholar]
- Jourdain P, Bergersen LH, Bhaukaurally K, Bezzi P, Santello M, Domercq M, Matute C, Tonello F, Gundersen V, Volterra A. Glutamate exocytosis from astrocytes controls synaptic strength. Nat Neurosci 10: 331–339, 2007 [DOI] [PubMed] [Google Scholar]
- Kemp JA, McKernan RM. NMDA receptor pathways as drug targets. Nat Neurosci 5, Suppl: 1039–1042, 2002 [DOI] [PubMed] [Google Scholar]
- Kew JN, Trube G, Kemp JA. A novel mechanism of activity-dependent NMDA receptor antagonism describes the effect of ifenprodil in rat cultured cortical neurones. J Physiol 497: 761–772, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohr G, De Koninck Y, Mody I. Properties of NMDA receptor channels in neurons acutely isolated from epileptic (kindled) rats. J Neurosci 13: 3612–3627, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs C, Fernandes HB, Sheldon C, Raymond LA, Baimbridge KG. Functional NMDA receptor subtype 2B is expressed in astrocytes after ischemia in vivo and anoxia in vitro. J Neurosci 23: 3364–3372, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalo U, Pankratov Y, Kirchhoff F, North RA, Verkhratsky A. NMDA receptors mediate neuron-to-glia signaling in mouse cortical astrocytes. J Neurosci 26: 2673–2683, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Meur K, Galante M, Angulo MC, Audinat E. Tonic activation of NMDA receptors by ambient glutamate of non-synaptic origin in the rat hippocampus. J Physiol 580: 373–383, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D, Zhang X, O'Brien R, Ehlers MD, Huganir RL. Regulation of morphological postsynaptic silent synapses in developing hippocampal neurons. Nat Neurosci 2: 37–43, 1999 [DOI] [PubMed] [Google Scholar]
- Lin SY, Constantine-Paton M. Suppression of sprouting: an early function of NMDA receptors in the absence of AMPA/kainate receptor activity. J Neurosci 18: 3725–3737, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA. Pathologically activated therapeutics for neuroprotection. Nat Rev Neurosci 8: 803–808, 2007 [DOI] [PubMed] [Google Scholar]
- Loscher W. Pharmacology of glutamate receptor antagonists in the kindling model of epilepsy. Prog Neurobiol 54: 721–741, 1998 [DOI] [PubMed] [Google Scholar]
- McCool BA, Lovinger DM. Ifenprodil inhibition of the 5-hydroxytryptamine3 receptor. Neuropharmacology 34: 621–629, 1995 [DOI] [PubMed] [Google Scholar]
- McKinney RA, Luthi A, Bandtlow CE, Gahwiler BH, Thompson SM. Selective glutamate receptor antagonists can induce or prevent axonal sprouting in rat hippocampal slice cultures. Proc Natl Acad Sci USA 96: 11631–11636, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12: 529–540, 1994 [DOI] [PubMed] [Google Scholar]
- Mori H, Mishina M. Structure and function of the NMDA receptor channel. Neuropharmacology 34: 1219–1237, 1995 [DOI] [PubMed] [Google Scholar]
- Morris RG, Anderson E, Lynch GS, Baudry M. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-d-aspartate receptor antagonist, AP5. Nature 319: 774–776, 1986 [DOI] [PubMed] [Google Scholar]
- Muir KW, Lees KR. Clinical experience with excitatory amino acid antagonist drugs. Stroke 26: 503–513, 1995 [DOI] [PubMed] [Google Scholar]
- Neyton J, Paoletti P. Relating NMDA receptor function to receptor subunit composition: limitations of the pharmacological approach. J Neurosci 26: 1331–1333, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien RJ, Kamboj S, Ehlers MD, Rosen KR, Fischbach GD, Huganir RL. Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron 21: 1067–1078, 1998 [DOI] [PubMed] [Google Scholar]
- Olney JW, Labruyere J, Price MT. Pathological changes induced in cerebrocortical neurons by phencyclidine and related drugs. Science 244: 1360–1362, 1989 [DOI] [PubMed] [Google Scholar]
- Palmer GC. Neuroprotection by NMDA receptor antagonists in a variety of neuropathologies. Curr Drug Targets 2: 241–271, 2001 [DOI] [PubMed] [Google Scholar]
- Parsons CG, Danysz W, Quack G. Memantine is a clinically well tolerated N-methyl-d-aspartate (NMDA) receptor antagonist—a review of preclinical data. Neuropharmacology 38: 735–767, 1999 [DOI] [PubMed] [Google Scholar]
- Rao A, Craig AM. Activity regulates the synaptic localization of the NMDA receptor in hippocampal neurons. Neuron 19: 801–812, 1997 [DOI] [PubMed] [Google Scholar]
- Sah P, Hestrin S, Nicoll RA. Tonic activation of NMDA receptors by ambient glutamate enhances excitability of neurons. Science 246: 815–818, 1989 [DOI] [PubMed] [Google Scholar]
- Scherzer CR, Landwehrmeyer GB, Kerner JA, Counihan TJ, Kosinski CM, Standaert DG, Daggett LP, Velicelebi G, Penney JB, Jr, Young AB. Expression of N-methyl-d-aspartate receptor subunit mRNAs in the human brain: hippocampus and cortex. J Comp Neurol 390: 75–90, 1998 [DOI] [PubMed] [Google Scholar]
- Schiavo G, Benfenati F, Poulain B, Rossetto O, Polverino de Laureto P, DasGupta BR, Montecucco C. Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature 359: 832–835, 1992 [DOI] [PubMed] [Google Scholar]
- Semyanov A, Walker MC, Kullmann DM, Silver RA. Tonically active GABA A receptors: modulating gain and maintaining the tone. Trends Neurosci 27: 262–269, 2004 [DOI] [PubMed] [Google Scholar]
- Sheng M, Cummings J, Roldan LA, Jan YN, Jan LY. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature 368: 144–147, 1994 [DOI] [PubMed] [Google Scholar]
- Shimamoto K, Lebrun B, Yasuda-Kamatani Y, Sakaitani M, Shigeri Y, Yumoto N, Nakajima T. dl-Threo-beta-benzyloxyaspartate, a potent blocker of excitatory amino acid transporters. Mol Pharmacol 53: 195–201, 1998 [DOI] [PubMed] [Google Scholar]
- Sjostrom PJ, Turrigiano GG, Nelson SB. Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron 39: 641–654, 2003 [DOI] [PubMed] [Google Scholar]
- Standaert DG, Landwehrmeyer GB, Kerner JA, Penney JB, Jr, Young AB. Expression of NMDAR2D glutamate receptor subunit mRNA in neurochemically identified interneurons in the rat neostriatum, neocortex and hippocampus. Mol Brain Res 42: 89–102, 1996 [DOI] [PubMed] [Google Scholar]
- Stocca G, Vicini S. Increased contribution of NR2A subunit to synaptic NMDA receptors in developing rat cortical neurons. J Physiol 507: 13–24, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods 37: 173–182, 1991 [DOI] [PubMed] [Google Scholar]
- Suzuki T, Kodama S, Hoshino C, Izumi T, Miyakawa H. A plateau potential mediated by the activation of extrasynaptic NMDA receptors in rat hippocampal CA1 pyramidal neurons. Eur J Neurosci 28: 521–534, 2008 [DOI] [PubMed] [Google Scholar]
- Sveinbjornsdottir S, Sander JW, Upton D, Thompson PJ, Patsalos PN, Hirt D, Emre M, Lowe D, Duncan JS. The excitatory amino acid antagonist d-CPP-ene (SDZ EAA-494) in patients with epilepsy. Epilepsy Res 16: 165–174, 1993 [DOI] [PubMed] [Google Scholar]
- Tammaro P, Ashcroft FM. A mutation in the ATP-binding site of the Kir6.2 subunit of the KATP channel alters coupling with the SUR2A subunit. J Physiol 584: 743–753, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci 19: 4180–4188, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bockstaele EJ, Colago EE. Selective distribution of the NMDA-R1 glutamate receptor in astrocytes and presynaptic axon terminals in the nucleus locus coeruleus of the rat brain: an immunoelectron microscopic study. J Comp Neurol 369: 483–496, 1996 [DOI] [PubMed] [Google Scholar]
- Wang XM, Bausch SB. Effects of distinct classes of N-methyl-d-aspartate receptor antagonists on seizures, axonal sprouting and neuronal loss in vitro: suppression by NR2B-selective antagonists. Neuropharmacology 47: 1008–1020, 2004 [DOI] [PubMed] [Google Scholar]
- Woodhall G, Evans DI, Cunningham MO, Jones RS. NR2B-containing NMDA autoreceptors at synapses on entorhinal cortical neurons. J Neurophysiol 86: 1644–1651, 2001 [DOI] [PubMed] [Google Scholar]
- Yamakura T, Shimoji K. Subunit- and site-specific pharmacology of the NMDA receptor channel. Prog Neurobiol 59: 279–298, 1999 [DOI] [PubMed] [Google Scholar]