

Abstract
Diabetes is a major worldwide problem. Despite some progress in the development of new antidiabetic agents, the ability to maintain tight glycemic control in order to prevent renal, retinal, and neuropathic complications of diabetes without adverse complications still remains a challenge. Recent evidence suggests, however, that in addition to playing a key role in the regulation of energy homeostasis, the adiposity hormone leptin also plays an important role in the control of glucose metabolism via its actions in the brain. This review examines the role of leptin action in the central nervous system and the mechanisms whereby leptin mediates its effects to regulate glucose metabolism. These findings suggest that defects or dysfunction in leptin signaling may contribute to the etiology of diabetes and raise the possibility that either leptin or downstream targets of leptin may have therapeutic potential for the treatment of diabetes.
Keywords: Brain, diabetes, glucose, insulin, leptin
INTRODUCTION
The incidence and prevalence of diabetes is increasing globally.[1] More than 250 million people have diabetes worldwide and this number is expected to exceed 400 million by 2030.[2] This is a major health concern given that diabetes is associated with increased risk of cardiovascular disease and both macro- and micro-vascular diseases including blindness, amputation, and renal disease.[1,2] The burden of diabetes leads to more than a doubling of individual medical expenses, with a total concomitant economic cost of $174 billion in the USA alone.[3] Given its considerable health and financial costs, a better understanding of diabetes pathogenesis is needed in order to develop new strategies for the safe and effective treatment of this disease.
Diabetes is a metabolic disease characterized by the chronic elevation of blood sugar levels (i.e. hyperglycemia) resulting from defects in insulin secretion, insulin resistance, or both. In general, there are two broad categories of diabetes.[4] The first category, type 1 diabetes, accounts for ~5–10% of those with diabetes and is an autoimmune disease that results in the destruction of pancreatic β-cells, causing an absolute insulin deficiency. The other, most common form is type 2 diabetes which accounts for >90% of those with diabetes and is caused by a combination of insulin resistance, with a relative, but not absolute insulin deficiency.[4] Thus, insulin secretion is impaired in these individuals and is insufficient to compensate for the insulin resistance in peripheral tissues.[5]
The control and management of blood glucose levels in both forms of diabetes is important for preventing ketoacidosis and diabetes-related complications. Given that people with type 1 diabetes produce little or no endogenous insulin, administration of exogenous insulin is necessary for survival.[6] However, while insulin therapy improves glycemic control and protects against diabetes-related complications, it also increases the risk of hypoglycemia and weight gain.[7–9] Unfortunately, the tighter the control of blood glucose levels, the greater the risk and severity of these untoward consequences.[7–9] However, because of the essential role of insulin for the treatment of type 1 diabetes, the only real advances in insulin therapy have occurred through modification of the insulin molecule to change its plasma half-life, absorption into the circulation, tissue distribution, and potency, and alternative approaches to type 1 diabetes treatment that are insulin independent have yet to emerge.[10]
For people with type 2 diabetes, exogenous treatment with insulin is generally not required unless lifestyle modifications including better nutrition and increased physical activity or pharmaceutical interventions with oral antidiabetic agents are not sufficient to manage blood glucose levels.[11,12] Currently, most strategies for treating type 2 diabetes have focused on either increasing insulin secretion from pancreatic β-cells or improving insulin sensitivity in peripheral tissues such as liver, muscle, and adipose tissue.[13] However, a growing body of evidence suggests that the central nervous system (CNS) plays a key role in the control of glucose metabolism.[14,15] The goal of this review is to discuss the literature that supports a role for the adiposity hormone, leptin, in the regulation of glucose metabolism and examine whether dysfunction in this system contributes to the pathogenesis of diabetes.
LEPTIN REGULATION OF GLUCOSE HOMEOSTASIS
Leptin is a polypeptide hormone produced and secreted by white adipose tissue (WAT)[16] that circulates in proportion to body fat mass,[17] enters the CNS in proportion to its plasma level,[18] and interacts with its receptor expressed in key brain areas that regulate food intake, energy expenditure, and autonomic function.[19] A large body of evidence suggests that leptin plays a vital role in the regulation of energy homeostasis as conditions characterized by leptin deficiency promote hyperphagia and weight gain,[16] whereas administration of leptin leads to reduced food intake, increased energy expenditure, and weight loss.[20–22] However, recent evidence implicates leptin not only in the regulation of energy balance but glucose homeostasis as well.[14]
While the effect of leptin to reduce food intake and body adiposity can improve insulin sensitivity in peripheral tissues via indirect mechanisms, several observations suggest that leptin can directly affect glucose metabolism independent of its effects on energy balance. Early studies suggested that the insulin resistance and diabetes phenotype of genetic leptin-deficient (ob/ob mouse) or leptin receptor-deficient (db/db mouse or fak/fak rat) rodent models could not be fully accounted for by their hyperphagia and obesity. Not only does caloric restriction fail to improve insulin sensitivity or prevent diabetes in models of either genetic leptin or leptin receptor deficiency,[23,24] but also leptin administration lowers blood glucose and plasma insulin levels in ob/ob mice even when differences in energy intake are controlled for.[25] Leptin deficiency has also been implicated to play a key role in the severe insulin resistance and diabetes phenotype of genetic disorders that impair adipogenesis, such as lipodystrophy.[26] Consistent with this, transplantation of body fat from wild-type, but not leptin-deficient, ob/ob mice improves glycemia in lipodystrophic mice,[27,28] while the diabetes phenotype of lipodystrophic mice[29,30] and humans[31,32] is ameliorated with leptin treatment. These data suggest that deficient leptin signaling has severe consequences for glucose metabolism, which are remedied by leptin replacement in a manner that is independent of, and additive to, its effects on energy intake and body fat content.
Another model of acquired leptin deficiency is that which occurs in uncontrolled insulin-deficient diabetes (uDM), a model of type 1 diabetes.[33] Because insulin action on adipocytes is required for both lipogenesis and inhibition of lipolysis, absolute insulin deficiency leads to uncontrolled mobilization of stored triglyceride and depletion of body fat stores. Progressive loss of adipose tissue is accompanied by a pronounced decrease of leptin levels, resulting in a deficiency of all known adiposity signals.[33] Consequently, uDM is characterized by diabetic hyperglycemia and hyperphagia,[34] and marked reductions of both plasma leptin and insulin levels have been implicated in these responses.[33,35] Leptin deficiency has also been implicated in the development of insulin resistance in uDM.[36] A physiological replacement dose of leptin administered systemically prevented the development of insulin resistance in a rat model of uDM via a mechanism independent of changes in food intake or body weight.[37] However, while systemic administration of exogenous leptin at doses to maintain physiological plasma leptin levels only lowered blood glucose levels slightly, it normalized the hyperglucagonemia and hypercorticosteronemia characteristic of uDM. However, in contrast to physiological replacement doses of leptin, hyperleptinemia induced by either pharmacological doses of leptin[38,39] or an adenoviral gene therapy approach[40] ameliorates hyperglycemia in rodent models of uDM, despite very low insulin levels. Thus, leptin deficiency plays a fundamental role in the pathogenesis of insulin resistance and related endocrine disorders in uDM.
CENTRAL NERVOUS SYSTEM LEPTIN IN THE REGULATION OF GLUCOSE HOMEOSTASIS
Since most of the effects of leptin on energy homeostasis are mediated by the brain, a similar mechanism has been invoked for leptin's effects on glucose metabolism. Consistent with this hypothesis, intracerebroventricular (ICV) administration of low doses of leptin ameliorates the insulin resistance and diabetes phenotype of both ob/ob and lipodystrophic mice[20,41] to the same extent as much higher doses of leptin given systemically. In a similar manner, leptin administration directly into the brain normalizes blood glucose levels in rodent models of uDM[42–46] at doses that are ineffective when administered systemically. Furthermore, in non-diabetic rats, central leptin gene therapy blocks high fat diet-induced weight gain, hyperleptinemia, and hyperinsulinemia,[47] while acute infusion of leptin directly into the brain reverses diet-induced hepatic insulin resistance in non-diabetic rats exposed to a high fat diet for 3 days.[48] Taken together, these data provide compelling evidence that the CNS mediates key effects of leptin on glucose metabolism.
SITE OF LEPTIN ACTION IN THE CENTRAL NERVOUS SYSTEM
While leptin receptors are expressed in several hypothalamic[49,50] and extrahypothalamic areas[51,52] involved in the control of energy balance and autonomic function, several observations implicate the hypothalamic arcuate nucleus (ARC) as an important site for leptin-mediated effects on glucose metabolism. Using a combination of gene targeting and gene therapy techniques, Coppari and colleagues found that unilateral restoration of leptin receptors to the ARC of leptin receptor-deficient mice only had a modest effect on food intake and body weight, but a marked effect to lower plasma insulin and blood glucose levels.[53] In a complementary approach, we used adenoviral gene therapy to express leptin receptors in the ARC of Koletsky (fak/fak) rats that develop severe hyperphagia, obesity, and insulin resistance due to genetic absence of leptin receptors. Here, selective rescue of leptin receptor signaling to the ARC of Koletsky rats dramatically improved peripheral insulin sensitivity independent of changes in food intake and body weight,[54] providing further support that the hypothalamic ARC plays a key role in mediating leptin's effects on glucose metabolism.
Growing evidence, however, suggests that brain areas outside the ARC are also likely involved in leptin's effects on glucose metabolism. Several lines of evidence implicate a role for the ventromedial nucleus of the hypothalamus (VMH) as neurons in this brain area express the leptin receptor,[50] are activated by leptin (as measured by the induction of pSTAT3, a marker of leptin activation),[55] and administration of leptin to the VMH increases insulin-independent glucose uptake in muscle, brown adipose tissue (BAT), and heart via the sympathetic nervous system (SNS).[56,57] In addition, selective inactivation of suppressor of cytokine signaling (SOCS-3; an inhibitor of leptin signaling) in VMH neurons improves glucose homeostasis without affecting body weight,[58] while deletion of leptin receptors from VMH neurons results in an obese, insulin-resistant phenotype.[59,60] Collectively, these findings support a role for both the ARC and VMH in the regulation of glucose metabolism. However, leptin receptors are also expressed in the paraventricular nucleus (PVN) and the dorsomedial nucleus of the hypothalamus (DMH),[49,50] as well as outside the hypothalamus,[51,52] and future studies examining the role of leptin signaling in these brain areas on glucose metabolism are warranted.
CENTRAL NERVOUS SYSTEM MECHANISMS OF LEPTIN ACTION
One key area of research has been to understand how leptin signaling in the brain improves peripheral insulin sensitivity. In short-term high-fat fed rats, ICV infusion of leptin reverses diet-induced insulin resistance via a suppression of hepatic glucose production, by reducing both glycogenolysis and gluconeogenesis.[48] Moreover, rescue of leptin receptor signaling to the ARC of leptin receptor-deficient Koletsky rats improved insulin sensitivity via enhanced insulin-induced suppression of hepatic glucose production, rather than an increase in glucose uptake.[61] In addition, this effect was associated with improved insulin-induced activation of the insulin signal transduction pathway selectively in liver, relative to muscle and WAT, and was associated with reduced hepatic expression of the gluconeogenic genes, glucose-6-phosphatase (G6Pase) and phosphoenolpyruvate kinase (Pepck).[61] Thus, leptin action in the ARC improves peripheral insulin sensitivity primarily through an action in the liver.
One mechanism to explain how the brain communicates to the liver is via autonomic outflow through the vagus nerve. Recent studies suggest that the effect of intrahypothalamic infusion of insulin[62] or free fatty acids[63] to improve hepatic insulin sensitivity requires intact vagal input to the liver. Consistent with the hypothesis that a similar mechanism mediates the effect of leptin in the brain to suppress hepatic glucose production, the effect of restored hypothalamic leptin signaling to improve hepatic insulin sensitivity in obese Koletsky rats was blocked by selective hepatic vagotomy.[61] These data support a model whereby leptin activates a neural signal between the brain and the liver, which regulates hepatic insulin action.
In addition to regulating hepatic glucose production, leptin also has the capacity to enhance glucose uptake in peripheral tissues through insulin-independent mechanisms.[64] Leptin stimulation of glucose utilization involves the CNS, as either ICV or intrahypothalamic administration of leptin increases non-insulin–mediated glucose uptake in skeletal muscle, BAT, and heart (but not in WAT) via a mechanism involving the SNS.[56,57] Moreover, leptin-induced increases in glucose uptake in muscle and heart are mediated via the VMH, while leptin signaling in both the ARC and VMH regulates glucose uptake in BAT.[65] Based on these observations, we examined the mechanism whereby leptin action in the brain normalizes diabetic hyperglycemia in uDM. This glucose-lowering effect of leptin occurs via a mechanism that is independent of reduced food intake, increased urinary glucose loss, or recovery of pancreatic β-cells. Instead, using tracer dilution techniques, leptin was demonstrated to activate a previously unrecognized, insulin-independent mechanism for potently inhibiting hepatic glucose production, while increasing tissue glucose utilization, a combination that fully normalizes blood glucose levels in diabetic animals[43] [Figure 1]. These data establish that the brain has the previously unrecognized ability to normalize diabetic hyperglycemia, and we emphasize that this effect is distinct from the previously reported action of leptin to improve hepatic insulin sensitivity.
Figure 1.
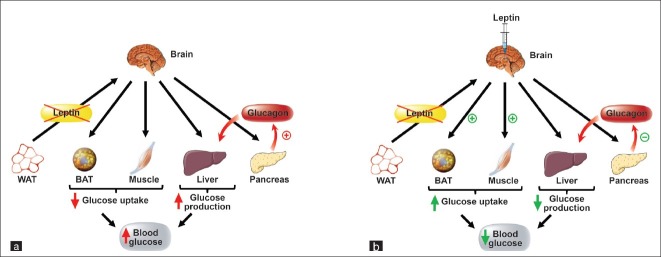
Leptin normalization of blood glucose levels in uDM. (a) Type 1 diabetes is characterized by diabetic hyperglycemia and both insulin and leptin deficiency due to the loss of pancreatic β-cells and the depletion of adipose tissue stores, respectively. This diabetic hyperglycemia is due to both reduced glucose uptake in peripheral tissues and increased rates of hepatic glucose production, in part due to increased glucagon secretion from pancreatic β-cells. (b) Leptin administration directly into the brain normalizes diabetic hyperglycemia in uDM by both potently suppressing hepatic glucose production, as well as increasing glucose uptake despite persistent severe insulin deficiency, an effect associated with normalization of elevated plasma glucagon levels.[79]
Besides its actions on liver and peripheral tissues, leptin is also suggested to regulate glucose homeostasis via the islet. One mechanism that has been hypothesized to contribute to the antidiabetic effects of leptin in uDM is the normalization of increased plasma glucagon secretion from pancreatic α-cells.[40] Hyperglucagonemia is thought to contribute to diabetic hyperglycemia in uDM, in part, by activating expression of the gluconeogenic genes, G6Pase and Pepck in the liver.[66,67] Consistent with this hypothesis, the glucose-lowering effect of leptin in uDM was accompanied by a normalization of hyperglucagonemia.[39,40] The CNS is implicated in this effect as the glucose-lowering effects of leptin in uDM are mediated via a direct action of leptin in the brain and are accompanied by a normalization of increased plasma glucagon levels.[43,68] Consistent with this, key brain areas including the VMH participate in the control of glucagon secretion[69] via activation of the autonomic nervous system[69,70] during hypoglycemia.[71,72]
Leptin receptors are also expressed on pancreatic β-cells[73] and systemic administration of leptin has been shown to decrease glucose-stimulated insulin secretion in a dose-dependent manner in vivo.[74] Subsequent studies, however, demonstrated that the acute effects of leptin on insulin secretion are mediated through its actions in the CNS via the melanocortin pathway.[75] Consistent with these observations, long-term infusion of ICV leptin decreases glucose-stimulated insulin secretion, an effect that is overcome by an improvement in insulin sensitivity in both normal and diabetic rats (90% pancreatectomy).[76] Moreover, this leptin-induced reduction in insulin secretion is independent of changes in either pancreatic β-cell area or mass and is mediated mainly through the SNS.[77] In addition, CNS leptin-transgene expression suppressed plasma insulin levels and improved insulin sensitivity in ob/ob mice fed either chow or a high-fat diet.[78] Collectively, these data suggest that intact CNS leptin signaling to the islet may also play an important role in preventing both type 1 and type 2 diabetes.[79]
THE HYPOTHALAMIC ARCUATE NUCLEUS
Identifying the hypothalamic neurons that transduce leptin signal into changes of energy homeostasis and glucose metabolism has been the focus of much research. Two well-characterized leptin-sensitive neuronal populations implicated in the control of both energy- and glucose-homeostasis are expressed in the hypothalamic ARC. One of these neuronal subsets expresses pro-opiomelanocortin (POMC) and these cells are stimulated by leptin[80,81] to release alpha-melanocyte stimulating hormone (α-MSH), a peptide that acts on melanocortin receptors (Mc3r/Mc4r) to promote weight loss[82] and improve insulin sensitivity.[83] Adjacent to these cells is a neuronal subset that expresses neuropeptide Y (NPY) and a melanocortin receptor blocker, agouti-related peptide (AgRP).[84] Both NPY and AgRP promote weight gain[85,86] and insulin resistance[87,88] and, in contrast to POMC neurons, these NPY/AgRP neurons are inhibited by leptin.[25] Therefore, in conditions of reduced leptin signaling such as in ob/ob mice or in uDM, NPY/AgRP neurons are activated whereas POMC neurons are inhibited, a combination of responses that promote weight gain and insulin resistance.[19] Moreover, NPY/AgRP neurons inhibit POMC neurons through release of the inhibitory neurotransmitter, γ-aminobutyric acid (GABA).[89] Thus, in addition to activating POMC neurons directly, leptin also hyperpolarizes NPY/AgRP neurons, thereby reducing the release of GABA onto POMC neurons, therefore disinhibiting POMC neurons[89,90] [Figure 2].
Figure 2.
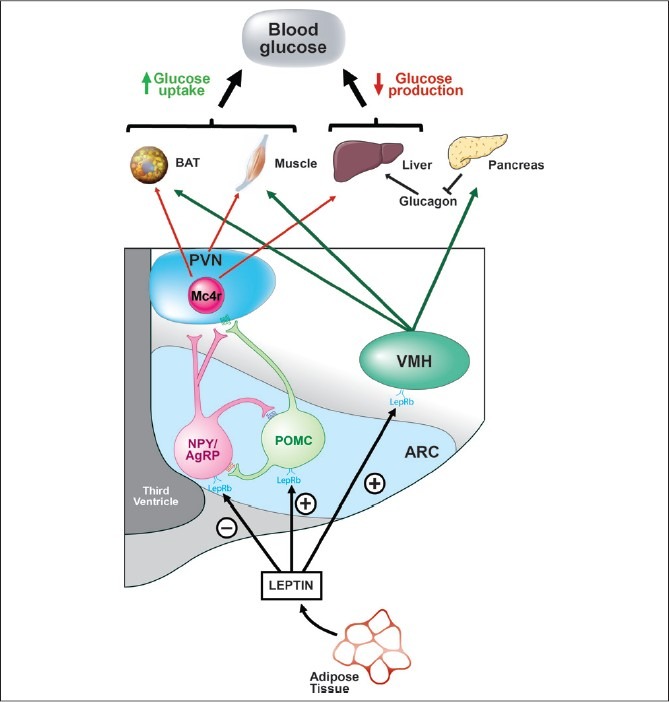
Model of CNS leptin regulation of glucose metabolism. Leptin is secreted by adipocytes, enters the CNS, and acts on its receptor expressed in key brain areas that regulate metabolism. Leptin inhibits neuropeptide and agouti-related peptide (NPY/AgRP) neurons and stimulates pro-opiomelanocortin (POMC) neurons in the ARC, responses that promote glucose uptake in peripheral tissues and the suppression of glucose production from the liver. In addition, leptin action in the VMH stimulates glucose uptake in peripheral tissues and this brain area is also implicated in the regulation of glucagon secretion. ARC, arcuate nucleus; VMH, ventromedial hypothalamus; PVN, paraventricular nucleus; Mc4r; melanocortin-4 receptor; LepRb, leptin receptor; BAT, brown adipose tissue.[104]
Both pharmacological and mouse genetic strategies have been employed to determine the role of leptin signaling in each of these ARC neuronal populations. Using a Cre-loxP approach, deletion of leptin receptors from both NPY/AgRP and POMC neurons results in hyperinsulinemia and an obesity phenotype that is nearly additive with respect to the increase of body weight following deletion of leptin receptors from individual neurons.[91] In contrast, reactivation of leptin signaling in all POMC neurons[92] or selectively in POMC neurons that express the leptin receptor[93] in mice that otherwise lack leptin receptors normalizes blood glucose levels and ameliorates hepatic insulin resistance independent of changes in energy homeostasis.[92,93] Conversely, deletion of leptin receptors from just POMC neurons results in a mild-obesity phenotype and only a small effect on glucose homeostasis.[94] Taken together, these data suggest that leptin action on POMC neurons in the ARC has important effects in the control of glucose metabolism.
Pharmacological studies have also supported a role for the melanocortin pathway in mediating leptin's effects on glucose metabolism. In non-diabetic animals, the effects of ICV leptin to stimulate hepatic gluconeogenesis are blocked by central administration of the Mc3/4r antagonist, SHU9119, while leptin-induced suppression of glycogenolysis remains intact, suggesting that leptin stimulation of gluconeogenesis is mediated via a melanocortin-dependent pathway while leptin-inhibition of glycogenolysis is melanocortin independent.[95] In addition, the effect of leptin administration to the VMH to stimulate glucose uptake is blocked by the Mc3/4r antagonist, SHU9119, suggesting that this leptin effect is also dependent on activation of melanocortin receptors.[65] Moreover, the antidiabetic effects of leptin in uDM require melanocortin signaling as co-infusion of the Mc3/4r antagonist, SHU9119, directly into the brain blocked the glucose-lowering effect of ICV leptin in uDM.[96] However, this effect of the Mc3/4r antagonist could block leptin action in either two ways – by blocking the increased release of α-MSH from POMC neurons or by mimicking the effect of increased release of the endogenous Mc3/4r antagonist, AgRP, from NPY/AgRP neurons, or both. In contrast, activation of the Mc3/4r alone was not sufficient to mimic the glucose-lowering effects of leptin in uDM.[96] These data suggest that stimulation of POMC neurons alone cannot fully explain the actions of leptin in uDM, implying an important role for leptin inhibition of NPY/AgRP neurons as well.
THERAPEUTIC IMPLICATIONS
The therapeutic potential of leptin for the treatment of obesity has been dampened thus far by its reduced effectiveness to induce weight loss in obese individuals.[97] Except for rare cases, obesity is not caused by leptin deficiency as most obese humans and rodents have elevated levels of circulating plasma leptin levels.[17] Moreover, in rodent models of diet-induced obesity, the ability of leptin to suppress food intake and induce weight loss and to activate its signal transduction pathway (pSTAT3) in the CNS is impaired.[55,98] This phenomenon is commonly referred to as “leptin resistance” and is thought to be due to impaired leptin receptor signaling in the hypothalamus, the impaired ability of leptin to cross the blood-brain barrier, or both.[99,100] Identifying the mechanisms contributing to the development of leptin resistance is an active line of research and recently reviewed elsewhere.[99,100] Experiments investigating whether leptin administration improves glucose metabolism in type 2 diabetic individuals seem warranted given evidence from rodent studies suggesting that leptin has beneficial effects on glucose metabolism at doses that are ineffective at reducing food intake and body weight. A beneficial role for leptin in the treatment of type 2 diabetes is further supported by a recent study demonstrating that systemic administration of leptin improves insulin sensitivity and normalizes fasting plasma glucose levels in University of California, Davis, type 2 diabetes mellitus (UCD-T2DM) rats, independent of energy intake.[101] Given that rodent models of type 1 diabetes treated with leptin are much more sensitive to the effects of insulin,[37,39,102] it also raises the therapeutic possibility that supplementing insulin treatment with leptin may be a useful adjunct in the management of type 1 diabetes.
CONCLUSIONS
In conclusion, in addition to its well-known effects on energy homeostasis, leptin is a hormone that also directly regulates glucose metabolism through its actions via the CNS. Identification of the specific neuronal subsets downstream of leptin action, which link communication between the brain and peripheral tissues to control both hepatic glucose production and glucose uptake, will help facilitate the development of new approaches to diabetes treatment. While there are several hurdles to overcome for targeting the CNS,[103] it nonetheless has untapped potential for the treatment of diabetes.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health (NIH) to GJM (DK-089056), the Nutrition Obesity Research Unit (NORC, P30 DK-035816) to GJM, the Diabetes and Metabolism Training Grant (T32 DK-0007247) to THM, at the University of Washington, and an American Heart Association Scientist Development Grant to GJM.
Footnotes
Source of Support: This work was supported by grants from the National Institutes of Health (NIH) to GJM (DK-089056), the Nutrition Obesity Research Unit (NORC, P30 DK-035816) the Diabetes and Metabolism Training Grant (T32 DK-0007247) at the University of Washington, and an American Heart Association Scientist Development Grant to GJM
Conflict of Interest: No
REFERENCES
- 1.Smyth S, Heron A. Diabetes and obesity: The twin epidemics. Nat Med. 2006;12:75–80. doi: 10.1038/nm0106-75. [DOI] [PubMed] [Google Scholar]
- 2.Chen L, Magliano DJ, Zimmet PZ. The worldwide epidemiology of type 2 diabetes mellitus-present and future perspectives. Nat Rev Endocrinol. 2011;8:228–36. doi: 10.1038/nrendo.2011.183. [DOI] [PubMed] [Google Scholar]
- 3.Economic costs of diabetes in the U.S. in 2007. Diabetes Care. 2008;31:596–615. doi: 10.2337/dc08-9017. [DOI] [PubMed] [Google Scholar]
- 4.Standards of medical care in diabetes—2011. Diabetes Care. 2011;34(Suppl 1):S11–61. doi: 10.2337/dc11-S011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kahn SE, Prigeon RL, McCulloch DK, Boyko EJ, Bergman RN, Schwartz MW, et al. Quantification of the relationship between insulin sensitivity and beta-cell function in human subjects.Evidence for a hyperbolic function. Diabetes. 1993;42:1663–72. doi: 10.2337/diab.42.11.1663. [DOI] [PubMed] [Google Scholar]
- 6.Devendra D, Liu E, Eisenbarth GS. Type 1 diabetes: Recent developments. BMJ. 2004;328:750–4. doi: 10.1136/bmj.328.7442.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weight gain associated with intensive therapy in the diabetes control and complications trial. The DCCT research group. The DCCT research group. Diabetes Care. 1988;11:567–73. doi: 10.2337/diacare.11.7.567. [DOI] [PubMed] [Google Scholar]
- 8.The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The diabetes control and complications trial research group. N Engl J Med. 1993;329:977–86. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 9.Hypoglycemia in the diabetes control and complications trial. the diabetes control and complications trial research group. Diabetes. 1997;46:271–86. [PubMed] [Google Scholar]
- 10.Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature. 2010;464:1293–300. doi: 10.1038/nature08933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yki-Jarvinen H. Combination therapies with insulin in type 2 diabetes. Diabetes Care. 2001;24:758–67. doi: 10.2337/diacare.24.4.758. [DOI] [PubMed] [Google Scholar]
- 12.Riddle MC, Rosenstock J, Gerich J. The treat-to-target trial: Randomized addition of glargine or human NPH insulin to oral therapy of type 2 diabetic patients. Diabetes Care. 2003;26:3080–6. doi: 10.2337/diacare.26.11.3080. [DOI] [PubMed] [Google Scholar]
- 13.Lebovitz HE. Type 2 diabetes mellitus-current therapies and the emergence of surgical options. Nat Rev Endocrinol. 2011;7:408–19. doi: 10.1038/nrendo.2011.10. [DOI] [PubMed] [Google Scholar]
- 14.Schwartz MW, Porte D., Jr Diabetes, obesity, and the brain. Science. 2005;307:375–9. doi: 10.1126/science.1104344. [DOI] [PubMed] [Google Scholar]
- 15.Morton GJ, Schwartz MW. Leptin and the central nervous system control of glucose metabolism. Physiol Rev. 2011;91:389–411. doi: 10.1152/physrev.00007.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–32. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 17.Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996;334:292–5. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- 18.Schwartz MW, Peskind E, Raskind M, Boyko EJ, Porte D., Jr Cerebrospinal fluid leptin levels: Relationship to plasma levels and to adiposity in humans. Nat Med. 1996;2:589–93. doi: 10.1038/nm0596-589. [DOI] [PubMed] [Google Scholar]
- 19.Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–71. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 20.Campfield LA, Smith FJ, Guisez Y, Devos R, Burn P. Recombinant mouse OB protein: Evidence for a peripheral signal linking adiposity and central neural networks. Science. 1995;269:546–9. doi: 10.1126/science.7624778. [DOI] [PubMed] [Google Scholar]
- 21.Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, et al. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–6. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- 22.Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, et al. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. 1995;269:540–3. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- 23.Dubuc PU. The development of obesity, hyperinsulinemia, and hyperglycemia in ob/ob mice. Metabolism. 1976;25:1567–74. doi: 10.1016/0026-0495(76)90109-8. [DOI] [PubMed] [Google Scholar]
- 24.Wyse BM, Dulin WE. The influence of age and dietary conditions on diabetes in the db mouse. Diabetologia. 1970;6:268–73. doi: 10.1007/BF01212237. [DOI] [PubMed] [Google Scholar]
- 25.Schwartz MW, Baskin DG, Bukowski TR, Kuijper JL, Foster D, Lasser G, et al. Specificity of leptin action on elevated blood glucose levels and hypothalmic neuropeptide Y gene expression in ob/ob mice. Diabetes. 1996;45:531–5. doi: 10.2337/diab.45.4.531. [DOI] [PubMed] [Google Scholar]
- 26.Joffe BI, Panz VR, Raal FJ. From lipodystrophy syndromes to diabetes mellitus. Lancet. 2001;357:1379–81. doi: 10.1016/S0140-6736(00)04616-X. [DOI] [PubMed] [Google Scholar]
- 27.Colombo C, Cutson JJ, Yamauchi T, Vinson C, Kadowaki T, Gavrilova O, et al. Transplantation of adipose tissue lacking leptin is unable to reverse the metabolic abnormalities associated with lipoatrophy. Diabetes. 2002;51:2727–33. doi: 10.2337/diabetes.51.9.2727. [DOI] [PubMed] [Google Scholar]
- 28.Gavrilova O, Marcus-Samuels B, Graham D, Kim JK, Shulman GI, Castle AL, et al. Surgical implantation of adipose tissue reverses diabetes in lipoatrophic mice. J Clin Invest. 2000;105:271–8. doi: 10.1172/JCI7901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gavrilova O, Marcus-Samuels B, Leon LR, Vinson C, Reitman ML. Leptin and diabetes in lipoatrophic mice. Nature. 2000;403:850. doi: 10.1038/35002663. discussion 850-1. [DOI] [PubMed] [Google Scholar]
- 30.Shimomura I, Hammer RE, Ikemoto S, Brown MS, Goldstein JL. Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature. 1999;401:73–6. doi: 10.1038/43448. [DOI] [PubMed] [Google Scholar]
- 31.Oral EA, Simha V, Ruiz E, Andewelt A, Premkumar A, Snell P, et al. Leptin-replacement therapy for lipodystrophy. N Engl J Med. 2002;346:570–8. doi: 10.1056/NEJMoa012437. [DOI] [PubMed] [Google Scholar]
- 32.Petersen KF, Oral EA, Dufour S, Befroy D, Ariyan C, Yu C, et al. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest. 2002;109:1345–50. doi: 10.1172/JCI15001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Havel PJ, Uriu-Hare JY, Liu T, Stanhope KL, Stern JS, Keen CL, et al. Marked and rapid decreases of circulating leptin in streptozotocin diabetic rats: Reversal by insulin. Am J Physiol. 1998;274:R1482–91. doi: 10.1152/ajpregu.1998.274.5.R1482. [DOI] [PubMed] [Google Scholar]
- 34.Leedom LJ, Meehan WP. The psychoneuroendocrinology of diabetes mellitus in rodents. Psychoneuroendocrinology. 1989;14:275–94. doi: 10.1016/0306-4530(89)90030-9. [DOI] [PubMed] [Google Scholar]
- 35.Sindelar DK, Havel PJ, Seeley RJ, Wilkinson CW, Woods SC, Schwartz MW. Low plasma leptin levels contribute to diabetic hyperphagia in rats. Diabetes. 1999;48:1275–80. doi: 10.2337/diabetes.48.6.1275. [DOI] [PubMed] [Google Scholar]
- 36.Gelling RW, Morton GJ, Morrison CD, Niswender KD, Myers MG, Jr, Rhodes CJ, et al. Insulin action in the brain contributes to glucose lowering during insulin treatment of diabetes. Cell Metab. 2006;3:67–73. doi: 10.1016/j.cmet.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 37.German JP, Wisse BE, Thaler JP, Oh-IS, Sarruf DA, Ogimoto K, et al. Leptin deficiency causes insulin resistance induced by uncontrolled diabetes. Diabetes. 2010;59:1626–34. doi: 10.2337/db09-1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chinookoswong N, Wang JL, Shi ZQ. Leptin restores euglycemia and normalizes glucose turnover in insulin-deficient diabetes in the rat. Diabetes. 1999;48:1487–92. doi: 10.2337/diabetes.48.7.1487. [DOI] [PubMed] [Google Scholar]
- 39.Wang MY, Chen L, Clark GO, Lee Y, Stevens RD, Ilkayeva OR, et al. Leptin therapy in insulin-deficient type I diabetes. Proc Natl Acad Sci U S A. 2010;107:4813–9. doi: 10.1073/pnas.0909422107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu X, Park BH, Wang MY, Wang ZV, Unger RH. Making insulin-deficient type 1 diabetic rodents thrive without insulin. Proc Natl Acad Sci U S A. 2008;105:14070–5. doi: 10.1073/pnas.0806993105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Asilmaz E, Cohen P, Miyazaki M, Dobrzyn P, Ueki K, Fayzikhodjaeva G, et al. Site and mechanism of leptin action in a rodent form of congenital lipodystrophy. J Clin Invest. 2004;113:414–24. doi: 10.1172/JCI19511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.da Silva AA, Tallam LS, Liu J, Hall JE. Chronic antidiabetic and cardiovascular actions of leptin: Role of CNS and increased adrenergic activity. Am J Physiol Regul Integr Comp Physiol. 2006;291:R1275–82. doi: 10.1152/ajpregu.00187.2006. [DOI] [PubMed] [Google Scholar]
- 43.German JP, Thaler JP, Wisse BE, Oh-IS, Sarruf DA, Matsen ME, et al. Leptin activates a novel CNS mechanism for insulin-independent normalization of severe diabetic hyperglycemia. Endocrinology. 2011;152:394–404. doi: 10.1210/en.2010-0890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hidaka S, Yoshimatsu H, Kondou S, Tsuruta Y, Oka K, Noguchi H, et al. Chronic central leptin infusion restores hyperglycemia independent of food intake and insulin level in streptozotocin-induced diabetic rats. Faseb J. 2002;16:509–18. doi: 10.1096/fj.01-0164com. [DOI] [PubMed] [Google Scholar]
- 45.Kojima S, Asakawa A, Amitani H, Sakoguchi T, Ueno N, Inui A, et al. Central leptin gene therapy, a substitute for insulin therapy to ameliorate hyperglycemia and hyperphagia, and promote survival in insulin-deficient diabetic mice. Peptides. 2009;30:962–6. doi: 10.1016/j.peptides.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 46.Lin CY, Higginbotham DA, Judd RL, White BD. Central leptin increases insulin sensitivity in streptozotocin-induced diabetic rats. Am J Physiol Endocrinol Metab. 2002;282:E1084–91. doi: 10.1152/ajpendo.00489.2001. [DOI] [PubMed] [Google Scholar]
- 47.Dube MG, Beretta E, Dhillon H, Ueno N, Kalra PS, Kalra SP. Central leptin gene therapy blocks high-fat diet-induced weight gain, hyperleptinemia, and hyperinsulinemia: Increase in serum ghrelin levels. Diabetes. 2002;51:1729–36. doi: 10.2337/diabetes.51.6.1729. [DOI] [PubMed] [Google Scholar]
- 48.Pocai A, Morgan K, Buettner C, Gutierrez-Juarez R, Obici S, Rossetti L. Central leptin acutely reverses diet-induced hepatic insulin resistance. Diabetes. 2005;54:3182–9. doi: 10.2337/diabetes.54.11.3182. [DOI] [PubMed] [Google Scholar]
- 49.Baskin DG, Breininger JF, Schwartz MW. Leptin receptor mRNA identifies a subpopulation of neuropeptide Y neurons activated by fasting in rat hypothalamus. Diabetes. 1999;48:828–33. doi: 10.2337/diabetes.48.4.828. [DOI] [PubMed] [Google Scholar]
- 50.Elmquist JK, Ahima RS, Elias CF, Flier JS, Saper CB. Leptin activates distinct projections from the dorsomedial and ventromedial hypothalamic nuclei. Proc Natl Acad Sci U S A. 1998;95:741–6. doi: 10.1073/pnas.95.2.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Figlewicz DP, Evans SB, Murphy J, Hoen M, Baskin DG. Expression of receptors for insulin and leptin in the ventral tegmental area/substantia nigra (VTA/SN) of the rat. Brain Res. 2003;964:107–15. doi: 10.1016/s0006-8993(02)04087-8. [DOI] [PubMed] [Google Scholar]
- 52.Grill HJ, Schwartz MW, Kaplan JM, Foxhall JS, Breininger J, Baskin DG. Evidence that the caudal brainstem is a target for the inhibitory effect of leptin on food intake. Endocrinology. 2002;143:239–46. doi: 10.1210/endo.143.1.8589. [DOI] [PubMed] [Google Scholar]
- 53.Coppari R, Ichinose M, Lee CE, Pullen AE, Kenny CD, McGovern RA, et al. The hypothalamic arcuate nucleus: A key site for mediating leptin's effects on glucose homeostasis and locomotor activity. Cell Metab. 2005;1:63–72. doi: 10.1016/j.cmet.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 54.Morton GJ, Gelling RW, Niswender KD, Morrison CD, Rhodes CJ, Schwartz MW. Leptin regulates insulin sensitivity via phosphatidylinositol-3-OH kinase signaling in mediobasal hypothalamic neurons. Cell Metab. 2005;2:411–20. doi: 10.1016/j.cmet.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 55.Munzberg H, Flier JS, Bjorbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology. 2004;145:4880–9. doi: 10.1210/en.2004-0726. [DOI] [PubMed] [Google Scholar]
- 56.Haque MS, Minokoshi Y, Hamai M, Iwai M, Horiuchi M, Shimazu T. Role of the sympathetic nervous system and insulin in enhancing glucose uptake in peripheral tissues after intrahypothalamic injection of leptin in rats. Diabetes. 1999;48:1706–12. doi: 10.2337/diabetes.48.9.1706. [DOI] [PubMed] [Google Scholar]
- 57.Minokoshi Y, Haque MS, Shimazu T. Microinjection of leptin into the ventromedial hypothalamus increases glucose uptake in peripheral tissues in rats. Diabetes. 1999;48:287–91. doi: 10.2337/diabetes.48.2.287. [DOI] [PubMed] [Google Scholar]
- 58.Zhang R, Dhillon H, Yin H, Yoshimura A, Lowell BB, Maratos-Flier E, et al. Selective inactivation of Socs3 in SF1 neurons improves glucose homeostasis without affecting body weight. Endocrinology. 2008;149:5654–61. doi: 10.1210/en.2008-0805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bingham NC, Anderson KK, Reuter AL, Stallings NR, Parker KL. Selective loss of leptin receptors in the ventromedial hypothalamic nucleus results in increased adiposity and a metabolic syndrome. Endocrinology. 2008;149:2138–48. doi: 10.1210/en.2007-1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dhillon H, Zigman JM, Ye C, Lee CE, McGovern RA, Tang V, et al. Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron. 2006;49:191–203. doi: 10.1016/j.neuron.2005.12.021. [DOI] [PubMed] [Google Scholar]
- 61.German JP, Kim F, Schwartz GJ, Havel PJ, Rhodes CJ, Schwartz MW, et al. Hypothalamic leptin signaling regulates hepatic insulin sensitivity via a neurocircuit involving the vagus nerve. Endocrinology. 2009;150:4502–11. doi: 10.1210/en.2009-0445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pocai A, Lam TK, Gutierrez-Juarez R, Obici S, Schwartz GJ, Bryan J, et al. Hypothalamic K (ATP) channels control hepatic glucose production. Nature. 2005;434:1026–31. doi: 10.1038/nature03439. [DOI] [PubMed] [Google Scholar]
- 63.Pocai A, Obici S, Schwartz GJ, Rossetti L. A brain-liver circuit regulates glucose homeostasis. Cell Metab. 2005;1:53–61. doi: 10.1016/j.cmet.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 64.Kamohara S, Burcelin R, Halaas JL, Friedman JM, Charron MJ. Acute stimulation of glucose metabolism in mice by leptin treatment. Nature. 1997;389:374–7. doi: 10.1038/38717. [DOI] [PubMed] [Google Scholar]
- 65.Toda C, Shiuchi T, Lee S, Yamato-Esaki M, Fujino Y, Suzuki A, et al. Distinct effects of leptin and a melanocortin receptor agonist injected into medial hypothalamic nuclei on glucose uptake in peripheral tissues. Diabetes. 2009;58:2757–65. doi: 10.2337/db09-0638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dobbs R, Sakurai H, Sasaki H, Faloona G, Valverde I, Baetens D, et al. Glucagon: Role in the hyperglycemia of diabetes mellitus. Science. 1975;187:544–7. doi: 10.1126/science.1089999. [DOI] [PubMed] [Google Scholar]
- 67.Muller WA, Faloona GR, Unger RH. The effect of experimental insulin deficiency on glucagon secretion. J Clin Invest. 1971;50:1992–9. doi: 10.1172/JCI106691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fujikawa T, Chuang JC, Sakata I, Ramadori G, Coppari R. Leptin therapy improves insulin-deficient type 1 diabetes by CNS-dependent mechanisms in mice. Proc Natl Acad Sci U S A. 2010;107:17391–6. doi: 10.1073/pnas.1008025107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shimazu T. Neuronal regulation of hepatic glucose metabolism in mammals. Diabetes Metab Rev. 1987;3:185–206. doi: 10.1002/dmr.5610030109. [DOI] [PubMed] [Google Scholar]
- 70.Shimazu T. Central nervous system regulation of liver and adipose tissue metabolism. Diabetologia. 1981;20(Suppl):343–56. [PubMed] [Google Scholar]
- 71.Nonogaki K, Iguchi A. Role of central neural mechanisms in the regulation of hepatic glucose metabolism. Life Sci. 1997;60:797–807. doi: 10.1016/s0024-3205(96)00596-6. [DOI] [PubMed] [Google Scholar]
- 72.Thorens B. Glucose sensing and the pathogenesis of obesity and type 2 diabetes. Int J Obes (Lond) 2008;32(Suppl 6):S62–71. doi: 10.1038/ijo.2008.208. [DOI] [PubMed] [Google Scholar]
- 73.Kieffer TJ, Heller RS, Habener JF. Leptin receptors expressed on pancreatic beta-cells. Biochem Biophys Res Commun. 1996;224:522–7. doi: 10.1006/bbrc.1996.1059. [DOI] [PubMed] [Google Scholar]
- 74.Cases JA, Gabriely I, Ma XH, Yang XM, Michaeli T, Fleischer N, et al. Physiological increase in plasma leptin markedly inhibits insulin secretion in vivo. Diabetes. 2001;50:348–52. doi: 10.2337/diabetes.50.2.348. [DOI] [PubMed] [Google Scholar]
- 75.Muzumdar R, Ma X, Yang X, Atzmon G, Bernstein J, Karkanias G, et al. Physiologic effect of leptin on insulin secretion is mediated mainly through central mechanisms. Faseb J. 2003;17:1130–2. doi: 10.1096/fj.02-0991fje. [DOI] [PubMed] [Google Scholar]
- 76.Park S, Hong SM, Sung SR, Jung HK. Long-term effects of central leptin and resistin on body weight, insulin resistance, and beta-cell function and mass by the modulation of hypothalamic leptin and insulin signaling. Endocrinology. 2008;149:445–54. doi: 10.1210/en.2007-0754. [DOI] [PubMed] [Google Scholar]
- 77.Park S, Ahn IS, Kim da S. Central infusion of leptin improves insulin resistance and suppresses beta-cell function, but not beta-cell mass, primarily through the sympathetic nervous system in a type 2 diabetic rat model. Life Sci. 2010;86:854–62. doi: 10.1016/j.lfs.2010.03.021. [DOI] [PubMed] [Google Scholar]
- 78.Boghossian S, Dube MG, Torto R, Kalra PS, Kalra SP. Hypothalamic clamp on insulin release by leptin-transgene expression. Peptides. 2006;27:3245–54. doi: 10.1016/j.peptides.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 79.Kalra SP. Central leptin gene therapy ameliorates diabetes type 1 and 2 through two independent hypothalamic relays; a benefit beyond weight and appetite regulation. Peptides. 2009;30:1957–63. doi: 10.1016/j.peptides.2009.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cheung CC, Clifton DK, Steiner RA. Proopiomelanocortin neurons are direct targets for leptin in the hypothalamus. Endocrinology. 1997;138:4489–92. doi: 10.1210/endo.138.10.5570. [DOI] [PubMed] [Google Scholar]
- 81.Schwartz MW, Seeley RJ, Woods SC, Weigle DS, Campfield LA, Burn P, et al. Leptin increases hypothalamic pro-opiomelanocortin mRNA expression in the rostral arcuate nucleus. Diabetes. 1997;46:2119–23. doi: 10.2337/diab.46.12.2119. [DOI] [PubMed] [Google Scholar]
- 82.Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature. 1997;385:165–8. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 83.Obici S, Feng Z, Tan J, Liu L, Karkanias G, Rossetti L. Central melanocortin receptors regulate insulin action. J Clin Invest. 2001;108:1079–85. doi: 10.1172/JCI12954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hahn TM, Breininger JF, Baskin DG, Schwartz MW. Coexpression of Agrp and NPY in fasting-activated hypothalamic neurons. Nat Neurosci. 1998;1:271–2. doi: 10.1038/1082. [DOI] [PubMed] [Google Scholar]
- 85.Rossi M, Kim MS, Morgan DG, Small CJ, Edwards CM, Sunter D, et al. A C-terminal fragment of Agouti-related protein increases feeding and antagonizes the effect of alpha-melanocyte stimulating hormone in vivo. Endocrinology. 1998;139:4428–31. doi: 10.1210/endo.139.10.6332. [DOI] [PubMed] [Google Scholar]
- 86.Stanley BG, Kyrkouli SE, Lampert S, Leibowitz SF. Neuropeptide Y chronically injected into the hypothalamus: A powerful neurochemical inducer of hyperphagia and obesity. Peptides. 1986;7:1189–92. doi: 10.1016/0196-9781(86)90149-x. [DOI] [PubMed] [Google Scholar]
- 87.Marks JL, Waite K. Intracerebroventricular neuropeptide Y acutely influences glucose metabolism and insulin sensitivity in the rat. J Neuroendocrinol. 1997;9:99–103. doi: 10.1046/j.1365-2826.1997.00554.x. [DOI] [PubMed] [Google Scholar]
- 88.Adage T, Scheurink AJ, de Boer SF, de Vries K, Konsman JP, Kuipers F, et al. Hypothalamic, metabolic, and behavioral responses to pharmacological inhibition of CNS melanocortin signaling in rats. J Neurosci. 2001;21:3639–45. doi: 10.1523/JNEUROSCI.21-10-03639.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, et al. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–4. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- 90.Pinto S, Roseberry AG, Liu H, Diano S, Shanabrough M, Cai X, et al. Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science. 2004;304:110–5. doi: 10.1126/science.1089459. [DOI] [PubMed] [Google Scholar]
- 91.van de Wall E, Leshan R, Xu AW, Balthasar N, Coppari R, Liu SM, et al. Collective and individual functions of leptin receptor modulated neurons controlling metabolism and ingestion. Endocrinology. 2008;149:1773–85. doi: 10.1210/en.2007-1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Huo L, Gamber K, Greeley S, Silva J, Huntoon N, Leng XH, et al. Leptin-dependent control of glucose balance and locomotor activity by POMC neurons. Cell Metab. 2009;9:537–47. doi: 10.1016/j.cmet.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Berglund ED, Vianna CR, Donato J, Jr, Kim MH, Chuang JC, Lee CE, et al. Direct leptin action on POMC neurons regulates glucose homeostasis and hepatic insulin sensitivity in mice. J Clin Invest. 2012;122:1000–9. doi: 10.1172/JCI59816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Balthasar N, Coppari R, McMinn J, Liu SM, Lee CE, Tang V, et al. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron. 2004;42:983–91. doi: 10.1016/j.neuron.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 95.Gutierrez-Juarez R, Obici S, Rossetti L. Melanocortin-independent effects of leptin on hepatic glucose fluxes. J Biol Chem. 2004;279:49704–15. doi: 10.1074/jbc.M408665200. [DOI] [PubMed] [Google Scholar]
- 96.da Silva AA, do Carmo JM, Freeman JN, Tallam LS, Hall JE. A functional melanocortin system may be required for chronic CNS-mediated antidiabetic and cardiovascular actions of leptin. Diabetes. 2009;58:1749–56. doi: 10.2337/db08-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Heymsfield SB, Greenberg AS, Fujioka K, Dixon RM, Kushner R, Hunt T, et al. Recombinant leptin for weight loss in obese and lean adults: A randomized, controlled, dose-escalation trial. JAMA. 1999;282:1568–75. doi: 10.1001/jama.282.16.1568. [DOI] [PubMed] [Google Scholar]
- 98.El-Haschimi K, Pierroz DD, Hileman SM, Bjorbaek C, Flier JS. Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J Clin Invest. 2000;105:1827–32. doi: 10.1172/JCI9842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Myers MG, Jr, Leibel RL, Seeley RJ, Schwartz MW. Obesity and leptin resistance: Distinguishing cause from effect. Trends Endocrinol Metab. 2010;21:643–51. doi: 10.1016/j.tem.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kalra SP. Central leptin insufficiency syndrome: An interactive etiology for obesity, metabolic and neural diseases and for designing new therapeutic interventions. Peptides. 2008;29:127–38. doi: 10.1016/j.peptides.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cummings BP, Bettaieb A, Graham JL, Stanhope KL, Dill R, Morton GJ, et al. Subcutaneous administration of leptin normalizes fasting plasma glucose in obese type 2 diabetic UCD-T2DM rats. Proc Natl Acad Sci U S A. 2011;108:14670–5. doi: 10.1073/pnas.1107163108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Miyanaga F, Ogawa Y, Ebihara K, Hidaka S, Tanaka T, Hayashi S, et al. Leptin as an adjunct of insulin therapy in insulin-deficient diabetes. Diabetologia. 2003;46:1329–37. doi: 10.1007/s00125-003-1193-6. [DOI] [PubMed] [Google Scholar]
- 103.Sandoval DA, Obici S, Seeley RJ. Targeting the CNS to treat type 2 diabetes. Nat Rev Drug Discov. 2009;8:386–98. doi: 10.1038/nrd2874. [DOI] [PubMed] [Google Scholar]
- 104.Barsh GS, Schwartz MW. Genetic approaches to studying energy balance: Perception and integration. Nat Rev Genet. 2002;3:589–600. doi: 10.1038/nrg862. [DOI] [PubMed] [Google Scholar]