

Abstract
There are an estimated 36 million dementia patients worldwide. The anticipated tripling of this number by year 2050 will negatively impact the capacity to deliver quality health care. The epidemic in diabetes is particularly troubling, because diabetes is a substantial risk factor for dementia independently of cerebrovascular disease. There is an urgent need to elucidate the pathogenesis of progressive brain atrophy, the cause of dementia, to allow rational design of new therapeutic interventions. This review summarizes recent tests of the hypothesis that the concomitant loss of insulin and insulin-like growth factors (IGFs) is the dominant cause for age-dependent, progressive brain atrophy with degeneration and cognitive decline. These tests are the first to show that insulin and IGFs regulate adult brain mass by maintaining brain protein content. Insulin and IGF levels are reduced in diabetes, and replacement of both ligands can prevent loss of total brain protein, widespread cell degeneration, and demyelination. IGF alone prevents retinal degeneration in diabetic rats. It supports synapses and is required for learning and memory. Replacement doses in diabetic rats can cross the blood-brain barrier to prevent hippocampus-dependent memory impairment. Insulin and IGFs are protective despite unabated hyperglycemia in diabetic rats, severely restricting hyperglycemia and its consequences as dominant pathogenic causes of brain atrophy and impaired cognition. These findings have important implications for late-onset alzheimer's disease (LOAD) where diabetes is a major risk factor, and concomitant decline in insulin and IGF activity suggest a similar pathogenesis for brain atrophy and dementia.
Keywords: Diabetes mellitus, insulin, brain atrophy, cognitive impairment, insulin-like growth factors, Alzheimer's disease
INTRODUCTION
Both type 1 diabetes (T1D) and type 2 diabetes (T2D) are associated with brain atrophy and cognitive impairments. Progressive age-dependent accelerated brain atrophy with cell degeneration is the cause of cognitive impairments that may progress to dementia, particularly in elderly T2D patients, independently of cerebrovascular disease. Brain atrophy and cognitive disturbances are not correlated with poor glycemic control or hypoglycemic episodes. This review will discuss the relationship between brain atrophy and concomitant decline in insulin and insulin-like growth factor (IGF) levels in diabetes independently of hyperglycemia. Age-dependent concomitant loss of insulin and IGF is also associated with brain atrophy and dementia in Late Onset Alzheimer's Disease (LOAD).
BRAIN ATROPHY AND COGNITIVE IMPAIRMENTS IN TYPE 1 DIABETES
Pancreatic beta-cell degeneration results in a defect in insulin production in T1D. Children with early onset and long duration T1D exhibit greater impairments in intellectual and reading skills compared with age-matched nondiabetic subjects.[1] Performance is poorer in auditory verbal learning tests involving verbal mastery and longer disease duration predicts poorer learning over time.[2] In tests requiring sustained attention, rapid analysis of visuospatial detail, and hand-eye coordination, T1D patients perform significantly worse; poor performance may not be correlated with a history of hypoglycemic episodes.[3] The incidence of anxiety, depression, and antisocial personality disorders is higher[4–7] and increased prevalence of dementia is reported among elderly T1D patients.[3,8]
Autopsy studies show structural lesions in the CNS of long-term T1D subjects[9] and brain atrophy is detected with magnetic resonance imaging (MRI). As many as 70% of young T1D patients have enlarged brain lateral ventricles and subarachnoidal spaces.[10] Sub-cortical and brain stem lesions[11] as well as “high-intensity rounded lesions and cortical atrophy are found in long duration T1D patients, suggestive of premature aging of the brain”.[12]
BRAIN ATROPHY AND COGNITIVE IMPAIRMENTS IN TYPE 2 DIABETES
T2D is characterized by insulin resistance with partially diminished capacity to produce insulin. Compared with age-matched control subjects, T2D patients perform significantly worse when tested for effective attention-concentration-working memory or when faced with tasks requiring mathematical problem solving, constructional abilities, verbal fluency, psychomotor speed, mental flexibility, complex psychomotor functioning, abstract reasoning, or storage and retrieval of new information.[13–16] Population-based studies show that T2D patients have significantly greater risk for dementia.[17,18] A longitudinal, prospective, cross-sectional population-based study with over 6300 elderly type II diabetic subjects showed a near doubling of the risk for dementia after adjusting for age, sex, cerebrovascular disease, hypertension, and other confounders.[19] The risk of dementia is independent of cerebrovascular disease, and diabetes is a separate and major risk factor.
Electrophysiological abnormalities in the CNS, including slowing of saltatory conduction, occur in both T1D and T2D.[20,21] MRI studies demonstrate brain atrophy in T2D patients to be more frequent, particularly in the 6th and 8th decades of life.[22] Like dementia, brain atrophy occurs independently of vascular disease. In a study controlled for age, sex, hypertension, and level of education, impairments in attention, executive functioning, information processing, and memory in T2D patients were associated with brain atrophy and white matter hyperintensities (lesions characterized by partial loss of myelin, axons, and oligodendrocytes).[23] Computerized tomography[24] and postmortem autopsy investigations[25] confirm these findings.
These data show that brain atrophy in both types of diabetes is associated with cognitive, behavioral, and electrophysiological impairments. There is increased risk for dementia, principally among the elderly. Glycemic control is not correlated with impaired cognition in diabetes[8] and cognitive function in T1D patients was found to be unaffected by hyperglycemia.[26] Repeated episodes of severe hypoglycemia were correlated with neither cognitive impairment nor brain atrophy in young T1D patients.[27]
BRAIN INSULIN AND INSULIN-LIKE GROWTH FACTOR ACTIVITY IS DIMINISHED IN DIABETES
Diabetes is a complex disorder, and various hypotheses have been proposed to account for neurological dysfunction. These mainly include, but are not limited to, pathogenic effects of hyperglycemia, polyol accumulation, protein glycation, accumulation of advanced glycation end products, ischemia, and reactive oxygen species. The focus of our research has been to elucidate the normal neurobiological actions of insulin and IGFs, as well as to test the hypothesis that diminished levels of these ligands is pathogenic for neural dysfunction.[28,29]
Availability of insulin to the brain in T1D is diminished due to a decline in circulating levels of insulin. T2D, on the other hand, is characterized by insulin resistance with partial failure of insulin secretion. Insulin crosses the blood-brain barrier (BBB), and such transport is inhibited by a high fat diet which also causes insulin resistance. Low cerebrospinal fluid (CSF) insulin levels are found in T2D patients.[30]
IGF-I and IGF-II are members of the insulin gene family that structurally resemble pro-insulin. Both IGFs bind to and activate the type 1 IGF receptors that are ubiquitously present in brain. IGF levels gradually decline with aging[31] and are partially reduced even in the prediabetic state prior to frank hyperglycemia. Compared with age-matched nondiabetic subjects, IGF levels are reduced by approximately half in both T1D and T2D diabetic populations.[32] This decline is reported to be greater in diabetic patients with progressive peripheral neuropathy.[33] IGF availability is diminished even further due to altered binding to IGF binding proteins (IGFBPs), which normally regulate IGF bioavailability.[34] The predominant IGF in adult mammalian brain is IGF-II, and we have shown that IGF-II mRNA levels decline in both T1D and T2D rats.[35] Depending on duration and severity of diabetes, circulating IGF-II levels may also be reduced.[36,37]
SOURCE OF BRAIN INSULIN AND PROPERTIES OF NEURONAL INSULIN RECEPTORS
Brain insulin is primarily derived from the circulation by saturable receptor-mediated transport across the BBB.[38,39] Perhaps the best understood physiological role of insulin in the brain is the regulation of feeding and satiety through the hypothalamus. The insulin receptor (IR) is widespread in brain[40] and several attributes distinguish the neuronal IR from its peripheral counterpart, suggesting special functions. The neuronal IR exhibits reduced glycosylation and smaller subunit size[41–43] and, unlike its peripheral counterpart, it does not down-regulate in response to excess insulin.[44,45] Physiological concentrations of insulin are sufficient to elicit half-maximal response of the receptor.[41,46] The IRs are localized to neuronal soma, neurites, and nerve terminals and their roles may include electrophysiological modulation of neurons. Interestingly, glia display the peripheral tissue-type IR.
Physiological doses of insulin do not significantly influence global brain glucose utilization. Intravenous infusion of insulin under steady-state normoglycemic conditions does not increase brain glucose utilization in humans[47] nor rats,[48] except at the hypothalamic satiety centers.[49] In a key study, selective inactivation of the neuronal IR in mice has no effect on brain glucose utilization,[50] indicating that insulin is not the principal mediator of glucose uptake in brain.
INSULIN-LIKE GROWTH FACTORS REGULATE LEARNING AND MEMORY
Liver is the principal source of brain IGF-I.[51] To show that IGFs cross the BBB, 125I-labeled IGF was injected into rat carotid artery. Subsequent autoradiography of brain slices revealed radioactivity of ambiguous molecular identity in the brain parenchyma.[52] A more conclusive study in our lab sampled CSF and showed using protein electrophoresis that some of the radioactivity co-migrated with authentic IGF.[53] The dose-dependent saturable uptake of IGF-I from blood into CSF is neither IGF-I receptor nor IGFBP-dependent.[54] A multicargo endocytic receptor, megalin, is believed to mediate the uptake of both insulin and IGF-I into brain.[55] As is the case with insulin, most of the IGF-I present in CSF is derived from blood.[53]
IGF gene expression is closely correlated with synaptogenesis[56] and establishment of polyneuronal innervation during nerve regeneration.[57,58] Likewise, IGF treatment increases synaptic spine density in brain slices.[59] Consequently, our laboratory tested the hypothesis that brain IGF normally supports Learning and Memory (L&M) in adult healthy rats. Infusion of an IGF antiserum into the lateral brain ventricle caused the impairment of L&M in a passive-avoidance test, whereas preimmune serum had no effect.[60] Others have found that injection of a cDNA viral vector encoding a dominant-negative IGF-I receptor into healthy adult rat brain lateral ventricle resulted in disruption of receptor activity whereupon the animals exhibited cognitive impairments.[61]
Diminished serum IGF levels in normal aging and disease may disturb cognitive function as a consequence of reduced IGF flux across the BBB. Human cognitive performance correlates with serum IGF-I levels and with age;[62,63] elderly subjects with low circulating IGF-I levels perform more poorly on standard cognitive tests. The hypothesis that diminished IGF levels in diabetes may be a cause of impaired L&M was tested (see below).
THE STREPTOZOTOCIN-DIABETIC RAT AS A MODEL FOR BRAIN ATROPHY WITH IMPAIRED LEARNING AND MEMORY
Streptozotocin (STZ) is a structural glucose analogue that is selectively taken up by beta-pancreatic cells, resulting in destruction of insulin-producing capacity. Peripherally administered STZ does not cross the BBB and is not expected to directly affect the CNS. Depending on dose and route of STZ administration, circulating insulin levels can decline by 40–80% or more.[64,65] IGF levels are also reduced, as hepatic IGF-I mRNA content decreases rapidly over a period of 3 days following STZ injection.[66,67] Circulating IGF-I levels are lowered 50% to more than 80%.[68–70]
The STZ-diabetic rat shares with clinical diabetes both brain atrophy and L&M impairments. STZ-injected rodents exhibit cell degeneration and loss of brain mass,[71,72] affecting a wide range of cell populations including neurons,[73,74] astrocytes,[75] oligodendrocytes,[76] and microglia.[77] There is evidence of impaired synaptic plasticity and myelination, reduced neurite arborization, and increased cell death, as well as electrophysiological impairments.[78] STZ-diabetic animals perform significantly worse on L&M tests compared with nondiabetic littermates and advancing age exacerbates that deficit.[60,78]
SUBCUTANEOUS INSULIN-LIKE GROWTH FACTOR-I REPLACEMENT DOSES PREVENT LEARNING AND MEMORY DEFICITS IN STREPTOZOTOCIN-DIABETIC RATS INDEPENDENTLY OF HYPERGLYCEMIA
Is hyperglycemia or the decreased levels of IGFs the primary cause of cognitive impairment in diabetes? IGF levels are lower in diabetes, and IGFs regulate synapses. Synaptic spine density and arborization are reduced in the brains of diabetic rats.[73] The hypothesis was tested that IGF-I administered from a subcutaneous site can cross the BBB and prevent hippocampus-dependent spatial orientation deficits in STZ-diabetic rats irrespective of persisting hyperglycemia.[60] Nondiabetic and diabetic rat groups were subjected to Morris water maze testing 11 weeks after the onset of diabetes, where diabetic rats were implanted with subcutaneous minipumps that continuously administered either vehicle or a replacement dose of IGF-I during the last 6 weeks of diabetes. Compared with nondiabetic rats, diabetic rats performed more poorly in the water maze task. On the other hand, IGF-I treatment prevented L&M impairment in diabetic rats, irrespective of uncorrected hyperglycemia and ongoing catabolic state.
All rats irrespective of test group were capable of swimming with the same velocity, showing that diabetes did not cause motor or proprioceptive disturbances sufficient to impair swim performance. However, when tests were performed using a visible platform, greater latency among diabetic rats receiving vehicle was uncovered, suggesting mild impairment in comprehension, motivation, executive function, or vision. Biessels et al., did not detect hippocampus-dependent L&M impairments in STZ-diabetic rats after 4 weeks of disease progression,[79] suggesting that they arise between 4 and 11 weeks after disease onset.
BRAIN ATROPHY IS LARGELY UNCORRECTED BY INSULIN-LIKE GROWTH FACTOR ADMINISTRATION
To assess the effect of IGF-I treatment on brain atrophy, the same animals discussed in the foregoing section were euthanized and brains collected for analysis 12 weeks after STZ injection.[72] Brain wet weight was significantly reduced in diabetic rats. Brains were homogenized in buffer and lyophilized to determine water and dry weights. Aliquots of homogenate were used to measure total mRNA transcripts, 18S rRNA, and total protein content. Brain total DNA content was measured using a fluorometric protocol. All measured parameters were significantly reduced in diabetic animals, showing a severely catabolic state that involves perturbations in the protein regulatory pathway as well as significant cell loss.
Among the earliest biochemical changes in brain is a reduction in total mRNA content in whole brain, hippocampus, and hypothalamus as well as a selective decrease in IGF-II mRNA content per total mRNA that is observed after 2 weeks of STZ-induced diabetes.[35] Interestingly, peripheral administration of IGF-I can prevent these decreases independently of hyperglycemia, showing that brain IGF-II gene expression is regulated by IGF-I. A loss of brain weight is not yet evident 2 weeks after STZ injection, and this loss of transcripts precedes brain atrophy and impaired cognition. After 10 weeks, total brain protein content was reduced, although cell loss was not yet evident (unpublished observations). Significant DNA loss appears to emerge only after approximately 12 weeks of unabated diabetes. Thus, there is progressive loss of total brain mRNA and protein in diabetes. These perturbations in the protein regulatory pathway, when sufficiently severe and prolonged, most likely lead to loss of cells.
It is interesting that subcutaneous IGF-I administration in diabetic rats prevented impairment of L&M, but not brain atrophy.[72] IGF may have selective effects on L&M by influencing synaptic spine density, spine plasticity, synaptic protein turnover, and/or on long-term potentiation. However, these IGF experiments did not reveal whether the cause of brain atrophy was hyperglycemia or loss of insulin and IGF activities.
INSULIN-LIKE GROWTH FACTORS PREVENT RETINAL DEGENERATION IN DIABETES
While IGF treatment did not by itself prevent gross brain atrophy in diabetic rats, it is possible that it may prevent cell degeneration in limited regions of the CNS. To test this, the eye was studied. It is formed developmentally as an outpocketing of the brain, and is a CNS tissue. Diabetes is a prominent cause of vision loss and blindness.[80,81] Retinal degeneration is observed in both clinical and experimental diabetes[82,83] and the progression of diabetic retinopathy is not correlated with glycemic control.[84,85]
Armed with the knowledge that IGF-I and certain of its structural analogues can cross the BBB,[53,54] it was tested whether IGF can cross the blood–retinal barrier and prevent predegenerative changes in retina in diabetic rats. Adult rats rendered diabetic with STZ were implanted with subcutaneous minipumps that released either vehicle or des(1-3)IGF-I, an IGF-I analogue that lacks the N-terminal tripeptide and whose sequestration by IGFBPs is consequently greatly diminished. After 2 weeks of diabetes, animals were euthanized, eyes were collected, and processed for immunohistochemistry. There was a significant increase in IGF-I receptor immunoreactivity in the inner nuclear and ganglion cell layers in STZ diabetic rat retina compared with nondiabetic controls,[86] consistent with observations by others.[87] Such increase may be the result of a homeostatic mechanism up-regulating the IGF receptor as a consequence of decreased IGF-I levels. This up-regulation was prevented in diabetic rats treated with des(1-3)IGF-I despite hyperglycemia.
Abnormalities in the apoptosis-stress response enzyme Akt were observed in the same two retinal layers. Akt phosphorylation at residue Thr308 was significantly increased in diabetic compared with nondiabetic controls, showing preapoptotic degenerative changes.[86] Treatment with des(1-3)IGF-I prevented this increase in Akt phosphorylation. This study establishes that an IGF-I analogue crossed the blood–retinal barrier and prevented biochemical abnormalities associated with predegenerative changes in the retina of diabetic rats.
In apoptotic cell death there is fragmentation of DNA detectable by the terminal deoxyuridine nick end labeling TUNEL method. In long-term diabetic rats there is an increase in TUNEL-positive cells in the retina after 4 weeks of diabetes.[82] In our experiments, there was a 6-fold increase in TUNEL staining in the photoreceptor cell layer and an 8-fold increase in the inner nuclear layer after 12 weeks of STZ-diabetes. Subcutaneous IGF-I administration crossed the blood–retinal barrier and prevented such increases.[88] Qualitative results showed that pro-apoptotic caspase-3 and BAD immunoreactivities were also elevated, and these abnormalities were likewise prevented with the IGF-I treatment.
Taken together, these data show that predegenerative and degenerative changes in the retina of diabetic rats can be prevented by replacement of IGF-I or des(1-3)IGF-I despite persisting hyperglycemia. Although IGF-I does not prevent the loss of bulk brain mass in diabetes,[72] it does selectively prevent the loss of certain types of neurons within the adult CNS.
INSULIN AND INSULIN-LIKE GROWTH FACTOR ARE NEUROTROPHIC FACTORS THAT ACTIVATE A COMMON BIOCHEMICAL PATHWAY
IGFs were discovered to be neurotrophic factors based on their capacity to increase the expression of axonal cytoskeletal genes, stimulate neurite outgrowth, increase RNA synthesis, and support cell survival.[89–91] Early studies with extremely large doses of impure insulin reported its actions on cultured neural tissues,[90,91] however, the first unequivocal demonstration that physiological concentrations of insulin had neurotrophic activity came when highly purified biosynthetic insulin became available.[89,90] Our laboratory was able to show that insulin and IGF, each through its own receptor on the same cloned cell, could activate a common biochemical pathway regulating neurotrophic activity.[89,92] This is highly pertinent to understanding how they prevent brain atrophy.
IGF-I treatment alone does not prevent the loss of bulk brain mass in diabetic rats.[72] Interestingly, conditional inactivation of the neuronal IR in mice not only has no effect on brain glucose utilization, but also there is no sign of neurodegeneration in brain.[50,93] It would initially appear that neither insulin nor IGF play a substantial role in adult brain atrophy. However, because both IGF and insulin activate a common biochemical pathway, loss of one ligand may be compensated by the other. We proposed that the risk for brain atrophy is greatest when there is concomitant decline in insulin and IGF activities, such as in diabetes.[94]
INSULIN IS A POTENT REGULATOR OF ADULT BRAIN MASS AND ITS COMBINATION WITH INSULIN-LIKE GROWTH FACTOR-I PREVENTS BRAIN ATROPHY IN DIABETES
We tested the inter-related hypotheses that (1) small replacement doses of both insulin and IGF can prevent brain atrophy in diabetes, (2) the combination of these ligands is more effective than insulin alone, and (3) insulin and IGF can prevent brain atrophy and degeneration irrespective of ongoing hyperglycemia.[94] Adult rats were rendered diabetic with STZ and implanted with osmotic minipumps to deliver either artificial CSF, a small dose of insulin, or insulin in combination with IGF-I directly into brain lateral ventricles over a period of 12 weeks. Tiny ligand doses were used that did not prevent hyperglycemia, and that were sufficiently small that cross-occupancy of insulin or IGF receptors was highly unlikely. Brains were homogenized in buffer and aliquots were removed for analysis of total water and dry mass, DNA content, and relative abundance of cell-type specific proteins using western blots. All results were calculated on a per brain basis. Other brains were fixed and sliced for immunohistochemical examination.
This study is notable in that a loss of 9% of total brain DNA was found in diabetic rats, showing the remarkable extent of cell loss. Virtually all previous studies measured cell degeneration in a few brain tissue sections, and such data do not reveal the global magnitude of cell death. Magnitude is important, because due to massive parallel processing, loss of a few brain cells may not lead to functional significance.
Administration of insulin alone prevented the loss of brain wet, water, and dry weights per brain in diabetic rats, revealing insulin as a substantial regulator of brain mass.[94] Insulin's effect on brain water content is of considerable interest with regard to brain edema, which is known to sometimes develop during overly aggressive insulin treatment in uncontrolled diabetic patients. Insulin also significantly prevented the loss of proteins unique to glia. This included loss of glial fibrillary acidic protein in astrocytes and loss of myelin basic protein and proteolipid protein in oligodendrocytes. The latter two polypeptides together comprise 80% of myelin proteins, and myelin constitutes a significant fraction of brain mass. On the other hand, insulin treatment had no significant impact on loss of the particular neuronal marker proteins selected for study, nor did insulin alone have a detectible influence on loss of total brain DNA.
In addition to preventing the loss of brain wet, water, and dry weight, insulin in combination with IGF-I completely prevented the loss of total brain DNA in diabetic rats. The combination treatment was clearly more efficacious than insulin alone. Prevention of loss of brain dry weight included preserving levels of highly abundant proteins such as actin and alpha and beta tubulins. In addition to preventing loss of proteins selectively expressed in astrocytes and oligodendrocytes, the combination prevented the loss of neuron-specific neurofilament proteins (regulate diameter of axons) and class III beta-tubulin (localized to dendrites).
Immunohistochemical studies on brain slices showed that visible loss of glia- and neuron-specific proteins was prevented by treatment with the combination of ligands. This was studied in two brain regions of importance to cognition; the cortex and the hippocampus.
Thus, progressive cognitive impairment in diabetes is proposed to be primarily a consequence of age- and disease-dependent loss of insulin and IGF activities followed by progressive decline in protein levels, including those necessary for synaptic plasticity, synapses, axonal maintenance, myelin, neurotransmitter levels, and cell survival. Not all brain proteins are expected to be regulated by insulin and IGFs, however, and selective susceptibility of particular brain regions to atrophy is well to be expected.
INSULIN AND INSULIN-LIKE GROWTH FACTOR REGULATE BRAIN MASS INDEPENDENTLY OF HYPERGLYCEMIA
All of the above described effects of insulin and its combination with IGF in diabetic rats were independent of persisting plasma and CSF hyperglycemia. Body weight is highly sensitive to metabolic disturbances, and the treatments did not prevent the severe decrease in body weights. This revealed that the combined loss of insulin and IGF, rather than hyperglycemia, is the main risk factor for brain atrophy in diabetes. Because hyperglycemia was unabated for 12 weeks in these rats, it is expected that there would be elevated levels of polyols, protein glycation, advanced glycation end products, and other disturbances linked to hyperglycemia. This study severely constrains the importance of these factors in the pathogenesis of diabetic neurological complications.
INSULIN PREVENTS BRAIN ATROPHY IN MICE
In agreement with our findings in diabetic rats, using MRI Francis et al. found that intranasal insulin administration averted brain atrophy in diabetic mice.[95] The study also demonstrated presence of white matter hyperintensities in brains of diabetic mice and insulin administration prevented such hyperintensities independently of hyperglycemia. These results, taken together with our findings that insulin prevents loss of myelin basic protein and proteolipid protein,[94] provide strong experimental evidence that insulin is a major regulator of myelin in adult brain. These results are directly relevant to the clinical observation by MRI of white matter hyperintensities in diabetic patients.[11,96,97]
EFFECT OF INSULIN ON LEARNING AND MEMORY
Others have shown that subcutaneous treatment of STZ-diabetic rats with insulin reverses or prevents L&M deficits as well as electrophysiological impairments.[79,98] However, since insulin treatment alleviated hyperglycemia in these studies, it remains unclear whether insulin directly prevents impaired L&M. In other studies, where high doses of insulin were used to prevent cognitive disturbances, adequate controls are needed to exclude cross-occupancy of IGF receptors as a potential mechanism for enhancing cognitive function. Conditional knock-out of the neuronal IR has no effect on L&M in mice.[50]
CONCOMITANT DECLINE OF INSULIN AND INSULIN-LIKE GROWTH FACTOR ACTIVITIES IN ALZHEIMER's DISEASE
LOAD comprises approximately 95% of nongenetic Alzheimer's cases where progressive brain atrophy is the cause of dementia. White matter hyperintensities are present[99,100] and dementia correlates best with synapse loss.[101] Neuritic plaques and neurofibrillary tangles were shown to emerge with aging independently of dementia status[102–104] and immunization against amyloid-beta successfully reduces or eliminates neuritic plaque burden, yet has no effect on dementia nor life expectancy.[105]
There is significant evidence for concomitant decline of insulin and IGF activities in LOAD. The dominant risk factors for LOAD are advancing age and a mid-life clinical history of either diabetes or impaired fasting glucose (insulin resistance).[106] Elderly type II diabetic patients are at a significantly higher risk for developing LOAD compared with age-matched controls.[19,107] Compared with nondemented subjects, LOAD patients exhibit perturbations in insulin activity and glucose disposal, a pattern consistent with insulin resistance observed in type II diabetes.[108] Serum IGF-I levels are reduced in patients with LOAD as well[109,110] and progressive stages of LOAD are associated with greater decline in mRNA levels of brain insulin, IGF, IR, and IGF-I receptor in postmortem frontal lobe.[111] Thus, diminished levels of insulin and IGFs in brain may contribute to the pathogenesis for brain atrophy in LOAD as well.
CONCLUSIONS
Our studies have shown that insulin is a potent regulator of adult brain mass and that IGF is essential for L&M. We have also demonstrated that IGF acts protectively in the retina. These findings were made in the context of the STZ-diabetic rat which exhibits progressive CNS degeneration and concomitant decline of both insulin and IGF, which allows for investigating the roles of one peptide independently of the other. Importantly, our findings were made despite persisting hyperglycemia and body weight loss characteristic of unabated diabetes. Insulin and IGFs clearly potentiate one another's effects, which may be mediated in part through activation of a shared intracellular signaling pathway (Path B) that is distinct from hyperglycemia (Path A) [Figure 1]. Path A represents the classical hypothesis implicating hyperglycemia and its secondary consequences such as accumulation of polyols and generation of advanced glycation end products as main culprits for brain atrophy. Path B, on the other hand, represents the alternative hypothesis that insulin and IGFs are master switches regulating numerous proteins necessary for synaptic plasticity, synapses, myelination, neurites, and cell survival. We have tested and confirmed the Path B hypothesis while keeping Path A constant. These findings have important implications for treatment and prevention of brain atrophy and dementia in diabetes and possibly in LOAD as well.
Figure 1.
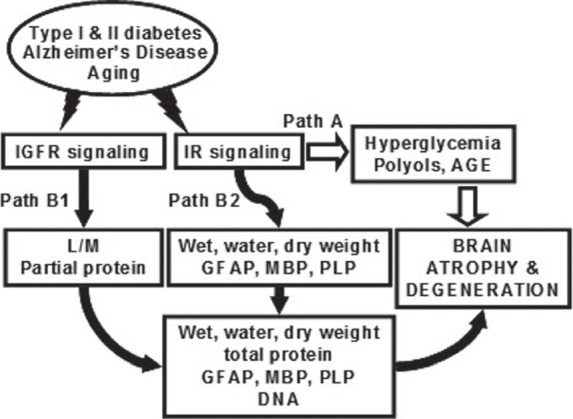
The two Paths show alternative hypotheses for the pathogenesis of brain atrophy predisposing to dementia in diabetes and possibly in Alzheimer's disease
Footnotes
Source(s) of Support: Centers for Disease Control and Prevention Grant R49/CCR811509 and the Colorado Commission on Higher Education Technology Advancement Grant TAG 06-01
Conflict of Interest: Dr. Ishii holds patents and patent applications for the use of insulin and IGFs to treat various neurodegenerative disorders. He is shareholder, Founder, and President of Aurogen Inc. a spinout biotechnology company from Colorado State University that has licensed these patents
REFERENCES
- 1.Holmes CS, Richman LC. Cognitive profiles of children with insulin-dependent diabetes. J Dev Behav Pediatr. 1985;6:323–6. [PubMed] [Google Scholar]
- 2.Fox MA, Chen RS, Holmes CS. Gender differences in memory and learning in children with insulin-dependent diabetes mellitus (IDDM) over a 4-year follow-up interval. J Pediatr Psychol. 2003;28:569–78. doi: 10.1093/jpepsy/jsg047. [DOI] [PubMed] [Google Scholar]
- 3.Ryan CM, Williams TM, Finegold DN, Orchard TJ. Cognitive dysfunction in adults with type 1 (insulin-dependent) diabetes mellitus of long duration: Effects of recurrent hypoglycaemia and other chronic complications. Diabetologia. 1993;36:329–34. doi: 10.1007/BF00400236. [DOI] [PubMed] [Google Scholar]
- 4.Anderson RJ, Freedland KE, Clouse RE, Lustman PJ. The prevalence of comorbid depression in adults with diabetes: A meta-analysis. Diabetes Care. 2001;24:1069–78. doi: 10.2337/diacare.24.6.1069. [DOI] [PubMed] [Google Scholar]
- 5.Peyrot M, Rubin RR. Levels and risks of depression and anxiety symptomatology among diabetic adults. Diabetes Care. 1997;20:585–90. doi: 10.2337/diacare.20.4.585. [DOI] [PubMed] [Google Scholar]
- 6.Popkin MK, Callies AL, Lentz RD, Colon EA, Sutherland DE. Prevalence of major depression, simple phobia, and other psychiatric disorders in patients with long-standing type I diabetes mellitus. Arch Gen Psychiatry. 1988;45:64–8. doi: 10.1001/archpsyc.1988.01800250078010. [DOI] [PubMed] [Google Scholar]
- 7.Roy M, Collier B, Roy A. Excess of depressive symptoms and life events among diabetics. Compr Psychiatry. 1994;35:129–31. doi: 10.1016/0010-440x(94)90057-o. [DOI] [PubMed] [Google Scholar]
- 8.Prescott JH, Richardson JT, Gillespie CR. Cognitive function in diabetes mellitus: The effects of duration of illness and glycaemic control. Br J Clin Psychol. 1990;29:167–75. doi: 10.1111/j.2044-8260.1990.tb00866.x. [DOI] [PubMed] [Google Scholar]
- 9.Reske-Nielsen E, Lundbaek K. Diabetic Encephalopathy.Diffuse and focal lesions of the brain in long-term diabetes. Acta Neurol Scand Suppl. 1963;39(suppl 4):273–90. [PubMed] [Google Scholar]
- 10.Lunetta M, Damanti AR, Fabbri G, Lombardo M, Di Mauro M, Mughini L. Evidence by magnetic resonance imaging of cerebral alterations of atrophy type in young insulin-dependent diabetic patients. J Endocrinol Invest. 1994;17:241–5. doi: 10.1007/BF03348967. [DOI] [PubMed] [Google Scholar]
- 11.Dejgaard A, Gade A, Larsson H, Balle V, Parving A, Parving HH. Evidence for diabetic encephalopathy. Diabet Med. 1991;8:162–7. doi: 10.1111/j.1464-5491.1991.tb01564.x. [DOI] [PubMed] [Google Scholar]
- 12.Perros P, Deary IJ, Sellar RJ, Best JJ, Frier BM. Brain abnormalities demonstrated by magnetic resonance imaging in adult IDDM patients with and without a history of recurrent severe hypoglycemia. Diabetes Care. 1997;20:1013–8. doi: 10.2337/diacare.20.6.1013. [DOI] [PubMed] [Google Scholar]
- 13.Stewart R, Liolitsa D. Type 2 diabetes mellitus, cognitive impairment and dementia. Diabet Med. 1999;16:93–112. doi: 10.1046/j.1464-5491.1999.00027.x. [DOI] [PubMed] [Google Scholar]
- 14.Perlmuter LC, Hakami MK, Hodgson-Harrington C, Ginsberg J, Katz J, Singer DE, et al. Decreased cognitive function in aging non-insulin-dependent diabetic patients. Am J Med. 1984;77:1043–8. doi: 10.1016/0002-9343(84)90186-4. [DOI] [PubMed] [Google Scholar]
- 15.Reaven GM, Thompson LW, Nahum D, Haskins E. Relationship between hyperglycemia and cognitive function in older NIDDM patients. Diabetes Care. 1990;13:16–21. doi: 10.2337/diacare.13.1.16. [DOI] [PubMed] [Google Scholar]
- 16.Tun PA, Nathan DM, Perlmuter LC. Cognitive and affective disorders in elderly diabetics. Clin Geriatr Med. 1990;6:731–46. [PubMed] [Google Scholar]
- 17.Leibson CL, Rocca WA, Hanson VA, Cha R, Kokmen E, O’Brien PC, et al. Risk of dementia among persons with diabetes mellitus: A population-based cohort study. Am J Epidemiol. 1997;145:301–8. doi: 10.1093/oxfordjournals.aje.a009106. [DOI] [PubMed] [Google Scholar]
- 18.Peila R, Rodriguez BL, Launer LJ. Honolulu-Asia Aging Study.Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes. 2002;51:1256–62. doi: 10.2337/diabetes.51.4.1256. [DOI] [PubMed] [Google Scholar]
- 19.Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology. 1999;53:1937–42. doi: 10.1212/wnl.53.9.1937. [DOI] [PubMed] [Google Scholar]
- 20.Brands AM, Kessels RP, de Haan EH, Kappelle LJ, Biessels GJ. Cerebral dysfunction in type 1 diabetes: Effects of insulin, vascular risk factors and blood-glucose levels. Eur J Pharmacol. 2004;490:159–68. doi: 10.1016/j.ejphar.2004.02.053. [DOI] [PubMed] [Google Scholar]
- 21.McCall AL. The impact of diabetes on the CNS. Diabetes. 1992;41:557–70. doi: 10.2337/diab.41.5.557. [DOI] [PubMed] [Google Scholar]
- 22.Araki Y, Nomura M, Tanaka H, Yamamoto H, Yamamoto T, Tsukaguchi I, et al. MRI of the brain in diabetes mellitus. Neuroradiology. 1994;36:101–3. doi: 10.1007/BF00588069. [DOI] [PubMed] [Google Scholar]
- 23.Manschot SM, Brands AM, van der Grond J, Kessels RP, Algra A, Kappelle LJ, et al. Brain magnetic resonance imaging correlates of impaired cognition in patients with type 2 diabetes. Diabetes. 2006;55:1106–13. doi: 10.2337/diabetes.55.04.06.db05-1323. [DOI] [PubMed] [Google Scholar]
- 24.Soininen H, Puranen M, Helkala EL, Laakso M, Riekkinen PJ. Diabetes mellitus and brain atrophy: A computed tomography study in an elderly population. Neurobiol Aging. 1992;13:717–21. doi: 10.1016/0197-4580(92)90095-f. [DOI] [PubMed] [Google Scholar]
- 25.Patrick AW, Campbell IW. Fatal hypoglycaemia in insulin-treated diabetes mellitus: Clinical features and neuropathological changes. Diabet Med. 1990;7:349–54. doi: 10.1111/j.1464-5491.1990.tb01403.x. [DOI] [PubMed] [Google Scholar]
- 26.Draelos MT, Jacobson AM, Weinger K, Widom B, Ryan CM, Finkelstein DM, et al. Cognitive function in patients with insulin-dependent diabetes mellitus during hyperglycemia and hypoglycemia. Am J Med. 1995;98:135–44. doi: 10.1016/S0002-9343(99)80397-0. [DOI] [PubMed] [Google Scholar]
- 27.Ferguson SC, Blane A, Perros P, McCrimmon RJ, Best JJ, Wardlaw J, et al. Cognitive ability and brain structure in type 1 diabetes: Relation to microangiopathy and preceding severe hypoglycemia. Diabetes. 2003;52:149–56. doi: 10.2337/diabetes.52.1.149. [DOI] [PubMed] [Google Scholar]
- 28.Ishii DN. Implication of insulin-like growth factors in the pathogenesis of diabetic neuropathy. Brain Res Brain Res Rev. 1995;20:47–67. doi: 10.1016/0165-0173(94)00005-a. [DOI] [PubMed] [Google Scholar]
- 29.Ishii DN, Lupien SB. Insulin-like growth factor replacement therapy for diabetic neuropathy: Experimental basis. Exp Diabesity Res. 2003;4:257–69. doi: 10.1155/EDR.2003.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kern W, Benedict C, Schultes B, Plohr F, Moser A, Born J, et al. Low cerebrospinal fluid insulin levels in obese humans. Diabetologia. 2006;49:2790–2. doi: 10.1007/s00125-006-0409-y. [DOI] [PubMed] [Google Scholar]
- 31.Hall K, Sara VR. Somatomedin levels in childhood, adolescence and adult life. Clin Endocrinol Metab. 1984;13:91–112. doi: 10.1016/s0300-595x(84)80010-9. [DOI] [PubMed] [Google Scholar]
- 32.Tan K, Baxter RC. Serum insulin-like growth factor I levels in adult diabetic patients: The effect of age. J Clin Endocrinol Metab. 1986;63:651–5. doi: 10.1210/jcem-63-3-651. [DOI] [PubMed] [Google Scholar]
- 33.Migdalis IN, Kalogeropoulou K, Kalantzis L, Nounopoulos C, Bouloukos A, Samartzis M. Insulin-like growth factor-I and IGF-I receptors in diabetic patients with neuropathy. Diabet Med. 1995;12:823–7. doi: 10.1111/j.1464-5491.1995.tb02086.x. [DOI] [PubMed] [Google Scholar]
- 34.Arner P, Sjöberg S, Gjötterberg M, Skottner A. Circulating insulin-like growth factor I in type 1 (insulin-dependent) diabetic patients with retinopathy. Diabetologia. 1989;32:753–8. doi: 10.1007/BF00274537. [DOI] [PubMed] [Google Scholar]
- 35.Wuarin L, Namdev R, Burns JG, Fei ZJ, Ishii DN. Brain insulin-like growth factor-II mRNA content is reduced in insulin-dependent and non-insulin-dependent diabetes mellitus. J Neurochem. 1996;67:742–51. doi: 10.1046/j.1471-4159.1996.67020742.x. [DOI] [PubMed] [Google Scholar]
- 36.Bowsher RR, Lee WH, Apathy JM, O’Brien PJ, Ferguson AL, Henry DP. Measurement of insulin-like growth factor-II in physiological fluids and tissues. I. An improved extraction procedure and radioimmunoassay for human and rat fluids. Endocrinology. 1991;128:805–14. doi: 10.1210/endo-128-2-805. [DOI] [PubMed] [Google Scholar]
- 37.Amiel SA, Sherwin RS, Hintz RL, Gertner JM, Press CM, Tamborlane WV. Effect of diabetes and its control on insulin-like growth factors in the young subject with type I diabetes. Diabetes. 1984;33:1175–9. doi: 10.2337/diab.33.12.1175. [DOI] [PubMed] [Google Scholar]
- 38.Steffens AB, Scheurink AJ, Porte D, Jr, Woods SC. Penetration of peripheral glucose and insulin into cerebrospinal fluid in rats. Am J Physiol. 1988;255:R200–4. doi: 10.1152/ajpregu.1988.255.2.R200. [DOI] [PubMed] [Google Scholar]
- 39.Woods SC, Seeley RJ, Baskin DG, Schwartz MW. Insulin and the blood-brain barrier. Curr Pharm Des. 2003;9:795–800. doi: 10.2174/1381612033455323. [DOI] [PubMed] [Google Scholar]
- 40.Havrankova J, Roth J, Brownstein M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature. 1978;272:827–9. doi: 10.1038/272827a0. [DOI] [PubMed] [Google Scholar]
- 41.Gammeltoft S, Fehlmann M, Van Obberghen E. Insulin receptors in the mammalian central nervous system: Binding characteristics and subunit structure. Biochimie. 1985;67:1147–53. doi: 10.1016/s0300-9084(85)80113-9. [DOI] [PubMed] [Google Scholar]
- 42.Lowe WL, Jr, Boyd FT, Clarke DW, Raizada MK, Hart C, LeRoith D. Development of brain insulin receptors: Structural and functional studies of insulin receptors from whole brain and primary cell cultures. Endocrinology. 1986;119:25–35. doi: 10.1210/endo-119-1-25. [DOI] [PubMed] [Google Scholar]
- 43.Heidenreich KA, Brandenburg D. Oligosaccharide heterogeneity of insulin receptors.Comparison of N-linked glycosylation of insulin receptors in adipocytes and brain. Endocrinology. 1986;118:1835–42. doi: 10.1210/endo-118-5-1835. [DOI] [PubMed] [Google Scholar]
- 44.Boyd FT, Jr, Raizada MK. Effects of insulin and tunicamycin on neuronal insulin receptors in culture. Am J Physiol. 1983;245:C283–7. doi: 10.1152/ajpcell.1983.245.3.C283. [DOI] [PubMed] [Google Scholar]
- 45.Zahniser NR, Goens MB, Hanaway PJ, Vinych JV. Characterization and regulation of insulin receptors in rat brain. J Neurochem. 1984;42:1354–62. doi: 10.1111/j.1471-4159.1984.tb02795.x. [DOI] [PubMed] [Google Scholar]
- 46.Gammeltoft S, Van Obberghen E. Protein kinase activity of the insulin receptor. Biochem J. 1986;235:1–11. doi: 10.1042/bj2350001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seaquist ER, Damberg GS, Tkac I, Gruetter R. The effect of insulin on in vivo cerebral glucose concentrations and rates of glucose transport/metabolism in humans. Diabetes. 2001;50:2203–9. doi: 10.2337/diabetes.50.10.2203. [DOI] [PubMed] [Google Scholar]
- 48.Lucignani G, Namba H, Nehlig A, Porrino LJ, Kennedy C, Sokoloff L. Effects of insulin on local cerebral glucose utilization in the rat. J Cereb Blood Flow Metab. 1987;7:309–14. doi: 10.1038/jcbfm.1987.68. [DOI] [PubMed] [Google Scholar]
- 49.Schwartz MW, Figlewicz DP, Baskin DG, Woods SC, Porte D., Jr Insulin in the brain: A hormonal regulator of energy balance. Endocr Rev. 1992;13:387–414. doi: 10.1210/edrv-13-3-387. [DOI] [PubMed] [Google Scholar]
- 50.Schubert M, Gautam D, Surjo D, Ueki K, Baudler S, Schubert D, et al. Role for neuronal insulin resistance in neurodegenerative diseases. Proc Natl Acad Sci U S A. 2004;101:3100–5. doi: 10.1073/pnas.0308724101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murphy LJ, Bell GI, Friesen HG. Tissue distribution of insulin-like growth factor I and II messenger ribonucleic acid in the adult rat. Endocrinology. 1987;120:1279–82. doi: 10.1210/endo-120-4-1279. [DOI] [PubMed] [Google Scholar]
- 52.Reinhardt RR, Bondy CA. Insulin-like growth factors cross the blood-brain barrier. Endocrinology. 1994;135:1753–61. doi: 10.1210/endo.135.5.7525251. [DOI] [PubMed] [Google Scholar]
- 53.Armstrong CS, Wuarin L, Ishii DN. Uptake of circulating insulin-like growth factor-I into the cerebrospinal fluid of normal and diabetic rats and normalization of IGF-II mRNA content in diabetic rat brain. J Neurosci Res. 2000;59:649–60. doi: 10.1002/(SICI)1097-4547(20000301)59:5<649::AID-JNR8>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 54.Pulford BE, Ishii DN. Uptake of circulating insulin-like growth factors (IGFs) into cerebrospinal fluid appears to be independent of the IGF receptors as well as IGF-binding proteins. Endocrinology. 2001;142:213–20. doi: 10.1210/endo.142.1.7894. [DOI] [PubMed] [Google Scholar]
- 55.Carro E, Spuch C, Trejo JL, Antequera D, Torres-Aleman I. Choroid plexus megalin is involved in neuroprotection by serum insulin-like growth factor I. J Neurosci. 2005;25:10884–93. doi: 10.1523/JNEUROSCI.2909-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ishii DN. Relationship of insulin-like growth factor II gene expression in muscle to synaptogenesis. Proc Natl Acad Sci U S A. 1989;86:2898–902. doi: 10.1073/pnas.86.8.2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Glazner GW, Morrison AE, Ishii DN. Elevated insulin-like growth factor (IGF) gene expression in sciatic nerves during IGF-supported nerve regeneration. Brain Res Mol Brain Res. 1994;25:265–72. doi: 10.1016/0169-328x(94)90162-7. [DOI] [PubMed] [Google Scholar]
- 58.Ishii DN, Glazner GW, Pu SF. Role of insulin-like growth factors in peripheral nerve regeneration. Pharmacol Ther. 1994;62:125–44. doi: 10.1016/0163-7258(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 59.Nieto-Bona MP, Garcia-Segura LM, Torres-Alemán I. Transynaptic modulation by insulin-like growth factor I of dendritic spines in Purkinje cells. Int J Dev Neurosci. 1997;15:749–54. doi: 10.1016/s0736-5748(97)00021-x. [DOI] [PubMed] [Google Scholar]
- 60.Lupien SB, Bluhm EJ, Ishii DN. Systemic insulin-like growth factor-I administration prevents cognitive impairment in diabetic rats, and brain IGF regulates learning/memory in normal adult rats. J Neurosci Res. 2003;74:512–23. doi: 10.1002/jnr.10791. [DOI] [PubMed] [Google Scholar]
- 61.Carro E, Trejo JL, Spuch C, Bohl D, Heard JM, Torres-Aleman I. Blockade of the insulin-like growth factor I receptor in the choroid plexus originates Alzheimer's-like neuropathology in rodents: New cues into the human disease? Neurobiol Aging. 2006;27:1618–31. doi: 10.1016/j.neurobiolaging.2005.09.039. [DOI] [PubMed] [Google Scholar]
- 62.Rollero A, Murialdo G, Fonzi S, Garrone S, Gianelli MV, Gazzerro E, et al. Relationship between cognitive function, growth hormone and insulin-like growth factor I plasma levels in aged subjects. Neuropsychobiology. 1998;38:73–9. doi: 10.1159/000026520. [DOI] [PubMed] [Google Scholar]
- 63.Dik MG, Pluijm SM, Jonker C, Deeg DJ, Lomecky MZ, Lips P. Insulin-like growth factor I (IGF-I) and cognitive decline in older persons. Neurobiol Aging. 2003;24:573–81. doi: 10.1016/s0197-4580(02)00136-7. [DOI] [PubMed] [Google Scholar]
- 64.Kim E, Sohn S, Lee M, Jung J, Kineman RD, Park S. Differential responses of the growth hormone axis in two rat models of streptozotocin-induced insulinopenic diabetes. J Endocrinol. 2006;188:263–70. doi: 10.1677/joe.1.06501. [DOI] [PubMed] [Google Scholar]
- 65.Arroba AI, Lechuga-Sancho AM, Frago LM, Argente J, Chowen JA. Cell-specific expression of X-linked inhibitor of apoptosis in the anterior pituitary of streptozotocin-induced diabetic rats. J Endocrinol. 2007;192:215–27. doi: 10.1677/joe.1.06985. [DOI] [PubMed] [Google Scholar]
- 66.Fagin JA, Roberts CT, Jr, LeRoith D, Brown AT. Coordinate decrease of tissue insulinlike growth factor I posttranscriptional alternative mRNA transcripts in diabetes mellitus. Diabetes. 1989;38:428–34. doi: 10.2337/diab.38.4.428. [DOI] [PubMed] [Google Scholar]
- 67.Yang H, Scheff AJ, Schalch DS. Effects of streptozotocin-induced diabetes mellitus on growth and hepatic insulin-like growth factor I gene expression in the rat. Metabolism. 1990;39:295–301. doi: 10.1016/0026-0495(90)90050-m. [DOI] [PubMed] [Google Scholar]
- 68.Scheiwiller E, Guler HP, Merryweather J, Scandella C, Maerki W, Zapf J, et al. Growth restoration of insulin-deficient diabetic rats by recombinant human insulin-like growth factor I. Nature. 1986;323:169–71. doi: 10.1038/323169a0. [DOI] [PubMed] [Google Scholar]
- 69.Pao CI, Farmer PK, Begovic S, Goldstein S, Wu GJ, Phillips LS. Expression of hepatic insulin-like growth factor-I and insulin-like growth factor-binding protein-1 genes is transcriptionally regulated in streptozotocin-diabetic rats. Mol Endocrinol. 1992;6:969–77. doi: 10.1210/mend.6.6.1379675. [DOI] [PubMed] [Google Scholar]
- 70.Busiguina S, Fernandez AM, Barrios V, Clark R, Tolbert DL, Berciano J, et al. Neurodegeneration is associated to changes in serum insulin-like growth factors. Neurobiol Dis. 2000;7:657–65. doi: 10.1006/nbdi.2000.0311. [DOI] [PubMed] [Google Scholar]
- 71.Jakobsen J, Sidenius P, Gundersen HJ, Osterby R. Quantitative changes of cerebral neocortical structure in insulin-treated long-term streptozocin-induced diabetes in rats. Diabetes. 1987;36:597–601. doi: 10.2337/diab.36.5.597. [DOI] [PubMed] [Google Scholar]
- 72.Lupien SB, Bluhm EJ, Ishii DN. Effect of IGF-I on DNA, RNA, and protein loss associated with brain atrophy and impaired learning in diabetic rats. Neurobiol Dis. 2006;21:487–95. doi: 10.1016/j.nbd.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 73.Magariños AM, McEwen BS. Experimental diabetes in rats causes hippocampal dendritic and synaptic reorganization and increased glucocorticoid reactivity to stress. Proc Natl Acad Sci U S A. 2000;97:11056–61. doi: 10.1073/pnas.97.20.11056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Martínez-Tellez R, Gómez-Villalobos Mde J, Flores G. Alteration in dendritic morphology of cortical neurons in rats with diabetes mellitus induced by streptozotocin. Brain Res. 2005;1048:108–15. doi: 10.1016/j.brainres.2005.04.048. [DOI] [PubMed] [Google Scholar]
- 75.Lechuga-Sancho AM, Arroba AI, Frago LM, Pañeda C, García-Cáceres C, Delgado Rubín de Célix A, et al. Activation of the intrinsic cell death pathway, increased apoptosis and modulation of astrocytes in the cerebellum of diabetic rats. Neurobiol Dis. 2006;23:290–9. doi: 10.1016/j.nbd.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 76.Dheen ST, Tay SS, Wong WC. Ultrastructure of the cuneate nucleus in the streptozotocin-induced diabetic rat. J Hirnforsch. 1994;35:253–62. [PubMed] [Google Scholar]
- 77.Luo Y, Kaur C, Ling EA. Neuronal and glial response in the rat hypothalamus-neurohypophysis complex with streptozotocin-induced diabetes. Brain Res. 2002;925:42–54. doi: 10.1016/s0006-8993(01)03258-9. [DOI] [PubMed] [Google Scholar]
- 78.Kamal A, Biessels GJ, Urban IJ, Gispen WH. Hippocampal synaptic plasticity in streptozotocin-diabetic rats: Impairment of long-term potentiation and facilitation of long-term depression. Neuroscience. 1999;90:737–45. doi: 10.1016/s0306-4522(98)00485-0. [DOI] [PubMed] [Google Scholar]
- 79.Biessels GJ, Kamal A, Urban IJ, Spruijt BM, Erkelens DW, Gispen WH. Water maze learning and hippocampal synaptic plasticity in streptozotocin-diabetic rats: Effects of insulin treatment. Brain Res. 1998;800:125–35. doi: 10.1016/s0006-8993(98)00510-1. [DOI] [PubMed] [Google Scholar]
- 80.Porta M. Diabetic retinopathy and metabolic control. Eur J Ophthalmol. 1993;3:207–15. doi: 10.1177/112067219300300406. [DOI] [PubMed] [Google Scholar]
- 81.Tang J, Mohr S, Du YD, Kern TS. Non-uniform distribution of lesions and biochemical abnormalities within the retina of diabetic humans. Curr Eye Res. 2003;27:7–13. doi: 10.1076/ceyr.27.2.7.15455. [DOI] [PubMed] [Google Scholar]
- 82.Barber AJ, Lieth E, Khin SA, Antonetti DA, Buchanan AG, Gardner TW. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest. 1998;102:783–91. doi: 10.1172/JCI2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zeng XX, Ng YK, Ling EA. Neuronal and microglial response in the retina of streptozotocin-induced diabetic rats. Vis Neurosci. 2000;17:463–71. doi: 10.1017/s0952523800173122. [DOI] [PubMed] [Google Scholar]
- 84.Alder VA, Su EN, Yu DY, Cringle SJ, Yu PK. Diabetic retinopathy: Early functional changes. Clin Exp Pharmacol Physiol. 1997;24:785–8. doi: 10.1111/j.1440-1681.1997.tb02133.x. [DOI] [PubMed] [Google Scholar]
- 85.Zhang L, Krzentowski G, Albert A, Lefebvre PJ. Risk of developing retinopathy in Diabetes Control and Complications Trial type 1 diabetic patients with good or poor metabolic control. Diabetes Care. 2001;24:1275–9. doi: 10.2337/diacare.24.7.1275. [DOI] [PubMed] [Google Scholar]
- 86.Kummer A, Pulford BE, Ishii DN, Seigel GM. Des(1-3)IGF-1 treatment normalizes type 1 IGF receptor and phospho-Akt (Thr 308) immunoreactivity in predegenerative retina of diabetic rats. Int J Exp Diabesity Res. 2003;4:45–57. doi: 10.1080/15438600303729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Spoerri PE, Ellis EA, Tarnuzzer RW, Grant MB. Insulin-like growth factor: Receptor and binding proteins in human retinal endothelial cell cultures of diabetic and non-diabetic origin. Growth Horm IGF Res. 1998;8:125–32. doi: 10.1016/s1096-6374(98)80102-0. [DOI] [PubMed] [Google Scholar]
- 88.Seigel GM, Lupien SB, Campbell LM, Ishii DN. Systemic IGF-I treatment inhibits cell death in diabetic rat retina. J Diabetes Complications. 2006;20:196–204. doi: 10.1016/j.jdiacomp.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 89.Recio-Pinto E, Ishii DN. Effects of insulin, insulin-like growth factor-II and nerve growth factor on neurite outgrowth in cultured human neuroblastoma cells. Brain Res. 1984;302:323–34. doi: 10.1016/0006-8993(84)90246-4. [DOI] [PubMed] [Google Scholar]
- 90.Recio-Pinto E, Rechler MM, Ishii DN. Effects of insulin, insulin-like growth factor-II, and nerve growth factor on neurite formation and survival in cultured sympathetic and sensory neurons. J Neurosci. 1986;6:1211–9. doi: 10.1523/JNEUROSCI.06-05-01211.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Recio-Pinto E, Ishii DN. Insulin and insulinlike growth factor receptors regulating neurite formation in cultured human neuroblastoma cells. J Neurosci Res. 1988;19:312–20. doi: 10.1002/jnr.490190306. [DOI] [PubMed] [Google Scholar]
- 92.Wang C, Li Y, Wible B, Angelides KJ, Ishii DN. Effects of insulin and insulin-like growth factors on neurofilament mRNA and tubulin mRNA content in human neuroblastoma SH-SY5Y cells. Brain Res Mol Brain Res. 1992;13:289–300. doi: 10.1016/0169-328x(92)90212-t. [DOI] [PubMed] [Google Scholar]
- 93.Brüning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, et al. Role of brain insulin receptor in control of body weight and reproduction. Science. 2000;289:2122–5. doi: 10.1126/science.289.5487.2122. [DOI] [PubMed] [Google Scholar]
- 94.Serbedzija P, Madl JE, Ishii DN. Insulin and IGF-I prevent brain atrophy and DNA loss in diabetes. Brain Res. 2009;1303:179–94. doi: 10.1016/j.brainres.2009.09.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Francis GJ, Martinez JA, Liu WQ, Xu K, Ayer A, Fine J, et al. Intranasal insulin prevents cognitive decline, cerebral atrophy and white matter changes in murine type I diabetic encephalopathy. Brain. 2008;131:3311–34. doi: 10.1093/brain/awn288. [DOI] [PubMed] [Google Scholar]
- 96.Jongen C, van der Grond J, Kappelle LJ, Biessels GJ, Viergever MA, Pluim JP, et al. Automated measurement of brain and white matter lesion volume in type 2 diabetes mellitus. Diabetologia. 2007;50:1509–16. doi: 10.1007/s00125-007-0688-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.van Harten B, Oosterman JM, Potter van Loon BJ, Scheltens P, Weinstein HC. Brain lesions on MRI in elderly patients with type 2 diabetes mellitus. Eur Neurol. 2007;57:70–4. doi: 10.1159/000098054. [DOI] [PubMed] [Google Scholar]
- 98.Flood JF, Mooradian AD, Morley JE. Characteristics of learning and memory in streptozocin-induced diabetic mice. Diabetes. 1990;39:1391–8. doi: 10.2337/diab.39.11.1391. [DOI] [PubMed] [Google Scholar]
- 99.Brun A, Englund E. A white matter disorder in dementia of the Alzheimer type: A pathoanatomical study. Ann Neurol. 1986;19:253–62. doi: 10.1002/ana.410190306. [DOI] [PubMed] [Google Scholar]
- 100.Sjöbeck M, Haglund M, Englund E. Decreasing myelin density reflected increasing white matter pathology in Alzheimer's disease—a neuropathological study. Int J Geriatr Psychiatry. 2005;20:919–26. doi: 10.1002/gps.1384. [DOI] [PubMed] [Google Scholar]
- 101.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer's disease: Synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–80. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 102.Xuereb JH, Brayne C, Dufouil C, Gertz H, Wischik C, Harrington C, et al. Neuropathological findings in the very old.Results from the first 101 brains of a population-based longitudinal study of dementing disorders. Ann N Y Acad Sci. 2000;903:490–6. doi: 10.1111/j.1749-6632.2000.tb06404.x. [DOI] [PubMed] [Google Scholar]
- 103.Schmitt FA, Davis DG, Wekstein DR, Smith CD, Ashford JW, Markesbery WR. “Preclinical” AD revisited: Neuropathology of cognitively normal older adults. Neurology. 2000;55:370–6. doi: 10.1212/wnl.55.3.370. [DOI] [PubMed] [Google Scholar]
- 104.Neuropathology Group. Medical Research Council Cognitive Function and Aging Study. Pathological correlates of late-onset dementia in a multicentre, community-basedpopulation in England and Wales. Neuropathology Group of the Medical ResearchCouncil Cognitive Function and Ageing Study (MRC CFAS) Lancet. 2001;357:169–75. doi: 10.1016/s0140-6736(00)03589-3. [DOI] [PubMed] [Google Scholar]
- 105.Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, et al. Long-term effects of Abeta42 immunisation in Alzheimer's disease: Follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372:216–23. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- 106.Janson J, Laedtke T, Parisi JE, ’Brien P, Petersen RC, Butler PC. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes. 2004;53:474–81. doi: 10.2337/diabetes.53.2.474. [DOI] [PubMed] [Google Scholar]
- 107.Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol. 2004;61:661–6. doi: 10.1001/archneur.61.5.661. [DOI] [PubMed] [Google Scholar]
- 108.Craft S, Asthana S, Schellenberg G, Cherrier M, Baker LD, Newcomer J, et al. Insulin metabolism in Alzheimer's disease differs according to apolipoprotein E genotype and gender. Neuroendocrinology. 1999;70:146–52. doi: 10.1159/000054469. [DOI] [PubMed] [Google Scholar]
- 109.Murialdo G, Barreca A, Nobili F, Rollero A, Timossi G, Gianelli MV, et al. Relationships between cortisol, dehydroepiandrosterone sulphate and insulin-like growth factor-I system in dementia. J Endocrinol Invest. 2001;24:139–46. doi: 10.1007/BF03343833. [DOI] [PubMed] [Google Scholar]
- 110.Watanabe T, Miyazaki A, Katagiri T, Yamamoto H, Idei T, Iguchi T. Relationship between serum insulin-like growth factor-1 levels and Alzheimer's disease and vascular dementia. J Am Geriatr Soc. 2005;53:1748–53. doi: 10.1111/j.1532-5415.2005.53524.x. [DOI] [PubMed] [Google Scholar]
- 111.Rivera EJ, Goldin A, Fulmer N, Tavares R, Wands JR, de la Monte SM. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer's disease: Link to brain reductions in acetylcholine. J Alzheimers Dis. 2005;8:247–68. doi: 10.3233/jad-2005-8304. [DOI] [PubMed] [Google Scholar]