

Abstract
Cushing's disease (CD), caused by an ACTH-secreting pituitary corticotroph adenoma, is the commonest cause of Cushing syndrome in children over 5 years of age. It is rare in the pediatric age range and presents difficult diagnostic and therapeutic challenges. Key presenting features include weight gain, growth failure and change in facial appearance. Most pediatric endocrinologists have limited experience managing children or adolescents with CD and thus benefit from close consultation with adult colleagues. We describe a diagnostic protocol which broadly follows the model for adult patients. Treatment strategies are examined and appraised. The management of pediatric CD patients after cure is also discussed.
Keywords: Cushing's disease, Cushing's syndrome, pediatrics, transsphenoidal surgery, radiotherapy
INTRODUCTION
Cushing's syndrome (CS) is a clinical syndrome which comprises symptoms and signs reflecting excessive circulating glucocorticoid (GC) concentrations. It is very rare in childhood and adolescence and can be classified into two groups of adrenocorticotrophic hormone (ACTH)-independent and ACTH-dependent causes [Table 1]. Iatrogenic exogenous GC administration is the most common cause in pediatric patients but these cases are rarely referred for endocrine review and will not be discussed further.
Table 1.
Classification of pediatric Cushing's syndrome

Cushing's disease (CD), caused by an ACTH-secreting pituitary corticotroph adenoma, is the commonest cause of CS in children over 5 years of age.[1,2] CD accounts for 75-80% of pediatric CS cases compared to 49-71% of adult cases.[1,3] Some aspects of pediatric CD differ from those in adults. Examples are the increased frequency in prepubertal males compared to females, frequent absence of radiological evidence of a corticotroph adenoma on pituitary MRI scanning and higher incidence of lateralization of ACTH secretion demonstrated by inferior petrosal sinus sampling. Children also have a more exuberant cortisol response to IV CRH and a more rapid response to external beam pituitary radiotherapy.[3] Clinically children can present differently from adults, most notably with growth failure associated with weight gain.
Clinical and diagnostic aspects of pediatric Cushing's disease
Epidemiology
CS can occur throughout childhood and adolescence, however, different aetiologies are commonly associated with particular age groups [Figure 1], with CD being the commonest cause after the pre-school years.[1] The peak incidence of pediatric CD is during adolescence; in 182 cases taken from the literature the median age of presentation was 14.1 years.
Figure 1.
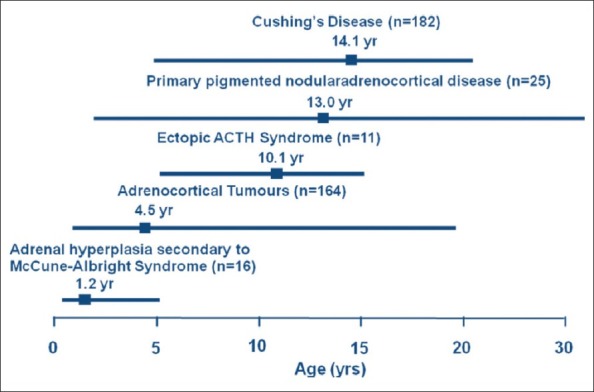
Different aetiologies of pediatric Cushing's syndrome from the literature (n = 398 cases) shown at ages of peak incidence (boxes).
Pediatric CD is almost always caused by a pituitary microadenoma with diameter < 5 mm.[1] We have seen one macroadenoma in 37 pediatric cases.[4] We analyzed gender distribution in 50 CD patients aged from 6 to 30 years and found a significant predominance of males in the pre-pubertal patients.[5] In our current series of 37 cases aged from 5.8 to 17.8 years there are 24 males and 13 females. The large series from the NIH[2] also revealed the same phenomenon of male predominance in young patients.
Clinical features in CD
The recognition of the features of CD is crucial to allow early diagnosis and effective treatment. Key presenting features include growth failure, weight gain and a change in facial appearance. Most children and adolescents have a typical Cushingoid appearance but parents and general practitioners frequently fail to recognize the pathological nature of the change of the child's appearance. The mean length of symptoms prior to diagnosis in our patients was 2.5 ± 1.7 years (range 0.3-6.6 years).
Striae were present in 50% of our patients, more frequently in the older patients. The young child with CD often presented with only obesity and growth failure, without features of plethora, hirsutism, acne and striae. Additional features included emotional lability, hypertension and fatigue. Short stature (height less than -2.0 SD) was present in 43% of our patients and growth velocity when available was subnormal.
Height SDS, which was almost always below the mean, contrasted strongly with BMI SDS, which was consistently above it [Figure 2].[1] Bone age at diagnosis of CD was delayed in 15 of 17 patients (mean delay 2.0 years; range -0.5 to 4.1 years) and correlated negatively with height SDS, duration of symptoms and age at diagnosis.[6]
Figure 2.
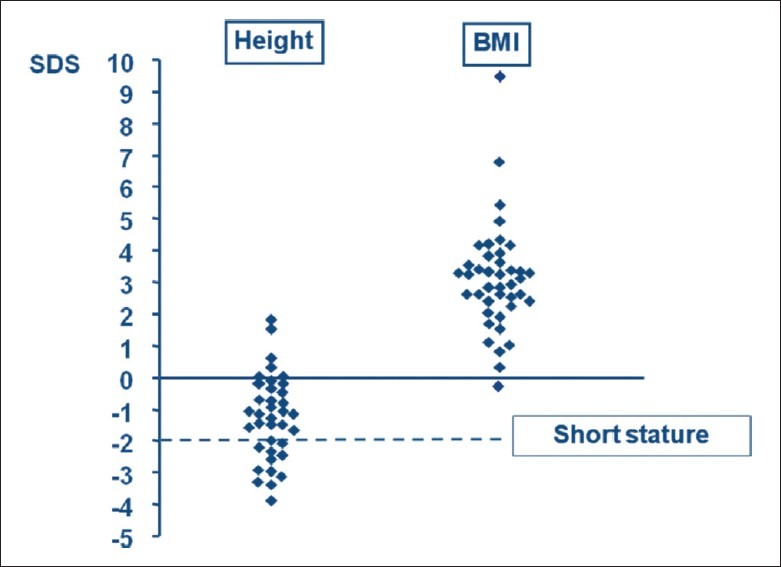
Height and body mass index (BMI) SDS values in 37 pediatric patients with Cushing's disease. The dotted line indicates the SDS value below which patients are significantly shorter than average.
True precocious puberty in CD is unusual, however, virilization with pseudo-precocious puberty is an important presenting feature.[1,7] Abnormal virilization, defined as unusual advance of Tanner pubic hair stage compared to testicular volume or breast development, was identified in 12 of 27 patients.[7] Values of serum androstenedione, DHEAS and testosterone SDS were higher (P = 0.03, 0.008, 0.03 respectively) than in subjects without abnormal virilization and SHBG SDS values were lower (P = 0.006). Gonadotrophin levels were subnormal in the patients who had commenced true puberty, suggesting a suppressive effect of chronic hypercortisolemia.
Investigation of CD
Guidelines for the diagnosis of pediatric CD have been published.[1] The algorithm for testing in children consists initially of confirmation or exclusion of CS followed by investigations to determine the aetiology. This scheme is listed in Table 2. The initial screening investigations have a high sensitivity; if the initial test results are normal the patient is very unlikely to have CS.
Table 2.
Scheme of investigation for patients with suspected Cushing's syndrome

Confirmation or exclusion of cushing's syndrome
Our initial screening procedure includes three consecutive 24 h urine collections for urinary free cortisol (UFC) which provides an integrated assessment of cortisol secretion over a 24 h period. If there is doubt about these values, we proceed to admit the child for measurement of serum cortisol at 3 time-points (09.00 h, 18.00 h and midnight [sleeping]) to assess circadian rhythm. Determination of midnight cortisol in the sleeping child gives the highest sensitivity for the diagnosis of CS.[8] The value in normal subjects is < 50 nmol/L (< 1.8 μg/dl), although some young children may reach their cortisol nadir earlier than midnight. Midnight serum cortisol was measurable in all the patients with Cushing's syndrome. Late night salivary cortisol may also be used as a screening test.
Following assessment of midnight cortisol, we perform a low-dose dexamethasone suppression test (LDDST), using the regimen of 0.5 mg every 6 h (at 09.00, 15.00, 21.00 and 03.00 h) for 48 h, unless the child weighs < 40 kg when we use the dose of 30 μg/kg/day.[3] In the LDDST, blood is taken for serum cortisol at 0 and at 48 h, 6 h after the last dose, when it is undetectable (< 50 nmol/L, < 1.8 μg/dl) in normal subjects. These tests performed have a high sensitivity for CS and an even higher specificity for the exclusion of this diagnosis. The 1 mg overnight dexamethasone test has been used as a screening test in children but there are no available data on its interpretation or reliability.[8]
Confirmation of cushing's disease
Following confirmation of CS, the cause of the hypercortisolism is determined. CD is most easily confirmed by determination of basal plasma ACTH. In all of our patients with CD, ACTH was detectable, ranging from 12 to 128 ng/L (normal 10-50 ng/L) [Figure 3]. In ACTH-independent CS, ACTH is always low and usually undetectable. We then perform a CRH test using human sequence CRH (1 μg/kg IV) and in 27 CD patients serum cortisol increased by > 20% (range 106-554%). Ectopic ACTH syndrome is so rare in children that the need for a CRH test is questionable, however we find an increased cortisol response contributes to the diagnosis of CD. We no longer routinely perform a high dose dexamethasone suppression test (HDDST) because a decrease of cortisol during the LDDST strongly supports the diagnosis of CD and negates the need for a HDDST.
Figure 3.
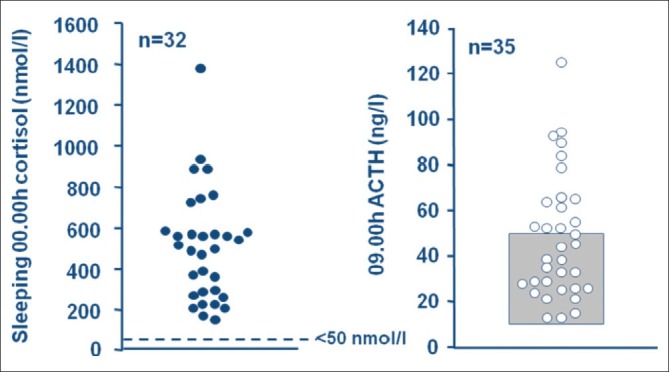
Midnight cortisol was > 50 nmol/l with detectable ACTH, ranging from 12-128 ng/L, in all patients with Cushing's disease.
Imaging studies
Most ACTH-secreting pituitary tumors in children are microadenomas and the majority have hypointense signal on MRI, which fails to enhance with gadolinium.[2,3] In the NIH series, approximately 50% of microadenomas were visible on pituitary MRI.[3] In our experience pituitary imaging was relatively unhelpful, showing a normal appearance in approximately half of the patients, with a low predictive value of the position of the adenoma as identified at surgery [Table 3].[3] Therefore, although pituitary imaging using MRI is an important step towards the successful treatment of Cushing's disease by transsphenoidal surgery (TSS), results should be interpreted together with bilateral inferior petrosal sinus sampling (BIPSS) for ACTH (see below).
Table 3.
Pituitary imaging, surgical identification of microadenoma and cure by TSS

In some cases, the distinction between CD and ectopic ACTH syndrome may be in doubt. Here, a CT scan of the chest using 0.5 cm cuts will usually exclude a bronchial carcinoid tumor.
Bilateral inferior petrosal sinus sampling for ACTH (BIPSS)
BIPSS was initially piloted at the NIH and has now become routine in adult practice unless the MRI unequivocally shows a pituitary adenoma. In children, ectopic ACTH secretion is extremely rare so the primary aim of BIPSS is to localize the microadenoma by demonstrating lateral or midline ACTH secretion. The first pediatric data were reported in the large NIH series where a predictive value of lateralization was 75-80%.[2]
BIPSS is a highly specialized technique and in our unit is performed by the same radiologist who regularly studies adult patients. In the majority of cases we do not use general anesthesia (GA) to avoid potential alteration of ACTH secretion. However in two children aged 5.8 and 6.2 years GA was used. We have now performed BIPSS in 28 pediatric CD patients without complications. Our results shown in Table 4 suggest that ACTH sampling gives a better prediction of the site of the microadenoma than pituitary imaging.[3]
Table 4.
BIPSS results, surgical identification of adenoma and cure by TSS

Treatment of cushing's disease
CD in childhood requires urgent evaluation, diagnosis and expert treatment. Treatment options have advanced over the last 50 years. Initially bilateral adrenalectomy was widely practiced however, the pituitary adenoma remained in situ and there was an appreciable risk of post-adrenalectomy Nelson's syndrome. In addition, patients required lifelong glucocorticoid and mineralocorticoid replacement. In the management of 37 cases, we have performed adrenalectomy twice, when the patients were extremely unwell and not fit to undergo pituitary surgery. In one of these patients, the hypercortisolemia was uncontrollable by oral metyrapone and treatment was given with IV etomidate which successfully controlled the cortisol levels prior to adrenalectomy. Medical therapy to lower cortisol using metyrapone or ketoconazole is a useful short-term option prior to surgery or radiotherapy but cannot be recommended as a long-term definitive therapy for CD.
Transsphenoidal surgery
Definitive cure of CD can be achieved by transsphenoidal pituitary surgery (TSS) or external pituitary radiotherapy. TSS is regarded as a safe and effective procedure in children[3,9] and is now considered first-line therapy as it involves selective removal of the adenoma maximizing the potential for normal pituitary tissue to remain in situ. However, selective microadenomectomy can be technically very difficult in children. As discussed above, the microadenomas may be very small and an appreciable rate of failure exists even in the hands of the most experienced transsphenoidal surgeons.
The variable surgical success rates reported depend on which definition of cure is adopted in that unit. We define successful treatment, i.e., cure, as undetectable postoperative serum cortisol (< 50 nmol/L , < 1.8 μg/dl), which is consistent with our adult endocrine unit.[10] The overall cure rate from TSS in 33 pediatric patients with microadenomas treated from 1982 to 2010 was 61%. Since 1986, the cure rate has been 75% in the 28 of these patients who were treated since routine BIPSS was introduced as preoperative preparation [Table 4]. Other pediatric TSS series report cure rates varying from 45% to 78%, but very few report rates of > 90%.[3,9] We have not seen recurrence of CD after cure by TSS, but as many patients are referred from other centers, this has not been formally studied.
Pituitary radiotherapy
Pituitary radiotherapy (RT) is a therapeutic option for pediatric CD. In our center, external beam RT is used as second-line therapy, following unsuccessful TSS. We usually proceed to RT within 2-4 weeks of TSS, when it is clear from circulating cortisol levels that complete cure has not been achieved.[11] The RT protocol we follow consists of delivering 45 Gy in 25 fractions over 35 days[11] reflecting evidence demonstrating that children with CD respond more rapidly than adults. We have treated 13 patients during the past 26 years with a successful cure rate of 85%, which occurred at a mean interval of 0.8 years (range 0.3-2.9) following completion of therapy.[11]
Post-cure growth and development and pituitary function
Growth failure and short stature are almost always seen at diagnosis.[1] Virilization may lead to acceleration of bone age and further compromise growth potential. A key article from the NIH described the abnormalities of height and GH secretion[12] together with a poor outcome for post-treatment catch-up growth and adult height.[13] We also reported disappointing post-cure catch-up, which we attributed to continuing GH deficiency, occurring either from TSS, pituitary RT or the long-standing effects of chronic hypercortisolemia.[1]
Our approach now is to test for GH deficiency 3 months after TSS or completion of RT. If GH therapy is required, we start GH at a dose of 0.025 mg/kg/day. GnRH analogue therapy may be added to delay epiphyseal closure. Results demonstrate that this regime usually enables adequate catch-up growth and adult height within range of target height. Many patients however remain obese and BMI SDS was elevated (P < 0.01) at a mean interval of 3.9 years after cure in 14 patients.[14] We have analyzed pituitary function in 6 patients at intervals of 6.6 to 16.5 years after receiving RT and have shown that although GH deficiency was frequent initially, some recovery may occur in adult life. Gonadotrophin secretion was generally preserved and TSH and ACTH deficiency was minimal.[15]
CONCLUSIONS
Pediatric CD has a number of features distinct from adult CD, and early diagnosis remains a challenge. Once suspected, the patient requires investigation using a formal protocol and the choice and interpretation of tests is most productively discussed with an adult specialist with experience of CD. Referral should be considered to a center combining pediatric and adult endocrinology, TSS and pituitary RT. In addition, choosing a neurosurgeon experienced in TSS in children is likely to significantly improve the chance of effective and curative therapy.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared
REFERENCES
- 1.Storr HL, Chan LF, Grossman AB, Savage MO. Paediatric Cushing's Disease: Epidemiology, Investigation and Therapeutic Advances. Trends Endocrinol Metab. 2007;18:267–74. doi: 10.1016/j.tem.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 2.Magiakou MA, Mastorakos G, Oldfield EH, Gomes MT, Doppman JL, Cutler GB, et al. Cushing's syndrome in children and adolescents. Presentation, diagnosis and therapy. N Engl J Med. 1994;331:629–36. doi: 10.1056/NEJM199409083311002. [DOI] [PubMed] [Google Scholar]
- 3.Storr HL, Alexandraki KI, Martin L, Isidori AM, Kaltsas G, Monson JP, et al. Comparisons in the epidemiology, diagnostic features and cure rate by transsphenoidal therapy between paediatric and adult Cushing's disease. Eur J Endocrinol. 2011;164:667–74. doi: 10.1530/EJE-10-1120. [DOI] [PubMed] [Google Scholar]
- 4.Storr HL, Afshar F, Matson M, Sabin I, Savage KM, Evanson J, et al. Factors influencing cure by transsphenoidal selective adenomectomy in paediatric Cushing's disease. Eur J Endocrinol. 2005;152:825–33. doi: 10.1530/eje.1.01921. [DOI] [PubMed] [Google Scholar]
- 5.Storr HL, Isidori AM, Monson JP, Besser GM, Grossman AB, Savage MO. Prepubertal Cushing's disease is more common in males, but there is no increase in severity at diagnosis. J Clin Endocrinol Metab. 2004;89:3818–20. doi: 10.1210/jc.2003-031531. [DOI] [PubMed] [Google Scholar]
- 6.Peters CJ, Ahmed ML, Storr HL, Davies KM, Martin LJ, Allgrove J, et al. Factors influencing skeletal maturation at diagnosis of paediatric Cushing's disease. Horm Res. 2007;68:231–5. doi: 10.1159/000101336. [DOI] [PubMed] [Google Scholar]
- 7.Dupuis CC, Storr HL, Perry LA, Ho JT, Ahmed L, Ong KK, et al. Abnormal puberty in paediatric Cushing's disease: relationship with adrenal androgen, sex hormone binding globulin and gonadotrophin concentrations. Clin Endocrinol. 2007;66:838–43. doi: 10.1111/j.1365-2265.2007.02822.x. [DOI] [PubMed] [Google Scholar]
- 8.Newell-Price J, Trainer P, Besser GM, Grossman AB. The diagnosis and differential diagnosis of Cushing's syndrome and pseudo-Cushing's states. Endocr Rev. 1998;19:647–72. doi: 10.1210/edrv.19.5.0346. [DOI] [PubMed] [Google Scholar]
- 9.Devoe DJ, Miller WL, Conte FA, Kaplan SL, Grumbach MM, Rosenthal SM, et al. Long-term outcome in children and adolescents after transsphenoidal surgery for Cushing's disease. J Clin Endocrinol Metab. 1997;82:3196–202. doi: 10.1210/jcem.82.10.4290. [DOI] [PubMed] [Google Scholar]
- 10.Trainer PJ, Lawrie HS, Verhelst J, Howlett TA, Lowe DG, Grossman AB. Transsphenoidal resection in Cushing's disease: Undetectable serum cortisol as the definition of successful treatment. Clin Endocrinol. 1993;38:73–8. doi: 10.1111/j.1365-2265.1993.tb00975.x. [DOI] [PubMed] [Google Scholar]
- 11.Storr HL, Plowman PN, Carroll PV, Francois I, Krassas G, Afshar F, et al. Clinical and endocrine responses to pituitary radiotherapy in pediatric Cushing's disease: An effective second-line treatment. J Clin Endocrinol Metab. 2003;88:34–7. doi: 10.1210/jc.2002-021032. [DOI] [PubMed] [Google Scholar]
- 12.Magiakou MA, Mastorakos G, Gomez MT, Rose SR, Chrousos GP. Suppressed spontaneous and stimulated growth hormone secretion in patients with Cushing's disease before and after surgical cure. J Clin Endocrinol Metab. 1994;78:131–7. doi: 10.1210/jcem.78.1.7507118. [DOI] [PubMed] [Google Scholar]
- 13.Magiakou MA, Mastorakos G, Chrousos GP. Final stature in patients with endogenous Cushing's syndrome. J Clin Endocrinol Metab. 1994;79:1082–5. doi: 10.1210/jcem.79.4.7962277. [DOI] [PubMed] [Google Scholar]
- 14.Davies JH, Storr HL, Davies K, Monson JP, Besser GM, Afshar F, et al. Final adult height and body mass index after cure of paediatric Cushing's disease. Clin Endocrinol. 2005;62:466–72. doi: 10.1111/j.1365-2265.2005.02244.x. [DOI] [PubMed] [Google Scholar]
- 15.Chan LF, Storr HL, Plowman PN, Perry LA, Besser GM, Grossman AB, et al. Long-term anterior pituitary function in patients with paediatric Cushing's disease treated with pituitary radiotherapy. Eur J Endocrinol. 2007;156:477–82. doi: 10.1530/EJE-06-0588. [DOI] [PubMed] [Google Scholar]