

Abstract
Asymptomatic reversible pituitary hyperplasia is common in patients with untreated primary hypothyroidism. Occurrence of empty sella (ES) in this scenario is extremely rare (only three reports till the date) and panhypopituitarism has not been reported in such patients. We report a 27 year man with severe short stature (height-133 cm; standard deviation score-7.36) and delayed puberty who had symptoms suggestive of hypothyroidism along with chronic persistent headache since 6 years of age. Pituitary imaging done for headache at 11 years age showed pituitary hyperplasia. He was diagnosed of primary hypothyroidism for the 1st time at 21 year age, a diagnosis which was likely missed for 15 years. Levothyroxine therapy leads to resolution of all symptoms and a height gain of 28 cm over last 6 years. Evaluation for lack of progression of puberty along with chronic nausea, vomiting, fatigue and weight loss for the last 1 year revealed secondary hypocortisolism (9 am cortisol-4.8 mcg/dl; ACTH-3.2 pg/ml), growth hormone deficiency (IGF-1: 65 ng/ml; normal: 117-325 ng/ml) and hypogonadotrophic hypogonadism (9 am testosterone: 98 ng/dl; [280-1500] LH-0.01 mIU/L [1.14-5.75]) with ES on magnetic resonance imaging (MRI) brain. Uncontrolled thyrotroph hyperplasia due to chronic untreated primary hypothyroidism for 15 years may have been damaging the adjacent corticotrophs, somatotrophs and gonadotrophs resulting in panhypopituitarism and empty sella. This is perhaps the first report of panhypopituitarism with empty sella syndrome developing in a patient with pituitary hyperplasia, a sequel of chronic untreated primary hypothyroidism.
Keywords: Empty sella, feedback adenoma, panhypopituitarism, pituitary hyperplasia, primary hypothyroidism
INTRODUCTION
Empty sella (ES) is a rare disorder which can be due to pituitary surgery for tumours, spontaneous infarction in pituitary tumours, benign intracranial hypertension, lymphocytic hypophysitis, Sheehan's syndrome, cranial irradiation, trauma or idiopathic[1] Patients with ES may be asymptomatic or present with features of one or more pituitary hormone deficiencies. Presence of three or more anterior pituitary hormone deficiency is known as panhypopituitarism.
Asymptomatic pituitary hyperplasia in patients with primary hypothyroidism varies from 25% to 81%,[2] and is especially common in patients with thyroid stimulating hormone (TSH) levels ≥50 μIU/ml (70%),[2] which is usually reversible within weeks to months of levothyroxine replacement. Symptoms of raised intracranial tension (ICT) due to growing sellar/suprasellar mass are uncommon in these patients and their usual presenting feature is overt symptoms of primary hypothyroidism. Occasionally pituitary adenomas have been described in patients with primary hypothyroidism, which resolved with levothyroxine therapy.[2]
We present a man with severe short stature due to chronic untreated hypothyroidism who initially had features of raised ICT and subsequently over 21 years developed ES along with multiple pituitary hormone deficiency (MPHD).
CASE REPORT
A 27-year-old man, born of non-consanguineous marriage, with normal perinatal history and milestones, complained of recurrent episodes of vomiting with headache since 6 years of age along with generalized weakness, fatigue ability, malaise and swelling of the body. There was history of cold intolerance, constipation and proximal weakness. However, he did not seek formal medical advice at that time due to lack of awareness and poverty. Evaluation for the persisting headache at 11 years of age (1995) lead to a Computerized tomography (CT) brain, which showed a homogenously enhancing midline mass in the pituitary fossa suggestive of pituitary hyperplasia [Figure 1]. However, he did not come back for further evaluation and treatment.
Figure 1.
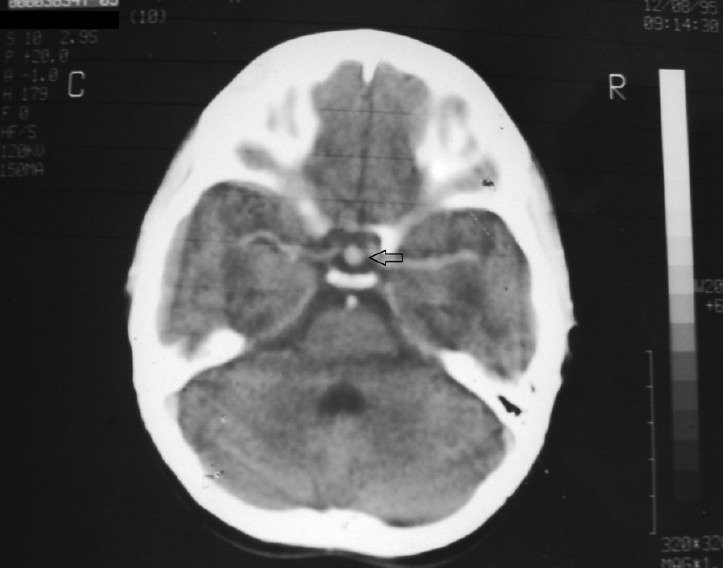
Computerized tomography brain with contrast showing homogenously enhancing mass in the sella suggestive of pituitary hyperplasia (11 years age; 1995)
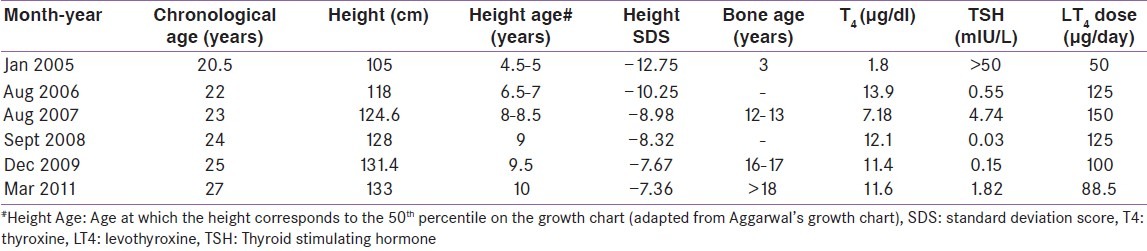
He presented to us for the first time 6 years back (in 2005, 21 years age) with persistence of the previous complaints. Examination at that time was significant for Grade-2 soft goitre, depressed nasal bridge, absent axillary and pubic hair, and bilateral testicular volume of 8 ml each. Biochemical evaluation revealed severe primary hypothyroidism, a diagnosis which was likely missed all these years [Table 1]. Levothyroxine replacement was started at 50 mcg/day. Patient responded dramatically to treatment, with rapid resolution of all symptoms within months along with a height gain of 28 cm in 6 years (from 105 cm to 133 cm) [Table 1].
Table 1.
Changes in height, bone age and thyroid function with levothyroxine replacement over 6 years
He now complained of progressively worsening nausea, vomiting and fatigue for the last 1 year, along with weight loss. Examination was significant for severe short stature (height-133 cm; standard deviation score-7.36), normal blood pressure (90/60 mm Hg), lack of facial hair and signs of masculinization, normal phallus, Tanner-II stage public hair and bilateral testicular volume of 8 ml [Figure 2]. Hormonal evaluation this time was significant for secondary hypocortisolism, low IGF-1 and hypogonadotropic hypogonadism [Table 2]. T1 Weighted (T1W) MRI brain (after 6 years of levothyroxine therapy) revealed an ES with intact posterior pituitary signal [Figure 3].
Figure 2.
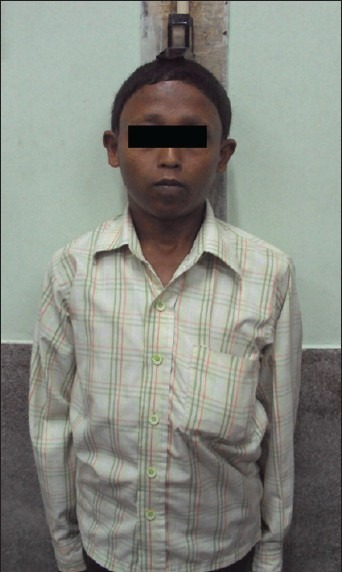
Profile of the patient showing short stature, with pre-pubertal facial features; Stadiometer showing sever short stature (height 133 years at 27 years age)
Table 2.
Hormonal analysis showing multiple pituitary hormone deficiency
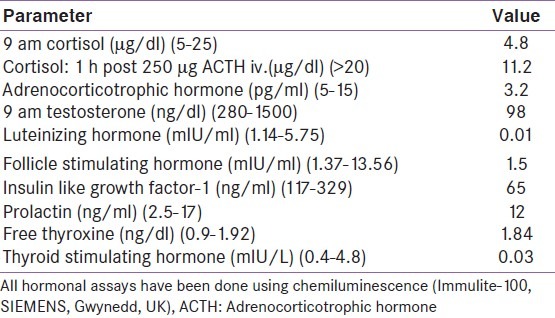
Figure 3.
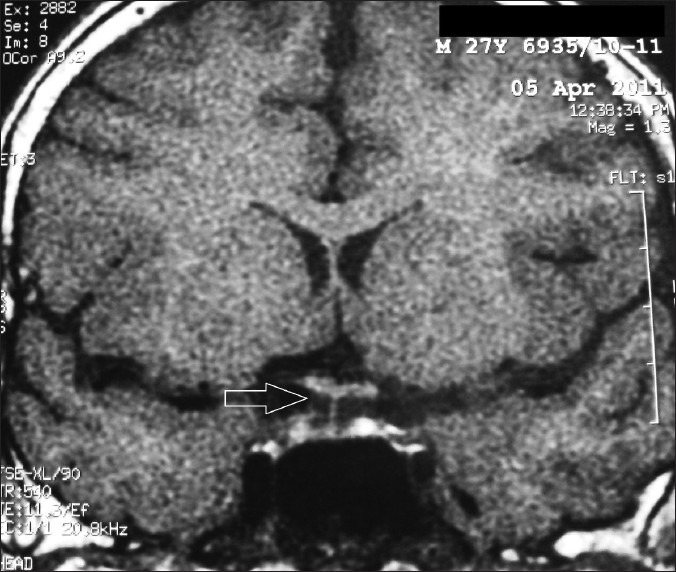
T1W MRI brain (saggital section) showing empty sella (arrow); Coronal section showing ES with stalk visible till the floor of sella (arrow) (27 years age, 2011)
Hydrocortisone supplementation was started at 15 mg per day. Levothyroxine replacement was continued. Testosterone propionate 20 mg, testosterone phenlypropionate 40 mg and testosterone isocaprote 40 mg injection (Sustanon-100, Organon, India) was started every 3 weekly. He could not afford growth hormone replacement.
DISCUSSION
The loss of thyroxine feedback inhibition and the subsequent overproduction of thyrotropin-releasing hormone (TRH) in a patient with long-standing primary hypothyroidism results in hyperplasia of the thyrotroph cells and subsequent enlargement of the pituitary gland.[1] Regression of pituitary enlargement following thyroxine therapy in these patients is the usual phenomenon. Occurrence of ES following treatment of primary hypothyroidism is extremely uncommon and only three cases reported to the best of our knowledge.[3–5] Panhypopituitarism as a sequel of pituitary hyperplasia secondary to untreated primary hypothyroidism has not been reported to the best of our knowledge.
Our patient likely had primary hypothyroidism since 6 years of age, a diagnosis which was missed until 21 years of age. Pituitary hyperplasia [Figure 1] was a result of this lack of feedback inhibition of TRH mediated TSH release. Treatment of primary hypothyroidism even at 21 years of age leads to significant height improvement (28 cm) [Table 1]. Severely delayed bone age with open epiphyses facilitated this height gain. The protracted phase of untreated hypothyroidism (15 years) may have led to uncontrolled thyrotroph hyperplasia, damaging the adjacent somatotrophs, corticotrophs and gonadotrophs, leading to MPHD and ES.
This is perhaps the first report of panhypopituitarism developing in a patient of primary hypothyroidism who had pituitary hyperplasia. Evaluation of anterior pituitary function is advisable in patients of chronic untreated primary hypothyroidism, especially those who have headache or visual disturbances.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared
REFERENCES
- 1.Melmed S, Kleinberg D, Ho K. William's Textbook of Endocrinology. 12th ed. Saunders Elsevier; 2011. Pituitary evaluation and diagnostic evaluation; p. 216. [Google Scholar]
- 2.Khawaja NM, Taher BM, Barham ME, Naser AA, Hadidy AM, Ahmad AT, et al. Pituitary enlargement in patients with primary hypothyroidism. Endocr Pract. 2006;12:29–34. doi: 10.4158/EP.12.1.29. [DOI] [PubMed] [Google Scholar]
- 3.Jawadi MH, Ballonoff LB, Stears JC, Katz FH. Primary hypothyroidism and pituitary enlargement. Radiological evidence of pituitary regression. Arch Intern Med. 1978;138:1555–7. [PubMed] [Google Scholar]
- 4.Semple CG, Beastall GH, Teasdale G, Thomson JA. Hypothyroidism presenting with hyperprolactinaemia. Br Med J (Clin Res Ed) 1983;286:1200–1. doi: 10.1136/bmj.286.6372.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kelestimur F, Selçuklu A, Ozcan N. Empty sella developing during thyroxine therapy in a patient with primary hypothyroidism and hyperprolactinaemia. Postgrad Med J. 1992;68:589–91. doi: 10.1136/pgmj.68.801.589. [DOI] [PMC free article] [PubMed] [Google Scholar]