

Abstract
The PHD finger protein 1 (PHF1) is essential in epigenetic regulation and genome maintenance. Here, we demonstrate that the Tudor domain of human PHF1 binds to histone H3 trimethylated at Lys36 (H3K36me3). We report a 1.9 Å resolution crystal structure of the Tudor domain in complex with H3K36me3 and describe the molecular mechanism of H3K36me3 recognition using NMR analysis. Binding of PHF1 to H3K36me3 inhibits the ability of the Polycomb PRC2 complex to methylate H3K27 in vitro and in vivo. Laser micro-irradiation data reveal that PHF1 is transiently recruited to DNA double-strand breaks (DSBs), and PHF1 mutants impaired in the H3K36me3 interaction exhibit reduced retention at DSB sites. Together, our findings suggest that PHF1 can mediate deposition of the repressive H3K27me3 mark and acts as an early DNA damage response cofactor.
The human Polycomblike (PCL) protein PHF1 associates with pivotal nuclear complexes including PRC2 (Polycomb repressive complex 2) and Ku70-Ku80 and is involved in transcription regulatory and DNA damage repair pathways. The PRC2 complex possesses intrinsic histone lysine methyltransferase (HKMT) activity1–3. It methylates Lys27 of histone H3 generating H3K27me2/3 marks of transcriptionally inactive chromatin4–7. PRC2 consists of four core subunits, including the catalytic Enhancer of zeste homolog 2 (EZH2), Suppressor of zeste 12 (SUZ12), Embryonic ectoderm development (EED) and the histone-binding proteins RBAP46/48, also known as RBBP7/4. Additionally, three associating proteins, PHF1, JARID2 and AEBP2, have recently been identified. These proteins are not always present in the complex, which implies that variations in the composition of PRC2 may be important for modulating its activity8. Indeed, the association of PHF1 with PRC2 has been shown to affect the enzymatic activity and expression of PRC2 target genes8–10. More recently, PHF1 has been implicated in DNA damage repair. It physically interacts with Ku70-Ku80, which binds to DSB ends, and may play a role in nonhomologous end joining (NHEJ) and maintaining genomic stability11.
PHF1 is a ~50 kDa protein that contains an N-terminal single Tudor domain followed by two plant homeodomain (PHD) fingers. Only a few residues part the Tudor domain and the first PHD finger, whereas the second PHD finger is separated by a linker of ~50 amino acids. The region encompassing the two PHD fingers of PHF1 has been shown to directly interact with the catalytic subunit of PRC2, EZH2 (ref. 12). This function of the PHD finger region is conserved in Pcl, the Drosophila homologue of PHF1, which binds E(z), the Drosophila homologue of EZH2 (ref. 12). The Tudor domain and the region following the second PHD finger are involved in the Ku70-Ku80-dependent recruitment to DNA damage sites11, however the precise function of the Tudor domain in this process remains unclear.
Tudor is a member of the Royal superfamily of protein modules, which can exist as a single domain or in tandem. While no binding of a single Tudor to a methylated lysine has been reported, several tandem Tudor domains (TTD) are known to recognize methylated lysine residues in histone tails and p53. The canonical TTD of 53BP1 associates with H4K20me2 and p53K382me2, whereas the hybrid TTD of JMJD2A binds H3K4me3 and H4K20me3 (refs. 13–16). In contrast, a single Tudor domain has been characterized as a reader of methylated arginine. The SMN and SPF30 Tudor domains bind symmetrically and asymmetrically dimethylated arginines present in their target proteins, and the TDRD3 Tudor recognizes H3R17me2a and H4R3me2a17,18. Two modules, a chromo-barrel domain and PWWP associate with H3K36me3, a mark linked to primarily active chromatin and transcription elongation19–21, however these modules interact very weakly exhibiting binding affinities in the 2–4 mM range22,23.
In this study, we demonstrate that the Tudor domain of PHF1 binds to histone H3K36me3 with high specificity and affinity, providing the first example of a robust reader of this posttranslational modification (PTM). It is also the first single Tudor module capable of recognizing methylated lysine. We found that interaction of Tudor with H3K36me3 inhibits PRC2-mediated H3K27 methylation. This is in line with recent reports that H3K36me3 antagonizes H3K27 methylation by PRC224,25,26. Furthermore, our results show Tudor-dependent accumulation of PHF1 at irradiation-induced DNA damage sites, suggesting a novel role of this interaction in DNA repair.
RESULTS
Structure of the H3K36me3-bound Tudor domain of PHF1
To elucidate the molecular mechanism of the H3K36me3 recognition, we obtained a 1.9 Å resolution crystal structure of the PHF1 Tudor domain in complex with an H3K36me3 peptide and established the determinants of specificity toward this epigenetic mark. In the complex, the PHF1 Tudor domain folds into a five-stranded β-barrel, whereas the H3K36me3 peptide adopts an extended conformation (Fig. 1). The peptide is bound across one of the open edges of the β-barrel, formed by the twisted β2 and β4 strands. Overall the binding interface is extensive with 14 residues of the Tudor domain and nine residues of the peptide (Thr32-Arg40) being involved in direct contacts. The complex buries an accessible surface area of 437 Å in the protein and 535 Å in the peptide. The H3K36me3 binding site consists of three well-defined regions: a central aromatic cage, a hydrophobic patch and an acidic groove, shown in brown, green and blue, respectively, in Figure 1.
Figure 1.
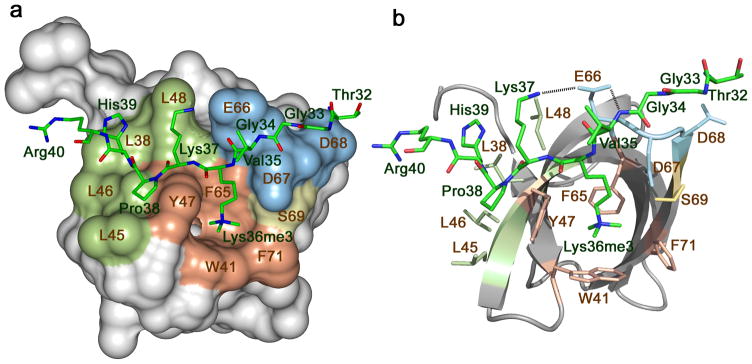
The crystal structure of the Tudor domain of PHF1 in complex with the H3K36me3 peptide. The Tudor domain is depicted as a solid surface (a) and a ribbon diagram (b) with the peptide shown as a stick model. The aromatic cage residues involved in the interaction with the methylated lysine residue are colored brown, those in the hydrophobic patch and the acidic groove are colored green and blue, respectively. H3 residues are labeled in green and Tudor residues are labeled in brown. Dashed lines represent intermolecular hydrogen bonds. For clarity, here and throughout the text, individual residues of the Tudor domain are denoted using a single-letter code, whereas individual residues of the histone peptide are denoted using a three-letter code.
The extended side chain of trimethylated Lys36 of the peptide occupies the aromatic cage formed by the Y47, W41, F65 and F71 residues of the Tudor domain. The aromatic moieties of Y47, W41 and F65 are positioned orthogonally to each other and are engaged in cation-π and hydrophobic interactions with the trimethylammonium group of Lys36, whereas the aromatic moiety of F71 is slightly rotated, most likely contributing more to the hydrophobic contact and less to the cation-π interaction. A similar mode of trimethylated lysine recognition via an aromatic cage has been found in other histone binding modules, including chromodomain, PHD finger and TTD (reviewed in27,28). In contrast to many of these modules, the aromatic cage of PHF1 Tudor is not pre-formed in the free state, even though the secondary structure elements of PHF1 Tudor in complex with the peptide and in the apo-state (PDB 2E5P) superimpose well (root mean square deviation (rmsd) of 1 Å), indicating that binding induces conformational changes in the aromatic cage.
The hydrophobic side chain of Pro38 and the neutral side chain of His39 of the peptide are bound in the hydrophobic patch composed of four solvent exposed leucine residues of the PHF1 Tudor domain, L38, L45, L46 and L48. Additionally, the intermolecular hydrogen bond formed between the backbone NH group of His39 and the carbonyl group of L46 restrains His39. The Thr32, Gly33 and Gly34 residues of the peptide lay in a shallow acidic groove where the backbone HN of Val35 is hydrogen bonded to one of the oxygen atoms of the carboxyl moiety of E66. Another oxygen atom of this carboxyl group makes a hydrogen bond and a salt bridge with the ammonium group of Lys37.
The PHF1 Tudor domain is specific for H3K36me3
We used three approaches to define the physiological ligand of the Tudor domain of PHF1 (Fig. 2, Fig. 3 and Supplementary Fig. 1). Initially, we carried out a pull-down experiment with biotinylated histone peptides corresponding to unmodified H3 and singly modified H3K4me1/2/3, H3K9me1/2/3, H3K27me1/2/3 and H3K36me1/2/3. As shown in Supplementary Figure 1a, the PHF1 Tudor domain associated most strongly with H3K36me3 and weaker with other modified histone peptides. To gain mechanistic insight into this interaction, we tested binding of PHF1 Tudor to the untagged histone peptides by NMR spectroscopy. Titration of unlabeled H3K36me3 induced large chemical shift perturbations in the 15N-labeled Tudor domain (Fig. 2a). In agreement with the crystallographic data, in solution the residues of PHF1 comprising the aromatic cage, the hydrophobic patch and the acidic groove were most perturbed (Fig. 2b). H3K36me2 and H3K36me1 peptides caused smaller resonance perturbations and were bound five and 25 times weaker than H3K36me3 as measured by isothermal titration calorimetry (ITC) and NMR (Fig. 2c, Fig. 3a and Supplementary Fig. 1b). This interaction was essentially abolished by removing all methyl groups at Lys36 (Kd for H3K36me0 ~2.5 mM).
Figure 2.

The PHF1 Tudor domain recognizes H3K36me3. (a) Superimposed 1H,15N Heteronuclear single quantum coherence (HSQC) spectra of the Tudor domain collected upon titration of the H3K36me3 peptide. Spectra are color coded according to the protein:peptide molar ratio. (b) The normalized chemical shift changes observed in the corresponding (a) spectra of the Tudor domain as a function of residue. Residues showing large chemical shift differences are labeled. Differences greater than the average plus one standard deviation (SD), the average plus one-half SD, and the average are shown in red, orange and yellow, respectively. (c) 1H,15N HSQC overlays of the PHF1 Tudor domain in the presence of increasing concentrations of dimethylated, monomethylated or unmodified H3K36 peptides. Spectra are color coded by the protein:peptide molar ratio (legend at left). (d) 1H,15N HSQC overlays of the PHF1 Tudor domain in the free state (black) and in the presence of a 1:10 molar ratio of H3K36me3 (red), H3K4me3 (orange), H3K9me3 (green), H3K27me3 (dark blue) and H4K20me3 (light blue). The resonances of S69 and Q70 are omitted in the right panel for clarity.
Figure 3.

Interaction of the PHF1 Tudor domain with H3K36me3 is specific. (a) Binding affinities of PHF1 Tudor for H3 and H4 peptides as measured by ITC a or NMR b. ND-not detected, NB-no binding. (b) Conservation of the aromatic cage residues across PHF1 homologues. Structural overlay of H3K36me3-bound PHF1 Tudor (histone peptide is not shown) with the NMR structure of the apo-state Drosophila homologue Pcl (PDB code 2XK0). The aromatic cage residues of PHF1 are colored salmon, and corresponding Pcl residues are colored blue. (c) Sequence alignment of PHF1 homologues shows that all four aromatic residues (marked in salmon), necessary for binding to methylated lysine, are conserved only in PHF19.
The histone peptides trimethylated at other lysine residues, including H4K20me3, H3K4me3, H3K9me3 and H3K27me3, induced slightly different patterns of chemical shift changes and associated ~5, 8, 9 and 13 times, respectively, weaker than H3K36me3 (Fig. 2d, Fig. 3a and Supplementary Figs. 1c and 2). Whereas binding of either peptide caused resonance perturbations in the aromatic cage residues, the hydrophobic patch and the acidic groove were perturbed more in the case of H3K36me3. This suggests that residues adjacent to Lys36 are necessary for the interaction. A comparison of the amino acid sequences of the histone peptides reveals that only H3K36 contains a basic residue (Lys37) followed by a proline and a histidine residue C-terminal to Lys36me, and a Gly33-Gly34 tandem N-terminal to Lys36me (Supplementary Fig. 2c). The cyclic pyrrolidine ring of Pro38 and the imidazole ring of His39 cover the extensive hydrophobic patch, whereas the hydrogen bond between the side chains of Lys37 and E66 is essential for reinforcing the extended conformation of the peptide. The small Gly33 and Gly34 residues of the peptide precisely fit the narrow acidic groove. We concluded that the GGxKme3KPH motif directs the specificity of the PHF1 Tudor domain.
Alignment of the sequences of the Tudor domain from PHF1, MTF2 and PHF19, the three human homologues of Drosophila Pcl, reveals that all four aromatic residues in the cage are conserved only in PHF19, suggesting that it may also recognize H3K36me3 (Fig. 3). Conversely, the Drosophila Pcl Tudor domain, which lacks the two aromatic residues corresponding to W41 and F71 of PHF1, does not bind methylated histone peptides29. The importance of the aromatic residues in PHF1 was underscored by the fact that substitution of either W41 or Y47 with an alanine disrupted binding to H3K36me3 even as the structure of the protein remained intact (Fig. 3a and Supplementary Fig. 3). Like TTD of 53BP1, which is specific for a dimethylated lysine residue13,16, the single PHF1 Tudor domain contains an acidic residue in close proximity to the aromatic cage. In the 53BP1 complex, the aspartate forms a hydrogen bond and an ionic contact with the dimethylammonium group, and these interactions account for the preference for dimethylated over trimethylated Lys. A much longer distance between the carboxylic group of D66 and the K36me3 group in the PHF1 complex (5.6 Å in PHF1 versus 2.8 Å in 53BP1) precludes such interactions and the more hydrophobic nature of the aromatic cage favors binding to the trimethylated species. The binding mode of the PHF1 Tudor also differs from the binding mode of the BRPF1 PWWP domain and the EAF3 chromo-barrel module that have been shown to weakly associate with H3K36me3/2 (refs. 22,23) (Supplementary Fig. 4). Consequently, our findings reveal that the PHF1 Tudor domain is, by far, the strongest reader of this imperative transcription-linked PTM.
PHF1 Tudor-H3K36me3 recognition inhibits PRC2 activity
The human PHF1-PRC2 complexes were purified from HEK293T cells using wild type or mutated Flag-PHF1. Mutations that disrupt the PHF1 Tudor domain aromatic cage had no apparent effect on the association with EZH2, in agreement with previous observations that the PHD fingers region of PHF1 (ref. 12) but not the Tudor domain is responsible for the interaction with EZH2 (Fig. 4a). Enzymatic activity of PHF1-PRC2 was tested on purified native short oligonucleosomes (SON) by histone methyltransferase (HMT) assays (Fig. 4b). We used native chromatin from yeast Saccharomyces cerevisiae because it contains a high level of H3K36me3 due to high transcription activity and gene density30. When compared to the wild type PHF1-PRC2 complex, the PRC2 complexes containing PHF1 mutants impaired in H3K36me3 binding (W41A or Y47A) displayed a substantial increase in methyltransferase activity (blue columns). Interestingly, a similar marked increase in the methyltransferase activity of wild type PHF1-PRC2 was detected on chromatin purified from Δset2 mutant cells, which completely lack H3K36 methylation (Set2 is the sole H3K36-specific methyltransferase in yeast31) (Fig. 4b, compare first pair of blue and red columns). As evidenced by liquid counts and autoradiogram (Supplementary Fig. 5), loss of the H3K36me mark on chromatin resulted in more robust catalytic activity of wild type PHF1-PRC2. Importantly, in contrast to normal chromatin, the PRC2 complexes harboring H3K36me3-binding defective PHF1 mutants did not show increased methyltransferase activity compared to wild type PHF1-PRC2 when using Δset2 chromatin (Fig. 4b, compare red columns). These results confirm that H3K36me3 is inhibitory to PRC2 (refs. 24,25,26) and now clearly link PHF1 binding to this functional crosstalk, as both wild type PHF1 and H3K36me3 are required to block PRC2. Altogether these data indicate that recognition of H3K36me3 by the PHF1 Tudor domain negatively regulates the enzymatic activity of PRC2.
Figure 4.

Recognition of H3K36me3 by PHF1 inhibits PRC2 methyltransferase activity. (a) Western analysis of wild type and mutated Flag-PHF1 and EZH2 in the PRC2 complexes. (b) HMT assays with PHF1-PRC2 complexes purified from HEK293T cells on native wild type chromatin (wt SON, blue) and chromatin lacking the H3K36me mark (Δset2 SON, red). Error bars represent SD based on three experiments. (c) Western analysis of whole cell extract from HEK293T cells 48 hours after transfection with wild type HA-PHF1 or W41A or Y47A mutants. Empty vector is control. (d) Western analysis of K562 cells stably expressing Flag-PHF1.
Binding of the PHF1 Tudor domain to H3K36me3 decreases H3K27me3 levels in vivo. To examine the effect of this interaction in vivo we overexpressed wild type or mutated HA-tagged PHF1 in HEK293T cells and assessed the global levels of H3K27me3 and H3K36me3 by Western blot analysis (Fig. 4c). Overexpression of wild type PHF1 led to a decrease in the global level of H3K27me3 as compared to the endogenous level of this mark seen in the empty vector control. In contrast, exogenous PHF1 W41A or Y47A mutants had no effect on the H3K27me3 level. Importantly the levels of EZH2, total H3 and H3K36me3 were equal regardless if wild type or mutated PHF1 was tested. Furthermore, stable overexpression of wild type PHF1 in K562 cells resulted in a similar reduction of the H3K27me3 levels (Fig. 4d). These data support the idea that PHF1 inhibits PRC2 methyltransferase activity in response to H3K36me3, as overexpression of PHF1 oversensitizes PRC2 to H3K36me3 causing a decrease in H3K27me3 levels, an effect not seen upon overexpression of mutants unable to bind H3K36me3.
To determine the role of H3K36me3 recognition by PHF1 in the PHF1-PRC2 HKMT activity at target loci, we tested the occupancy of HA-PHF1 (wild type and mutants) and the levels of H3K27me3 and H3K36me3 at the promoter of MYT1 (ref. 32) in HEK293T cells (Fig. 5). We performed chromatin immunoprecipitation (ChIP) analyses on chromatin that was sheared to ~500 bp after fixing and assayed using primer probes specific for the MYT1 gene, spanning from −2000 to +1200 bp relative to the transcription start site. At all locations tested, the occupancy of exogenous wild type PHF1 was considerably higher than the occupancy of the PHF1 W41A or Y47A mutants impaired in H3K36me3 binding, whereas the H3K36me3 levels remained unchanged (Fig. 5a). These results indicate that binding of the Tudor domain to H3K36me3 is important for targeting of PHF1 to the MYT1 gene. However, as expected, the H3K27me3 levels were substantially reduced upon overexpression of wild type PHF1. Mutation of PHF1 Tudor domain residues critical for H3K36me3 binding partially rescued this effect and restored the methyltransferase activity of PRC2. The inverse correlation between H3K36me3-dependent PHF1 occupancy and the H3K27me3 level reinforced the notion of PHF1 inhibiting PRC2 activity in response to H3K36me3.
Figure 5.

Binding of PHF1 to H3K36me3 decreases PRC2-mediated deposition of H3K27me3. (a) ChIP assays on chromatin from HEK293T cells fixed and harvested 48 hours after transfection with either empty vector, wild-type HA-PHF1 or W41A or Y47A mutants. The levels of HA-PHF1, H3K27me3, H3K36me3 and H3 were probed across the MYT1 promoter using 7 primer sets. The top panel shows occupancy of wild type and mutant HA-PHF1 and the bottom two panels show H3K27me3 and H3K36me3 levels normalized to H3 (H3K27me3:H3). All data are relative to the PCLB4 non-target control gene. Error bars represent SD based on three experiments. (b) ChIP assays on chromatin harvested from mouse ES cells transduced with empty vector (pRTF, blue) or vector containing Flag-PHF1 (PHF1, red). Using quantitative PCR (qPCR), the levels of H3K27me3 and H3 were probed at the Oct4, Nanog, HoxA4 and HoxA11 promoters. Data are presented as the ratio of H3K27me3 to total H3 signal to correct for possible differences in nucleosome density at the different loci examined. Error bars represent SD based on two experiments. (c) A model for inhibition of EZH2-PRC2 activity through recognition of H3K36me3 by the Tudor domain of PHF1.
To investigate the importance of the PHF1-H3K36me3 interaction in PRC2-mediated deposition of H3K27me3 in a more physiological context, we generated mouse embryonic stem (ES) cells stably expressing Flag-PHF1. Flag IP and mass spectrometry analysis, which identified PHF1, EZH2, Suz12, EED and RBBP4, indicated that transduced Flag-PHF1 was assembled in a normal PRC2 complex in ES cells (data not shown). We performed ChIP assays using primers for genes known to be either transcriptionally active or silenced in ES cells. For example, the HoxA genes are not transcribed in ES cells and do not contain the H3K36me3 mark33. In agreement, ES cells stably expressing Flag-PHF1 or transduced with an empty vector had a comparable H3K27me3 level at the HoxA4 and HoxA11 clusters (Fig. 5b). In contrast, transcriptionally active genes Oct4 and Nanog, which typically contain a high level of H3K36me3 and we detected a reproducible decrease of the H3K27me3 signal at these loci in cells expressing Flag-PHF1. These results suggest that PHF1 Tudor-mediated inhibition of the HKMT activity of PRC2 is specific to the genomic sites that contain H3K36me3.
Functional Tudor domain is required for PHF1 at DSB sites
The H3K36me2 level increases rapidly and drastically at the DNA damage sites after irradiation and decreases shortly after34. Analysis of histone methylation reveals that generation of this PTM is the major immediate methylation event at DSBs, which also correlates with the DNA repair efficiency34. H3K36me2 recruits and stabilizes DNA repair components including Ku70, Ku80 and NBS1 at DSBs, enhancing NHEJ repair34. Recently, PHF1 was shown to localize to the sites of DNA damage and directly interact with Ku70-Ku80 (ref. 11). Although the PHF1 Tudor domain is not involved in the interaction with Ku70-Ku80, it plays a role in targeting of PHF1 to DSBs11. We sought to examine whether the PHF1 localization at DSBs depends on the ability of the Tudor domain to recognize methylated H3K36. U2OS cells expressing GFP-fusion PHF1, wild type or W41A and Y47A mutants impaired in binding to H3K36me3, were micro-irradiated with a 405 nm laser to induce DNA DSBs and the GFP-tagged proteins were visualized by fluorescence microscopy. As shown in Figure 6, Supplementary Figure 6a, and Supplementary Movie 1, wild type GFP-PHF1 was recruited to the DSB sites one minute after irradiation and dissociated within six minutes. In contrast, the W41A and Y47A mutants defective in H3K36me3 binding displayed a substantial reduction in their retention at the DSB sites, dissociating immediately after the initial accumulation at the DSBs. Accumulation of wild type and mutant proteins was completely abrogated in the presence of a Poly (ADP-ribose) polymerase 1 (PARP1) inhibitor and reduced by an ataxia telangiectasia mutated (ATM)/ataxia telangiectasia and Rad3 related (ATR) kinase inhibitor (Supplementary Fig. 6b–d and Supplementary Movie 2), revealing that the recruitment of PHF1 to DNA lesions strongly depends on activation of PARP1 in addition to requiring Ku70-Ku80 (ref. 11). Together, these data suggest a role for the H3K36me recognition by the Tudor domain in stabilization of PHF1 at the DSB sites in the PARP1- and Ku70-Ku80-initiated DNA damage response pathways.
Figure 6.
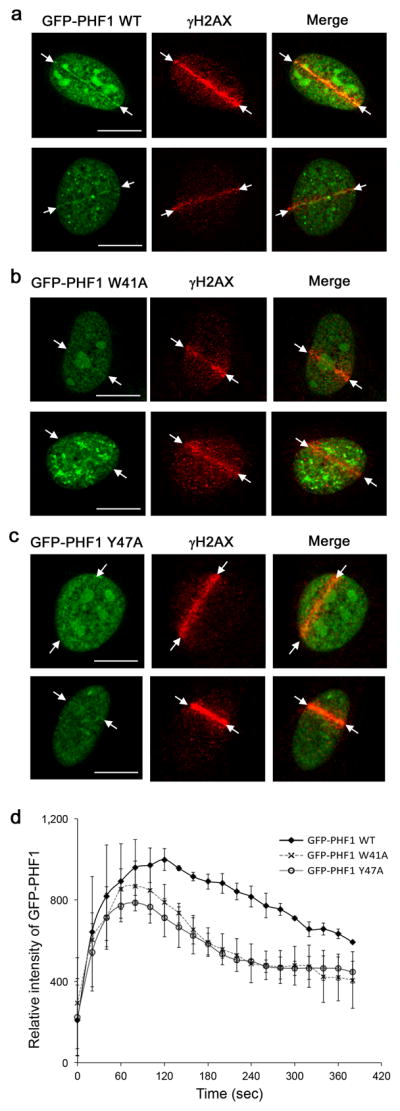
Tudor-dependent accumulation and retention of PHF1 at laser-irradiated sites of DSBs. Co-localization of GFP-PHF1 wild type (a), GFP-PHF1 W41A (b) and GFP-PHF1 Y47A (c) at the DSB sites in U2OS cells 3 min after irradiation. Scale bars correspond to 10 μm. (d) Kinetic analysis of GFP-PHF1 intensity at laser irradiated sites. GFP-PHF1 wild type (closed diamond), GFP-PHF1 W41A (X) and GFP-PHF1 Y47A (open circle). Error bars represent SD based on three experiments.
DISCUSSION
In this study, we demonstrate that the Tudor domain of PHF1 binds strongly and specifically to histone H3K36me3 and that this interaction is required for the multifaceted function of PHF1. We found that recognition of H3K36me3 by PHF1 inhibits the ability of the PRC2 complex to methylate H3K27. Binding of PHF1 to H3K36me3 may represent a mechanism for regulation of PRC2, which can be particularly important for delineating transcriptionally active and repressed regions. H3K27me3 and H3K36me3 are known to be antagonistic marks. Previous studies have demonstrated the inhibition of PRC2 by H3K36me3 (ref. 24,25,26) and it has been proposed that this PTM acts as a barrier to prevent deposition of H3K27 methylation at transcriptionally active genes (refs. 24,25). It has been shown that knockdown of the mouse homolog of PHF1 results in down-regulation of several Hox genes in NIH 3T3 cells8. Our data reveal that it is the PHF1 component of the PRC2 complex that is responsible for reading H3K36me3 and inhibiting PRC2 activity in the presence of this PTM.
Binding of the PHF1 Tudor domain to H3K36me3 may sterically preclude the EZH2 priming at H3K27 (Fig. 5c), however further work is needed to define the precise mechanism and to explore the effect of additional components that modulate PRC2 function. EED and JARID2, two other subunits of the PRC2 complex, have been found to both activate and reduce PRC2 activity35–41. The WD40 repeat of EED increases methyltransferase activity in response to H3K27me3 but decreases activity in response to H1K26me3 (ref. 40). Furthermore, in the absence of H3K36me3, PHF1 has also been shown to increase methyltransferase activity of PRC2 (ref. 8, 9), which could be due to possible interactions of the PHD fingers of PHF1 with other PTMs (for example, unmodified H3 or H3K4me3, hence no steric collision with H3K27me3), supporting the idea that PHF1 senses the local epigenetic environment through distinct modules. The presence of H3K36me3 and its recognition by the Tudor domain is required for inhibiting the PRC2 activity, and distinctive distributions of this PTM in different cells may lead to opposite outcomes, for example as seen in regulation of the Hox genes in the NIH 3T3 and GC1Spg cells8. Thus the exact composition of the PRC2 complex and the level and distribution of PTMs, which are known to fluctuate spatially and temporally1,42, may fine-tune the function of this complex, making it a modulator of transcription from the early stages of development to adulthood.
Our findings also point to the role of the Tudor-H3K36me interaction in transient localization of PHF1 at DNA damage sites. PHF1 has been shown to interact and co-localize with Ku70-Ku80 at DSBs, and two regions necessary for targeting were identified as Tudor and a region C-terminal to the PHD fingers of PHF1 (ref. 11). PHF1 can utilize the Ku70-Ku80-and/or PARP1-dependent mechanisms for accumulation, which may differentially contribute to anchoring and retention of PHF1. Rapid accumulation and stabilization of PHF1 could be essential in the early response to DNA damage involving not only Ku70-Ku80 and PARP1 but also PRC2, since other components of PRC2, such as EZH2, are found to co-localize with damaged DNA43,44. A dynamic regulation of these complexes through varying their composition can lead to greater sensitivity to the cellular environment and in response to extrinsic stimuli, providing a mechanism for differentiating the local epigenetic states.
ONLINE METHODS
DNA constructs, mutagenesis, and protein expression and purification
The wild-type PHF1 Tudor domain (residues 14–87 and residues 28–87) was cloned from full length hPHF1 (obtained from Open Biosystems). Point mutants (W41A and Y47A) were generated by site-directed mutagenesis using the Stratagene QuickChange XL kit. Wild type and mutant proteins were expressed in E. coli BL21(DE3) pLysS cells grown in LB and in 15NH4Cl- or 15NH4Cl- and 13C-glucose-supplemented M9-minimal media and induced with 0.5 mM IPTG. Bacteria were harvested by centrifugation and lysed by sonication. The unlabeled, 15N-labeled and 15N/13C-labeled GST-fusion proteins were purified using glutathione Sepharose 4B beads (GE Healthcare). The GST tag was either cleaved with Prescission protease, or left for the purposes of western blot analysis, in which case the GST-fusion protein was eluted off the glutathione Sepharose beads using 0.05 M reduced L-glutathione (Sigma Aldrich). Proteins were further purified by size exclusion chromatography.
X-ray crystallography
The solution of 3mg/mL PHF1 Tudor (residues 28–87) was incubated overnight with H3K36me3 peptide (residues 31–40) in a 1:1.5 molar ratio prior to crystallization. Crystals of the complex were grown using the sitting drop vapor diffusion method at 4°C by mixing 1 μl of the protein-peptide solution with 1 μl of precipitant solution containing 0.1 M HEPES pH 7.5, 20% PEG 10000. Crystals grew in a monoclinic space group C2 with two molecules per asymmetric unit. The complete data sets were collected at 100 K on a home source Rigaku/MSC Ru-H3R X-ray generator and Raxis IV++ area detectors system. The data were processed with D*TREK45. The molecular replacement solution was generated using the program Phaser46 and the solution structure of PHF1 (PDB 2E5P) as a search model. The initial models were built with COOT and refined using the program Phenix47. Ramachandran plot statistics showed 96.8% of residues in the allowed regions, 3.2% in the generously allowed regions and none in the disallowed regions. Remaining statistics are shown in Table 1. Coordinates and structure factors have been deposited to the Protein Data Bank with accession number 4HCZ.
Table 1.
Data collection and refinement statistics
| PHF1 Tudor-H3K36me3 | |
|---|---|
| Data collection | |
| Space group | C2 |
| Cell dimensions | |
| a, b, c (Å) | 88.03, 29.67, 62.00 |
| α, β, γ(°) | 90, 116.41, 90 |
| Resolution (Å) | 23.41-1.85 (1.92-1.85) |
| Rmerge | 0.024 (0.13) |
| I / σI | 19.4 (4.3) |
| Completeness (%) | 96.4 (81.1) |
| Redundancy | 2.2 (1.7) |
| Refinement | |
| Resolution (Å) | 1.85 |
| No. reflections | 12073 |
| Rwork / Rfree | 0.2038/0.2439 |
| No. atoms | |
| Protein | 467 |
| Ligand/ion | 76 |
| Water | 161 |
| B-factors | |
| Protein | 27.32 |
| Ligand/ion | 37.37 |
| Water | 39.6 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.007 |
| Bond angles (°) | 1.06 |
A single crystal was used for solving the structure. Values in parentheses are for highest-resolution shell.
Pulldown assays
GST-fusion PHF1 Tudor (residues 14–87) was incubated with biotinylated at the C-terminus peptides (Upstate Biotechnology) corresponding to the unmodified H3 (residues 1–21) and singly modified H3K4me1/2/3 (residues 1–21), H3K9me1/2/3 (residues 1–21), H3K27me1/2/3 (residues 21–44) and H3K36me1/2/3 (residues 21–44) histone tails in the presence of streptavidin Sepharose beads (GE Healthcare) in binding buffer containing 50 mM Tris (pH 7.5), 150 mM NaCl, and 0.05% Nonidet P-40. The beads were collected via centrifugation and washed five times with the peptide binding buffer. Bound protein was detected by western blot using anti-GST HRP conjugate monoclonal antibodies (GE Healthcare). Negative control using GST-fusion Tudor in the absence of the peptides was run in parallel to ensure that the protein does not bind to the streptavidin beads.
NMR Spectroscopy
NMR experiments were collected on a Varian INOVA 600 MHz spectrometer equipped with a cryogenic probe. Backbone assignments were obtained through analysis of the triple-resonance HNCACB and CBCA(CO)NH experiments carried out on uniformly 15N/13C-labeled PHF1 Tudor (residues 14–87) in 20 mM Tris pH 6.8 and 150 mM NaCl. Initial assignments were obtained using the PINE program and were verified and completed in CcpNMR.
Chemical shift perturbation experiments were carried out using uniformly 15N-labeled wild type or mutant Tudor (residues 14–87). 1H,15N heteronuclear single quantum coherence (HSQC) spectra were recorded in the presence of increasing concentrations of 12-mer histone tail peptides (synthesized by the University of Colorado Denver Biophysics Core Facility). Kd values were calculated by a nonlinear least-squares analysis in Kaleidagraph using the equation:
where [L] is concentration of the peptide, [P] is concentration of the protein, Δδ is the observed normalized chemical shift change and Δδmax is the normalized chemical shift change at saturation, calculated as
where δ is the chemical shift in parts per million (ppm).
Isothermal Titration Calorimetry (ITC)
ITC experiments were carried out at 25°C on a VP-ITC calorimeter (MicroCal). Protein and peptide were in identical buffer of 20 mM Tris pH 6.8 and 150 mM NaCl. 30 sequential injections of 10 μl of peptide (1.5–3 mM) were made into 1.7 ml of PHF1 Tudor (0.04 mM) at 180 second intervals. Control experiments measuring the heat of dilution were performed by injecting the peptide into buffer and subtracted from the raw data before the fitting process. Binding isotherms were analyzed by non-linear least-squares fitting of the data using Microcal ORIGIN software (Microcal) and a one-site binding model.
PHF1-PRC2 complex purification
Small amounts of plasmids encoding wild-type or mutant 3xFlag-PHF1 were transfected into HEK293T cells using calcium phosphate. 48 hrs post-transfection, cells were harvested and whole cell extracts were prepared. Flag-PHF1 was purified by incubating the extracts overnight with 3xFLAG agarose resin (Sigma-Aldrich), which was then extensively washed. Complex composition was examined by SDS-PAGE and silver staining, and by western blotting with antibodies against Flag (Sigma-Aldrich) or EZH2 (Santa Cruz Biotechnology). PRC2 complexes were also purified similarly but after co-transfection of vectors expressing all subunits (HA-tagged EZH2, SUZ12, EED, RbAp48, and Flag-PHF1), with similar results.
Histone methyltransferase assays
Purified protein complexes were incubated with 500 ng of short oligonucleosomes (purified from yeast cells as described in48) or 300 ng of indicated H3 peptides in 15 μl final volume using 1 mCi of [3H]AdoMet (S-adenosyl-L-[methyl-3H] methionine, 80 Ci/mmol; SAM) in HMT buffer (20 mM Tris/HCl [pH 8], 50 mM KCl, 0.1 mM EDTA, 5% glycerol, 1 mM DTT, 1 mM phenylmethylsulfonyl fluoride) for 45 min at 30°C. Each reaction was spotted onto p81 filters, washed three times with 50 mM Na Carbonate (pH 9.2) and processed for scintillation counting, or loaded on gel and processed for fluorography. In results from liquid counting, error bars represent standard deviation of independent reactions. Oligonucleosomes were purified from yeast cells because of very high content in H3K36me3 and the possibility to use yeast Δset2 mutant cells that completely lack this mark.
Cell culture, transfection and antibodies for Western Analysis
Full length hPHF1 was obtained from Open Biosystems (Thermo Scientific) and cloned into the pIRES2-EGFP plasmid to produce the protein with a C-terminal HA-tag. The mutant constructs W41A and Y47A were generated using the Stratagene QuickChange XL Site Directed Mutagenesis kit according to the manufacturer’s instructions. HEK293T cells were propagated in DMEM containing 10% Fetal Bovine Serum and penicillin streptomycin and PHF1 (wild-type or mutant) or an empty plasmid (negative control) was transiently transfected using Polyethylenimine. Cells were harvested 48 hours post-transfection and the whole cell lysate collected for Western analysis. Alternatively Western analysis was performed on acid extracted histones from stably transfected K562 cells. Antibodies used included rabbit polyclonal to HA tag (abcam ab9110), rabbit polyclonal to histone H3 (abcam ab1791), rabbit polyclonal to H3K27me3 (Millipore 17–622), rabbit monoclonal to H3K36me3 (cell signaling D5A7), rabbit monoclonal to EZH2 (cell signaling D2C9), and normal Rabbit IgG (abcam ab37415).
Chromatin Immunoprecipitation (ChIP)
1×107 transiently transfected cells were fixed with 1% formaldehyde for 14 minutes at room temperature and collected. Chromatin was sonicated to fragments ~500 bp in length. Immunoprecipitations (IPs) were performed in 167 mM NaCl, 0.01% SDS, 1.1% Triton X-100, 16.7 mM Tris pH 8.1, 1 mM EDTA containing protease inhibitor cocktail with 4 μg antibody and Protein A-coupled magnetic beads (Millipore). Samples were assayed by quantitative PCR using 7 primer sets for the MYT1 gene corresponding to positions −1680–−1555(1), −1280–−1211(2), −807–−725(3), −442–−297(4), −35–83(5), 292–397(6) and 761–866(7) around the transcription start site, and a primer set for the non-target PCLB4 gene as a control. 1% of each of the input chromatin DNA and the ChIP DNA samples were assayed by qPCR in triplicate using a Roche Light Cycler 480. Fold-enrichments of histone marks were calculated as IgG-subtracted %Input divided by the IgG-subtracted %Input of the reference gene PCLB4. Data is shown as an average and standard deviation over three biological replicates.
Mouse Embryonic Stem Cells
Mouse embryonic stem cells (ww6) were cultured following standard protocol on fibroblasts or in the presence of recombinant Leukemia Inhibitory Factor. Clones expressing Flag-tagged PHF1 were selected for after transduction with virus containing the pRTF vector (empty or PHF1). ChIP analysis was performed using 500 μg of cross-linked chromatin and anti-H3 (Abcam) or anti-H3K27me3 (ActiveMotif). Primers used for qPCR are available upon request and are located in the proximity of the transcription start site. The mean values and s.e.m. are computed between two distinct ChIP experiments.
Fluorescence microscopy and UVA-laser irradiation
Fluorescence images were obtained and processed using a confocal scanning laser microscopy system (FV-500, Olympus). UVA-laser irradiation was used to induce DSBs in U2OS cells as described previously11. Briefly, U2OS cells in glass-bottomed dishes were micro-irradiated with a 405 nm pulse laser (Olympus) along a user-defined path. Laser was focused through a 40X objective lens and the treatment dose was controlled by number of scans used. A single laser scan at full power delivers about 1600 nW. Cells were treated with 0.1 μM 8-MOP. The irradiation dose was fixed in the experiments as 50 scans. Experiments with inhibitors were carried out by pre-treating cells with 1.0 μM PARP1 inhibitor (AZD2281/KU0059436) for 4 hours or 1 mM ATM/ATR kinase inhibitor (#118501, CALBIOCHEM) for 4 hours prior to irradiation. The efficacy of the PARP1 inhibitor (AZD2281) was confirmed through the loss of accumulation of the PARP1 co-factor XRCC1 to single stranded breaks (induced by low doses of radiation) as well as a decrease in poly (ADP-ribose) PAR polymer as detected by Western analysis.
Supplementary Material
Acknowledgments
We thank B. Phillips, J. Gupta, D. Maranon and A. Morris and for helping with experiments. This research is supported by grants from the NIH GM096863 and CA113472 (T.G.K.) and GM084020 (J.N.) and CIHR (MOP-64289) (J.C.). C.A.M. is supported as an NIH NRSA postdoctoral fellow (F32 HL096399).
Footnotes
Accession codes. Atomic coordinates for the structure have been deposited in Protein Data Bank under accession code 4HCZ.
Note: Supplementary information is available in the online version of the paper.
AUTHOR CONTRIBUTIONS
C.A.M. and T.G.K. designed the study. C.A.M., N.A., R.W., C.G.A., M-E.L., S.R. and J.K.N. performed experiments and together with Z.H., C.A., J.N., C.A.K., A.Y., J.C and T.G.K. analyzed the data. T.G.K. and C.A.M. wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–9. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Varambally S, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–9. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- 3.Morin RD, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nature genetics. 2010;42:181–5. doi: 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cao R, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–43. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 5.Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002;16:2893–905. doi: 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Czermin B, et al. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell. 2002;111:185–96. doi: 10.1016/s0092-8674(02)00975-3. [DOI] [PubMed] [Google Scholar]
- 7.Muller J, et al. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell. 2002;111:197–208. doi: 10.1016/s0092-8674(02)00976-5. [DOI] [PubMed] [Google Scholar]
- 8.Cao R, et al. Role of hPHF1 in H3K27 methylation and Hox gene silencing. Mol Cell Biol. 2008;28:1862–72. doi: 10.1128/MCB.01589-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sarma K, Margueron R, Ivanov A, Pirrotta V, Reinberg D. Ezh2 requires PHF1 to efficiently catalyze H3 lysine 27 trimethylation in vivo. Mol Cell Biol. 2008;28:2718–31. doi: 10.1128/MCB.02017-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nekrasov M, et al. Pcl-PRC2 is needed to generate high levels of H3-K27 trimethylation at Polycomb target genes. The EMBO journal. 2007;26:4078–88. doi: 10.1038/sj.emboj.7601837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hong Z, et al. A polycomb group protein, PHF1, is involved in the response to DNA double-strand breaks in human cell. Nucleic acids research. 2008;36:2939–47. doi: 10.1093/nar/gkn146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O’Connell S, et al. Polycomblike PHD fingers mediate conserved interaction with enhancer of zeste protein. The Journal of Biological Chemistry. 2001;276:43065–73. doi: 10.1074/jbc.M104294200. [DOI] [PubMed] [Google Scholar]
- 13.Botuyan MV, et al. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127:1361–73. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang Y, Fang J, Bedford MT, Zhang Y, Xu RM. Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A. Science. 2006;312:748–51. doi: 10.1126/science.1125162. [DOI] [PubMed] [Google Scholar]
- 15.Lee J, Thompson JR, Botuyan MV, Mer G. Distinct binding modes specify the recognition of methylated histones H3K4 and H4K20 by JMJD2A-tudor. Nature structural & molecular biology. 2008;15:109–11. doi: 10.1038/nsmb1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roy S, et al. Structural insight into p53 recognition by the 53BP1 tandem Tudor domain. J Mol Biol. 2010;398:489–96. doi: 10.1016/j.jmb.2010.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tripsianes K, et al. Structural basis for dimethylarginine recognition by the Tudor domains of human SMN and SPF30 proteins. Nature structural & molecular biology. 2011;18:1414–20. doi: 10.1038/nsmb.2185. [DOI] [PubMed] [Google Scholar]
- 18.Yang Y, et al. TDRD3 is an effector molecule for arginine-methylated histone marks. Molecular cell. 2010;40:1016–23. doi: 10.1016/j.molcel.2010.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wagner EJ, Carpenter PB. Understanding the language of Lys36 methylation at histone H3. Nature reviews Molecular cell biology. 2012;13:115–26. doi: 10.1038/nrm3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krogan NJ, et al. Methylation of histone H3 by Set2 in Saccharomyces cerevisiae is linked to transcriptional elongation by RNA polymerase II. Molecular and cellular biology. 2003;23:4207–18. doi: 10.1128/MCB.23.12.4207-4218.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris SA, et al. Histone H3 K36 methylation is associated with transcription elongation in Schizosaccharomyces pombe. Eukaryotic cell. 2005;4:1446–54. doi: 10.1128/EC.4.8.1446-1454.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vezzoli A, et al. Molecular basis of histone H3K36me3 recognition by the PWWP domain of Brpf1. Nat Struct Mol Biol. 2010;17:617–9. doi: 10.1038/nsmb.1797. [DOI] [PubMed] [Google Scholar]
- 23.Xu C, Cui G, Botuyan MV, Mer G. Structural basis for the recognition of methylated histone H3K36 by the Eaf3 subunit of histone deacetylase complex Rpd3S. Structure. 2008;16:1740–50. doi: 10.1016/j.str.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmitges FW, et al. Histone methylation by PRC2 is inhibited by active chromatin marks. Molecular Cell. 2011;42:330–41. doi: 10.1016/j.molcel.2011.03.025. [DOI] [PubMed] [Google Scholar]
- 25.Yuan W, et al. H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. The Journal of biological chemistry. 2011;286:7983–9. doi: 10.1074/jbc.M110.194027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Voigt P, et al. Asymmetrically modified nucleosomes. Cell. 2012;151:181–92. doi: 10.1016/j.cell.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–40. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kutateladze TG. SnapShot: Histone Readers. Cell. 2011;146:842–842. e1. doi: 10.1016/j.cell.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Friberg A, Oddone A, Klymenko T, Muller J, Sattler M. Structure of an atypical Tudor domain in the Drosophila Polycomblike protein. Protein science : a publication of the Protein Society. 2010;19:1906–16. doi: 10.1002/pro.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pokholok DK, et al. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell. 2005;122:517–27. doi: 10.1016/j.cell.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 31.Kizer KO, et al. A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Molecular and cellular biology. 2005;25:3305–16. doi: 10.1128/MCB.25.8.3305-3316.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kirmizis A, et al. Silencing of human polycomb target genes is associated with methylation of histone H3 Lys 27. Genes Dev. 2004;18:1592–605. doi: 10.1101/gad.1200204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mikkelsen TS, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–60. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fnu S, et al. Methylation of histone H3 lysine 36 enhances DNA repair by nonhomologous end-joining. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:540–5. doi: 10.1073/pnas.1013571108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng JC, et al. Jarid2/Jumonji coordinates control of PRC2 enzymatic activity and target gene occupancy in pluripotent cells. Cell. 2009;139:1290–302. doi: 10.1016/j.cell.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shen X, et al. Jumonji modulates polycomb activity and self-renewal versus differentiation of stem cells. Cell. 2009;139:1303–14. doi: 10.1016/j.cell.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pasini D, et al. JARID2 regulates binding of the Polycomb repressive complex 2 to target genes in ES cells. Nature. 2010;464:306–10. doi: 10.1038/nature08788. [DOI] [PubMed] [Google Scholar]
- 38.Li G, et al. Jarid2 and PRC2, partners in regulating gene expression. Genes Dev. 2010;24:368–80. doi: 10.1101/gad.1886410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Margueron R, et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature. 2009;461:762–7. doi: 10.1038/nature08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu C, et al. Binding of different histone marks differentially regulates the activity and specificity of polycomb repressive complex 2 (PRC2) Proc Natl Acad Sci U S A. 2010 doi: 10.1073/pnas.1008937107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hansen KH, et al. A model for transmission of the H3K27me3 epigenetic mark. Nat Cell Biol. 2008;10:1291–300. doi: 10.1038/ncb1787. [DOI] [PubMed] [Google Scholar]
- 42.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 43.Chou DM, et al. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:18475–80. doi: 10.1073/pnas.1012946107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gieni RS, Ismail IH, Campbell S, Hendzel MJ. Polycomb group proteins in the DNA damage response: a link between radiation resistance and “stemness”. Cell cycle. 2011;10:883–94. doi: 10.4161/cc.10.6.14907. [DOI] [PubMed] [Google Scholar]
- 45.Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Crystallogr D Biol Crystallogr. 1999;55:1718–25. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 46.McCoy AJ, Storoni LC, Read RJ. Simple algorithm for a maximum-likelihood SAD function. Acta Crystallogr D Biol Crystallogr. 2004;60:1220–8. doi: 10.1107/S0907444904009990. [DOI] [PubMed] [Google Scholar]
- 47.Adams PD, et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–54. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 48.Altaf M, et al. Interplay of chromatin modifiers on a short basic patch of histone H4 tail defines the boundary of telomeric heterochromatin. Molecular cell. 2007;28:1002–14. doi: 10.1016/j.molcel.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.