

Abstract
Cardiovascular diseases are the most prevalent cause of morbidity and mortality among patients with type 1 or type 2 diabetes. The proposed mechanisms that can link accelerated atherosclerosis and increased cardiovascular risk in this population are poorly understood. It has been suggested that an association between hyperglycemia and intracellular metabolic changes can result in oxidative stress, low-grade inflammation, and endothelial dysfunction. Recently, epigenetic factors by different types of reactions are known to be responsible for the interaction between genes and environment and for this reason can also account for the association between diabetes and cardiovascular disease. The impact of clinical factors that may coexist with diabetes such as obesity, dyslipidemia, and hypertension are also discussed. Furthermore, evidence that justify screening for subclinical atherosclerosis in asymptomatic patients is controversial and is also matter of this review. The purpose of this paper is to describe the association between poor glycemic control, oxidative stress, markers of insulin resistance, and of low-grade inflammation that have been suggested as putative factors linking diabetes and cardiovascular disease.
1. Introduction
Diabetes is an important chronic disease which incidence is globally increasing and though considered as an epidemic [1]. The World Health Organization (WHO) estimated there were 30 million people who had diabetes worldwide in 1985. This number increased to 135 million by 1995 and reached 217 million in 2005. By the year 2030 WHO predicts this number will increase to at least 366 million [1]. This growth in diabetes prevalence, driven principally by an increased prevalence of type 2 diabetes (T2D), is occurring in both developing and developed countries [1]. The incidence of type 1 diabetes (T1D) is also increasing in parallel to that of T2D worldwide [2–4].
Individuals with diabetes and with chronically poor metabolic control can experience microvascular and macrovascular complications leading to a significant burden for the individual and for the society. This burden includes direct costs of medical care and indirect costs, such as loss of productivity, which result from diabetes-related morbidity and premature mortality [5, 6]. Health care expenses for people with diabetes is more than double of that for people without diabetes; the direct and indirect expenditures attributable to diabetes in 2007 in the USA were conservatively estimated at $174 billion, with slightly more spent on chronic complications attributable to diabetes than on diabetes care itself [6]. The International Diabetes Federation (IDF) estimated that diabetes accounts for 5–10% of the total healthcare budget in many countries [3]. The outpatient costs of T2D in Brazil were estimated by the ESCUDI study in 2011 [7]. The total costs were US$ 2,108 per patient/year, which consisted mostly of direct costs (63.3%) [7].
Cardiovascular diseases (CVD) are the most prevalent cause of mortality and morbidity among people with T2D and T1D [8–10]. In 2004, in the USA the presence of CVD and stroke was found in 68% and 16% of deaths related to diabetes among people older than 65 years, respectively [11]. Adult people with diabetes present rates of mortality due to heart disease and stroke from two to four times higher than those without diabetes [11]. It has been stated that patients with T2D without a previous history of myocardial infarction have the same risk of coronary artery disease (CADs) as nondiabetic subjects with a history of myocardial infarction [12]; this has led the National Cholesterol Education Program to consider diabetes as a coronary heart disease risk equivalent [13]. However, there is still some uncertainty as to whether the cardiovascular risk conferred by diabetes is truly equivalent to that of a previous myocardial infarction [14]. In general, patients with diabetes aggregate other comorbidities such as obesity, hypertension, and dyslipidemia which also contribute to increase the risk for CVD [15]. In the period of 2005 to 2008, the American Diabetes Association (ADA) estimated that 67% of people with diabetes older than 20 years presented blood pressure levels ≥140/90 mmHg or were using antihypertensive drugs [16]. Although there is strong evidence that supports both the efficacy and cost effectiveness of programs directed towards an improvement of glycemic control and other cardiovascular risk factors in patients with T2D [17] and T1D [18], the majority of these patients [19, 20] never achieve the goals established by guidelines issued by diabetes societies [16, 21].
The underlying mechanisms that cause accelerated atherosclerosis in patients with diabetes and consequently an increased prevalence of CVD are poorly understood. The purpose of this paper is to describe the association between poor glycemic control, oxidative stress, markers of insulin resistance, and of low-grade inflammation that have been suggested as putative factors linking these two conditions.
2. The Role of Glycemic Control
In recent decades, several clinical trials have investigated the effect of intensive treatment of hyperglycemia on cardiovascular risk reduction, in both T2D [22–25] and T1D [26] presenting conflicting results. The clinical characteristics of the studied populations regarding the presence of CVD and the duration of diabetes as well as the type of intensive intervention performed and the goals to be achieved partly explain the differences in the results.
In the United Kingdom Prospective Diabetes Study (UKPDS) [22], in newly diagnosed T2D patients, the early intensive treatment of hyperglycemia within the first five years of disease resulted in a long-term cardiovascular benefit, compared with patients in the conventional treatment group. This benefit was observed even after the loss of difference in glycemic control between the groups that occurred during the further five years of observational followup. However, the same was not observed in the three other large clinical trials conducted in patients with T2D. In the Veterans Affairs Diabetes Trial (VADT) [23], older patients with a 10 years mean duration of diabetes had no cardiovascular benefit when submitted to an intensive glycemic control regimen. This population comprised 40% of patients with a previous history of cardiovascular disease. Similar results were obtained from the Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified Release Controlled Evaluation (ADVANCE) trial [24] which aimed to achieve an A1c of 6.5% through intensive treatment with gliclazide plus other drugs. This strategy did not reduce the rate of major macrovascular events or death, despite a reduction in the incidence of diabetic nephropathy. As in VADT, patients were older (mean age of 66 years) and had a longer duration of diabetes (8 years) than UKPDS patients when the intensive treatment was started. In contrast, more strict intensive treatment aiming to reduce HbA1c below 6% in T2D patients, as occurred in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial [25], in addition to showing no benefit in reducing macrovascular events resulted in increased mortality, weight gain, and risk of hypoglycemia. Patients in this study presented a high cardiovascular risk profile when first initiated intensive glycemic treatment. Table 1 presents the main differences between these trials.
Table 1.
Differences of the effects of glycemic control in cardiovascular risk reduction in type 2 diabetes.
| UKPDS-10 years | VADT | ADVANCE | ACCORD | |
|---|---|---|---|---|
| Sample size | 5,102* | 1,791 | 11,140 | 10,251 |
| Followup (years) | 10 | 5.6 | 5 | 3.4 |
| Baseline characteristics | ||||
| Age (years) | 54 | 60.4 | 58 | 62.2 |
| Duration of diabetes (years) | Recently diagnosed | 11.5 | 8 | 10 |
| Presence of cardiovascular disease | 9% | 40% | 32% | 35% |
| Presence of microvascular complications | 18% | 62% | 10% | Albuminuria 14.0 (6.9–45.8)# |
| A1c levels | 6.2% | 8.3% | 7.5% | 8.3% |
| Effects of intensive treatment | ||||
| Difference in A1c levels (intensive/conventional) | 7.0/7.9% | 6.9/8.4% | 6.5/7.3% | 6.4/7.5% |
| Reduction in macrovascular events | Sulfa/insulin group ↓15% MI, ↓13% death Metformin group ↓33% MI, ↓27% death |
NS | NS | ↓Nonfatal MI ↑Death |
| Reduction in microvascular events (diabetic retinopathy, nephropathy, or neuropathy) |
↓24% (combined) |
NS | ↓incident nephropathy | — |
NS: nonstatistically significant.
A1c: glycated hemoglobin; MI: myocardial infarction.
*3,277 posttrial monitoring.
#The percentage of subjects with microvascular complications is not available. Ratio of urinary albumin (mg) to creatinine (g); median (Interquartile range).
In T1D patients, the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) study [26] showed the cardiovascular benefits of an intensive glycemic control after a followup of 17 years. The patients in this study were treated intensively for about 6.5 years and followed for 10 years observationally. Even after losing the strict glycemic control, represented by a glycated hemoglobin level below 7%, during the observational period, the group previously intensively treated presented a reduction of any cardiovascular event by 42%.
The main lesson learned from these results is that intensive treatment of hyperglycemia, targeting glycated hemoglobin levels below 7%, when initiated early in patients with short duration of diabetes and low cardiovascular risk, results in cardiovascular benefits. The same is not true when looking up tighter glycemic targets in older patients exposed to hyperglycemia for years before and with a higher cardiovascular risk profile.
This early protection is postulated to result from a mechanism known as “metabolic memory,” which means that the effect of the early glycemic exposure environment is remembered later in target organs [27] resulting in long-term deleterious or protective effects. The mechanisms involved in this process appear to comprehend epigenetic changes and intracellular metabolic changes that result in oxidative stress, low-grade inflammation, and endothelial dysfunction (Figure 1). These topics will be discussed below.
Figure 1.
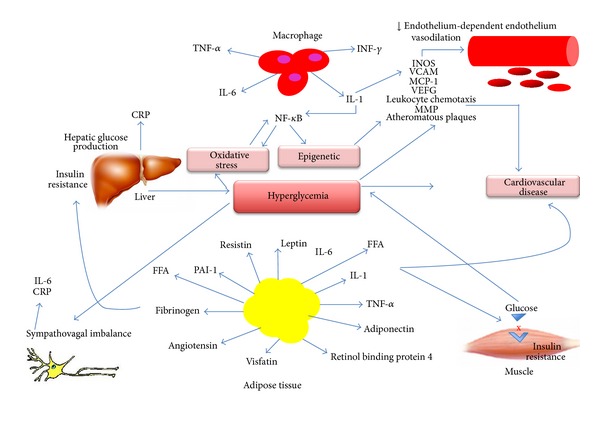
Pathogenesis of cardiovascular disease in diabetes. The mechanisms involved in the pathogenesis of cardiovascular disease in diabetes comprehend epigenetic changes and intracellular metabolic changes that result in oxidative stress, low-grade inflammation, and endothelial dysfunction. CRP: C-reactive protein; FFA: free fatty acids; INOS: inducible nitric oxide synthase; IL-1: interleukin 1; IL-6: interleukin 6; MCP-1: monocyte chemoattractant molecule 1; MMP: matrix metalloproitenase; NF-κB: nuclear factor kappa-β; PAI-1: plasminogen activator inhibitor-1; VCAM-1; vascular cell adhesion molecule-1; VEFG: vascular endothelial growth factor; TNF-α: Tumor necrosis factor-α; INF-γ: Interferon-γ.
3. Obesity
Obesity, which prevalence is also increasing worldwide [28] is becoming a major public health issue due to its association with chronic diseases such as diabetes mellitus, hypertension, dyslipidemia, sleep apnea, osteoarticular disease, and cardio and cerebrovascular diseases [28]. According to data from the WHO in 2008, the global prevalence of obesity (body mass index (BMI) ≥ 30 kg/m2) was 10% in men and 14% in women. Data from the National Health and Nutrition Examination Survey (NHANES) showed that the prevalence of overweight and obesity in adults increased from 55.9% to 64.5% and from 22.9% to 30.5%, from 1988–1994 to 1999-2000, respectively [28].
Obesity, especially with visceral fat deposition, is associated with low-grade inflammation, which plays a role in the pathogenesis of diabetes, and both diseases are associated with significant increase in morbidity and mortality due to CVD [28–30].
The main determinants for the onset of diabetes are beyond genetic factors, obesity and sedentary lifestyle [31]. Several studies have shown decreased incidence of diabetes by nonpharmacologic treatments, lifestyle changes, and body weight reduction. The Finish Diabetes Prevention Study Group showed that the incidence of diabetes was reduced in 58% in the group with only intensive lifestyle changes [31]. The Diabetes Prevention Program (DPP), diabetes incidence was reduced by 58% with intensive lifestyle intervention when compared to placebo and remained reduced by 34% after 10 years of followup [32]. Therefore, efforts should be made to encourage the adoption of healthy lifestyle and thus to combat the obesity epidemic.
4. Dyslipidemia
Dyslipidemia in T2D worsens cardiovascular risk due to the peculiar atherogenic profile composed by increased very low-density lipoprotein (VLDL) cholesterol, triglycerides and small and dense LDL cholesterol levels and decreased high-density lipoprotein (HDL) cholesterol levels. With such lipoproteins modified by oxidation and glycosylation there is a reduction on vascular compliance predisposing to early and aggressive atherosclerosis [33]. This may also occur in T1D, even though they are young patients and seldom present lipid abnormalities, but in this case, the atherogenic profile is not caused exclusively by increased lipid levels, and hyperglycemia per se is also pivotal in this process [34]. This was evidenced in an experimental study which concluded that either diabetic hyperlipidemia or hyperglycemia accelerates distinct phases of atherogenesis in diabetes [35]. In this study, it was shown that the dyslipidemia associated with diabetes is not sufficient to initiate the atherosclerotic lesion, because the progression of atherosclerosis process could be normalized after intensive glycemic control with insulin in mice [35].
In many interventional studies, the reduction of LDL cholesterol and triglycerides and increase of HDL cholesterol have been proved to be effective in reducing macrovascular disease and mortality in patients with T2D, especially in those with previous CAD.
The Collaborative Atorvastatin Diabetes Study (CARDS) was the first trial that studied T2D patients without previous CVD. Intervention with atorvastatin 10 mg showed 37% reduction in Cardiovascular events and 48% reduction in stroke when compared to placebo [36]. In the HDL Atherosclerosis Treatment Study (HATS), the combined use of low doses of simvastatin (10 to 20 mg/day) with high doses of niacin (2 to 4 g/day) showed a reduction in absolute risk of 13% for cardiovascular outcomes when HDL reached the target [37]. The study TNT (treatment to new targets) studied patients with T2D with previous CVD and compared the use of Atorvastatin 10 mg (conventional group) with Atorvastatin 80 mg (intensive group), and the goals were 100 mg and 80 mg for LDL cholesterol, respectively. The aggressive target achieved in this study (1.9 mmol/L) showed the most reduced rates of mortality due to cardiovascular events among all studies with statins [38].
Although decreasing LDL cholesterol has brought enough and established evidence on reducing cardiovascular mortality in T2D, if the treatment of dyslipidemia starts too late it may not be effective in avoiding atherosclerosis progression. According to the Deutsche Diabetes Dialyze Study (4D) that studied 1,255 T2D patients with end-stage renal disease which were randomized to Atorvastatin 20 mg/day or matching placebo during four years, there was no significant reduction in cardiovascular events with the intervention when compared to placebo [39]. Concerning cholesterol goals for diabetics, as far as we know, we should get as lower cholesterol levels as possible as stated by National Cholesterol Education Program Adult Treatment Panel III Guidelines (NCEP ATP III) [40]. It is well established that diabetic subjects are considered to belong to a high-risk category, thus their benefit from LDL-lowering therapy appears when LDL-C goal of 1.8 mmol/L is achieved [40].
In a recent meta-analysis which reviewed 22 trials with statins versus control, it was showed that statin use could be associated with an increased incidence of diabetes [40]. Despite the fact that an immediate doubling in cardiovascular risk in individuals with 5-year risk of major vascular events lower than 10%, such an effect is more than 50-times smaller than the absolute benefit observed with statin therapy in such individuals (about 11 fewer major vascular events per 1,000 treated over 5 years per 1.0 mmol/L reduction in LDL cholesterol) [41].
Considering hypertriglyceridemia, there is little evidence to support the benefits the goals to be achieved can bring. Fibrates are recommended to reduce pancreatitis risk in patients with triglycerides levels above 4.5 mmol/L when lifestyle modification does not succeed [42]. Until recently, there were no data that support that the combined use of statins and fibrates could reduce cardiovascular mortality. The Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study was a multinational randomized trial conducted with 9,795 patients with T2D which showed that fenofibrate did not significantly reduce the risk of the primary outcomes of coronary events. Instead, it reduced the number of total cardiovascular events (fewer nonfatal myocardial infarctions and revascularizations) [43]. Another message from this study was that fibrate confers microvascular protection, because it reduced the need for laser treatment for diabetic retinopathy [44].
5. Hypertension
Hypertension is a highly prevalent disease worldwide and very common among patients with diabetes. Approximately from 10 to 30% of T1D and 60% of T2D patients have hypertension [45, 46].
The coexistence of these two conditions increase the risk of developing macrovascular complications (myocardial infarction, stroke) and also microvascular complications (nephropathy and retinopathy) [47]. The vigorous treatment of hypertension may reduce the progression of these complications.
The time hypertension starts differs in different types of diabetes. In patients with T1D, hypertension develops years after diagnosis usually already reflecting the development of diabetic nephropathy [46]. Blood pressure (BP) tends to increase three years after the onset of microalbuminuria [48]. In patients with T2D, hypertension may be present at diagnosis or even before the elevation of blood glucose levels [49]. The association between hypertension and obesity is well established leading to a higher rate of cardiovascular morbidity and mortality in patients with these two conditions [49].
The recommended target blood pressure for patients with diabetes, according to the ADA [50] is characterized by BP < 130/80 mmHg [51]. Although, the European Society of Hypertension Task Force [52] states that BP goals traditionally recommended in diabetes are not supported by outcomes evidence from trials. They also reinforce only to pursue a reasonable BP reduction without indicating a goal which is unproven, since it has also been very difficult to achieve blood pressure goals in the majority of the patients [53].
According to the ADVANCE study in diabetic patients at high cardiovascular risk lower BP levels should be reached [53]. One should always take into consideration the individualization of treatment and its correlation with response to therapy, drug tolerance, and individual characteristics. However, randomized clinical trials have demonstrated that the established therapeutic target (BP < 130/80 mmHg) own benefits in reducing CHD, stroke, and kidney disease [52, 54]. In patients with renal insufficiency and proteinuria above 1 to 2 g per day, the target BP should approach 120/75 mmHg [53].
The treatment of hypertension in diabetic patients aims at the prevention of CVD, minimizing the progression of renal disease and diabetic retinopathy. According to the UKPDS, patients with T2D may benefit more from tight control of BP than with strict control of blood glucose levels [55]. Initial treatment should include nonpharmacological measures such as weight reduction (in overweight and obesity), regular exercising, reducing salt intake (<1500 mg per day), avoiding excessive alcohol consumption (no more than two servings per day in men and no more than one serving per day in women), and smoking cessation. Pharmacological therapy should be initiated in all diabetics who persist with BP > 130/80 mmHg, when a change in lifestyle has already been implemented for 3 months or when the maximum BP levels are already higher than 140/90 mmHg at diagnosis [50, 56].
Pharmacological therapy can be accomplished with various classes of antihypertensive agents. Diuretics, angiotensin converting enzyme inhibitors, angiotensin II antagonists, beta blockers, calcium channel blockers, alpha blockers, and combination of blockers of the renin-angiotensin have shown to be effective in reducing cardiovascular events. In most cases, the association of two or three drugs may be necessary in order to achieve the goals of the treatment.
The ACCORD-BP study, evaluating more intensive treatment of blood pressure (systolic blood pressure reduction aiming at levels lower than 120 mmHg) in patients with T2D and CVD or at least two cardiovascular risk factors, showed no reduction in cardiovascular events rates (myocardial infarction, CHF, and cardiovascular death), although it was observed a reduction in the number of strokes [57].
6. Oxidative Stress
Increased intracellular glucose concentrations result in the activation of alternative pathways of metabolism such as the hexosamine and the aldose reductase pathways, both involved in the pathophysiology of chronic complications of diabetes. These pathways trigger an increased production of reactive oxygen species (ROS) and depletes substrates for important antioxidant enzymes. Additionally, increased intracellular glucose leads to the formation of advanced glycation end products (AGES) and the activation of protein kinase C (PKC). All these mechanisms lead to a common effect, an increased oxidative stress state.
Oxidative stress results from an imbalance between the production of ROS and the antioxidant defense. The ROSs are chemically instable and highly reactive molecules [58] continuously produced by aerobic organisms that function as second messengers regulating the expression of redox signal sensitive genes (e.g., nuclear factor kappa-β (NFκ-B) gene) and in the production of inflammatory mediators. They are generated from enzymes that use oxygen as electron acceptor including the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, nitric oxide synthase (NOS), xanthine oxidase, the mitochondrial chain electron transport, lipoxygenase, cyclooxygenase, and cytochrome P450. The first three are the main sources of ROS in the vascular wall [59].
The active form of NADPH oxidase is responsible for the reduction of the molecular oxygen resulting in the formation of superoxide anion [NAD(P)H + 2O2 → NAD(P)+ + H+ + 2O2 −] [58]. This enzyme could act as a sensor of the concentration of oxygen in the vasculature modulating the vascular tone [60]. Components of the NADPH oxidase were demonstrated in vascular and renal cells in animals and humans [61–67].
The ROSs produced in the vascular wall are involved in various cellular events such as mitosis, apoptosis, migration, hypertrophy and extracellular matrix modification, and changes in gene transcription and protein synthesis [68]. They may also function as mediators of the metabolic memory to hyperglycemia. Human retinal endothelial cells exposed to hyperglycemia in vitro, maintained high levels of oxidative stress markers such as PKC and β subunits of NADPH oxidase p47phox, even after normalization of blood glucose levels [69].
Another important source of ROS in diabetes is the mitochondria. It is postulated that the mitochondrial O2 − anion acts as a factor initiating a cascade of events that result in increased production of ROS and reactive nitrogen species (RNS) through activation of NFκ-B. This results in the production of inflammatory cytokines, activation of PKC and NADPH oxidase. In addition, NOS can divert the production of nitric oxide (NO) to generate O2 − in conditions of deficiency of L-arginine or tetrahydropterin in the endothelium of diabetic patients [70]. When both are produced the formation of peroxynitrite (NOO−) occurs, causing damage to cellular structures such as DNA, lipids, and proteins [71].
Under normal conditions, the presence of ROS induces the expression of antioxidant enzymes as a defense mechanism. This is not a rule under diabetes condition. For instance, in fibroblasts from T1D patients with overt nephropathy, the exposure to hyperglycemia led to an increase in lipid peroxidation without a compensatory increase in the level of the antioxidant enzyme Cu-Zn superoxide dismutase, catalase, and glutathione peroxidase [72]. Even patients with a short diabetes duration and without chronic complications present less antioxidant plasma capacity and uric acid levels suggesting that the oxidative stress occurs early in the disease [73].
Nonenzymatic extracellular antioxidants include α-tocopherol, vitamin A, β-carotene, ascorbic acid, albumin, and uric acid. The lipid solubility properties of α-tocopherol, vitamin A, and β-carotene are particularly important to protect against lipid peroxidation. The role of uric acid in the pathogenesis of CVD and endothelial dysfunction is still conflicting [74–76]. Another important component of the antioxidant defense in diabetes is haptoglobin [77–79]. This plasma protein binds free hemoglobin resulting in the inhibition of iron-induced oxidative damage, since hemoglobin released in the blood after hemolysis of senescent erythrocytes is a potent oxidant.
7. Epigenetics
Nowadays, there is compelling evidence linkingepigenetic factors to many human diseases including diabetes and CVD [80]. Epigenetic factors, by different types of reactions, could mediate the interplay between genes and environment resulting in activation or repression of genetic transcription, or even silencing the genetic transcription. The most important epigenetic reactions affecting genetic transcription are acetylation and methylation. These reactions occur mainly in the tail of histones that are proteins where DNA is wrapped. Brownlee et al. [81] have demonstrated in human aortic endothelial cells that excess ROS resulting from hyperglycemia can induce monomethylation of lysine from histone 3 increasing the expression of the subunit p65 of NFκ-B. This reaction is responsible for the increased transcription of vascular cell adhesion molecule 1 (VCAM-1), monocyte chemoattractant molecule 1 (MCP-1), and some inflammatory proteins like interleukin 6 (IL-6), intercellular adhesion molecule 1 (ICAM-1), and NOS that are related tohyperglycemia-induced arterial pathology. Moreover, this reaction persisted after asix-day period of subsequent normoglycemia, supporting the concept of metabolic memory. Epigenetic reactions could be an important mediator between diabetes, CVD, and chronic inflammatory response. Besides, some comorbidities associated with diabetes have also been associated with epigenetics like hypertension [82] and obesity [83]. The epigenetic modifications associated with hypertension are related to intrauterine environmental factors which can limit the development of the nephrons and to other factors that are related to autonomic responsiveness, vessel remodeling, salt sensitivity, and to the renin-angiotensin system. The mechanisms involved in these associations are mainly methylation of histones and of DNA. The relationship between epigenetics and obesity is more complex and is related to genomic imprinting, epigenetic mosaicism, and nonimprinted gene which through different pathways can influence energy balance, body weight, and fat mass.
8. Inflammatory Cascade, Diabetes, and Atherosclerosis
Diabetes, obesity, and insulin resistance are associated with subclinical inflammation characterized by overexpression of cytokines produced by adipose tissue, activated macrophages, and other cells [84, 85]. Inflammatory mediators, such as TNF-α, interleukin-1 (IL-1), IL-6, leptin, resistin, MCP-1, plasminogen activator inhibitor-1 (PAI-1), C-reactive protein (CRP), fibrinogen, angiotensin, visfatin, retinol binding protein-4, and adiponectin are involved in signaling pathways, in insulin action, and perpetuation of inflammatory response [85]. These cytokines are involved in the chronic inflammatory process of the vessels wall, promoting lipid accumulation with consequent development of atherosclerosis and CVD [86].
Atherosclerosis is a complex multifactorial disease, and the acceleration of atherosclerosis in diabetes may be explained by several conditions including hyperglycemia, increased oxidative stress, advanced glycation end products (AGE), dyslipidemia, autonomic imbalance, hyperinsulinemia, inflammatory markers excess, and genetic variables [35, 87, 88].
It is assumed that the adipose tissue initiates obesity-induced inflammation and leads to the recruitment of immune cells which contributes to the maintenance of inflammatory response [84], besides leading to endothelial dysfunction with increased expression of adhesion molecules (ICAM-1, VCAM-1, P-selectin, and E-selectin), migration of monocytes, neutrophils, and T lymphocytes [89].
Insulin resistance induces chronic elevation in FFA plasma concentrations leading to increased storage of triglycerides in muscle, promoting reduction of muscle glucose uptake and liver, and increased hepatic glucose production, that have been shown to impair insulin action and promote hyperinsulinemia [90]. Hyperinsulinemia can, per se, induce cardiomyocyte hypertrophy through myocyte growth induced by an activation of PI3 K/Akt-1 pathway and also by enhancing FFA levels. FFA are also implicated in the development of myocardial contractile dysfunction [91].
Several cytokines described to be related with insulin resistance are also involved with the development of atherosclerosis and CVD. TNF-α and other cytokines, FFA and ROS, activate inflammatory pathways and promote the expression of numerous genes involved in insulin resistance [84, 85, 89].
IL-1 is another cytokine produced as a consequence of stress or cell injury mainly by macrophages that modulate key events in the process of atherosclerosis such as vessels wall inflammation, leukocyte chemotaxis and adhesion by increasing expression of VCAM-1 and MCP-1, angiogenesis (through vascular endothelial growth factor—(VEFG) induction), upregulation of matrix metalloproteinases (MMP), and destabilization of atheromatous plaques, that can lead to plaque rupture and thrombosis [86].
CRP is an acute phase protein and is primarily derived from IL-6 hepatic biosynthesis [92]. Atherogenic mechanisms of CRP include impaired production of endothelial NO and prostacyclin; increased production of endothelin-1 and other cell adhesion molecules, monocyte chemoattractant protein-1, IL-8, and PAI-1; ROS and proinflammatory macrophage production; monocyte adhesion and chemotaxis; uptake of oxidized low-density lipoprotein (LDL); CRP also stimulates the expression of metalloproteinases, activates NF-κB, and promotes cell proliferation in vascular smooth muscle cells due to upregulation of the angiotensin type 1 receptor [93].
Adiponectin has many protective actions in the atherosclerosis process due to its inhibition of LDL oxidation, activation of macrophages (via TNF-α), reduction of adhesion molecule (VCAM and ICAM), inhibition of proliferation and migration, of smooth cells, and an increased production of NO in endothelial cells [87]. Adiponectin is markedly reduced with increased obesity, and in diabetes [85] and hypoadiponectinemia is associated with an increase in CVD rates [94].
Leptin is a hormone secreted by adipose tissue and primarily involved in the regulation of energy expenditure and food intake. Plasma leptin concentrations are increased in obese and diabetic patients [95]. Leptin has been shown to participate in the development of atherosclerosis in several ways: inducing oxidative stress; increasing the production of MCP-1, endotelin-1 (ET-1) which leads to cardiomyocyte hypertrophy; promoting migration, proliferation, hypertrophy of vascular smooth muscle cells (VSMC), and vascular cell wall calcification; stimulating platelet aggregation; attenuating cardiomyocyte contractility through increased nitric oxide production, reduction of intracellular calcium, and decreased β-adrenergic response [95].
Therefore, evidences suggest that the hypothesis that is low-grade inflammation would be the causal common factor between diabetes, insulin resistance, obesity, and CVD [32].
9. Endothelial Dysfunction
Endothelial vasodilation and vascular reactivity in diabetes are known to be impaired since its early phases [96, 97]. This is explained by the hypothesis that there are changes in endothelial cells function present in the early atherosclerosis lesion [98]. Thus, oxidative stress, inflammation, and endothelial dysfunction are closely correlated in diabetes, because the formers increase vascular endothelial permeability, generating leukocyte adhesion, which is coupled with impairment in endothelial signal transduction and redox-regulated transcription factors [99, 100]. Another possible mechanism to link these conditions is that the impaired endothelium-dependent vasodilation in diabetes is associated with reduced action of NO secondary to its inactivation, and this is a consequence of oxidative stress, rather than decreased NO production from endothelial cells. Moreover, the abnormal metabolism of NO is related to advanced diabetes microvascular complications [99]. Many factors can explain endothelial dysfunction in diabetes such as hyperlipidemia [96, 98], insulin resistance [86, 98, 101], hyperglycemia [98], hyperamylinemia [101], hypertension [101], and hyperhomocysteinemia [101].
10. Endothelial Dysfunction in T1D
Endothelial function of the macro- and microcirculation, which is usually evaluated through the vasodilator response to endothelium-dependent vasodilators or physiological stimuli, is characteristically impaired in patients with T1D [76, 102–104]. The endothelial response to acetylcholine is correlated with diabetes duration, glycemic control, triglycerides, and age [76, 105, 106].
Endothelial dysfunction in T1D is an important determinant of inflammatory activity regardless of the presence or absence of complications showing that it can be considered an early marker for CVD [107]. The disturbances in vascular responses can be seen even in children with T1D, as evidenced in studies that showed impaired flow-mediated dilation (FMD) responses and also association with increased carotid artery intima-media thickness in this group [97, 108]. And, as evidenced by Davi et al. [97], this alteration represents an early and, in some cases, a reversible event in the natural history of T1D in children and adolescents because it was noted that in approximately 45% of this population the tissue plasminogen activator (tPA) levels were reversed after 1 year.
Several markers of endothelial function in T1D have been described such as Von Willebrand factor, thrombomodulin, selectin, PAI-1, Type IV collagen, and tPA, that are so forth indicators of endothelial cell dysfunction when increased. VCAM-1 levels are more markedly increased in patients with T1D with retinopathy when compared with those with micro- or macroalbuminuria only [98]. It has been shown that the cellular adhesion molecule E-selectin may enhance CAD prediction beyond traditional risk factors in T1D [98, 107]. Other markers of low-grade inflammation levels are described to be elevated in this group such as of oxidized LDL [109], monocyte IL-6, superoxide anion, plasma CRP, sCD40L, and nitrotyrosine levels [110].
So, endothelial dysfunction in T1D represents a high risk for micro- and macroangiopathy and hyperglycemia, appears to be one of the main causes, that alone seems not to be sufficient to cause it, because other agents such as genes and environmental factors are likely to play a role [76, 98].
11. Endothelial Dysfunction in T2D
T2D is independently associated with impaired FMD, and endothelial dysfunction is considered to be the determinant factor for the vascular complications that is aggravated, rather than caused by hyperglycemia, because of the presence of many other risk factors such as obesity, hypertension, dislypidemia, and ageing as well [96, 101, 111]. One possible explanation for this is the increased calpain (calcium-dependent protease) activity in response to hyperglycemia. Hyperglycemic states can induce loss of NO via a calpain-dependent decrease in the association with endothelial NOS. Moreover, inhibition of calpain activity decreases endothelial cell surface expression of the proinflammatory adhesion molecules ICAM-1 and VCAM-1 during hyperglycemia [112]. Markers of endothelial dysfunction are early signs for the development of microangiopathy.
The hallmark of T2D is insulin resistance, therefore there is sufficient evidence pointing to the coexistence of endothelial dysfunction with this condition [101]. Elevated circulating levels of PAI-1 and ET-1 can be seen in obesity as well as the correlation between endothelial activation and acute-phase reaction with insulin resistance and obesity in T2D. Abnormalities in vascular reactivity and insulin resistance can also be seen in young first-degree relatives of T2D patients independent of the presence of classic cardiovascular risk factors [113].
12. Cardiovascular Autonomic Neuropathy (CAN)
CAN is one of the most common chronic complications of diabetes mellitus and has shown negative impact on survival and quality of life in patients with diabetes [114]. The prevalence of CAN ranges from 2.6% to 90% among subjects with diabetes, and the incidence of CAN increases with age, diabetes duration, and inadequate glycemic control [115].
Recent studies [116, 117] have shown that dysregulation of the Autonomic Nervous System (ANS) with increased sympathetic activity is associated with elevated inflammatory markers such as IL-6 and CRP, demonstrating a link between autonomic imbalance, inflammation, and CVD. The ANS is responsible for modulating the activity of the sinus node (heart rate), ventricular (end systolic and diastolic volume) and blood vessels (systemic vascular resistance), and the dysfunction of the ANS may contribute to the development of arterial stiffness, left ventricular hypertrophy, and ventricular diastolic dysfunction [118].
The clinical manifestations of CAN are described as resting tachycardia, postural hypotension, exercise intolerance, abnormal coronary vasomotor regulation (risk of silent myocardial ischemia and infarction), increased QT interval, perioperative instability, increased risk of renal disease, stroke, and sudden death [114].
CAN represents a strong indicator of cardiovascular risk in both T1D and T2D [119]. In the Detection of Silent Myocardial Ischemia in Asymptomatic Diabetic Subjects (DIAD) study, the strongest predictors for abnormal perfusion tests were abnormal Valsalva maneuver, male sex, and diabetes duration, demonstrating that CAN may have an important role in the screening of CVD [120]. Patients with diabetes and CAN have 5-year mortality rates ranging from 16 to 53%, depending on its severity [119]. The mortality rates from CVD in T1D and T2D are 4.2 and 10 times higher, respectively, than in healthy individuals without diabetes [120, 121].
13. Screening for Subclinical Atherosclerosis
The screening for the detection of subclinical atherosclerosis in asymptomatic diabetic patients is the subject of considerable controversy. There are no prospective studies that support its usefulness and that can modify the natural history of those patients [122, 123]. Even today, there is no consensus on which tests should be performed. Intensive medical therapy seems to provide equal outcomes to invasive revascularization [124]. Which raises questions on how screening results would change management? The clinical risk factors that indicate increased risk of CVD in diabetic patients are CAD, cerebrovascular or peripheral vascular disease, female sex, age greater than 40 years in men and greater than 50 years in women, long duration of diabetes (for every 10 years the risk increases 86% according to the Framingham study), presence of renal disease, autonomic neuropathy and classic risk factors such as hypertension, dyslipidemia, smoking, sedentary lifestyle, family early atherosclerotic disease, metabolic syndrome, and presence of atrial fibrillation [124, 125].
One of the major limitations of the routine screening for subclinical atherosclerosis is the different rates of coronary events in previous studies. The prevalence of silent myocardial ischemia (SMI) in diabetic population varies in different studies, ranging from 12% to almost 57% [126, 127]. This variability underlines the difficulty to have a cost-effective screening and the necessity to define the cardiovascular risk in the asymptomatic diabetic population who could benefit from this screening. The ADA does not recommend the detection of CVD in asymptomatic diabetic patients as a routine. Their recommendations for investigating SMI are very conservative, being the exercise testing in diabetic patients with typical (chest pain, dyspnea) or atypical cardiac symptoms and changes in baseline electrocardiogram. Asymptomatic patients with carotid or peripheral vascular disease or sedentary patients who want to start high-intensity exercise can also be investigated [50]. The DIAD study [120] accessed 1,123 asymptomatic diabetic patients in a randomized controlled trial. The patients were randomly assigned to be screened with adenosine-stress radionuclide myocardial perfusion imaging (MPI) or not to be screened. The cumulative cardiac event rate was 2.9% over a mean (SD) followup of 4.8 years for an average rate of 0.6% per year. A comparison of the cardiac event rates (0.6% per year) with those reported in ACCORD trial for the subgroup of patients with T2D without previous cardiac events (1.4% per year) which included a selection of older patients with specific additional risk factors for CVD would appear favorable and compatible in these two studies [25]. The data from these two studies show that there is no evidence that the complete survey of subclinical arterial disease may modify the natural history of CAD in asymptomatic diabetic patients with risk factors controlled by recommended goals.
Despite the controversy regarding the screening, several studies using various invasive and noninvasive cardiovascular examinations are being conducted. The presence of calcium in coronary arteries is a specific marker of atherosclerosis, independent of its etiology [128]. The presence of calcified plaques correlates with increasing age, especially after age 50 [129, 130]. Though the calcium score represents an estimate of the total amount of plaque present in an individual, it does not correspond directly to the degree of luminal narrowing of a given vessel [128]. The calcium score was higher than the scores of Framingham and UKPDS for the prediction of events [129]. According to the Patients with Renal Impairment and Diabetes undergoing Computed Tomography (PREDICT) study, the coronary artery calcium (CAC) score was taken as independent risk marker for incremental coronary events and stroke [130].
The US National Cholesterol Education Programme Adult Treatment Panel III (NCEP ATP III) recommends the use of calcium score in a selection of patients with intermediate risk by traditional methods (between 10 and 20% risk in 10 years), and when added to conventional methods, these patients may become high risk, and benefit of a therapy aimed to more restrictive treatment targets [13]. A recently meta-analysis showed that diabetic patients without a history of myocardial infarction had 43% less risk of developing coronary events when compared with patients without diabetes but with prior infarction [14]. Both coronary calcifications as average intimal thickness are increased in this population; however, the classification of these individuals to a higher category of risk is still controversial when using these methods [131]. Although being very promising the use of the calcium score for CVD in asymptomatic diabetic patients still needs further prospective studies and cost effectiveness to demonstrate its benefits.
Revascularization of asymptomatic T2D subjects is still polemic. The Bypass Angioplasty Revascularization Investigation 2 Diabetes Trial (BARI 2D) was a randomized study with 2,368 patients with T2D and SMI comparing revascularization versus intensive medical therapy, showed no differences on reducing rates of death and cardiovascular events among patients undergoing prompt revascularization and those undergoing medical therapy or between strategies insulin sensitizers or insulin provision [123].
According to the 2010 American College of Cardiology Foundation/American Heart Association (ACCF/AHA) Guideline for Assessment of Cardiovascular Risk in Asymptomatic Adults [132], in asymptomatic adults with diabetes, 40 years and older, measurement of CAC score is reasonable for cardiovascular risk assessment (Class IIa, evidence B). Stress MPI may be considered for advanced cardiovascular risk assessment in asymptomatic adults with diabetes or when previous risk assessment testing suggests a high risk of CHD, such as CAC score of 400 or greater (Class IIb, evidence C).
Carotid intima-media thickness (C-IMT) is considered one of the independent predictors of coronary artery disease, a marker of early atherosclerosis and vascular remodeling [133]. According to Irie et al. in an evaluation of 251 asymptomatic T2D patients, the addition of max-IMT (the greatest IMT in the observation-possible areas) to conventional risk factors improves the risk stratification for CAD [133]. In T1D patients, the DCCT/EDIC research group demonstrated that in 12 years after the DCCT intervention the C-IMT progression in the group that received intensive diabetes therapy was slower than the group that received conventional therapy from years 1 to 6. It could be assigned to a durable “metabolic memory” that exists for atherosclerosis. But, the similar C-IMT progression in the original treatment groups over EDIC from years 6 to 12 indicates a “metabolic memory amnesia” over time [134].
The ankle brachial pressure index (ABPI) is a simple method to evaluate the presence of peripheral vascular diseases [135]. A low ABPI (<0.9) was considered to be a marker of cardiovascular diseases risk. The AHA recommends the evaluation of ABPI as a diagnostic criterion for the prevalence of peripheral arterial diseases [136]. In the study of Doza et al. with 1,121 T2D patients in north India, the prevalence of low ABPI was 4.5% in men and 4.7% in woman. The results were similar to those found in studies with Chinese Korean and Brazilian populations [137].
Changing the natural history of silent coronary artery disease, without considering the control of classical risk factors, represents a major challenge facing the global epidemic of diabetes mellitus. More research is needed to identify appropriate screening strategies for diabetic asymptomatic patients with CHD.
14. Perspectives and Conclusions
The incidence of diabetes is sharply increasing worldwide which represents an important burden for patients and for the society as well due to micro- and macrovascular complications that people with this condition may experience and consequently cardiovascular diseases that are the most prevalent causes of morbidity and mortality among patients with diabetes.
The classical risk factors for the development of CVD in subjects with diabetes are the presence of poor glycemic control, obesity, dyslipidemia, and hypertension. In recent decades, several clinical trials have investigated the effect of intensive treatment of hyperglycemia on cardiovascular risk reduction, in both T1D and T2D, like the DCCT and UKPDS, and the main lesson learned from these trials is that intensive treatment of hyperglycemia initiated early in patients with short duration of diabetes and low cardiovascular risk, result in cardiovascular benefits. The same is not true for older patients exposed to hyperglycemia for a long time and with a high cardiovascular risk profile. This protection might result from a mechanism known as “metabolic memory,” which means that the effect of the early glycemic exposure environment is imprinted in target organs resulting in long-term deleterious or protective effects. Obesity, especially with visceral fat deposition, is associated with low-grade inflammation, which plays a role in the pathogenesis of diabetes, and both diseases are associated with significant increase in morbidity and mortality due to CVD. Dyslipidemia mainly that represented by high levels of LDL-cholesterol is also a risk factor for CVD because small increases in LDL-cholesterol levels increase the risk for CVD. The coexistence of hypertension and diabetes increase the risk of developing macrovascular complications (myocardial infarction, stroke) and also microvascular complications (nephropathy and retinopathy).
These clinical conditions might be associated with intracellular and mitochondrial metabolic changes that can result in oxidative stress, a state of low-grade inflammation characterized by overexpression of cytokines produced by adipose tissue, activated macrophages and other cells, and the presence of many inflammatory mediators that will finally cause a generalized endothelial dysfunction or even a cardiovascular autonomic neuropathy, an important cause of sudden death among subjects with diabetes.
The proposed mechanisms that can link accelerated atherosclerosis and increased cardiovascular risk in subjects with diabetes are still poorly understood. It has been suggested that an association between hyperglycemia and epigenetic factors by different types of reactions could be responsible for the interaction between genes and environment and for this reason account for the association between diabetes and cardiovascular disease. Many trials have shown that an early intervention in patients with short duration of diabetes could result in cardiovascular benefits, but there is no robust evidence that justify screening for subclinical atherosclerosis in asymptomatic patients with diabetes.
The purpose of this paper was to describe the association between poor glycemic control, oxidative stress, markers of insulin resistance and of low-grade inflammation that have been suggested as putative factors linking diabetes, and cardiovascular disease and to elucidate the mechanisms involved in the pathogenesis of CVD in this population.
Abbreviations
- ABPI:
Ankle brachial pressure index
- ACCORD:
Action to Control Cardiovascular Risk in Diabetes
- ACCF:
American College of Cardiology Foundation
- ADA:
American Diabetes Association
- ADVANCE:
Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified Release Controlled Evaluation
- AGE:
Advanced glycation end products
- AHA:
American Heart Association
- ANS:
Autonomic Nervous System
- BARI 2D:
Bypass Angioplasty Revascularization Investigation 2 Diabetes Trial
- BMI:
Body mass index
- BP:
Blood pressure
- CAC:
Coronary artery calcium
- CAD:
Coronary artery disease
- CAN:
Cardiac Autonomic Neuropathy
- CARDS
Collaborative Atorvastatin Diabetes Study
- CHF:
Congestive heart failure
- CIMT:
Carotid intima-media thickness
- CRP:
C-reactive protein
- CVD:
Cardiovascular disease
- DCCT:
Diabetes Control and Complications Trial
- DIAD:
Detection of Silent Myocardial Ischemia in Asymptomatic Diabetic Subjects
- DPP:
The Diabetes Prevention Program
- EDIC:
Epidemiology of Diabetes Interventions and Complications Trial
- ERK:
Extracellular signal regulated kinase
- ET-1:
Endotelin-1
- FFA:
Free fatty acids
- FIELD:
Fenofibrate Intervention and Event Lowering in Diabetes
- FMD:
flow-mediated dilation
- GSK-3:
Glycogen synthases kinase-3
- HATS:
HDL Atherosclerosis Treatment Study
- HbA1c;
Glycated hemoglobin
- HDL:
High-density lipoprotein
- ICAM-1
Intercellular adhesion molecule 1
- IDF:
International Diabetes Federation;
- IL-1:
Interleukin 1
- IL-6:
Interleukin 6
- IKKβ:
Inhibitor of nuclear factor kappa-B kinase subunit beta
- IRS-1:
Insulin receptor substrate-1
- LDL:
Low-density lipoprotein
- MCP-1:
Monocyte chemoattractant molecule 1
- MMP:
Matrix metalloproteinase
- MPI:
Myocardial perfusion imaging
- mTOR:
Mammalian target of rapamycin
- NADPH:
Nicotinamide adenine dinucleotide phosphate
- NCEP ATP III:
National Cholesterol Education Programme Adult Treatment Panel III
- NFAT-3:
Nuclear factor in activated lymphocytes
- NF-κB:
Nuclear factor kappa-β
- NCEP ATP III:
National Cholesterol Education Program Adult Treatment Panel III Guidelines
- NHANES:
National Health and Nutrition Examination Survey
- NO:
Nitric oxide
- NOO−:
Peroxynitrite
- NOS:
Nitric oxide synthase
- PAI-1:
Plasminogen activator inhibitor-1
- PDGF:
Platelet derived growth factor
- PKC:
Protein kinase C
- PPAR-α:
Peroxisome proliferator-activated receptor α
- PREDICT:
Patients with Renal Impairment and Diabetes undergoing Computed Tomography
- RNS
Reactive nitrogen species
- ROS:
Reactive oxygen species
- sBP:
Systolic blood pressure
- SMI:
Silent myocardial ischemia
- T1D:
Type 1 diabetes
- T2D:
Type 2 diabetes
- TGF-β:
Transforming growth factor-β
- TNT:
Treatment to new targets
- tPA:
Tissue plasminogen activator
- UKPDS:
United Kingdom Prospective Diabetes Study
- WHO:
World Health Organization
- VADT:
Veterans Affairs Diabetes Trial
- VCAM-1;
Vascular cell adhesion molecule-1
- VEFG:
Vascular endothelial growth factor
- VLDL:
Very low-density lipoprotein
- VSMC:
Vascular smooth muscle cells.
References
- 1.Wild S, Roglic G, Green A, et al. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 27(5):1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 2.American Diabetes Association. Standards of medical care for patients with diabetes mellitus. Diabetes Care. 2003;26(supplement 1):533–550. [Google Scholar]
- 3.IDF. International Diabetes Federation. Diabetes Atlas. 3rd edition. Brussels, Belgium: International Diabetes Federation Diabetes Atlas; 2006. [Google Scholar]
- 4.Negrato CA, Dias JP, Teixeira MF, et al. Temporal trends in incidence of Type 1 diabetes between 1986 and 2006 in Brazil. Journal of Endocrinology Investigation. 2010;33:373–377. doi: 10.1007/BF03346606. [DOI] [PubMed] [Google Scholar]
- 5.Harris MI. Diabetes in America. 2nd edition. Washington, DC, USA: Government Printing Office; 1995. Summary of national diabetes data group; pp. 1–13. (NIH Publication No. 95-1468). [Google Scholar]
- 6.American Diabetes Association. Economic costs of diabetes in the US in 2007. Diabetes Care. 2008;31:596–615. doi: 10.2337/dc08-9017. [DOI] [PubMed] [Google Scholar]
- 7.Bahia LR, Araújo DV, Schaan BD, et al. The costs of type 2 diabetes mellitus outpatient care in the Brazilian public health system. Value in Health. 2011;14:S137–S140. doi: 10.1016/j.jval.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 8.Orasanu G, Plutzky J. The pathologic continuum of diabetic vascular disease. Journal of the American College of Cardiology. 2009;53(5):S35–S42. doi: 10.1016/j.jacc.2008.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kemp TM, Barr ELM, Zimmet PZ, et al. Glucose, lipid, and blood pressure control in Australian adults with type 2 diabetes: the 1999–2000 AusDiab. Diabetes Care. 2005;28(6):1490–1492. doi: 10.2337/diacare.28.6.1490. [DOI] [PubMed] [Google Scholar]
- 10.Laing SP, Swerdlow AJ, Slater SD, et al. Mortality from heart disease in a cohort of 23,000 patients with insulin-treated diabetes. Diabetologia. 2003;46(6):760–765. doi: 10.1007/s00125-003-1116-6. [DOI] [PubMed] [Google Scholar]
- 11.Centers for Disease Control and Prevention. National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States. Atlanta, Ga, USA: 2011. [Google Scholar]
- 12.Haffner SM, Lehto S, Ronnemaa T, et al. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. The New England Journal of Medicine. 1998;339(4):229–234. doi: 10.1056/NEJM199807233390404. [DOI] [PubMed] [Google Scholar]
- 13.National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) Third report of the National Cholesterol Education Program (NCEP) expert panel on detection. Circulation. 2002;106:3143–3421. [PubMed] [Google Scholar]
- 14.Bulugahapitiya U, Siyambalapitiya S, Sithole J, Idris I. Is diabetes a coronary risk equivalent? Systematic review and meta-analysis. Diabetic Medicine. 2009;26(2):142–148. doi: 10.1111/j.1464-5491.2008.02640.x. [DOI] [PubMed] [Google Scholar]
- 15.Al Ghatrif M, Kuo YF, Al Snih S, Raji MA, Ray LA, Markides KS. Trends in Hypertension Prevalence, Awareness, Treatment and Control in Older Mexican Americans, 1993-2005. Annals of Epidemiology. 2011;21(1):15–25. doi: 10.1016/j.annepidem.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.American Diabetes Association. Standards of medical care in diabetes 2011. Diabetes Care. 2011;34(supplement 1):11–61. doi: 10.2337/dc11-0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stratton IM, Adler AI, Neil HAW, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. British Medical Journal. 2000;321(7258):405–412. doi: 10.1136/bmj.321.7258.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DCCT. The diabetes control and complications trial. The New England Journal of Medicine. 1993;329:977–986. doi: 10.1056/NEJM199309303291410. [DOI] [PubMed] [Google Scholar]
- 19.Gomes MB, Gianella D, Faria M, et al. Prevalence of patients with diabetes type 2 within the targets of care guidelines in daily clinical practice: a multicenter study of type 2 diabetes in Brazil. The Revision of Diabetic Studies. 2006;3:73–78. doi: 10.1900/RDS.2006.3.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gomes MB, Coral M, Cobas RA, et al. Prevalence of adults with type 1 diabetes who meet the goals of care in daily clinical practice: a nationwide multicenter study in Brazil Diabetes. Diabetes Research Clinical Practice. 2012;97(1):63–70. doi: 10.1016/j.diabres.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 21.Sociedade Brasileira de Diabetes: Diretrizes da SBD/2011. São Paulo, Brazil: AC Farmaceutica Ltda; 2011. [Google Scholar]
- 22.Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HAW. 10-Year follow-up of intensive glucose control in type 2 diabetes. The New England Journal of Medicine. 2008;359(15):1577–1589. doi: 10.1056/NEJMoa0806470. [DOI] [PubMed] [Google Scholar]
- 23.Duckworth W, Abraira C, Moritz T, et al. Glucose control and vascular complications in veterans with type 2 diabetes. The New England Journal of Medicine. 2009;360(2):129–139. doi: 10.1056/NEJMoa0808431. [DOI] [PubMed] [Google Scholar]
- 24.Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. The New England Journal of Medicine. 2008;358(24):2560–2572. doi: 10.1056/NEJMoa0802987. [DOI] [PubMed] [Google Scholar]
- 25.The Action to Control Cardiovascular Risk in Diabetes Study Group. Effects of Intensive Glucose Lowering in Type 2 Diabetes. The New England Journal of Medicine. 2008;358:2545–2559. doi: 10.1056/NEJMoa0802743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.The Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study Research Group. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. The New England Journal of Medicine. 2005;353:2643–2653. doi: 10.1056/NEJMoa052187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ceriello A, Ihnat MA, Thorpe JE. The "Metabolic memory": is more than just tight glucose control necessary to prevent diabetic complications? Journal of Clinical Endocrinology and Metabolism. 2009;94(2):410–415. doi: 10.1210/jc.2008-1824. [DOI] [PubMed] [Google Scholar]
- 28.Poirier P, Giles TD, Bray GA, et al. Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss. Circulation. 2006;113:898–918. doi: 10.1161/CIRCULATIONAHA.106.171016. [DOI] [PubMed] [Google Scholar]
- 29.Mokdad AH, Bowman BA, Ford ES, Vinicor F, Marks JS, Koplan JP. The continuing epidemics of obesity and diabetes in the United States. Journal of the American Medical Association. 2001;286(10):1195–1200. doi: 10.1001/jama.286.10.1195. [DOI] [PubMed] [Google Scholar]
- 30.Duncan BB, Schmidt MI, Pankow JS, et al. Low-grade systemic inflammation and the development of type 2 diabetes: the atherosclerosis risk in communities study. Diabetes. 2003;52(7):1799–1805. doi: 10.2337/diabetes.52.7.1799. [DOI] [PubMed] [Google Scholar]
- 31.Tuomilehto J, Lindström J, Eriksson JG, et al. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. The New England Journal of Medicine. 2001;344(18):1343–1350. doi: 10.1056/NEJM200105033441801. [DOI] [PubMed] [Google Scholar]
- 32.Knowler WC, Fowler SE, Hamman RF, et al. 10-year follow-up of diabetes incidence and weight loss in the Diabetes Prevention Program Outcomes Study. The Lancet. 2009;374:1677–1686. doi: 10.1016/S0140-6736(09)61457-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watts GF, Playford DA. Dyslipoproteinaemia and hyperoxidative stress in the pathogenesis of endothelial dysfunction in non-insulin dependent diabetes mellitus: an hypothesis. Atherosclerosis. 1998;141(1):17–30. doi: 10.1016/s0021-9150(98)00170-1. [DOI] [PubMed] [Google Scholar]
- 34.Matheus ASDM, Cobas RA, Gomes MB. Dyslipidemias in type 1 diabetes: a current approach. Arquivos Brasileiros de Endocrinologia e Metabologia. 2008;52(2):334–339. doi: 10.1590/s0004-27302008000200021. [DOI] [PubMed] [Google Scholar]
- 35.Renard CB, Kramer F, Johansson F, et al. Diabetes and diabetes-associated lipid abnormalities have distinct effects on initiation and progression of atherosclerotic lesions. Journal of Clinical Investigation. 2004;114(5):659–668. doi: 10.1172/JCI17867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Colhoun HM, Betteridge DJ, Durrington PN, et al. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. The Lancet. 2004;364(9435):685–696. doi: 10.1016/S0140-6736(04)16895-5. [DOI] [PubMed] [Google Scholar]
- 37.Brown BG, Zhao XQ, Chait A, et al. Sinvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. The New England Journal of Medicine. 2001;345:1583–1592. doi: 10.1056/NEJMoa011090. [DOI] [PubMed] [Google Scholar]
- 38.LaRosa JC, Grundy SM, Waters DD, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. The New England Journal of Medicine. 2005;352(14):1425–1435. doi: 10.1056/NEJMoa050461. [DOI] [PubMed] [Google Scholar]
- 39.Wanner NC, Krane V, Marz W, et al. Atorvastatin in patients with type 2 diabetes mellitus undergoing hemodialysis. The New England Journal of Medicine. 2005;353:238–248. doi: 10.1056/NEJMoa043545. [DOI] [PubMed] [Google Scholar]
- 40.Grundy SM, Cleeman JI, Merz CNB, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III Guidelines. Circulation. 2004;110:227–239. doi: 10.1161/01.CIR.0000133317.49796.0E. [DOI] [PubMed] [Google Scholar]
- 41.Cholesterol Treatment Trialists' (CTT) Collaborators. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. The Lancet. 2012;380(9841):581–590. doi: 10.1016/S0140-6736(12)60367-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Furtado MV, Polanczyk CA. Prevenção cardiovascular em pacientes com diabetes: revisão baseada em evidências. Arquivos Brasileiros de Endocrinologia e Metabologia. 2007;51(2):312–318. doi: 10.1590/s0004-27302007000200022. [DOI] [PubMed] [Google Scholar]
- 43.The FIELD study investigators. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. The Lancet. 2005;366(9500):1849–11861. doi: 10.1016/S0140-6736(05)67667-2. [DOI] [PubMed] [Google Scholar]
- 44.Keech A, Mitchell P, Summanen P, et al. Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): a randomised controlled trial. The Lancet. 2007;370(9600):1687–1697. doi: 10.1016/S0140-6736(07)61607-9. [DOI] [PubMed] [Google Scholar]
- 45.Diabetes in America. 2nd edition. Bethesda, MD, USA: National Institutes of Health, National Institute of diabetes and Digestive and Kidney Diseases; 1995. [Google Scholar]
- 46.American Diabetes Association. Treatment of hypertension in diabetes. Diabetes Care. 1993;16:1394–1401. doi: 10.2337/diacare.16.10.1394. [DOI] [PubMed] [Google Scholar]
- 47.Curb JD, Pressel SL, Cutler JA, et al. Effect of diuretic-based antihypertensive treatment on cardiovascular disease risk in older diabetic patients with isolated systolic hypertension. Journal of the American Medical Association. 1996;276(23):1886–1892. [PubMed] [Google Scholar]
- 48.Mogensen CE, Hansen KW, Pedersen MM, Christensen CK. Renal factors influencing blood pressure threshold and choice of treatment for hypertension in IDDM. Diabetes Care. 1991;14(supplement 4):13–26. doi: 10.2337/diacare.14.4.13. [DOI] [PubMed] [Google Scholar]
- 49.Turner R, Stratton I, Fright V, Holman R, Manley S, Cull C. Hypertension in Diabetes Study (HDS): I. Prevalence of hypertension in newly presenting type 2 diabetic patients and the association with risk factors for cardiovascular and diabetic complications. Journal of Hypertension. 1993;11(3):309–317. doi: 10.1097/00004872-199303000-00012. [DOI] [PubMed] [Google Scholar]
- 50.American Diabetes Association. Standards of medical care in diabetes-2012. Diabetes Care. 2012;35(supplement 1):S11–S63. doi: 10.2337/dc12-s011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chobanian AV, Bakris GL, Black HR, et al. The seventh report of the joint national committee on prevention, detection, evaluation, and treatment of high blood pressure: the JNC 7 report. Journal of the American Medical Association. 2003;289(19):2560–2572. doi: 10.1001/jama.289.19.2560. [DOI] [PubMed] [Google Scholar]
- 52.Maneia G, Laurent S, Agabiti-Rosei E, et al. Reappraisal of European guidelines on hypertension management: a European Society of Hypertension Task Force document. Journal of Hypertension. 2009;27(11):2121–2158. doi: 10.1097/HJH.0b013e328333146d. [DOI] [PubMed] [Google Scholar]
- 53.Patel A. Effects of a fixed combination of perindopril and indapamide on macrovascular and microvascular outcomes in patients with type 2 diabetes mellitus (the ADVANCE trial): a randomised controlled trial. The Lancet. 2007;370(9590):829–840. doi: 10.1016/S0140-6736(07)61303-8. [DOI] [PubMed] [Google Scholar]
- 54.Adler AI, Stratton IM, Neil HAW, et al. Association of systolic blood pressure with macrovascular and microvascular complications of type 2 diabetes (UKPDS 36): prospective observational study. British Medical Journal. 2000;321(7258):412–419. doi: 10.1136/bmj.321.7258.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.UK Prospective Diabetes Study Group. Tight blood pressure control and risk of macrovascular and microvascular complications intype 2 diabetes: UKPDS 38. British Medical Journal. 1998;317:p. 703. [PMC free article] [PubMed] [Google Scholar]
- 56.Roccella EJ. The Sixth Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Archives of Internal Medicine. 1997;157(21):2413–2446. doi: 10.1001/archinte.157.21.2413. [DOI] [PubMed] [Google Scholar]
- 57.The ACCORD Study Group. Effects of intensive blood-pressure control in type 2 diabetes mellitus. The New England Journal of Medicine. 2010;362:1575–1585. doi: 10.1056/NEJMoa1001286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Berry C, Brosnan MJ, Fennell J, Hamilton CA, Dominiczak AF. Oxidative stress and vascular damage in hypertension. Current Opinion in Nephrology and Hypertension. 2001;10(2):247–255. doi: 10.1097/00041552-200103000-00014. [DOI] [PubMed] [Google Scholar]
- 59.Griendling KK. Novel NAD(P)H oxidases in the cardiovascular system. Heart. 2004;90(5):491–493. doi: 10.1136/hrt.2003.029397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goligorsky MS. Making sense out of oxygen sensor. Circulation Research. 2000;86(8):824–826. doi: 10.1161/01.res.86.8.824. [DOI] [PubMed] [Google Scholar]
- 61.Bayraktutan U, Blayney L, Shah AM. Molecular characterization and localization of the NAD(P)H oxidase components gp91-phox and p22-phox in endothelial cells. Arteriosclerosis, Thrombosis, and Vascular Biology. 2000;20(8):1903–1911. doi: 10.1161/01.atv.20.8.1903. [DOI] [PubMed] [Google Scholar]
- 62.Fukui T, Lassegue B, Kai H, et al. Cytochrome b558 alpha subunit cloning and expression in rat aortic smooth muscle cells. Biochimica et Biophysica Acta. 1995;231:215–219. doi: 10.1016/0005-2728(95)00098-4. [DOI] [PubMed] [Google Scholar]
- 63.Chabrashvili T, Tojo A, Onozato ML, et al. Expression and cellular localization of classic NADPH oxidase subunits in the spontaneously hypertensive rat kidney. Hypertension. 2002;39(2 I):269–274. doi: 10.1161/hy0202.103264. [DOI] [PubMed] [Google Scholar]
- 64.Azumi H, Inoue N, Takeshita S, et al. Expression of NADH/NADPH oxidase p22(phox) in human coronary arteries. Circulation. 1999;100(14):1494–1498. doi: 10.1161/01.cir.100.14.1494. [DOI] [PubMed] [Google Scholar]
- 65.Greiber S, Munzel T, Kastner S, et al. NAD(P)H oxidase activity in cultured human podocytes: effects of adenosine triphosphate. Kidney International. 1998;53:654–663. doi: 10.1046/j.1523-1755.1998.00796.x. [DOI] [PubMed] [Google Scholar]
- 66.Jones SA, Hancock JT, Jones OTG, Neubauer A, Topley N. The expression of NADPH oxidase components in human glomerular mesangial cells: detection of protein and mRNA for p47phox, p67phox, and p22phox. Journal of the American Society of Nephrology. 1995;5(7):1483–1491. doi: 10.1681/ASN.V571483. [DOI] [PubMed] [Google Scholar]
- 67.Inoue N, Kawashima S, Kanazawa K, Yamada S, Akita H, Yokoyama M. Polymorphism of the NADH/NADPH oxidase p22 phox gene in patients with coronary artery disease. Circulation. 1998;97(2):135–137. doi: 10.1161/01.cir.97.2.135. [DOI] [PubMed] [Google Scholar]
- 68.Van Heerebeek L, Meischl C, Stooker W, Meijer CJLM, Niessen HWM, Roos D. NADPH oxidase: new source of reactive oxygen species in the vascular system? Journal of Clinical Pathology. 2002;55(8):561–568. doi: 10.1136/jcp.55.8.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ihnat MA, Thorpe JE, Kamat CD, et al. Reactive oxygen species mediate a cellular “memory” of high glucose stress signaling. Diabetologia. 2007;50:1523–1531. doi: 10.1007/s00125-007-0684-2. [DOI] [PubMed] [Google Scholar]
- 70.Channon KM, Guzik TJ. Mechanisms of superoxide production in human blood vessels: relationship to endothelial dysfunction, clinical and genetic risk factors. Journal of Physiology and Pharmacology. 2002;53(4 I):515–524. [PubMed] [Google Scholar]
- 71.Ballinger SW, Patterson C, Yan CN, et al. Hydrogen peroxide- and peroxynitrite-induced mitochondrial DNA damage and dysfunction in vascular endothelial and smooth muscle cells. Circulation Research. 2000;86(9):960–966. doi: 10.1161/01.res.86.9.960. [DOI] [PubMed] [Google Scholar]
- 72.Ceriello A, Morocutti A, Mercuri F, et al. Defective intracellular antioxidant enzyme production in type 1 diabetic patients with nephropathy. Diabetes. 2000;49(12):2170–2177. doi: 10.2337/diabetes.49.12.2170. [DOI] [PubMed] [Google Scholar]
- 73.Marra G, Cotroneo P, Pitocco D, et al. Early increase of oxidative stress and reduced antioxidant defenses in patients with uncomplicated type 1 diabetes: a case for gender difference. Diabetes Care. 2002;25(2):370–375. doi: 10.2337/diacare.25.2.370. [DOI] [PubMed] [Google Scholar]
- 74.Strasak A, Ruttmann E, Brant L, et al. Serum uric acid and risk of cardiovascular mortality: a prospective long-term study of 83 683 Austrian men. Clinical Chemistry. 2008;54(2):273–284. doi: 10.1373/clinchem.2007.094425. [DOI] [PubMed] [Google Scholar]
- 75.Waring WS, McKnight JA, Webb DJ, Maxwell SRJ. Uric acid restores endothelial function in patients with type 1 diabetes and regular smokers. Diabetes. 2006;55(11):3127–3132. doi: 10.2337/db06-0283. [DOI] [PubMed] [Google Scholar]
- 76.Matheus ASM, Tibiriçá E, da Silva PB, et al. Uric acid levels are associated with microvascular endothelial dysfunction in patients with type 1 diabetes. Diabetic Medicine. 2011;28(10):1188–1193. doi: 10.1111/j.1464-5491.2011.03349.x. [DOI] [PubMed] [Google Scholar]
- 77.Costacou T, Ferrell RE, Orchard TJ. Haptoglobin genotype a determinant of cardiovascular complication risk in type 1 diabetes. Diabetes. 2008;57(6):1702–1706. doi: 10.2337/db08-0095. [DOI] [PubMed] [Google Scholar]
- 78.Vardi M, Blum S, Levy AP. Haptoglobin genotype and cardiovascular outcomes in diabetes mellitus-natural history of the disease and the effect of vitamin E treatment. Meta-analysis of the medical literature. European Journal of Internal Medicine. 2001;23(7):628–632. doi: 10.1016/j.ejim.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Levy AP, Hochberg I, Jablonski K, et al. Haptoglobin phenotype is an independent risk factor for cardiovascular disease in individuals with diabetes: the strong heart study. Journal of the American College of Cardiology. 2002;40(11):1984–1990. doi: 10.1016/s0735-1097(02)02534-2. [DOI] [PubMed] [Google Scholar]
- 80.Ling C, Groop L. Epigenetics: a molecular link between environmental factors and type 2 diabetes. Diabetes. 2009;58(12):2718–2725. doi: 10.2337/db09-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.El-Osta A, Brasacchio D, Yao D, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. The Journal of Experimental Medicine. 2008;205(10):2409–2417. doi: 10.1084/jem.20081188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Millis RM. Epigentics and hypertension. Current Hypertension Reports. 2011;13:21–28. doi: 10.1007/s11906-010-0173-8. [DOI] [PubMed] [Google Scholar]
- 83.Stoger R. Epigenetics and obesity. Pharmacogenomics. 2008;9:1851–1860. doi: 10.2217/14622416.9.12.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 85.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. Journal of Clinical Investigation. 2006;116(7):1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vicenova B, Vopalensky V, Burysek L, et al. Emerging role of interleukin-1 in cardiovascular disease. Physiology Research. 2009;58:481–498. doi: 10.33549/physiolres.931673. [DOI] [PubMed] [Google Scholar]
- 87.Ferrarezi DAF, Cheurfa N, Reis AF, Fumeron F, Velho G. Adiponectin gene and cardiovascular risk in type 2 diabetic patients: a review of evidences. Arquivos Brasileiros de Endocrinologia e Metabologia. 2007;51(2):153–159. doi: 10.1590/s0004-27302007000200003. [DOI] [PubMed] [Google Scholar]
- 88.Ait-Oufella H, Taleb S, Mallat Z, et al. Recent advances on the role of cytokines in atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2011;31:969–979. doi: 10.1161/ATVBAHA.110.207415. [DOI] [PubMed] [Google Scholar]
- 89.Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. Journal of Clinical Investigation. 2005;115(5):1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.DeFronzo RA. Pathogenesis of type 2 diabetes mellitus. Medical Clinics of North America. 2004;88(4):787–835. doi: 10.1016/j.mcna.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 91.Poornima IG, Parikh P, Shannon RP. Diabetic cardiomyopathy: the search for a unifying hypothesis. Circulation Research. 2006;98(5):596–605. doi: 10.1161/01.RES.0000207406.94146.c2. [DOI] [PubMed] [Google Scholar]
- 92.Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. Journal of the American Medical Association. 2001;286(3):327–334. doi: 10.1001/jama.286.3.327. [DOI] [PubMed] [Google Scholar]
- 93.Chait A, Chang YH, Oram JF, Heinecke JW. Lipoprotein-associated inflammatory proteins: markers or mediators of cardiovascular disease? Journal of Lipid Research. 2005;46(3):389–403. doi: 10.1194/jlr.R400017-JLR200. [DOI] [PubMed] [Google Scholar]
- 94.Yamauchi T, Kamon J, Minokoshi Y, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nature Medicine. 2002;8:1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 95.Yang R, Barouch LA. Leptin signaling and obesity: cardiovascular consequences. Circulation Research. 2007;101(6):545–559. doi: 10.1161/CIRCRESAHA.107.156596. [DOI] [PubMed] [Google Scholar]
- 96.Basha B, Samuel SM, Triggle CR, et al. Endothelial dysfunction in diabetes mellitus: possible involvement of endoplasmic reticulum stress? Experimental Diabetes Research. 2012;2012:14 pages. doi: 10.1155/2012/481840.481840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Romano M, Pomilio M, Vigneri S, et al. Endothelial perturbation in children and adolescents with type 1 diabetes: association with markers of the inflammatory reaction. Diabetes Care. 2001;24(9):1674–1678. doi: 10.2337/diacare.24.9.1674. [DOI] [PubMed] [Google Scholar]
- 98.Hadi HAR, Al Suwaidi JA. Endothelial dysfunction in diabetes mellitus. Vascular Health and Risk Management. 2007;3(6):853–876. [PMC free article] [PubMed] [Google Scholar]
- 99.Yan J, Tie G, Park B, Yan Y, Nowicki PT, Messina LM. Recovery from hind limb ischemia is less effective in type 2 than in type 1 diabetic mice: roles of endothelial nitric oxide synthase and endothelial progenitor cells. Journal of Vascular Surgery. 2009;50(6):1412–1422. doi: 10.1016/j.jvs.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hazel L, Kenneth A, Roebuck O. Oxidant stress and endothelial cell dysfunction. American Journal of Physiology. 2001;280:C719–C741. doi: 10.1152/ajpcell.2001.280.4.C719. [DOI] [PubMed] [Google Scholar]
- 101.Versari D, Daghini E, Virdis A, Ghiadoni L, Taddei S. Endothelial dysfunction as a target for prevention of cardiovascular disease. Diabetes care. 2009;32:S314–321. doi: 10.2337/dc09-S330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Skyrme-Jones RAP, O’Brien RC, Luo M, Meredith IT. Endothelial vasodilator function is related to low-density lipoprotein particle size and low-density lipoprotein vitamin E content in type 1 diabetes. Journal of the American College of Cardiology. 2000;35(2):292–299. doi: 10.1016/s0735-1097(99)00547-1. [DOI] [PubMed] [Google Scholar]
- 103.Gomes MB, Matheus ASM, Tibiriçá E. Evaluation of microvascular endothelial function in patients with type 1 diabetes using laser-Doppler perfusion monitoring: which method to choose? Microvascular Research. 2008;76(2):132–133. doi: 10.1016/j.mvr.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 104.Tibiriçá E, Rodrigues E, Cobas RA, et al. Endothelial function in patients with type 1 diabetes evaluated by skin capillary recruitment. Microvascular Research. 2007;73:107–112. doi: 10.1016/j.mvr.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 105.Koitka A, Abraham P, Bouhanick B, et al. Impaired pressure-induced vasodilation at the foot in young adults with type 1 diabetes. Diabetes. 2005;53:721–725. doi: 10.2337/diabetes.53.3.721. [DOI] [PubMed] [Google Scholar]
- 106.Chan NN, Vallance P, Colhoun HM. Endothelium-dependent and -independent vascular dysfunction in type 1 diabetes: role of conventional risk factors, sex, and glycemic control. Arteriosclerosis, Thrombosis, and Vascular Biology. 2003;23(6):1048–1054. doi: 10.1161/01.ATV.0000072968.00157.6B. [DOI] [PubMed] [Google Scholar]
- 107.Schram MT, Chaturvedi N, Schalkwijk C, et al. Vascular risk factors and markers of endothelial function as determinants of inflammatory markers in type 1 diabetes: the EURODIAB prospective complications study. Diabetes Care. 2003;26(7):2165–2173. doi: 10.2337/diacare.26.7.2165. [DOI] [PubMed] [Google Scholar]
- 108.Khan F, Elhadd TA, Greene SA, Belch JJF. Impaired skin microvascular function in children, adolescents, and young adults with type I diabetes. Diabetes Care. 2000;23(2):215–220. doi: 10.2337/diacare.23.2.215. [DOI] [PubMed] [Google Scholar]
- 109.Gomes MB, Cobas RA, Nunes E, et al. Plasma PAF-acetylhydrolase activity, inflammatory markers and susceptibility of LDL to in vitro oxidation in patients with type 1 diabetes mellitus. Diabetes Research and Clinical Practice. 2009;85:61–68. doi: 10.1016/j.diabres.2009.04.016. [DOI] [PubMed] [Google Scholar]
- 110.Devaraj S, Glaser N, Griffen S, Wang-Polagruto J, Miguelino E, Jialal I. Increased monocytic activity and biomarkers of inflammation in patients with type 1 diabetes. Diabetes. 2006;55(3):774–779. doi: 10.2337/diabetes.55.03.06.db05-1417. [DOI] [PubMed] [Google Scholar]
- 111.Kolluru GK, Bir SC, Kevil CG. Endothelial dysfunction and diabetes: effects on angiogenesis, vascular remodeling and wound healing. International Journal of Vascular Medicine. 2012;2012:30 pages. doi: 10.1155/2012/918267.918267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Stalker TJ, Skvarka CB, Scalia R. A novel role for calpains in the endothelial dysfunction of hyperglycemia. The FASEB Journal. 2003;17(11):1511–1513. doi: 10.1096/fj.02-1213fje. [DOI] [PubMed] [Google Scholar]
- 113.Balletshofer BM, Rittig K, Enderle MD, et al. Endothelial dysfunction is detectable in young normotensive first-degree relatives of subjects with type 2 diabetes in association with insulin resistance. Circulation. 2000;101(15):1780–1784. doi: 10.1161/01.cir.101.15.1780. [DOI] [PubMed] [Google Scholar]
- 114.Rolim LC, Sá JR, Chacra AR, Dib SA. Diabetic cardiovascular autonomic neuropathy: risk factors, clinical impact and early diagnosis. Arquivos brasileiros de cardiologia. 2008;90(4):e24–31. doi: 10.1590/s0066-782x2008000400014. [DOI] [PubMed] [Google Scholar]
- 115.Vinik AI, Ziegler D. Diabetic cardiovascular autonomic neuropathy. Circulation. 2007;115(3):387–397. doi: 10.1161/CIRCULATIONAHA.106.634949. [DOI] [PubMed] [Google Scholar]
- 116.Vinik AI, Maser RE, Ziegler D. Autonomic imbalance: prophet of doom or scope for hope? Diabetic Medicine. 2011;28(6):643–651. doi: 10.1111/j.1464-5491.2010.03184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lampert R, Bremner JD, Su S, et al. Decreased heart rate variability is associated with higher levels of inflammation in middle-aged men. American Heart Journal. 2008;156(4):759–767. doi: 10.1016/j.ahj.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Prince CT, Secrest AM, Mackey RH, Arena VC, Kingsley LA, Orchard TJ. Cardiovascular autonomic neuropathy, HDL cholesterol, and smoking correlate with arterial stiffness markers determined 18 years later in type 1 diabetes. Diabetes Care. 2010;33(3):652–657. doi: 10.2337/dc09-1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Schmid H. Impacto cardiovascular da neuropatia autonômica do diabetes mellitus. Arquivos Brasileiros de Endocrinologia e Metabologia. 2007;51:232–243. doi: 10.1590/s0004-27302007000200012. [DOI] [PubMed] [Google Scholar]
- 120.Young LH, Wackers FJT, Chyun DA, et al. Cardiac outcomes after screening for asymptomatic coronary artery disease in patients with type 2 diabetes the DIAD study: a randomized controlled trial. Journal of the American Medical Association. 2009;301(15):1547–1555. doi: 10.1001/jama.2009.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Frans JTH, Wackers FJT, Young LH, et al. Detection of silent myocardial ischemia in asymptomatic diabetic subjects. Diabetes Care. 2004;27:1954–1961. doi: 10.2337/diacare.27.8.1954. [DOI] [PubMed] [Google Scholar]
- 122.Boden WE, O’Rourke RA, Teo KK, et al. Optimal medical therapy with or without PCI for stable coronary disease. The New England Journal of Medicine. 2007;356:1503–1516. doi: 10.1056/NEJMoa070829. [DOI] [PubMed] [Google Scholar]
- 123.Frye RL, August P, Brooks MM, et al. A randomized trial of therapies for type 2 diabetes and coronary artery disease. The New England Journal of Medicine. 2009;360:2503–2515. doi: 10.1056/NEJMoa0805796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bax JJ, Young LH, Frye RL, Bonow RO, Steinberg HO, Barrett EJ. Screening for coronary artery disease in patients with diabetes. Diabetes Care. 2007;30(10):2729–2736. doi: 10.2337/dc07-9927. [DOI] [PubMed] [Google Scholar]
- 125.Fox CS, Sullivan L, D’Agostino RB, Wilson PWF. The significant effect of diabetes duration on coronary heart disease mortality: the framingham heart study. Diabetes Care. 2004;27(3):704–708. doi: 10.2337/diacare.27.3.704. [DOI] [PubMed] [Google Scholar]
- 126.Raggi P, Callister TQ, Cooil B, et al. Identification of patients at increased risk of first unheralded acute myocardial infarction by electron-beam computed tomography. Circulation. 2000;101(8):850–855. doi: 10.1161/01.cir.101.8.850. [DOI] [PubMed] [Google Scholar]
- 127.Raggi P, Cooil B, Callister TQ. Use of electron beam tomography data to develop models for prediction of hard coronary events. American Heart Journal. 2001;141(3):375–382. doi: 10.1067/mhj.2001.113220. [DOI] [PubMed] [Google Scholar]
- 128.Jasinowodolinski D, Szarf G. Calcium score in the cardiovascular evaluation of the diabetic patient. Arquivos Brasileiros de Endocrinologia e Metabologia. 2007;51(2):294–298. doi: 10.1590/s0004-27302007000200019. [DOI] [PubMed] [Google Scholar]
- 129.Hoff JA, Chomka EV, Krainik AJ, Daviglus M, Rich S, Kondos GT. Age and gender distributions of coronary artery calcium detected by electron beam tomography in 35,246 adults. American Journal of Cardiology. 2001;87(12):1335–1339. doi: 10.1016/s0002-9149(01)01548-x. [DOI] [PubMed] [Google Scholar]
- 130.Janowitz WR, Agatston AS, Kaplan G, Viamonte M. Differences in prevalence and extent of coronary artery calcium detected by ultrafast computed tomography in asymptomatic men and women. American Journal of Cardiology. 1993;72(3):247–254. doi: 10.1016/0002-9149(93)90668-3. [DOI] [PubMed] [Google Scholar]
- 131.Malik S, Budoff MJ, Katz R, et al. Impact of subclinical atherosclerosis on cardiovascular disease events in individuals with metabolic syndrome and diabetes. The multi-etnic study of atherosclerosis. Diabetes Care. 2011;4:2285–2290. doi: 10.2337/dc11-0816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Greenland P, Alpert JS, Beller GA, et al. 2010 ACCF/AHA guideline for assessment of cardiovascular risk in asymptomatic adults. Journal of the American College of Cardiology. 2010;56(25):e50–e103. doi: 10.1016/j.jacc.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 133.Irie Y, Katakami N, Kaneto H, et al. Maximum carotid intima-media thickness improves the prediction ability of coronary artery stenosis in type 2 diabetic patients without history of coronary artery disease. Atherosclerosis. 2012;221(2):438–444. doi: 10.1016/j.atherosclerosis.2012.01.022. [DOI] [PubMed] [Google Scholar]
- 134.Polak JF, Backlund JYC, Cleary PA, et al. Progression of carotid artery intima-media thickness during 12 years in the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) study. Diabetes. 2011;60(2):607–613. doi: 10.2337/db10-0296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mohler ER. Peripheral arterial disease: identification and implications. Archives of Internal Medicine. 2003;163(19):2306–2314. doi: 10.1001/archinte.163.19.2306. [DOI] [PubMed] [Google Scholar]
- 136.Hirsch AT, Haskal ZJ, Hertzer NR, et al. ACC/AHA 2005 guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): executive summary a collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional. Journal of the American College of Cardiology. 2006;47(6):1239–1312. doi: 10.1016/j.jacc.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 137.Doza B, Kaur M, Chopra S, et al. Cardiovascular risk factors and distributions of the ankle brachial index among type 2 diabetes mellitus patients. International Journal of Hypertension. 2012;2012:6 pages. doi: 10.1155/2012/485812.485812 [DOI] [PMC free article] [PubMed] [Google Scholar]