

TODAY'S PROBLEMS COME FROM YESTERDAY'S SOLUTIONS
Systems thinking is a discipline for seeing wholes. It is a framework for seeing interrelationships rather than isolated things and events, for seeing dynamic patterns of change rather than static snapshots.1 Senge's The Fifth Discipline emphasizes how systems thinking can help one understand the causes of and provide effective treatments for learning disabilities in organizations. The 11 laws of The Fifth Discipline are also applicable in many ways to problems of health and disease treatment and are recommended for problem solvers in this domain.
The challenges we face today for disease treatment are much different from those of the past. We are seeing growing incidences of autoimmune diseases, cancer, and neurodegenerative disorders. The most immediate factor in considering our current challenges is that we are simply living longer. This fact makes logical sense when considering cancer and diseases such as Alzheimer disease, but autoimmune diseases affect mostly the young and middle aged. The problem with autoimmune diseases is that we don't yet fully understand the problem; our current understanding is that the immune system mistakenly attacks the very tissues it is intended to protect, but we don't really understand the cause (the why) or the process. Some of the common factors proposed as potential contributors to the rise in autoimmune diseases include a decline in gut flora health,2 leaky gut,3 overuse of vaccines3 and antibiotics,4 environmental toxins such as pesticides,5 infections,6 reduced vitamin D,7 and environmental estrogen.8 Quite possibly, many of these factors are contributors or parts of a hidden bigger picture, but without an overall understanding of the cause and process of autoimmune diseases, we will continue to struggle to understand the problem and progress with a real solution.
This paper is based on published peer-reviewed literature, starting with the puzzle of chronic inflammation in disease. In an effort to solve this puzzle, the paper revisits a hormone system with capabilities that far exceed established expectations and explains how this system is corrupted in cancer to drive malignant processes and suppress the immune system. The cancer paradigm contributes to a greater understanding of how cells interact with their microenvironment and reveals a potentially clearer understanding of not only cancer but also many other diseases.
The purpose of this paper is to promote the importance of new ways of looking at things. Some people gain understanding by reading text, but many people gain understanding through pictures. Both of these approaches help facilitate the formation of mental models.
MENTAL MODELS DETERMINE UNDERSTANDING
Change in itself is not necessarily good or bad, but it is inevitable. Everything changes. In any system, the interaction and relationship between the system components and its environment are of supreme importance. In a system, things change due to time and circumstance as a result of interrelationships. Our minds, too, are systems, and perceptions are the result of numerous mental models we have built through our collective experience. These mental models help us to understand and interact successfully with our environment. Changes in thinking occur as a result of new knowledge or experience that expands and improves our understanding of how and why things work. During the process, the resultant changes in our mental models increase our success in life.
In 1900, Lord Kelvin famously stated, “There is nothing new to be discovered in physics now. All that remains is more and more precise measurement.”
While the accumulation of knowledge benefits from a reductionist approach, understanding of the why is only gained by considering the behavior of the whole in the context of its interaction with its environment. Five years after Lord Kelvin's statement, Albert Einstein published his paper on special relativity that challenged the very simple set of rules laid down by Newtonian mechanics that had been used to describe force and motion for more than 200 years. What Einstein had done was to provide a new way of looking at established data, new mental models that have revolutionized our understanding of the universe and our ability to interact with it.
In 1970, T. S. Kuhn, in The Structure of Scientific Revolutions, argued that scientists work by creating a comprehensive paradigm. He stated that one of the first signs that a paradigm is shifting is the discovery of facts that seem significant and indisputably true but that cannot be explained by the current model.9
Discovery commences with the awareness of anomaly, i.e. with the recognition that nature has somehow violated the paradigm-induced expectations that govern normal science. It then continues with a more or less extended exploration of the area of anomaly. And it closes only when the paradigm theory has been adjusted so that the anomalous has become the expected. Assimilating a new sort of fact demands a more than additive adjustment of theory, and until that adjustment is completed—until the scientist has learned to see nature in a different way—the new fact is not quite a scientific fact at all.p.52
One such case is the puzzling role of inflammation: Inflammation is regarded as a key component of the immune system that ensures tissues of the body are free from invading organisms and pathogens. When an area is infected, it becomes red, swollen, hot, and painful. Another recognized function of inflammation is to support the healing process by removing cells that have been damaged through injury or by infection. In disease conditions, the immune system somehow malfunctions (develops an aversion to self). Instead of attacking invaders and destroying damaged tissue, inflammation starts to destroy healthy tissue, causing biological dysfunction, immune suppression,10-13 and ultimately death. Chronic inflammation is a critical feature of most diseases. Regardless of the underlying cause (infection, cancer, aging, autoimmunity), it is the chronic inflammation that ultimately damages the body.
When we try to understand inflammation and why inflammation can be immune suppressive, a useful mental model is to recognize that the biological manifestation of inflammation is a tissue state, and when tissue is inflamed, the involved white blood cells are participating in a wound, or tissue remodeling, response. While the immune and wound response systems share many of the same components (cells—in particular white blood cells) and many interface messengers (such as cytokines, chemokines, hormones, and other mediators), categorizing cells such as white blood cells simply as immune cells limits our ability to understand what is taking place because their function changes over time and depends on their environment. Many cells, including white blood cells, are better described as multifunctional. Much the same can be said for many of the messengers—such as interleukin (IL)-1 or tumor necrosis factor-alpha (TNF-α) (both considered strong proinflammatory mediators)—because their message and corresponding action depend on the state of the cells that receive the message. Simply put, an inflammatory response is not necessarily an immune response; indeed one key message from this paper is that inflammation (the tissue state) results in the suppression of adaptive immune responses.
Some new mental models presented here through words and pictures will challenge established mental models and inevitably conflict with the current ways of thinking. The old models are not bad; however, the new mental models may provide a better understanding of how and why biological systems do what they do. Through an improved understanding of the big picture, we create opportunities to work in harmony with these systems and better treat disease by addressing the causes and not just the symptoms. This is the power of systems thinking.
INTRODUCING THE CLASSIC RENIN-ANGIOTENSIN SYSTEM
In 1898, Tigerstedt and Bergman14 published their observation that kidney extracts produce pressor effects. They partially characterized the substance and named it renin. Although this observation began our understanding of the role of the kidney in hypertension and the renin-angiotensin system (RAS), their discovery lay dormant for nearly 40 years. Around 1936, 2 independent groups of researchers, using the Goldblatt technique to produce experimental hypertension, demonstrated renal secretion of a pressor agent similar to renin. They eventually concluded that renin acted enzymatically on a plasma protein to produce a new substance: angiotensin.15 The current widely recognized view is that the main effector of the RAS (Figure 1) is angiotensin II (AngII). AngII is generated from the precursor protein angiotensinogen by the actions of renin, angiotensin-converting enzyme (ACE), chymases, and various carboxy- and amino-peptidases. AngII exerts its pressor effects on the cardiovascular and renal systems via interactions with its 2 receptor molecules, AngII type 1 receptor (AT1R) and AngII type 2 receptor (AT2R). The receptors are viewed as mutually antagonistic in the control of blood pressure, water, and electrolyte homeostasis.
Figure 1.
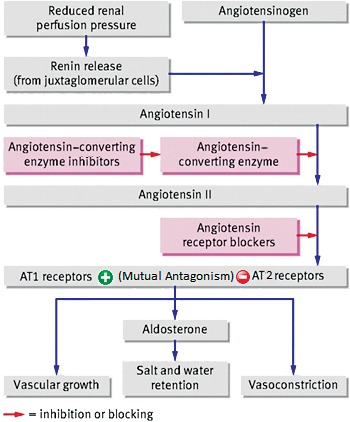
The classic renin-angiotensin system. AT1, angiotensin II type 1; AT2, angiotensin II type 2.
The angiotensin system is currently targeted primarily as a means of controlling blood pressure. The main drug targets are ACE inhibitors (ACEIs) or AT1R blockers (ARBs).16
REVEALING THE TRUE NATURE OF THE ELEPHANT
A contemporary model of the RAS is well described in a 2010 review of the emerging role of the RAS in cancer.17 This review also summarizes several aspects and new components of the system. The established view of the RAS being governed by the brain and the kidney is systemically being extended by the knowledge that many, if not all, components of the RAS can be produced as required by resident or infiltrating cells, such as mast cells, as a consequence of infection or injury. Also of great importance, an alternative pathway for angiotensin has been found whereby a new enzyme, ACE2, liberates angiotensin 1-7 (Ang1-7), which acts on the Mas receptor. Although the Mas receptor was originally identified in 1986 through the Mas protooncogene18 (protooncogenes code for proteins that help regulate cell growth and differentiation), only in 2007 did the ACE2/Ang1-7/Mas pathway come under consideration as a target outside of blood pressure control.19
Could the 21 years that separate these 2 papers indicate how difficult moving from one mental model to another can be? This situation may be an example of how inertia regarding paradigm shifts constrains understanding and progress. Resistance to change in mindsets is a key point; not only is the researcher's ability to make the initial mental jump important, but the mindsets of the people who make decisions about funding also play a vital role.
When the RAS was first studied and its potent blood control effects were discovered, a mental model was established that the RAS controlled blood pressure with complementary effects in controlling thirst, salt retention, and fluid homeostasis. As additional components of the RAS are identified and characterized with properties including inflammation, immune modulation, tissue development, and tissue repair, this model is being challenged. In the fable of the blind men who came across an elephant, each of them had a different perception of what the elephant was (Figure 2).
Figure 2.
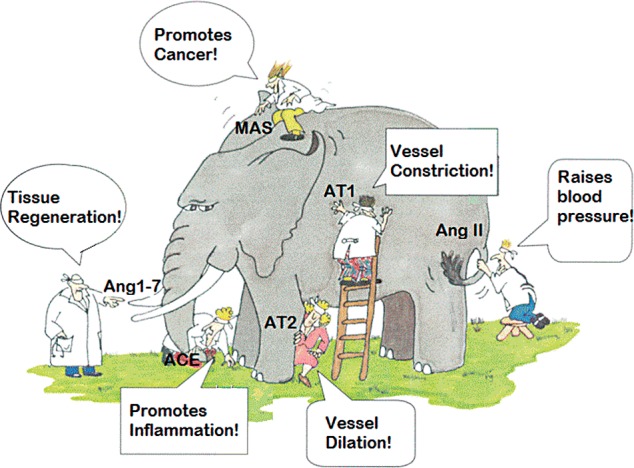
The elephant of understanding. (Adapted with permission from Himmelfarb et al.20) ACE, angiotensin-converting enzyme; Ang1-7, angiotensin 1-7; AngII, angiotensin II; AT1, angiotensin II type 1; AT2, angiotensin II type 2.
In the case of the RAS, when researchers first came across the system (and grasped the tail of the elephant), they concluded that it controlled blood pressure. Our continued exploration of the RAS is revealing new biologically critical, yet puzzling facets, but they are only puzzling when not placed in the context of the big picture. Through a synthesis of newly established knowledge, the emergent picture (mental model) is that in an established animal, the RAS controls and manages the body's response to injury and wounding.21 When looking overall at the big picture, we can see that the properties of the RAS in the control of blood pressure, salt retention, and fluid homeostasis are simply important aspects of this wider function (Figure 3). The implications of this new understanding could revolutionize our approach to treating many chronic inflammatory diseases, including cancers, autoimmune diseases, neurodegenerative disorders, and infections.
Figure 3.

Management of the wound response. ACE, angiotensin-converting enzyme; Ang1-7, angiotensin 1-7; AngII, angiotensin II; AT1, angiotensin II type 1; AT2, angiotensin II type 2.
The danger is that if we cannot adapt our thinking, then we will be slow, or worse still, will fail to take advantage of our new knowledge and understanding.
YES, THE RAS REALLY DOES HAVE A KEY ROLE IN HEALING
From a research perspective, the role of the RAS in healing has until recently perhaps been considered a peculiarity. However, with the discovery of the ACE2 enzyme and Ang1-7, commercial interest has increased and the role of the RAS in wound healing is becoming a hot topic. Several papers have now been published in the area of RAS and wound healing.
Inhibition of ACE has been found to promote healing in mouse models of bone fracture.22,23 Garcia et al23 found that ACE inhibition increased AT2 presentation. Meanwhile, an analog of Ang1-7 has been found to facilitate enhanced healing in diabetic wounds.24 A review of aging and cardiac fibrosis highlighted the important role of AngII and AT1 in mediating fibrotic remodeling of the aging heart.25
A recent study observed the change in the local level of AngII and the expression of AT1R and AT2R during wound healing. The results support the hypothesis that AngII participates in wound repair and remodeling in the late stage of wound healing through changes in the production of AngII and expression of AT1R and AT2R.26
In an investigation of the role of AngII receptor subtypes in subconjunctival injury in mice, knockouts of AT1a and AT2 were found to have contrasting effects in collagen deposition, cell infiltration, and expression of collagen and tissue inhibitor of metalloproteinase-1 (a mediator that in general promotes tissue formation).27 Similarly, another AT1a knockout model found reduced wound-induced angiogenesis and wound healing.28 Results in these models support similar findings in human skin tissue,29 in which immunohistochemically stained skin sections showed a stronger expression of AT2R than of AT1R within the area of scarring. Enhanced AT2R expression was detectable as early as 24 hours after injury and lasted for up to 3 months.
A rabbit model of deep alkaline-induced corneal burn injury found a significant increase in ACE activity in the tear and internal ocular tissue structures, promoting microcirculatory disorders and tissue inflammation. The local use of ACEIs as instillations substantially reduced inflammation and the incidence of deep and extensive corneal ulcers.30
Researchers modulating the response to local and systemic AngII in acutely injured skeletal muscle concluded that the clinical implications for the application of ARBs are potentially far reaching and include not only sports- and military-related injuries, but also diseases such as muscular dystrophy and trauma- and surgery-related injuries.31
A search on PubMed in March 2012 showed 260 papers from a search of “angiotensin” and “healing” and 522 papers from a search of “angiotensin” and “repair.” These results include multiple organs and diverse types of insults, including infections, physical and chemical injuries, and radiation. Collectively, these published studies suggest that interest in the role of the RAS in injury responses and healing is growing.
A summary of search results for “angiotensin” and “disease” in PubMed from March 2012 showed that the RAS has been noted as a participant in a great variety of diseases (Figure 4) and not simply as a participant in blood pressure control. When considering the overall picture, a central role for the RAS in most, if not all, chronic inflammatory diseases becomes evident. Readers are encouraged to explore this literature.
Figure 4.

Angiotensin and disease. AIDS, acquired immunodeficiency syndrome; CKD, chronic kidney disease; COPD, chronic obstructive pulmonary disease; SARS, severe acute respiratory syndrome.
FROM CELL STRESS TO WOUND RESPONSE
If the RAS plays a profound role in injury responses and wound healing, then we would expect to find that the RAS and its pathways are activated through sensing mechanisms that monitor damage or stress conditions. A review of the literature confirms this fact, with a locally derived RAS reported as active in a variety of disease conditions across many organs.32-39 A PubMed search in October 2012 found 4,684 papers in a search of “angiotensin” and “stress,” compared with 3,120 papers in a similar search of “stress” and “heme oxygenase,” a ubiquitous and well-documented stress-responsive protein.
In injury models, skeletal muscle myoblasts have been reported to possess a stretch-responsive local angiotensin signaling system, in which angiotensinogen was transcriptionally expressed and ACE, AngII, AT1, and AT2 were all locally evidenced.40 Renin transcripts were never detected. However, messenger RNA for the renin-like enzyme cathepsin D was observed, and AngI and AngII were identified in cell culture supernatants from proliferating myoblasts. The activation of a locally generated RAS has also been reported in hyperoxia (excess oxygen or higher than normal partial pressure of oxygen)-induced lung fibrosis in neonatal rats. In this study, RAS components including angiotensinogen, ACE, AngII, and the AT1 receptor were significantly upregulated by hyperoxia.41
AT1R expression would appear to be a fundamental reaction by all cells to tissue stress and injury (Figure 5), glycemic stress,42 hypoxia,43 shear stress,44-46 and oxidative stress via oxidized low-density lipoprotein acting on the LOX-1 receptor.47-50 These reactions all lead to the transcription and presentation of the AT1R on cells. A spectrum of important molecules involved in the cellular response to stress is induced by AT1R.21 These molecules include highly proinflammatory mediators such as IL-1β, TNF-α, IL-6, and cyclooxygenase-2 (COX-2), as well as other agents that promote the influx and migration of white blood cells (such as intercellular adhesion molecule 1 and vascular cell adhesion molecule 1), the growth of new blood vessels (notably through vascular endothelial growth factor expression and signaling), and tissue remodeling (notably through matrix metalloproteinases and transforming growth factor-beta [TGF-β]).
Figure 5.

Cells respond to stress through angiotensin II type 1 upregulation. COX-2, cyclooxygenase-2; IL-6, interkeukin-6; LDL, low-density lipoprotein; MCP-1, monocyte chemotactic protein-1; OPN, osteopontin; TNF-α, tumor necrosis factor alpha; VCAM-1, vascular cell adhesion protein; VEGF, vascular endothelial growth factor.
A key question is what is the purpose of the activity promoted by the RAS through the AT1 receptor? If cells are under stress and trigger changes in the environment of the cell, then the answer would seem to be a coordination of effort to remove or relieve the cause of the stress. In a healthy response to a wound, the damaged tissue is remodeled, dead and dying cells are cleared, a fibrous framework is laid down as the platform for tissue regeneration, and new blood vessels grow to support the new tissue. In disease conditions, the stress is not removed, the response continues unabated, and healthy tissue becomes subject to remodeling.
EXPLAINING THE ROLE OF INFLAMMATION IN CANCER
“The Hallmarks of Cancer,” the authoritative work by Hanahan and Weinberg,51 described a new systems approach that analyzed the evolved capabilities necessary for cancer cells to become life-threatening tumors. Figure 6, adapted from this work, highlights some of the common defects in growth, antigrowth, and death controls that are necessary for normal cells to become cancerous and for tumors to form.
Figure 6.
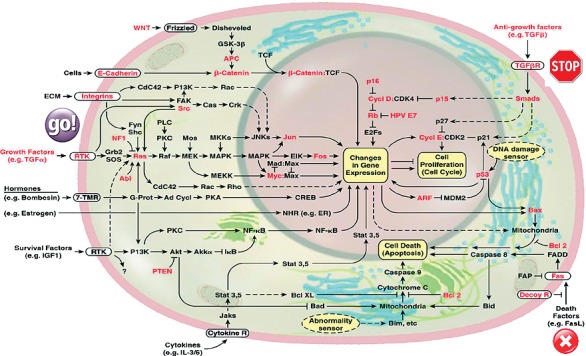
Cancers arise when growth and death controls fail. (Adapted with permission from Hanahan and Weinberg.51)
The key control signals that ensure that normal cells are maintained in relatively stable equilibrium (homeostasis) with their environment are presented in this cell circuitry. Figure 6 highlights some of the common genetic changes that must occur for cells to circumvent these controls and for carcinogenesis to happen. Inflammation pathways, such as radical oxygen species, overlap significantly with cancer circuits and increase the risk of genetic damage in targets that contribute to cancer initiation and progression. Inflammation from infection, injury, and stress or aggravators such as smoking or asbestos have been shown to induce cancers.52-56 What is more surprising, however, is the emerging critical role of inflammation in the progression of the disease to malignancy.57-61 As cancer cells evolve to ignore programmed cell death, the uncontrolled proliferation causes a microenvironmental stress that provokes an inflammatory response.62-64 The resultant influx of inflammatory cells62 and immune cells63 into the microenvironment is ineffective against the cancer because those cells have developed resistance to death signals. Instead, the inflammation exerts lethal effects (apoptosis and phagocytosis) on healthy tissue and promotes many essential environmental support processes (angiogenesis, cell motility, and immune suppression) necessary for the cancer to flourish and disseminate.64,65
This process is reflected in a direct relationship between systemic and chronic inflammation and patient mortality.66-68 Indeed, researchers have proposed that the reversal of these processes through microenvironment improvement could provide a new therapeutic approach.57,69
The effects of unresolved stress and activation of the local RAS are perhaps most clearly observed in cancer (Figure 7). While the origin of cancer involves unrestrained growth in the cancerous cells, the stress of this unrestrained growth on all of the cells—both cancerous and healthy—drives malignant processes through the AT1R.57
Figure 7.

Growth-induced microenvironmental stress drives malignancy in cancer. Expression of angiotensin II type 1 has been found to be associated with the aggressiveness of cancers and with decreased survival in patients.
As previously reviewed,21 knockout and other models demonstrate that AT1 expressed not only on tumor cells but also by the host stroma and on infiltrating white blood cells contributes to malignant processes. In knockout models, for instance, manipulation of various aspects of the angiotensin system can have significant consequences on cancer progression during tumorigenesis and malignancy.70-75 Additionally, what may appear to be conflicting influences are actually in keeping with the 2-phase view of the lifecycle of cancer. AT2 knockouts inhibit tumorigenesis but promote malignancy, while AT1 knockouts promote tumorigenesis (in cells that have already acquired prerequisite mutations) but inhibit malignancy.
ANGIOTENSIN MEDIATORS ARE CLINICALLY USEFUL IN CANCER
The previously reviewed promising prospects for the clinical use of ARBs and ACEIs in cancer prevention and treatment21 were challenged recently by a much-publicized metaanalysis of randomized controlled trials warning that ARBs were associated with a modestly increased risk of new cancer diagnosis.76 This publication resulted in a wave of press headlines—“ARBs Cause Cancer!” and “ARBs Increase the Risk of Cancer!”—and resulted in several commentaries and follow-up publications. Many of these papers advised caution in interpretation of the results.77-84 One solid finding from the metaanalysis was a small increase in the risk of cancer with an ARB/ACEI combination, but no increase in death.85 Many studies concluded that ARBs carry no significant risk of worsening cancer incidence or progression,86-93 and one highlighted a reduced risk of cancer with ARB use.94
What is interesting from a systems perspective is that all of these studies could be right, with the findings dependent on the circumstances and timing of the observations. From an understanding of the mechanistic perspective detailed so far in this paper, long-term (5+ years) ARB and ACEI use will progressively reduce cancer risk95 by reducing oxidative stress and genetic damage. Over fewer years, the drugs might slightly increase the risk, particularly when an ARB/ACEI combination is used in individuals such as smokers who already have precancerous nodules. The reason why ARBs and ACEIs have this effect is that they will push the RAS towards ACE2, Ang1-7, and Mas activation (the right-hand pathway in Figure 3) that will promote regenerative/growth processes and intracellular pathways that could facilitate the cell growth cycle. Mas activation by Ang1-7, for instance, might inhibit the downstream pathways of TGF-β receptor activators, such as the protein family that mediates intracellular TGF-β signaling (Smad), thus overriding TGF-β's growth inhibition effects.96,97 Note the importance of TGF-β and Smad pathways in Figure 6.
While understanding the circumstances of cancer risk is important, the effect of ARBs/ACEIs (or indeed Mas agonists98) from a mechanistic perspective should be of great significance in patient survival because these drugs would potentially block chronic inflammatory/tissue remodeling processes in the cancer microenvironment and thus reduce malignant processes. ARBs and ACEIs have many positive effects in advanced cancer, including increased survival,99-109 improved performance status,109 reduced reoccurrence,110-113 and reduced toxicity when used in conjunction with radiotherapy114,115 or chemotherapy.99,108,116 Additionally, synergistic benefits have been found when ARBs/ACEIs are used with surgery102,106,111,113 as well as with several new and existing agents under evaluation.101,104,105,109,110,112,114 Thus, it would be a great pity if the prospects for the use of angiotensin mediators as a therapy in cancer were dismissed as a result of a lack of system understanding.
CELL RESPONSE IN HEALTH AND DISEASE
The understanding that cells under stress promote a response intended to alleviate the stress led to the hypothesis that an overarching model taking into account other fundamental responses might be useful* in resolving puzzles such as inflammation.21 Smith and Missailidis21 introduced a mental model that provides an explanation of how cells interact with their tissue microenvironment (Figure 8). The model is based on 3 directional vectors that reflect 3 postulated adaptive cellular responses to injury, growth, and antigens. In addition, at the model center is an innate response that represents a very basic response to cell threat that evolved during the most ancient of times in single-cell organisms.
Figure 8.

The cell response model. AT1, angiotensin II type 1; COPD, chronic obstructive pulmonary disease; GFR, growth factor receptor; IGF-1, insulin-like growth factor; IL-2, interleukin-2; IL-10, interleukin-10; RAR, retinoic acid receptor.
The components of this innate response are hard-wired in the cell circuitry and not subject to tailoring or adaption through experience. This innate response forms the basic foundation for dealing with potential invaders and stress such as occurs with bacteriophages in bacteria.117 For multicellular organisms, these bacteriophages include the toll-like receptor and major histocompatibility complex recognition pathways.
In the cell response model, 3 adaptive responses are envisaged that provide specialist means whereby cells can respond to changes in their environment. Each of these responses involves a pressure, or force, that drives the response; an accelerator; and a brake for the process. Candidate drivers, accelerators, and brakes have been selected based on literature reviews using Occam's razor (ie, use the simplest model to explain a phenomenon) and a qualitative most-true-associations approach.
A basic premise for this model is that all cells have multifunctional capabilities. This fact is perhaps most strongly observed in leukocytes but is also applicable to other cells that are often perceived as more fixed in their activities, such as epithelial cells, nerve cells, etc. The model predicts that although cells are indeed multifunctional, their degree of focus to a particular response is determined by their environment. In the cell response model, “He who shouts loudest gets the most attention.”
Wound response has been the main subject of this paper and provoked the systems thinking that led to the cell response model. The pressure or force behind the wound response is cellular stress, but the governance of the response is provided by the RAS. Analysis of the literature indicates that the AT1 receptor appears primary to the acceleration of this response, whereas the Mas receptor acts as the brake.
In a normal healthy response to injury, tissue stress gradually leads to increased expression of AT1R. Then, as dead or dying tissue is cleared away and the new fiber is laid down, AT2R and then Mas receptor expression increase. Subsequently, over a period of weeks to a month, the activities of AT1R decline, tissue regeneration takes place, and AT2/Mas receptors are more gradually withdrawn.26,29 In disease conditions (a chronic wound response), the source of the cell stress is not resolved as a result of the wound response. Instead, tissue remodeling continues unabated through the dominant action of AngII on AT1R and is manifest as chronic inflammation of the tissue.
The second of the adaptive responses is the growth response. The pressure behind this response is provided by insulin-like growth factor-1 (IGF-1), which is mainly secreted by the liver as a result of stimulation by growth hormones. Importantly, IGF-1 is also expressed locally during wound healing and tissue regeneration.118 The accelerator for the growth response is tissue-specific growth factor receptors (glucocorticoid receptors and hormone receptors such as those for testosterone and estrogen) that steer the growth and replication of desired cells. A literature review suggests that the candidate brake to the growth response is the retinoic acid receptor. Teboul et al119 provide an interesting review of the role of nuclear hormone receptors, including key roles for glucocorticoid and retinoic acid receptors in the governance of the internal self-sustained circadian clocks present in virtually all cells. In a healthy growth response, the presentation of these receptors controls cell replication, but in disease conditions these processes fail and lead to uncontrolled proliferation.120,121
The adaptive immune response is driven by antigen presentation. This process has also been well described.122 The host cells express self-antigens. These antigens are different from those on the surface of bacteria or on the surface of virally infected host cells or cancer cells.
With the exception of nonnucleated cells (including erythrocytes), all cells are capable of presenting antigens and of activating the adaptive response. Some cells are specially equipped to present antigens and to prime naïve T cells. Dendritic cells, B cells, and to a lesser extent macrophages are equipped with special immunostimulatory receptors that allow for enhanced activation of T cells and are termed professional antigen-presenting cells (APC).
A key step in the adaptive immune response is the conditioning or maturing of the APC so it develops the ability to train T cells to recognize antigens in the lymph nodes. Several T-cell subgroups can be activated by professional APCs, and each type of T cell is specially equipped to deal with each unique toxin or bacterial and viral pathogen. The type of T cell activated and the type of response generated depend, in part, on the context in which the APC first encounters the antigen. Many cytokines have wide-ranging properties that steer either the early (innate) or the adaptive (antigen-derived) response. Candidate accelerators for the adaptive response included IL-4, interferon-alpha, IL-17, and IL-12, but these accelerators were specific for particular cell types. Because of its overarching presence in supporting immune cell population and function, whether inflammatory or regulatory,123 IL-2 was chosen as the accelerator. IL-10 was selected as the brake because it has an equally overarching suppressive role.
EXPLANATION, PREDICTION, AND GOALS
Established thinking has confined the RAS to the control of blood pressure, but through a process of experimentation and analysis of existing data we are discovering that the RAS is involved in cancer, chronic inflammatory diseases, autoimmune diseases, allergic diseases, and neurodegenerative disorders. Our existing mental models cannot accommodate this behavior of the RAS, nor the fact that inflammation is immunosuppressive. The cell response model explains that this otherwise puzzling behavior is caused by a failure mode of the wound response and unresolved tissue stress. Furthermore, the model predicts that this stress is caused by invaders such as cancer cells and infectious agents with the deliberate intent to avoid adaptive immune responses.
One cannot help but ask why such a chink in our biological defense exists. The logical answer would seem to be that in a healthy wound response, adaptive immunity is designed to be suppressed so when dead and dying tissue is phagocytized, the development of self-antigens is suppressed to avoid genuine autoimmune reactions. The model further predicts infection as a causative factor in autoimmune diseases and suggests that we should both look for and be more receptive to the wealth of evidence showing that dealing with the underlying ongoing chronic infection, rather than merely dealing with the symptoms, will lead to successful disease treatment.
Going beyond the ability of the cell response model to explain unexpected behavior and to make predictions about disease etiology, perhaps the model will become most useful in its ability to predict synergistic combinational approaches to disease treatment. Cancer is a complex disease; multiple processes at the cellular, tissue, organ, and super-system levels are at play, all changing over time. Opportunistic infections also lead to complications because strong growth and wound responses will further suppress the adaptive immune system and subject the patient to further attack and degradation. Defeating cancer will require more than just one silver bullet. Even if we identify intracellular approaches to overcoming one fault in the cell circuitry, the cancer cells will ultimately mutate to find an alternative pathway that will again allow the cancer cells to replicate uncontrollably. The cure for cancer will inevitably be a combinational approach that will necessitate restoring the body's ability to fight the cancer. The cell response model predicts that a combinational approach using ACEIs/ARBs or Mas agonists to suppress the wound response with retinoic acid agonists or hormone blockers in conjunction with immune stimulants such as low-dose IL-2 or dendritic cell vaccines could be synergistically effective.105 Finally, we must remember that we as living systems interact with our environment and with the microorganisms that both live within us and also sometimes incidentally prey on us. Changes to our environment and to these microorganisms have had unexpected effects in the past and will continue to do so until we are fully able to comprehend the big picture.
SYSTEMS THINKING AND ANGIOTENSIN
Established thinking has confined the RAS to the control of blood pressure, but through a process of synthesis of existing data we are on the cusp of recognizing its fuller role in maintaining organism, organ, and tissue integrity. One might say, “So what?” but this question is good to ask. The answer is that the fuller nature of the RAS is architecturally significant to the whole system; in other words, the RAS greatly influences the behavior of the whole. With a better understanding of the whole and of key effects such as inflammation and immune suppression, we significantly reduce the risk of unexpected and unwelcome side effects that occur because of interventions that do not take into account the systems context. Also with a better understanding of the whole, we significantly increase the probability of realizing the expected benefits of interventions by addressing the root causes of problems and not just the symptoms. Unexpected benefits and unexpected risks and problems are daily occurrences in the medical news, but these issues are only unexpected because we do not yet have a full understanding of the whole. In understanding the role of the RAS in its response to unrestrained stress in chronic inflammatory disease, we can be better prepared to embrace and explore the potential benefits of RAS manipulation, especially in synergy with other agents. Some might wonder if this review represents speculation, but when does speculation transition to established wisdom? One answer is when a consensus of opinion is established. From the perspective of the author and a growing number of peers in the angiotensin domain, many features of this paper are already established. If this new perspective is correct, then we need to hurry up and capitalize on it. Angiotensin is not just about blood pressure.
ACKNOWLEDGMENTS
For my dad Ian Brown Smith, who didn't have time for the new ways of thinking to become established. Also with thanks to my good friend Paul Jaep, who will also be greatly missed.
Footnotes
* There is a saying among modelers that all mental models are inherently wrong because they can only be visualizations of reality, but that some are useful.
REFERENCES
- 1.Senge PM. The Fifth Discipline: The Art and Practice of the Learning Organization. New York, NY: Doubleday/Currency;; 1990. [Google Scholar]
- 2.Rook GA. Hygiene hypothesis and autoimmune diseases. Clin Rev Allergy Immunol. 2012 Feb;42(1):5–15. doi: 10.1007/s12016-011-8285-8. [DOI] [PubMed] [Google Scholar]
- 3.Agmon-Levin N, Paz Z, Israeli E, Shoenfeld Y. Vaccines and autoimmunity. Nat Rev Rheumatol. 2009 Nov;5(11):648–652. doi: 10.1038/nrrheum.2009.196. [DOI] [PubMed] [Google Scholar]
- 4.Greer JB, O'Keefe SJ. Microbial induction of immunity, inflammation, and cancer. Front Physiol. 2011 Jan 26;1:168. doi: 10.3389/fphys.2010.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Corsini E, Liesivuori J, Vergieva T, Van Loveren H, Colosio C. Effects of pesticide exposure on the human immune system. Hum Exp Toxicol. 2008 Sep;27(9):671–680. doi: 10.1177/0960327108094509. [DOI] [PubMed] [Google Scholar]
- 6.Kivity S, Agmon-Levin N, Blank M, Shoenfeld Y. Infections and autoimmunity—friends or foes? Trends Immunol. 2009 Aug;30(8):409–414. doi: 10.1016/j.it.2009.05.005. Epub 2009 Jul 28. [DOI] [PubMed] [Google Scholar]
- 7.Hewison M. Vitamin D and innate and adaptive immunity. Vitam Horm. 2011;86:23–62. doi: 10.1016/B978-0-12-386960-9.00002-2. [DOI] [PubMed] [Google Scholar]
- 8.Chighizola C, Meroni PL. The role of environmental estrogens and autoimmunity. Autoimmun Rev. 2012 May;11(6-7):A493–A501. doi: 10.1016/j.autrev.2011.11.027. Epub 2011 Dec 4. [DOI] [PubMed] [Google Scholar]
- 9.Kuhn TS. The Structure of Scientific Revolutions. Vol. 52 Chicago, IL: University of Chicago Press;; 1962. [Google Scholar]
- 10.Ben-Baruch A. Inflammation-associated immune suppression in cancer: the roles played by cytokines, chemokines and additional mediators. Semin Cancer Biol. 2006 Feb;16(1):38–52. doi: 10.1016/j.semcancer.2005.07.006. Epub 2005 Aug 31. [DOI] [PubMed] [Google Scholar]
- 11.Sheng KC, Wright MD, Apostolopoulos V. Inflammatory mediators hold the key to dendritic cell suppression and tumor progression. Curr Med Chem. 2011;18(36):5507–5518. doi: 10.2174/092986711798347207. [DOI] [PubMed] [Google Scholar]
- 12.Gentile LF, Cuenca AG, Efron PA, et al. Persistent inflammation and immunosuppression: a common syndrome and new horizon for surgical intensive care. J Trauma Acute Care Surg. 2012 Jun;72(6):1491–1501. doi: 10.1097/TA.0b013e318256e000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kanterman J, Sade-Feldman M, Baniyash M. New insights into chronic inflammation-induced immunosuppression. Semin Cancer Biol. 2012 Aug;22(4):307–318. doi: 10.1016/j.semcancer.2012.02.008. Epub 2012 Feb 24. [DOI] [PubMed] [Google Scholar]
- 14.Phillips MI, Schmidt-Ott KM. The discovery of renin 100 years ago. News Physiol Sci. 1999 Dec;14:271–274. doi: 10.1152/physiologyonline.1999.14.6.271. [DOI] [PubMed] [Google Scholar]
- 15.Basso N, Terragno NA. History about the discovery of the renin-angiotensin system. Hypertension. 2001 Dec 1;38(6):1246–1249. doi: 10.1161/hy1201.101214. [DOI] [PubMed] [Google Scholar]
- 16.Ritter JM. Angiotensin converting enzyme inhibitors and angiotensin receptor blockers in hypertension. BMJ. 2011 doi: 10.1136/bmj.d1673. Apr 7;342:d1673. [DOI] [PubMed] [Google Scholar]
- 17.George AJ, Thomas WG, Hannan RD. The renin-angiotensin system and cancer: old dog, new tricks. Nat Rev Cancer. 2010 Nov;10(11):745–759. doi: 10.1038/nrc2945. Epub 2010 Oct 22. [DOI] [PubMed] [Google Scholar]
- 18.Young D, Waitches G, Birchmeier C, Fasano O, Wigler M. Isolation and characterization of a new cellular oncogene encoding a protein with multiple potential transmembrane domains. Cell. 1986 Jun 6;45(5):711–719. doi: 10.1016/0092-8674(86)90785-3. [DOI] [PubMed] [Google Scholar]
- 19.Chappell MC. Emerging evidence for a functional angiotensin-converting enzyme 2-angiotensin-(1-7)-MAS receptor axis: more than regulation of blood pressure? Hypertension. 2007 Oct;50(4):596–599. doi: 10.1161/HYPERTENSIONAHA.106.076216. Epub 2007 Sep 4. [DOI] [PubMed] [Google Scholar]
- 20.Himmelfarb J, Stenvinkel P, Ikizler TA, Hakim RM. The elephant in uremia: oxidant stress as a unifying concept of cardiovascular disease in uremia. Kidney Int. 2002 Nov;62(5):1524–1538. doi: 10.1046/j.1523-1755.2002.00600.x. [DOI] [PubMed] [Google Scholar]
- 21.Smith GR, Missailidis S. Learning from cancer: The adaptive growth, wound and immune responses. Gene Ther Mol Biol. 2009 Jun;13:158–185. [Google Scholar]
- 22.Zhao X, Wu ZX, Zhang Y, et al. Locally administrated perindopril improves healing in an ovariectomized rat tibial osteotomy model. PLoS One. 2012;7(3) doi: 10.1371/journal.pone.0033228. e33228. Epub 2012 Mar 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garcia P, Schwenzer S, Slotta JE, et al. Inhibition of angiotensin-converting enzyme stimulates fracture healing and periosteal callus formation - role of a local renin-angiotensin system. Br J Pharmacol. 2010 Apr;159(8):1672–1680. doi: 10.1111/j.1476-5381.2010.00651.x. Epub 2010 Mar 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodgers K, Verco S, Bolton L, Dizerega G. Accelerated healing of diabetic wounds by NorLeu(3)-angiotensin (1-7) Expert Opin Investig Drugs. 2011 Nov;20(11):1575–1581. doi: 10.1517/13543784.2011.619976. Epub 2011 Oct 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biernacka A, Frangogiannis NG. Aging and cardiac fibrosis. Aging Dis. 2011 Apr;2(2):158–173. [PMC free article] [PubMed] [Google Scholar]
- 26.Wu HJ, Liu HW, Cheng B, et al. [The change of angiotensin II production and its receptor expression during wound healing: possible role of angiotensin II in wound healing] Zhonghua Zheng Xing Wai Ke Za Zhi. 2011 Mar;27(2):124–128. Chinese. [PubMed] [Google Scholar]
- 27.Mizoue S, Iwai M, Ide A, et al. Role of angiotensin II receptor subtypes in conjunctival wound healing. Curr Eye Res. 2006 Feb;31(2):129–136. doi: 10.1080/02713680500507200. [DOI] [PubMed] [Google Scholar]
- 28.Kurosaka M, Suzuki T, Hosono K, et al. Reduced angiogenesis and delay in wound healing in angiotensin II type 1a receptor-deficient mice. Biomed Pharmacother. 2009 Nov;63(9):627–634. doi: 10.1016/j.biopha.2009.01.001. Epub 2009 Feb 14. [DOI] [PubMed] [Google Scholar]
- 29.Steckelings UM, Henz BM, Wiehstutz S, Unger T, Artuc M. Differential expression of angiotensin receptors in human cutaneous wound healing. Br J Dermatol. 2005 Nov;153(5):887–893. doi: 10.1111/j.1365-2133.2005.06806.x. [DOI] [PubMed] [Google Scholar]
- 30.Chesnokova NB, Kost OA. Nikol'skaia II, et al. [Mental rationale for local use of angiotensin-converting enzyme in the treatment of inflammatory processes in the eye] Vestn Oftalmol. 2008 Mar-Apr;124(2):16–19. Russian. [PubMed] [Google Scholar]
- 31.Bedair HS, Karthikeyan T, Quintero A, Li Y, Huard J. Angiotensin II receptor blockade administered after injury improves muscle regeneration and decreases fibrosis in normal skeletal muscle. Am J Sports Med. 2008 Aug;36(8):1548–1554. doi: 10.1177/0363546508315470. Epub 2008 Jun 11. Erratum in: Am J Sports Med. 2008 Dec;36(12):2465. [DOI] [PubMed] [Google Scholar]
- 32.Garrido-Gil P, Valenzuela R, Villar-Cheda B, Lanciego JL, Labandeira-Garcia JL. Expression of angiotensinogen and receptors for angiotensin and prorenin in the monkey and human substantia nigra: an intracellular renin-angiotensin system in the nigra. Brain Struct Funct. 2012 Mar 11; doi: 10.1007/s00429-012-0402-9. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thethi T, Kamiyama M, Kobori H. The link between the renin-angiotensin-aldosterone system and renal injury in obesity and the metabolic syndrome. Curr Hypertens Rep. 2012 Apr;14(2):160–169. doi: 10.1007/s11906-012-0245-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neo JH, Ager EI, Angus PW, et al. Changes in the renin angiotensin system during the development of colorectal cancer liver metastases. BMC Cancer. 2010 Apr 10;10:134. doi: 10.1186/1471-2407-10-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaschina E, Scholz H, Steckelings UM, et al. Transition from atherosclerosis to aortic aneurysm in humans coincides with an increased expression of RAS components. Atherosclerosis. 2009 Aug;205(2):396–403. doi: 10.1016/j.atherosclerosis.2009.01.003. Epub 2009 Jan 13. [DOI] [PubMed] [Google Scholar]
- 36.Beyazit Y, Aksu S, Haznedaroglu IC, et al. Overexpression of the local bone marrow renin-angiotensin system in acute myeloid leukemia. J Natl Med Assoc. 2007 Jan;99(1):57–63. [PMC free article] [PubMed] [Google Scholar]
- 37.Chalmers L, Kaskel FJ, Bamgbola O. The role of obesity and its bioclinical correlates in the progression of chronic kidney disease. Adv Chronic Kidney Dis. 2006 Oct;13(4):352–364. doi: 10.1053/j.ackd.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 38.Graciano ML, Cavaglieri Rde C, Dellê H, et al. Intrarenal Renin-Angiotensin system is upregulated in experimental model of progressive renal disease induced by chronic inhibition of nitric oxide synthesis. J Am Soc Nephrol. 2004 Jul;15(7):1805–1815. doi: 10.1097/01.asn.0000131528.00773.a9. [DOI] [PubMed] [Google Scholar]
- 39.Tsang SW, Cheng CH, Leung PS. The role of the pancreatic renin-angiotensin system in acinar digestive enzyme secretion and in acute pancreatitis. Regul Pept. 2004 Jul 15;119(3):213–219. doi: 10.1016/j.regpep.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 40.Johnston AP, Baker J, De Lisio M, Parise G. Skeletal muscle myoblasts possess a stretch-responsive local angiotensin signalling system. J Renin Angiotensin Aldosterone Syst. 2011 Jun;12(2):75–84. doi: 10.1177/1470320310381795. Epub 2010 Oct 4. [DOI] [PubMed] [Google Scholar]
- 41.Jiang JS, Lang YD, Chou HC, et al. Activation of the renin-angiotensin system in hyperoxia-induced lung fibrosis in neonatal rats. Neonatology. 2012;101(1):47–54. doi: 10.1159/000329451. Epub 2011 Jul 26. [DOI] [PubMed] [Google Scholar]
- 42.Takao T, Horino T, Kagawa T, et al. Possible involvement of intracellular angiotensin II receptor in high-glucose-induced damage in renal proximal tubular cells. J Nephrol. 2011 Mar-Apr;24(2):218–224. doi: 10.5301/jn.2010.5785. [DOI] [PubMed] [Google Scholar]
- 43.Krick S, Hänze J, Eul B, et al. Hypoxia-driven proliferation of human pulmonary artery fibroblasts: cross-talk between HIF-1alpha and an autocrine angiotensin system. FASEB J. 2005 May;19(7):857–859. doi: 10.1096/fj.04-2890fje. Epub 2005 Feb 17. [DOI] [PubMed] [Google Scholar]
- 44.Yasuda N, Akazawa H, Qin Y, Zou Y, Komuro I. A novel mechanism of mechanical stress-induced angiotensin II type 1-receptor activation without the involvement of angiotensin II. Naunyn Schmiedebergs Arch Pharmacol. 2008 Jun;377(4-6):393–399. doi: 10.1007/s00210-007-0215-1. Epub 2007 Nov 29. [DOI] [PubMed] [Google Scholar]
- 45.Hitomi H, Fukui T, Moriwaki K, et al. Synergistic effect of mechanical stretch and angiotensin II on superoxide production via NADPH oxidase in vascular smooth muscle cells. J Hypertens. 2006 Jun;24(6):1089–1095. doi: 10.1097/01.hjh.0000226199.51805.88. [DOI] [PubMed] [Google Scholar]
- 46.Delli Gatti C, Osto E, Kouroedov A et al. Pulsatile stretch induces release of angiotensin II and oxidative stress in human endothelial cells: effects of ACE inhibition and AT1 receptor antagonism. Clin Exp Hypertens. 2008 Oct;30(7):616–627. doi: 10.1080/10641960802443183. [DOI] [PubMed] [Google Scholar]
- 47.Watanabe T, Pakala R, Katagiri T, Benedict CR. Mildly oxidized low-density lipoprotein acts synergistically with angiotensin II in inducing vascular smooth muscle cell proliferation. J Hypertens. 2001 Jun;19(6):1065–1073. doi: 10.1097/00004872-200106000-00011. [DOI] [PubMed] [Google Scholar]
- 48.Li DY, Zhang YC, Philips MI, Sawamura T, Mehta JL. Upregulation of endothelial receptor for oxidized low-density lipoprotein (LOX-1) in cultured human coronary artery endothelial cells by angiotensin II type 1 receptor activation. Circ Res. 1999 May 14;84(9):1043–1049. doi: 10.1161/01.res.84.9.1043. [DOI] [PubMed] [Google Scholar]
- 49.Nickenig G, Sachinidis A, Seewald S, Böhm M, Vetter H. Influence of oxidized low-density lipoprotein on vascular angiotensin II receptor expression. J Hypertens Suppl. 1997 Dec;15(6):S27–S30. doi: 10.1097/00004872-199715066-00006. [DOI] [PubMed] [Google Scholar]
- 50.Hu C, Dandapat A, Sun L, et al. Modulation of angiotensin II-mediated hypertension and cardiac remodeling by lectin-like oxidized low-density lipoprotein receptor-1 deletion. Hypertension. 2008 Sep;52(3):556–562. doi: 10.1161/HYPERTENSIONAHA.108.115287. Epub 2008 Jul 21. [DOI] [PubMed] [Google Scholar]
- 51.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000 Jan 7;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 52.Anand P, Kunnumakkara AB, Sundaram C, et al. Cancer is a preventable disease that requires major lifestyle changes. Pharm Res. 2008 Sep;25(9):2097–2116. doi: 10.1007/s11095-008-9661-9. Epub 2008 Jul 15. Erratum in: Pharm Res. 2008 Sep;25(9):2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Azad N, Rojanasakul Y, Vallyathan V. Inflammation and lung cancer: roles of reactive oxygen/nitrogen species. J Toxicol Environ Health B Crit Rev. 2008 Jan;11(1):1–15. doi: 10.1080/10937400701436460. [DOI] [PubMed] [Google Scholar]
- 54.Munteanu I, Didilescu C. [Chemistry and toxicology of cigarette smoke in the lungs] Pneumologia. 2007;56(1):41, 43–46. Jan-Mar; Romanian. [PubMed] [Google Scholar]
- 55.Brody JS, Spira A. State of the art. Chronic obstructive pulmonary disease, inflammation, and lung cancer. Proc Am Thorac Soc. 2006 Aug;3(6):535–537. doi: 10.1513/pats.200603-089MS. [DOI] [PubMed] [Google Scholar]
- 56.Smith CJ, Perfetti TA, King JA. Perspectives on pulmonary inflammation and lung cancer risk in cigarette smokers. Inhal Toxicol. 2006 Aug;18(9):667–677. doi: 10.1080/08958370600742821. [DOI] [PubMed] [Google Scholar]
- 57.Smith GR, Missailidis S. Cancer, inflammation and the AT1 and AT2 receptors. J Inflamm (Lond) 2004 Sep 30;1(1):3. doi: 10.1186/1476-9255-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene. 2008 Oct 6;27(45):5904–5912. doi: 10.1038/onc.2008.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Menke MN, Menke NB, Boardman CH, Diegelmann RF. Biologic therapeutics and molecular profiling to optimize wound healing. Gynecol Oncol. 2008 Nov;111(2 Suppl):S87–S91. doi: 10.1016/j.ygyno.2008.07.052. Epub 2008 Oct 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dalgleish AG, O'Byrne K. Inflammation and cancer: the role of the immune response and angiogenesis. Cancer Treat Res. 2006;130:1–38. [PubMed] [Google Scholar]
- 61.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011 Mar 4;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 62.Sica A, Allavena P, Mantovani A. Cancer related inflammation: the macrophage connection. Cancer Lett. 2008 Aug 28;267(2):204–215. doi: 10.1016/j.canlet.2008.03.028. Epub 2008 Apr 29. [DOI] [PubMed] [Google Scholar]
- 63.Whiteside TL. The role of immune cells in the tumor microenvironment. Cancer Treat Res. 2006;130:103–124. doi: 10.1007/0-387-26283-0_5. [DOI] [PubMed] [Google Scholar]
- 64.Lee JM, Yanagawa J, Peebles KA, et al. Inflammation in lung carcinogenesis: new targets for lung cancer chemoprevention and treatment. Crit Rev Oncol Hematol. 2008 Jun;66(3):208–217. doi: 10.1016/j.critrevonc.2008.01.004. Epub 2008 Mar 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Peebles KA, Lee JM, Mao JT, et al. Inflammation and lung carcinogenesis: applying findings in prevention and treatment. Expert Rev Anticancer Ther. 2007 Oct;7(10):1405–1421. doi: 10.1586/14737140.7.10.1405. [DOI] [PubMed] [Google Scholar]
- 66.Erlinger TP, Muntner P, Helzlsouer KJ. WBC count and the risk of cancer mortality in a national sample of U.S. adults: results from the Second National Health and Nutrition Examination Survey mortality study. Cancer Epidemiol Biomarkers Prev. 2004 Jun;13(6):1052–1056. [PubMed] [Google Scholar]
- 67.Shankar A, Wang JJ, Rochtchina E, Yu MC, Kefford R, Mitchell P. Association between circulating white blood cell count and cancer mortality: a population-based cohort study. Arch Intern Med. 2006 Jan 23;166(2):188–194. doi: 10.1001/archinte.166.2.188. Erratum in: Arch Intern Med. 2006 Mar 27;166(6):681. [DOI] [PubMed] [Google Scholar]
- 68.Taranova AG, Maldonado D, 3rd, Vachon CM, et al. Allergic pulmonary inflammation promotes the recruitment of circulating tumor cells to the lung. Cancer Res. 2008 Oct 15;68(20):8582–8589. doi: 10.1158/0008-5472.CAN-08-1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ingber DE. Can cancer be reversed by engineering the tumor microenvironment? Semin Cancer Biol. 2008 Oct;18(5):356–364. doi: 10.1016/j.semcancer.2008.03.016. Epub 2008 Apr 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fujita M, Hayashi I, Yamashina S, et al. Angiotensin type 1a receptor signaling-dependent induction of vascular endothelial growth factor in stroma is relevant to tumor-associated angiogenesis and tumor growth. Carcinogenesis. 2005 Feb;26(2):271–279. doi: 10.1093/carcin/bgh324. Epub 2005 Jan 6. [DOI] [PubMed] [Google Scholar]
- 71.Imai N, Hashimoto T, Kihara M, et al. Roles for host and tumor angiotensin II type 1 receptor in tumor growth and tumor-associated angiogenesis. Lab Invest. 2007 Feb;87(2):189–198. doi: 10.1038/labinvest.3700504. Epub 2006 Dec 18. [DOI] [PubMed] [Google Scholar]
- 72.Hoshino K, Ishiguro H, Teranishi J, et al. Regulation of androgen receptor expression through angiotensin II type 1 receptor in prostate cancer cells. Prostate. 2011 Jun 15;71(9):964–975. doi: 10.1002/pros.21312. Epub 2010 Dec 28. [DOI] [PubMed] [Google Scholar]
- 73.Doi C, Egashira N, Kawabata A, et al. Angiotensin II type 2 receptor signaling significantly attenuates growth of murine pancreatic carcinoma grafts in syngeneic mice. BMC Cancer. 2010 Feb 24;10:67. doi: 10.1186/1471-2407-10-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Clere N, Corre I, Faure S, et al. Deficiency or blockade of angiotensin II type 2 receptor delays tumorigenesis by inhibiting malignant cell proliferation and angiogenesis. Int J Cancer. 2010 Nov 15;127(10):2279–2291. doi: 10.1002/ijc.25234. [DOI] [PubMed] [Google Scholar]
- 75.Shen XZ, Li P, Weiss D, Fuchs S, et al. Mice with enhanced macrophage angiotensin-converting enzyme are resistant to melanoma. Am J Pathol. 2007 Jun;170(6):2122–2134. doi: 10.2353/ajpath.2007.061205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sipahi I, Debanne SM, Rowland DY, Simon DI, Fang JC. Angiotensin-receptor blockade and risk of cancer: meta-analysis of randomised controlled trials. Lancet Oncol. 2010 Jul;11(7):627–636. doi: 10.1016/S1470-2045(10)70106-6. Epub 2010 Jun 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sica DA. Angiotensin receptor blockers and the risk of malignancy: a note of caution. Drug Saf. 2010 Sep 1;33(9):709–712. doi: 10.2165/11532450-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 78.Aalbers J. ARBs and possible cancer risk. Cardiovasc J Afr. 2010 Jul-Aug;21(4):232. [PMC free article] [PubMed] [Google Scholar]
- 79.Bloch MJ, Basile JN. Meta-analysis concludes angiotensin receptor blocker use increases the risk of developing cancer: concerns about the science and the message. J Clin Hypertens (Greenwich) 2010 Sep;12(9):661–663. doi: 10.1111/j.1751-7176.2010.00369.x. Epub 2010 Aug 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Volpe M, Azizi M, Danser AH, Nguyen G, Ruilope LM. Twisting arms to angiotensin receptor blockers/antagonists: the turn of cancer. Eur Heart J. 2011 Jan;32(1):19–22. doi: 10.1093/eurheartj/ehq382. Epub 2010 Oct 21. [DOI] [PubMed] [Google Scholar]
- 81.Madsen FF. [Angiotensin II type 1 receptor blockade and risk of cancer] Ugeskr Laeger. 2010;172(44) Nov 1; 3054; discussion 3054. Danish. [PubMed] [Google Scholar]
- 82.Lombard C, Nosworthy A, Albers J. Opinions in hypertension. ARBs and risk of cancer: international and South African expert comment. Cardiovasc J Afr. 2010 Nov-Dec;21(6):338–341. [PMC free article] [PubMed] [Google Scholar]
- 83.Marina I, Krakoff LR. Angiotensin receptor blockers, cancer, and smoking. J Clin Hypertens (Greenwich) 2010 Dec;12(12):945–948. doi: 10.1111/j.1751-7176.2010.00375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cohen DL, Townsend RR. Is there an increased cancer risk associated with the use of angiotensin receptor blockers and should it affect current prescribing? J Clin Hypertens (Greenwich) 2011 Feb;13(2):131–132. doi: 10.1111/j.1751-7176.2010.00403.x. Epub 2010 Dec 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bangalore S, Kumar S, Kjeldsen SE, et al. Antihypertensive drugs and risk of cancer: network meta-analyses and trial sequential analyses of 324,168 participants from randomised trials. Lancet Oncol. 2011 Jan;12(1):65–82. doi: 10.1016/S1470-2045(10)70260-6. Epub 2010 Nov 29. [DOI] [PubMed] [Google Scholar]
- 86.ARB Trialists Collaboration. Effects of telmisartan, irbesartan, valsartan, candesartan, and losartan on cancers in 15 trials enrolling 138,769 individuals. J Hypertens. 2011 Apr;29(4):623–635. doi: 10.1097/HJH.0b013e328344a7de. [DOI] [PubMed] [Google Scholar]
- 87.Pasternak B, Svanström H, Callréus T, Melbye M, Hviid A. Use of angiotensin receptor blockers and the risk of cancer. Circulation. 2011 Apr 26;123(16):1729–1736. doi: 10.1161/CIRCULATIONAHA.110.007336. Epub 2011 Apr 11. [DOI] [PubMed] [Google Scholar]
- 88.Yoon C, Yang HS, Jeon I, Chang Y, Park SM. Use of angiotensin-converting-enzyme inhibitors or angiotensin-receptor blockers and cancer risk: a meta-analysis of observational studies. CMAJ. 2011 Oct 4;183(14):E1073–E1084. doi: 10.1503/cmaj.101497. Epub 2011 Aug 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cassat M, Wuerzner G, Burnier M, Waeber B. [Angiotensin II receptor blockers (ARBs) and cancer: a reassuring balance] Rev Med Suisse. 2011 Sep 14;7(308):1757–1758. 1760. French. [PubMed] [Google Scholar]
- 90.No connection between ARBs and cancer. Harv Heart Lett. 2011 Aug;21(12):6. [PubMed] [Google Scholar]
- 91.Olin JL, Veverka A, Nuzum DS. Risk of cancer associated with the use of angiotensin II-receptor blockers. Am J Health Syst Pharm. 2011 Nov 15;68(22):2139–2146. doi: 10.2146/ajhp100570. [DOI] [PubMed] [Google Scholar]
- 92.Wuerzner G, Burnier M, Waeber B. Critical review of cancer risk associated with angiotensin receptor blocker therapy. Vasc Health Risk Manag. 2011;7:741–747. doi: 10.2147/VHRM.S13552. Epub 2011 Dec 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Singh A, Bangalore S. Which, if any, antihypertensive agents cause cancer? Curr Opin Cardiol. 2012 Jul;27(4):374–380. doi: 10.1097/HCO.0b013e328353bc4f. [DOI] [PubMed] [Google Scholar]
- 94.Huang CC, Chan WL, Chen YC, et al. Angiotensin II receptor blockers and risk of cancer in patients with systemic hypertension. Am J Cardiol. 2011 Apr 1;107(7):1028–1033. doi: 10.1016/j.amjcard.2010.11.026. Epub 2011 Jan 20. [DOI] [PubMed] [Google Scholar]
- 95.Wang KL, Liu CJ, Chao TF, et al. Long-term use of angiotensin II receptor blockers and risk of cancer: A population-based cohort analysis. Int J Cardiol. 2012 Jun 16; doi: 10.1016/j.ijcard.2012.05.096. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 96.Zeng W, Chen W, Leng X, He JG, Ma H. Chronic angiotensin-(1-7) administration improves vascular remodeling after angioplasty through the regulation of the TGF-beta/Smad signaling pathway in rabbits. Biochem Biophys Res Commun. 2009 Nov 6;389(1):138–144. doi: 10.1016/j.bbrc.2009.08.112. Epub 2009 Aug 26. [DOI] [PubMed] [Google Scholar]
- 97.Zeng WT, Chen WY, Leng XY, He JG, Ma H. [Angiotensin-(1-7) reduced postangioplasty vascular fibrosis in abdominal aorta of rabbits] Zhonghua Xin Xue Guan Bing Za Zhi. 2010 Jun;38(6):531–538. Chinese. [PubMed] [Google Scholar]
- 98.Krishnan B, Smith TL, Dubey P, et al. Angiotensin-(1-7) attenuates metastatic prostate cancer and reduces osteoclastogenesis. Prostate. [Epub ahead of print] 2012 May 29; doi: 10.1002/pros.22542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kim ST, Park KH, Oh SC, et al. How does inhibition of the renin-angiotensin system affect the prognosis of advanced gastric cancer patients receiving platinum-based chemotherapy? Oncology. 2012;83(6):354–360. doi: 10.1159/000337979. Epub 2012 Oct 8. [DOI] [PubMed] [Google Scholar]
- 100.Nakai Y, Isayama H, Sasaki T, et al. Clinical outcomes of chemotherapy for diabetic and nondiabetic patients with pancreatic cancer: better prognosis with statin use in diabetic patients. Pancreas. 2012 Sep 20; doi: 10.1097/MPA.0b013e31825de678. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 101.Nakai Y, Isayama H, Ijichi H, et al. Phase I trial of gemcitabine and candesartan combination therapy in normotensive patients with advanced pancreatic cancer: GECA1. Cancer Sci. 2012 Aug;103(8):1489–1492. doi: 10.1111/j.1349-7006.2012.02311.x. Epub 2012 Jun 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tanaka N, Miyajima A, Kikuchi E, et al. Prognonstic impact of renin-angiotensin system blockade in localised upper-tract urothelial carcinoma. Br J Cancer. 2012 Jan 17;106(2):290–296. doi: 10.1038/bjc.2011.565. Epub 2011 Dec 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mc Menamin ÚC, Murray LJ, Cantwell MM, Hughes CM. Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers in cancer progression and survival: a systematic review. Cancer Causes Control. 2012 Feb;23(2):221–230. doi: 10.1007/s10552-011-9881-x. Epub 2011 Nov 25. [DOI] [PubMed] [Google Scholar]
- 104.Keizman D, Huang P, Eisenberger MA, et al. Angiotensin system inhibitors and outcome of sunitinib treatment in patients with metastatic renal cell carcinoma: a retrospective examination. Eur J Cancer. 2011 Sep;47(13):1955–1961. doi: 10.1016/j.ejca.2011.04.019. Epub 2011 May 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tatokoro M, Fujii Y, Kawakami S, et al. Phase-II trial of combination treatment of interferon-α, cimetidine, cyclooxygenase-2 inhibitor and renin-angiotensin-system inhibitor (I-CCA therapy) for advanced renal cell carcinoma. Cancer Sci. 2011 Jan;102(1):137–143. doi: 10.1111/j.1349-7006.2010.01756.x. Epub 2010 Oct 26. [DOI] [PubMed] [Google Scholar]
- 106.Kaibori M, Ishizaki M, Matsui K, Kitade H, Matsui Y, Kwon AH. Evaluation of metabolic factors on the prognosis of patients undergoing resection of hepatocellular carcinoma. J Gastroenterol Hepatol. 2011 Mar;26(3):536–543. doi: 10.1111/j.1440-1746.2010.06439.x. [DOI] [PubMed] [Google Scholar]
- 107.Nakai Y, Isayama H, Ijichi H, et al. Inhibition of renin-angiotensin system affects prognosis of advanced pancreatic cancer receiving gemcitabine. Br J Cancer. 2010 Nov 23;103(11):1644–1648. doi: 10.1038/sj.bjc.6605955. Epub 2010 Oct 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wilop S, von Hobe S, Crysandt M, et al. Impact of angiotensin I converting enzyme inhibitors and angiotensin II type 1 receptor blockers on survival in patients with advanced non-small-cell lung cancer undergoing first-line platinum-based chemotherapy. J Cancer Res Clin Oncol. 2009 Oct;135(10):1429–1435. doi: 10.1007/s00432-009-0587-3. Epub 2009 Apr 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Uemura H, Hasumi H, Kawahara T, et al. Pilot study of angiotensin II receptor blocker in advanced hormone-refractory prostate cancer. Int J Clin Oncol. 2005 Dec;10(6):405–410. doi: 10.1007/s10147-005-0520-y. [DOI] [PubMed] [Google Scholar]
- 110.Chae YK, Valsecchi ME, Kim J, et al. Reduced risk of breast cancer recurrence in patients using ACE inhibitors, ARBs, and/or statins. Cancer Invest. 2011 Nov;29(9):585–593. doi: 10.3109/07357907.2011.616252. Epub 2011 Sep 21. [DOI] [PubMed] [Google Scholar]
- 111.Yuge K, Miyajima A, Tanaka N, et al. Prognostic value of renin-angiotensin system blockade in non-muscle-invasive bladder cancer. Ann Surg Oncol. 2012 Nov;19(12):3987–3993. doi: 10.1245/s10434-012-2568-z. Epub 2012 Aug 8. [DOI] [PubMed] [Google Scholar]
- 112.Yoshiji H, Noguchi R, Ikenaka Y, et al. Combination of branched-chain amino acids and angiotensin-converting enzyme inhibitor suppresses the cumulative recurrence of hepatocellular carcinoma: a randomized control trial. Oncol Rep. 2011 Dec;26(6):1547–1553. doi: 10.3892/or.2011.1433. Epub 2011 Aug 24. [DOI] [PubMed] [Google Scholar]
- 113.Ronquist G, Frithz G, Wang YH, Lindeborg T. Captopril may reduce biochemical (prostate-specific antigen) failure following radical prostatectomy for clinically localized prostate cancer. Scand J Urol Nephrol. 2009;43(1):32–36. doi: 10.1080/00365590802468875. [DOI] [PubMed] [Google Scholar]
- 114.Wedlake LJ, Silia F, Benton B, et al. Evaluating the efficacy of statins and ACE-inhibitors in reducing gastrointestinal toxicity in patients receiving radiotherapy for pelvic malignancies. Eur J Cancer. 2012 Sep;48(14):2117–2124. doi: 10.1016/j.ejca.2011.12.034. Epub 2012 Mar 3. [DOI] [PubMed] [Google Scholar]
- 115.Kharofa J, Cohen EP, Tomic R, Xiang Q, Gore E. Decreased risk of radiation pneumonitis with incidental concurrent use of angiotensin-converting enzyme inhibitors and thoracic radiation therapy. Int J Radiat Oncol Biol Phys. 2012 Sep 1;84(1):238–243. doi: 10.1016/j.ijrobp.2011.11.013. Epub 2012 Jan 31. [DOI] [PubMed] [Google Scholar]
- 116.Dessì M, Piras A, Madeddu C, et al. Long-term protective effects of the angiotensin receptor blocker telmisartan on epirubicin-induced inflammation, oxidative stress and myocardial dysfunction. Exp Ther Med. 2011 Sep;2(5):1003–1009. doi: 10.3892/etm.2011.305. Epub 2011 Jun 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Abedon ST. Bacterial 'immunity' against bacteriophages. Bacteriophage. 2012 Jan 1;2(1):50–54. doi: 10.4161/bact.18609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dai Z, Wu F, Yeung EW, Li Y. IGF-IEc expression, regulation and biological function in different tissues. Growth Horm IGF Res. 2010 Aug;20(4):275–281. doi: 10.1016/j.ghir.2010.03.005. Epub 2010 May 21. [DOI] [PubMed] [Google Scholar]
- 119.Teboul M, Guillaumond F, Gréchez-Cassiau A, Delaunay F. The nuclear hormone receptor family round the clock. Mol Endocrinol. 2008 Dec;22(12):2573–2582. doi: 10.1210/me.2007-0521. Epub 2008 Jul 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Duong V, Rochette-Egly C. The molecular physiology of nuclear retinoic acid receptors. From health to disease. Biochim Biophys Acta. 2011 Aug;1812(8):1023–1031. doi: 10.1016/j.bbadis.2010.10.007. Epub 2010 Oct 20. [DOI] [PubMed] [Google Scholar]
- 121.Pan D, Kocherginsky M, Conzen SD. Activation of the glucocorticoid receptor is associated with poor prognosis in estrogen receptor-negative breast cancer. Cancer Res. 2011 Oct 15;71(20):6360–6370. doi: 10.1158/0008-5472.CAN-11-0362. Epub 2011 Aug 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Reis e Sousa C. Activation of dendritic cells: translating innate into adaptive immunity. Curr Opin Immunol. 2004 Feb;16(1):21–25. doi: 10.1016/j.coi.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 123.Hoyer KK, Dooms H, Barron L, Abbas AK. Interleukin-2 in the development and control of inflammatory disease. Immunol Rev. 2008 Dec;226:19–28. doi: 10.1111/j.1600-065X.2008.00697.x. [DOI] [PubMed] [Google Scholar]