

Abstract
Background
We previously reported that IKAS are heterogeneously upregulated in failing rabbit ventricles and play an important role in arrhythmogenesis. This study goal is to test the hypothesis that subtype 2 of the small‐conductance Ca2+ activated K+ (SK2) channel and apamin‐sensitive K+ currents (IKAS) are upregulated in failing human ventricles.
Methods and Results
We studied 12 native hearts from transplant recipients (heart failure [HF] group) and 11 ventricular core biopsies from patients with aortic stenosis and normal systolic function (non‐HF group). IKAS and action potential were recorded with patch‐clamp techniques, and SK2 protein expression was studied by Western blotting. When measured at 1 μmol/L Ca2+ concentration, IKAS was 4.22 (median) (25th and 75th percentiles, 2.86 and 6.96) pA/pF for the HF group (n=11) and 0.98 (0.54 and 1.72) pA/pF for the non‐HF group (n=8, P=0.008). IKAS was lower in the midmyocardial cells than in the epicardial and the endocardial cells. The Ca2+ dependency of IKAS in HF myocytes was shifted leftward compared to non‐HF myocytes (Kd 314 versus 605 nmol/L). Apamin (100 nmol/L) increased the action potential durations by 1.77% (−0.9% and 7.3%) in non‐HF myocytes and by 11.8% (5.7% and 13.9%) in HF myocytes (P=0.02). SK2 protein expression was 3‐fold higher in HF than in non‐HF.
Conclusions
There is heterogeneous upregulation of IKAS densities in failing human ventricles. The midmyocardial layer shows lower IKAS densities than epicardial and endocardial layers of cells. Increase in both Ca2+ sensitivity and SK2 protein expression contributes to the IKAS upregulation.
Keywords: arrhythmia, calcium, heart failure, ion channels
Introduction
Heart failure (HF) is a major public health problem, with a prevalence of 5.8 million in the United States and over 23 million worldwide.1 Ventricular arrhythmia is a major cause of death in patients with HF.2 The mechanisms of arrhythmia in HF are attributed in part to the reduced repolarization reserve due to the upregulation of late INa and the downregulation of multiple major K+ currents (Ito, IKs, IKr, IK1, and IKATP),3–6 leading to increased risk of action potential duration (APD) prolongation, early afterdepolarization (EAD), triggered activity, and ventricular arrhythmias.7 Although APD prolongation is proarrhythmic, acute but reversible APD shortening after ventricular fibrillation (VF)–defibrillation episodes has been observed in a rabbit model of pacing‐induced HF.8 The APD shortening along with persistent intracellular Ca2+ (Cai) elevation can promote calcium transient‐triggered firing9 and late phase 3 EAD,10 leading to triggered activity and recurrent spontaneous VF. The mechanisms of acute postshock APD shortening in failing rabbit ventricles is due to VF‐induced Ca2+ accumulation11 and the upregulation of apamin‐sensitive K+ currents (IKAS).12 These studies suggest that IKAS upregulation in failing ventricles may be antiarrhythmic by preserving the repolarization reserve but may also be proarrhythmic by acutely and excessively shortening the APD during Cai overload. However, whether IKAS are upregulated in failing human ventricles remains unknown. The purpose of the present study is to test the hypothesis that subtype 2 of the small‐conductance Ca2+ activated K+ (SK2) channels and IKAS are upregulated in ventricular cells from patients with HF and that IKAS plays an important role in determining the APD in failing human ventricles.
Methods
Clinical Data Collection
The Human Heart Failure Tissue Collection Program is approved by the institutional review board at the Indiana University School of Medicine. The intraoperative atrial tissue collection is approved by the institutional review board of Medical College of Wisconsin and the University of Rome, La Sapienza. Written informed consents were obtained from all patients who participated in the study. Twelve native hearts from transplant recipients (HF group) and the apical tissues from 11 patients with aortic stenosis and normal systolic function who underwent apicoaortic conduit surgery (non‐HF group) were studied.13 Human atrial tissues were harvested during open heart surgery and preserved in liquid nitrogen. The atrial tissues were used for comparison because they are known to abundantly express SK2 proteins.14
Cell Isolation
Left ventricular myocytes were isolated using enzymatic digestion methods. The heart tissues were immersed in cardioplegic solution on collection. A branch of coronary arteries was cannulated for perfusion. The remaining arteries were ligated to prevent leaking of the perfusate. Tyrode solution, equilibrated with 95% O2 and 5% CO2, was then perfused for 10 minutes. The components of the Tyrode solution include (in mmol/L): NaCl, 140; KCl, 5.4; MgCl2, 1.2; NaH2PO4, 0.33; CaCl2, 1.8; glucose, 10; and HEPES, 5 (pH 7.4 with NaOH). After 10‐minute perfusion, digestion buffer was perfused for 15 minutes. The digestion buffer contained (in mmol/L, except described otherwise): NaCl, 125; KCl, 4.75; MgSO4, 1.18; KH2PO4, 1.2; EGTA, 0.02; glucose, 10; taurine, 58; and creatine, 25, in addition to BSA 1%, MEM amino acid 2%, MEM nonessential amino acid 1%, and MEM vitamin solution 1% (Invitrogen). The tissues were then digested with the same digestion buffer containing 150 U/mL type II collagenase (Worthington) for 15 to 20 minutes. After digestion, digestion buffer without collagenase was perfused to wash tissues for 5 minutes. All enzymatic isolation procedures were performed at 37°C, and the perfusion pressure was maintained at 70 to 90 cm H2O. The tissues from aortic stenosis patients were directly subjected to cutting and digestion without prior coronary perfusion. The success rate of isolating cells suitable for patch‐clamp experiments was ≈70%, and the cell viability was 30% to 70% depending on tissue condition. All chemicals were purchased from Sigma unless otherwise stated.
Whole‐Cell Patch‐Clamp Studies and Action Potential Recording
Patch‐clamp experiments were performed as previously described.12 Briefly, all experiments were performed at 36°C of chamber temperature, which was regulated by a PH‐1 heating platform, SH‐27B solution heater, and TC‐344B temperature controller (Warner Instruments). Axopatch 200B amplifier and pCLAMP‐10 software (Molecular Device/Axon) were used to generate pulse protocols and record data. Pipette electrodes were made from Corning 7056 glass capillaries (Warner Instruments). The pipette resistance was 3 to 5 MΩ in the bath solution. Whole‐cell patch‐clamp techniques were used to record IKAS. Extracellular NMG (N‐methylglucamine) solution contained (in mmol/L): NMG, 140; KCl, 4; MgCl2, 1; glucose, 5; and HEPES, 10 (pH 7.4 with HCl). Pipette solution contained (in mmol/L): potassium gluconate, 144; MgCl2, 1.15; EGTA, 1; and HEPES, 10 (pH 7.2 with KOH). Various concentrations of calcium chloride were used to generate different concentrations of free Ca2+ according to the calculation by Bers et al.15 All cells were superfused continuously with the extracellular NMG solution during recording. Current traces were analyzed with Clampfit software (Molecular Device/Axon). Action potentials were recorded with the whole‐cell perforated‐patch technique with the Tyrode solution as extracellular solution and the same pipette solution containing 120 μg/mL amphotericin‐B and 1 μmol/L free calcium. The extracellular solution without and with apamin 100 nmol/L was used to measure IKAS. All data were corrected for the junction potentials.
Western Blot Analysis
SK channels have 3 subtypes.16–17 Among them, SK2 is the most sensitive to apamin, whereas SK1 and SK3 are insensitive18 and moderately sensitive,19 respectively. We detected only a small amount of SK3 protein in the human ventricles with a commercial antibody (P0608; Sigma). However, the low signal‐to‐noise ratio on Western blots prevented proper analyses (data not shown). Therefore, we chose to focus our efforts in studying SK2 expression. For each well, 30 μg of homogenate of left atrial and ventricular tissues was loaded on an SDS–polyacrylamide gel and transferred to a nitrocellulose membrane. The blot was probed with the anti‐SK2 polyclonal antibody (1:600, Abcam). Antibody‐binding protein bands were visualized with 125I‐protein A and quantified with a Bio‐Rad Personal Fx PhosphorImager. Expression of SK2 for each sample were normalized to glyceraldehyde‐3‐phosphate‐dehydrogenase levels and expressed as arbitrary units (AUs). A right atrial appendage tissue from a 57‐year‐old male patient with paroxysmal atrial fibrillation, who received cardiac surgery, was used as a positive control.
Immunohistology and Confocal Imaging
Paraffin sections of 4‐μm thickness were incubated at 60°C for 15 minutes. Slides were then deparaffinized in 3 changes of xylene for 5 minutes each and rehydrated through graded ethanol to distilled water. After washing in PBS, the cells were permeabilized in 0.2% Triton X‐100 for 1 hour at room temperature. Then, slides were washed with PBS and blocked with 2% BSA plus 10% normal donkey serum (Jackson ImmunoResearch) for 1.5 hours. The cells were stained with affinity purified goat anti‐human KCNN2 polyclonal antibody (LifeSpan BioSciences) at a concentration of 4 μg/mL overnight. After washing, the cells were incubated with Alexa Fluor 555 labeled donkey anti‐goat IgG (Invitrogen) at 1:200 dilution for 1 hour. Finally, the slides were washed, stained with Hoechst and mounted in VectaShields medium. Samples were examined by scanning laser microscopy (Olympus FV1000) using a ×60 1.42 NA oil immersion objective and a pixel size of 138 nm. Images were obtained by sequential illumination with 405‐, 488‐, and 559‐nm laser light, while fluorescence was collected in the blue (425 to 475 nm), green (500 to 545 nm), and red (575 to 675 nm) ranges. Differential interference contrast images were also collected using the 488‐nm excitation light.
Statistical Analyses
Comparison of the continuous variables between 2 groups was performed using the Mann–Whitney–Wilcoxon test. Fisher exact test was used to compare categorical variables between the 2 groups. Kruskal–Wallis test was conducted to compare continuous variables among ≥3 groups, with post hoc Mann–Whitney–Wilcoxon test to compare differences between any 2 groups. Comparison of APD in the absence and presence of 100 nmol/L apamin was performed using Wilcoxon signed rank test. To compare Kd of calcium sensitivity of IKAS between the HF and the non‐HF groups, extra sum‐of‐square F test was used. All comparisons were performed to test 2‐tailed methods and P≤0.05 was considered statistically significant. Statistical analyses were performed using SPSS PASW Statistics 17 software (IBM), and Prism 5.0 (GraphPad). Data in the text and figures are presented as median [25th and 75th percentiles] or mean±SEM unless otherwise stated.
Results
Clinical Characteristics
The left ventricular ejection fraction was significantly lower in the HF group (19% [15% and 22%]) than in the non‐HF group (55% [51% and 64%], P<0.001). Nine (75%) patients in the HF group and none in the non‐HF group had a history of ventricular arrhythmias. In the HF group, 9 (75%) patients had an implantable cardioverter‐defibrillator and 9 (75%) had a left ventricular assist device (VAD) as a bridge to orthotopic heart transplantation. The 9 patients with a VAD waited for 208.5 days [80.75 and 319.5 days] before receiving a heart transplant. Table 1 summarizes the clinical characteristics. All 12 HF group and 11 non‐HF group patients were included in the single‐cell studies.
Table 1.
Clinical Characteristics
| HF (n=12) | Non‐HF (n=11) | P Value | |
|---|---|---|---|
| Age, y | 45 [38; 53] | 85 [77; 88] | <0.001 |
| Male gender, % | 11 (92) | 5 (45) | 0.027 |
| CAD, % | 4 (33) | 1 (9) | 0.317 |
| LVEF, % | 19 [15; 22] | 55 [51; 64] | <0.001 |
| Clinical VT/VF, % | 9 (75) | 0 (0) | <0.001 |
| ICD, % | 9 (75) | 0 (0) | <0.001 |
| VAD, % | 9 (75) | 0 (0) | <0.001 |
| ACEI or ARB, % | 6 (50) | 3 (27) | 0.400 |
| β‐Blocker, % | 5 (42) | 6 (54) | 0.684 |
| Antiarrhythmic agents, % | 5 (42) | 1 (9) | 0.155 |
| Inotropic agents, % | 4 (33) | 0 (0) | 0.093 |
Data were presented as median [25 percentile; 75 percentile] or number (percentage). HF indicates heart failure; CAD, coronary artery disease; LVEF, left ventricular ejection fraction; VT, ventricular tachycardia; device; ICD, implantable cardioverter‐debrillator; VAD, ventricular assist device; ACEI, angiotensin‐converting enzyme inhibitor; ARB, angiotensin receptor blocker.
IKAS in Cardiomyocytes of the HF and Non‐HF Groups
Characteristics of IKAS were studied in the ventricular myocytes isolated from HF and non‐HF hearts using patch‐clamp techniques. Figure 1A shows representative whole‐cell K+ current traces recorded in the absence and presence of 100 nmol/L apamin with an intrapipette free Ca2+ concentration of 1 μmol/L. IKAS were detected as 100 nmol/L apamin‐sensitive K+ currents (Figure 1A, right panels). A total of 22 cells from 11 hearts from the HF group and 16 cells from 8 hearts from the non‐HF group were studied. Myocytes from the HF group were found to have significantly higher IKAS values compared with cells from the non‐HF group. Current‐voltage relationship of IKAS demonstrates that both inward and outward IKAS were increased in cells from the HF group (Figure 1B). IKAS values measured at 0 mV were 4.22 [2.86 and 6.96] pA/pF for myocytes from the HF group (n=11 patients) and 0.98 [0.54 and 1.72] pA/pF for myocytes from the non‐HF group (n=8 patients, P=0.008).
Figure 1.
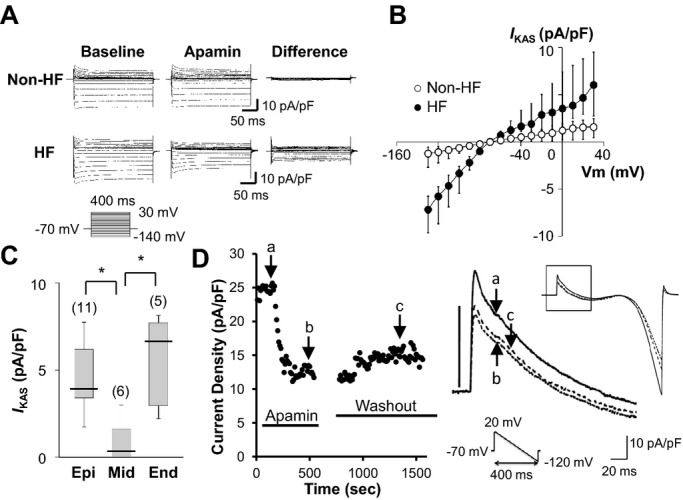
IKAS densities of isolated ventricular myocytes. A, Representative K+ current traces obtained from a non‐HF (top) and an HF (bottom) ventricular myocyte. The K+ currents were recorded with an intrapipette free‐Ca2+ of 1 μmol/L in the absence (IK‐baseline) and the presence of 100 nmol/L apamin (IK‐apamin). IKAS was calculated as the difference between IK‐apamin and IK‐baseline. B, Current‐voltage (I‐V) relationship of IKAS obtained from failing ventricles (n=22 cells from 11 patients) and nonfailing ventricles (n=16 cells from 8 patients). Current densities were presented as median [25th percentile; 75th percentile]. C, Transmural distribution of IKAS in failing ventricles. IKAS density at 0 mV with an intrapipette free‐Ca2+ of 1 μmol/L recorded from 3 different layers (Epi, epicardium; Mid, midmyocardium; Endo, endocardium). The midmyocardium had lower current density than the other 2 layers (P=0.005 for Kruskal–Wallis test; *P<0.05 for post hoc Mann–Whitney–Wilcoxon test). D, The graph shows the time course of total K+ currents measured at 0 mV with ramp‐pulse protocol (test pulse: between +20 and −140 mV; holding membrane potential: −80 mV; pulse frequency: every 6 seconds) in a cardiomyocyte isolated from a failing heart. The current density during apamin (b) and after washout (c) showed only slight differences, indicating incomplete washout of apamin. HF indicates heart failure.
To study transmural distributions of IKAS, cells were isolated from 3 different layers (endocardial, mid, and epicardial layers). IKAS were significantly lower in the cardiomyocytes from the mid layer of the HF group than those from the epicardial and the endocardial myocytes (Figure 1C, P=0.005 for Kruskal–Wallis test, P<0.05 for post hoc Mann–Whitney–Wilcoxon test between the midmyocardial layer versus the epicardial or the endocardial layers).
Figure 1D shows time course of recovery of IK from inhibition with apamin (100 nmol/L) obtained in a failing ventricular myocyte. IK were activated with 1 μmol/L intrapipette Ca2+ and repetitive ramp‐pulses. IK was rapidly inhibited with an application of apamin (100 nmol/L). However, IK was only partially reversible on washout.
Steady‐State Ca2+ Sensitivity of IKAS in HF and Non‐HF Cardiomyocytes
To further elucidate the underlying mechanisms of the IKAS upregulation in HF myocytes, a steady‐state Ca2+ sensitivity of IKAS was measured using various intrapipette Ca2+ concentrations. IKAS was normalized by the maximal currents and plotted against Cai. The data were fitted with the Hill equation, yielding significantly lower Kd values for the HF than for the non‐HF group (HF: 345±4 nmol/L versus non‐HF: 605±28 nmol/L). Hill coefficients were not significantly different between 2 groups (HF: 3.14±0.11 versus non‐HF: 3.79±0.61). As shown in Figure 2, the curve of Ca2+‐dependent IKAS was left‐shifted in the HF myocytes compared with the non‐HF myocytes (P<0.001). Each cell was exposed to only one Cai concentration. Therefore, the number in Figure 2 is equal to the number of cells studied.
Figure 2.

Steady‐state Ca2+ dependency of IKAS in non‐HF and HF ventricular myocytes. IKAS was normalized to the maximal IKAS with a free Ca2+ of 10 μmol/L and plotted as a function of Ca2+ concentration. The data were fitted with Hill equation: y=1/(1+[Kd/x]n), where y indicates the normalized IKAS and x is the intrapipette free calcium; Kd is the concentration of intrapipette free calcium at half‐maximal activation of IKAS; and n is the Hill coefficient. Error bars represent SEM. Numbers in parentheses indicate the number of cells patched. Normalized currents were presented as mean±SEM. HF indicates heart failure.
Measurement of Action Potential in HF and Non‐HF Cardiomyocytes
Next, effects of apamin on APD80 were studied using a perforated‐patch method. Although apamin (100 nmol/L) had only small effects on APD80 in the non‐HF myocytes, it significantly prolonged the APD80 in the HF myocytes by 11.8% (Figure 3). These results indicate that IKAS is important in maintaining the repolarization reserve in failing ventricles.
Figure 3.
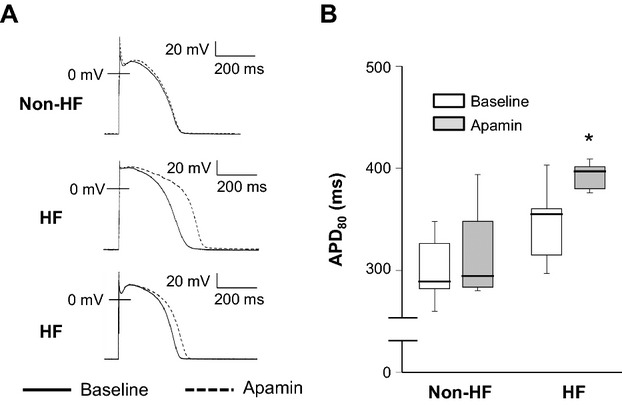
Effects of apamin on APD in ventricular cardiomyocytes. A, Action potentials recorded in 1 non‐HF and 2 HF ventricular myocytes at baseline (solid line) and in the presence of 100 nmol/L apamin (dotted line). B, The box plots depict APD80 in various conditions (*P<0.05 for Mann–Whitney–Wilcoxon test). APD80 were presented as median [25th percentile; 75th percentile]. HF indicates heart failure.
Western Blot Analysis
Because a large portion of IKAS is carried by SK2 channels,20 we examined expression levels of SK2 protein in the tissue homogenate. Using a commercial antibody raised against an immunogen peptide located between residues 450 to 560 of human SK2, we confirmed the previous observations that SK2 channels were expressed abundantly in nonfailing atria but weakly in nonfailing ventricles21 (Figure 4A, left, 66‐kDa bands). However, the failing ventricles had significantly higher expression of SK2 protein than the nonfailing ventricles (Figure 4A, right). Protein bands with molecular mass of 30 kDa were also significantly increased in the failing ventricles (Figure 4A, right). The SK3 is detected at low levels. The signal‐to‐noise ratio of SK3 was too low to accurately analyze the SK3 protein concentration. The low SK3 protein expression is consistent with that found by other investigators.19
Figure 4.

SK2 proteins in HF and non‐HF (NF) ventricles. A, Left, Western blot analysis of atrial and ventricular tissues from a 57‐year‐old male patient with paroxysmal atrial fibrillation and normal LVEF. Right, Western blot analysis of SK2 and glyceraldehyde‐3‐phosphate‐dehydrogenase (GAPDH) in non‐HF (n=5) and HF (n=6) human ventricles. B, Aggregated results of SK2 protein (66 kDa) and presumed SK2 short form protein (31 kDa) expression in non‐HF and HF groups. The signal intensity was normalized to the GAPDH. Data are presented as median [25th percentile; 75th percentile]. HF indicates heart failure; LVEF, left ventricular ejection fraction.
Immunohistochemical Staining Study
We performed immunofluorescence confocal imaging to determine the subcellular distribution of SK2 channel proteins. Representative examples from a nonfailing and a failing heart are shown in Figure 5. In non‐HF cardiomyocytes, anti‐SK2 immune reactivity (red signal; blue, nuclei) displayed a regular striated appearance, compatible with SK2 channel clusters being distributed along transverse‐tubular membranes (see also higher‐magnification views at the bottom). Spatial distribution of SK2 protein became more disorganized in cardiomyocytes from failing hearts, suggesting partial loss of SK2 channel clusters at striations. Both HF and non‐HF cardiomyocytes exhibited areas of unspecific yellow fluorescence, which were also present in the absence of the fluorescent secondary antibody and therefore result from the overlap of unspecific red and green autofluorescence in processed tissue. Small arteries in both nonfailing and failing hearts were found to express SK2, in agreement with previous observations by others.22
Figure 5.
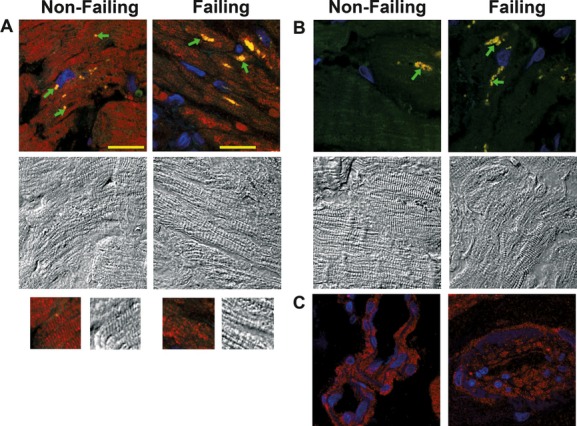
Anti‐KCNN2 immune reactivity in human cardiac tissue. A, Color and black‐and‐white panels show confocal fluorescence images and differential interference contrast (DIC) images, respectively, taken from nonfailing and failing hearts stained for KCNN2. Arrows denote areas of yellow fluorescence that were also detectable in the absence of the secondary antibody and result from the overlap of unspecific red and green autofluorescence in the tissue from nonfailing and failing hearts. B, Confocal fluorescence and DIC images taken from sections that were incubated with the secondary antibody only. C, Anti‐KCNN2 immunofluorescence in small‐artery walls. Scale bar, 20 μm.
Discussion
In this study, we report several novel findings: (1) IKAS was upregulated in failing human ventricles; (2) the distribution of IKAS was transmurally heterogeneous with the midmyocardial layer expressing significantly less IKAS than the epicardial and the endocardial layers; (3) the upregulated IKAS significantly contributes to the repolarization of failing ventricular myocytes; (4) both increased SK2 protein and increased Ca2+ sensitivity underlie the upregulation of IKAS; and (5) partial loss of SK2 channel clusters at striations in failing ventricles.
IKAS and Ventricular Arrhythmogenesis in HF
Abnormal Cai handling is one of the major underlying mechanisms of arrhythmias associated with HF. For example, diastolic Cai can be elevated in HF due to the increased Ca2+ leak from hyperphosphorylated ryanodine receptors23–24 and reduced Ca2+ uptake by sarcoplasmic reticulum Ca2+‐ATPase.24 The increased Cai can activate the Na+‐Ca2+ exchanger, which tends to prolong APDs, resulting in EADs and triggered activity.25 The APD‐prolonging effects of Cai accumulation are counterbalanced by the activation of various K+ currents in normal ventricular myocytes. However, because of the downregulation of multiple major K+ currents (Ito, IKs, IKr, IK1, and IKATP),3–6 the HF cardiomyocytes have reduced ability to shorten the APD. This reduced repolarization reserve may play an important role in proarrhythmia in failing ventricles. In our study, some of the midmyocardial layer myocytes in failing ventricles did not show increased IKAS. These cardiomyocytes might be particularly vulnerable to afterdepolarizations and triggered activities.
While the upregulation of IKAS may be antiarrhythmic, it can also be proarrhythmic. We showed that IKAS is upregulated in epicardial and endocardial myocytes in failing human ventricles. In these cells, IKAS is almost constitutively active due to the increased Ca2+ sensitivity. Although Cai in the range of 300 to 400 nmol/L activates only 10% of maximal IKAS in nonfailing myocytes, 50% of maximal IKAS can be activated in failing myocytes (Figure 3). The activation of IKAS can lead to excessive shortening of APDs in these cells during increased Cai states (such as during VF or during sustained ventricular tachycardia), which can promote late phase 3 EAD and triggered activities.12,23 Because elevated Cai may persist after successful defibrillation,11,26 the increased Cai can activate IKAS, leading to further shortening of APDs and spontaneously recurrent VF (electrical storm).8,12 Therefore, the upregulation of IKAS, by itself, can be one of the important underlying mechanisms of “VF begets VF.” In addition, due to the heterogeneous distributions of IKAS, an increase in Cai caused by tachycardia or sympathetic activation may cause different magnitudes of IKAS activation and thereby can increase transmural heterogeneity of APDs, which is known to be proarrhythmic. Taking these facts together, it is reasonable to speculate that the upregulation of IKAS can be both antiarrhythmic and proarrhythmic in HF.
Increased SK2 Proteins in Failing Ventricles
Macroscopic whole‐cell currents are determined by expression levels of channel proteins and ion channel functions such as channel open probability. The upregulation of IKAS can be achieved by an alteration of the expression of SK channels and/or the Ca2+‐sensitivity of IKAS. It has been reported that 3 different subtypes of SK channels (SK1, SK2, and SK3) are expressed in human hearts.27 Among these channel subtypes, SK2 is known to be the most sensitive to apamin and carry most of the IKAS.20 Our Western blot analyses using anti‐SK2 channel specific antibody showed that bands with a molecular mass of 66 kDa, corresponding to the known molecular size of SK2 variant 1 (NM_021614), were increased by 3‐fold in the failing ventricles compared with the nonfailing ventricles. Because we detected a similar band in the human atria, which is known to express SK2 channels, we believe that SK2 channels were overexpressed in the failing ventricles of these patients. All these findings are consistent with our hypothesis that increased SK2 channel protein expression gave rise to increased IKAS in failing ventricles. An intriguing finding is the appearance of protein bands at a molecular mass of 31 kDa in the failing ventricles but little or none in the nonfailing ones. Although the true identity of these smaller bands was unclear, a known isoform of human SK2, SK2‐s (NM_170775.1), might contribute to these 31‐kDa bands. There are no previous reports on the expression and function of SK2‐s in failing human hearts. Identification and characterization of this protein will be informative for further understanding the biophysical properties of IKAS in failing ventricles.
Subcellular Distribution of SK2 Protein
The subcellular distribution of SK2 protein in human ventricular myocytes is unknown. Previous immunohistochemical analyses have demonstrated that SK2 proteins are localized along the Z‐line in human and canine ventricular myocytes.14,17,28 Our immunofluorescence confocal imaging revealed SK2 protein localization along the Z‐line in nonfailing heart that is similar to that seen in murine atrial myocytes. On the contrary, in failing hearts, the distribution of SK2 proteins was more diffuse, compatible with the notion that the mechanism responsible for SK2 protein alignment in physiological conditions is disrupted in failing ventricular cardiomyocytes. Possible reasons include structural remodeling of the transverse‐axial tubular system29 and changes in targeting of the channel protein to or anchoring in the sarcolemma.30–31 The functional consequences of these spatial rearrangements will be the subject of future investigations.
SK2 Proteins and the Upregulation of IKAS
It has been reported that the gating of rat SK2 channels solely depends on Cai with a half‐maximal Kd of 500 nmol/L in Xenopus oocytes with an inside‐out patch technique.32 A similar Kd value was obtained in our whole‐cell current measurement in myocytes with intact regulatory proteins of SK channels. Interestingly, the Kd in the failing ventricular myocytes was significantly lower than the control (314±4 nmol/L versus 605±28 nmol/L). These results suggest that channel open probability is higher in failing ventricular cells than in control cells at the same Cai (ie, more sensitive to Cai; Figure 2). Recent studies identified several regulatory proteins that coassemble and regulate SK channels.33–34 These proteins include calmodulin, protein kinase CK2, and protein phosphatase (PP)2A. The study using a heterologous expression system demonstrated that CK2 can phosphorylate threonine 80 of calmodulin, resulting in a right‐shift of Ca2+ sensitivity of SK2 channels. This finding suggests that the left‐shift of Ca2+ sensitivity of IKAS observed in our study can be caused by a decrease in phosphorylation of calmodulin, by increased CK2 activity and/or increased PP2A activity. Unfortunately, limited availability of fresh human samples prevented us from making any conclusions regarding the role of these regulatory proteins in the current study. According to the study using canine heart failure models by Yeh et al,24 the protein expressions of PP1 and PP2A were not altered, but the activity of PP1 was significantly increased in failing atria. Although the role of CK2/PP1 in the regulation of SK channels in cardiomyocytes is not fully understood, there is a possibility that CK2/PP1 can alter the Ca2+ sensitivity of IKAS in human HF by modulating phosphorylation status of calmodulin, which requires further biochemical studies.
Study Limitations
There are several significant limitations in this study: (1) our control subjects had aortic stenosis and normal left ventricular ejection fraction because healthy hearts were not available for this study. These control patients are significantly older than the patients with HF, and there is a possibility that aging can reduce IKAS in normal human ventricles. However, studies using animal models and normal human samples agreed with the findings that IKAS was poorly expressed in the nonfailing ventricles.14,28 Therefore, it is reasonable to speculate that IKAS expression is not significantly affected solely by age. (2) Some of the patients with HF were taking antiarrhythmic agents. Whether those antiarrhythmic agents could be washed out during the cell isolation and patch‐clamp experiments remains unknown. It is possible that some of the antiarrhythmic agents may affect the function and expression of SK2 channels. (3) Because the number of patients was small and the study design was cross‐sectional (ie, done at the time of transplant), we cannot determine whether the upregulation of IKAS can alter the patients' mortality and incidence of arrhythmias. (4) Because the time window for the experiments was limited to no more than 10 hours after isolation, we were not able to study a large number of cells in each patient. As indicated in a recent study using human left ventricular wedge preparation,35 the distribution of M cells can be highly heterogeneous in human ventricles. Therefore, it is possible that we have studied both M cells and non‐M cells in the midmyocardial layer, leading to a large variation of the current densities.
Conclusions
Our study for the first time demonstrated the heterogeneous upregulation of IKAS in failing human ventricles. Both increased protein expression and increased Ca2+ sensitivity can contribute to the IKAS upregulation. Because all known K+ currents are downregulated in HF, the IKAS plays a unique role in maintaining repolarization reserve in failing human ventricles. We propose that IKAS plays an important role in ventricular repolarization in failing human ventricles and thereby pose as a new therapeutic target in patients with HF.
Sources of Funding
This study was supported in part by NIH grants P01HL78931, R01HL78932, and R01HL71140, Medtronic‐Zipes Endowment (P.‐S.C.), Methodist Research Institute Showalter Cardiovascular Research Awards (T.A.), and the Indiana University Health‐Indiana University School of Medicine Strategic Research Initiative (T.A., P.‐S.C.).
Disclosures
Our laboratory receives equipment donations from Medtronic Inc, St. Jude Inc, Cryocath Inc, and Cyberonics Inc. P.‐S.C. is a consultant to Cyberonics Inc.
Acknowledgments
The authors are grateful to all patients who participated in this study. The heart tissues and clinical data were provided by Thomas C. Wozniak, MD, John W. Brown, MD, Mark W. Turrentine, MD, Jacqueline A. O'Donnell, MD, Irmina Gradus‐Pizlo, MD, Adnan Malik, MD, and Azam Hadi, MD, at Indiana University School of Medicine. We also thank Jian Tan, MS, Jason Garlie, MD, Elizabeth Lopshire, BA, Lei Lin, David Wagner, and David Adams for their technical assistance; Susan Straka, RN, for coordinating the research protocol; and Changyu Shen, PhD, for statistical consultation.
References
- 1.Bui AL, Horwich TB, Fonarow GC. Epidemiology and risk profile of heart failure. Nat Rev Cardiol. 2011; 8:30-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McMurray J, Kober L, Robertson M, Dargie H, Colucci W, Lopez‐Sendon J, Remme W, Sharpe DN, Ford I. Antiarrhythmic effect of carvedilol after acute myocardial infarction: results of the carvedilol post‐infarct survival control in left ventricular dysfunction (CAPRICORN) trial. J Am Coll Cardiol. 2005; 45:525-530 [DOI] [PubMed] [Google Scholar]
- 3.Aiba T, Tomaselli GF. Electrical remodeling in the failing heart. Curr Opin Cardiol. 2010; 25:29-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nattel S, Maguy A, Le BS, Yeh YH. Arrhythmogenic ion‐channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev. 2007; 87:425-456 [DOI] [PubMed] [Google Scholar]
- 5.Hodgson DM, Zingman LV, Kane GC, Perez‐Terzic C, Bienengraeber M, Ozcan C, Gumina RJ, Pucar D, O'Coclain F, Mann DL, Alekseev AE, Terzic A. Cellular remodeling in heart failure disrupts K(ATP) channel‐dependent stress tolerance. EMBO J. 2003; 22:1732-1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shimokawa J, Yokoshiki H, Tsutsui H. Impaired activation of ATP‐sensitive K+ channels in endocardial myocytes from left ventricular hypertrophy. Am J Physiol Heart Circ Physiol. 2007; 293:H3643-H3649 [DOI] [PubMed] [Google Scholar]
- 7.Weiss JN, Garfinkel A, Karagueuzian HS, Chen PS, Qu Z. Early afterdepolarizations and cardiac arrhythmias. Heart Rhythm. 2010; 7:1891-1899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ogawa M, Morita N, Tang L, Karagueuzian HS, Weiss JN, Lin SF, Chen PS. Mechanisms of recurrent ventricular fibrillation in a rabbit model of pacing‐induced heart failure. Heart Rhythm. 2009; 6:784-792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patterson E, Po SS, Scherlag BJ, Lazzara R. Triggered firing in pulmonary veins initiated by in vitro autonomic nerve stimulation. Heart Rhythm. 2005; 2:624-631 [DOI] [PubMed] [Google Scholar]
- 10.Burashnikov A, Antzelevitch C. Reinduction of atrial fibrillation immediately after termination of the arrhythmia is mediated by late phase 3 early afterdepolarization‐induced triggered activity. Circulation. 2003; 107:2355-2360 [DOI] [PubMed] [Google Scholar]
- 11.Zaugg CE, Wu ST, Barbosa V, Buser PT, Wikman‐Coffelt J, Parmley WW, Lee RJ. Ventricular fibrillation‐induced intracellular Ca2+ overload causes failed electrical defibrillation and post‐shock reinitiation of fibrillation. J Moll Cell Cardiol. 1998; 30:2183-2192 [DOI] [PubMed] [Google Scholar]
- 12.Chua SK, Chang PC, Maruyama M, Turker I, Shinohara T, Shen MJ, Chen Z, Shen C, Rubart‐von der Lohe M, Lopshire JC, Ogawa M, Weiss JN, Lin SF, Ai T, Chen PS. Small‐conductance calcium‐activated potassium channel and recurrent ventricular fibrillation in failing rabbit ventricles. Circ Res. 2011; 108:971-979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown JW, Ruzmetov M, Fiore AC, Rodefeld MD, Girod DA, Turrentine MW. Long‐term results of apical aortic conduits in children with complex left ventricular outflow tract obstruction. Ann Thorac Surg. 2005; 80:2301-2308 [DOI] [PubMed] [Google Scholar]
- 14.Xu Y, Tuteja D, Zhang Z, Xu D, Zhang Y, Rodriguez J, Nie L, Tuxson HR, Young JN, Glatter KA, Vazquez AE, Yamoah EN, Chiamvimonvat N. Molecular identification and functional roles of a Ca(2+)‐activated K+ channel in human and mouse hearts. J Biol Chem. 2003; 278:49085-49094 [DOI] [PubMed] [Google Scholar]
- 15.Bers DM, Patton CW, Nuccitelli R. A practical guide to the preparation of Ca(2+) buffers. Methods Cell Biol. 2010; 99:1-26 [DOI] [PubMed] [Google Scholar]
- 16.Adelman JP, Maylie J, Sah P. Small‐conductance Ca(2+)‐activated K(+) channels: form and function. Annu Rev Physiol. 2011; 74:245-269 [DOI] [PubMed] [Google Scholar]
- 17.Tuteja D, Rafizadeh S, Timofeyev V, Wang S, Zhang Z, Li N, Mateo RK, Singapuri A, Young JN, Knowlton AA, Chiamvimonvat N. Cardiac small conductance Ca2+‐activated K+ channel subunits form heteromultimers via the coiled‐coil domains in the C termini of the channels. Circ Res. 2010; 107:851-859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ishii TM, Maylie J, Adelman JP. Determinants of apamin and D‐tubocurarine block in SK potassium channels. J Biol Chem. 1997; 272:23195-23200 [DOI] [PubMed] [Google Scholar]
- 19.Tuteja D, Xu D, Timofeyev V, Lu L, Sharma D, Zhang Z, Xu Y, Nie L, Vazquez AE, Young JN, Glatter KA, Chiamvimonvat N. Differential expression of small‐conductance Ca2+‐activated K+ channels SK1, SK2, and SK3 in mouse atrial and ventricular myocytes. Am J Physiol Heart Circ Physiol. 2005; 289:H2714-H2723 [DOI] [PubMed] [Google Scholar]
- 20.Kohler M, Hirschberg B, Bond CT, Kinzie JM, Marrion NV, Maylie J, Adelman JP. Small‐conductance, calcium‐activated potassium channels from mammalian brain. Science. 1996; 273:1709-1714 [DOI] [PubMed] [Google Scholar]
- 21.Li N, Timofeyev V, Tuteja D, Xu D, Lu L, Zhang Q, Zhang Z, Singapuri A, Albert TR, Rajagopal AV, Bond CT, Periasamy M, Adelman J, Chiamvimonvat N. Ablation of a Ca2+‐activated K+ channel (SK2 channel) results in action potential prolongation in atrial myocytes and atrial fibrillation. J Physiol. 2009; 587:1087-1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burnham MP, Bychkov R, Feletou M, Richards GR, Vanhoutte PM, Weston AH, Edwards G. Characterization of an apamin‐sensitive small‐conductance Ca(2+)‐activated K(+) channel in porcine coronary artery endothelium: relevance to EDHF. Br J Pharmacol. 2002; 135:1133-1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsuji Y, Hojo M, Voigt N, El‐Armouche A, Inden Y, Murohara T, Dobrev D, Nattel S, Kodama I, Kamiya K. Ca(2+)‐related signaling and protein phosphorylation abnormalities play central roles in a new experimental model of electrical storm. Circulation. 2011; 123:2192-2203 [DOI] [PubMed] [Google Scholar]
- 24.Yeh YH, Wakili R, Qi XY, Chartier D, Boknik P, Kaab S, Ravens U, Coutu P, Dobrev D, Nattel S. Calcium‐handling abnormalities underlying atrial arrhythmogenesis and contractile dysfunction in dogs with congestive heart failure. Circ Arrhythm Electrophysiol. 2008; 1:93-102 [DOI] [PubMed] [Google Scholar]
- 25.Hobai IA, O'Rourke B. Enhanced Ca(2+)‐activated Na(+)‐Ca(2+) exchange activity in canine pacing‐induced heart failure. Circ Res. 2000; 87:690-698 [DOI] [PubMed] [Google Scholar]
- 26.Koretsune Y, Marban E. Cell calcium in the pathophysiology of ventricular fibrillation and in the pathogenesis of postarrhythmic contractile dysfunction. Circulation. 1989; 80:369-379 [DOI] [PubMed] [Google Scholar]
- 27.Rimini R, Rimland JM, Terstappen GC. Quantitative expression analysis of the small conductance calcium‐activated potassium channels, SK1, SK2 and SK3, in human brain. Brain Res Mol Brain Res. 2000; 85:218-220 [DOI] [PubMed] [Google Scholar]
- 28.Nagy N, Szuts V, Horvath Z, Seprenyi G, Farkas AS, Acsai K, Prorok J, Bitay M, Kun A, Pataricza J, Papp JG, Nanasi PP, Varro A, Toth A. Does small‐conductance calcium‐activated potassium channel contribute to cardiac repolarization? J Mol Cell Cardiol. 2009; 47:656-663 [DOI] [PubMed] [Google Scholar]
- 29.Crossman DJ, Ruygrok PN, Soeller C, Cannell MB. Changes in the organization of excitation‐contraction coupling structures in failing human heart. PLoS ONE. 2011; 6:e17901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lyon AR, MacLeod KT, Zhang Y, Garcia E, Kanda GK, Lab MJ, Korchev YE, Harding SE, Gorelik J. Loss of t‐tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc Natl Acad Sci USA. 2009; 106:6854-6859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu L, Zhang Q, Timofeyev V, Zhang Z, Young JN, Shin HS, Knowlton AA, Chiamvimonvat N. Molecular coupling of a Ca2+‐activated K+ channel to L‐type Ca2+ channels via alpha‐actinin2. Circ Res. 2007; 100:112-120 [DOI] [PubMed] [Google Scholar]
- 32.Hirschberg B, Maylie J, Adelman JP, Marrion NV. Gating of recombinant small‐conductance Ca‐activated K+ channels by calcium. J Gen Physiol. 1998; 111:565-581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bildl W, Strassmaier T, Thurm H, Andersen J, Eble S, Oliver D, Knipper M, Mann M, Schulte U, Adelman JP, Fakler B. Protein kinase CK2 is coassembled with small conductance Ca(2+)‐activated K+ channels and regulates channel gating. Neuron. 2004; 43:847-858 [DOI] [PubMed] [Google Scholar]
- 34.Allen D, Fakler B, Maylie J, Adelman JP. Organization and regulation of small conductance Ca2+‐activated K+ channel multiprotein complexes. J Neurosci. 2007; 27:2369-2376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glukhov AV, Fedorov VV, Lou Q, Ravikumar VK, Kalish PW, Schuessler RB, Moazami N, Efimov IR. Transmural dispersion of repolarization in failing and nonfailing human ventricle. Circ Res. 2010; 106:981-991 [DOI] [PMC free article] [PubMed] [Google Scholar]