

Abstract
Exposure to traumatic events that produce extreme fear and horror is all too common in both military and civilian populations, but not all individuals develop posttraumatic stress disorder (PTSD) as a result of the exposure. What mediates risk and resilience in the development of PTSD and other stress-related psychopathology is of paramount importance to our further understanding of trauma-related psychopathology as well as the development of new treatment approaches. Biological factors, such as genotype and neurobiology, interact with environmental factors, such as childhood background and trauma load, to affect vulnerability and resilience in the aftermath of trauma exposure. One of the core symptoms of PTSD is the inability to control fear, which has led some investigators and clinicians to conceptualize PTSD as a disorder of fear or, more importantly, its inhibition. This review focuses on translational methods that have been used to examine fear conditioning and inhibition of fear in PTSD and summarizes genetic and neurobiological factors related to fear inhibition. The authors also discuss different pharmacological approaches that enhance fear inhibition and may improve treatment outcomes for patients with PTSD.
The popular expression “What does not kill you makes you stronger” points to the fact that some people respond resiliently to trauma. This statement may be true for highly resilient people. However, for those who are vulnerable, a more appropriate statement might be “What does not kill you can make you ill.” Such vulnerability is common. Approximately one-tenth of those who survive life-threatening events will develop mental health disorders such as posttraumatic stress disorder (PTSD) or depression or both (1, 2). One of the goals of modern psychiatry is to identify vulnerable individuals and intervene to prevent the development of these disorders by bolstering resiliency. The factors that contribute to resiliency encompass both biological and psychological aspects of the individual as well as the pre- and posttrauma environment (3). It has also been suggested that resiliency is a product of early stress—that is, that resiliency is an adaptive response that maintains homeostasis under stressful circumstances (4). However, this response is true only for some individuals; for others, traumatic stress can increase vulnerability.
Resiliency results from a combination of both biological factors (which are heritable) and environmental factors to which the individual is exposed (Figure 1). The environmental factors that promote resiliency, including social support after trauma, have been the focus of treatment for PTSD. The biological factors may be based on heritable genetic profiles, which may code for neurochemicals and neural mechanisms that promote resiliency. Recent studies have shown that specific gene alleles are associated with resilience, such that even severe levels of child abuse do not result in severe psychopathology (5, 6). The genetic profile may also code for associative learning mechanisms, such as fear conditioning, to enhance fear responses or to enhance fear extinction, which promotes suppression of fear responses to previously fearful stimuli (3).
FIGURE 1.
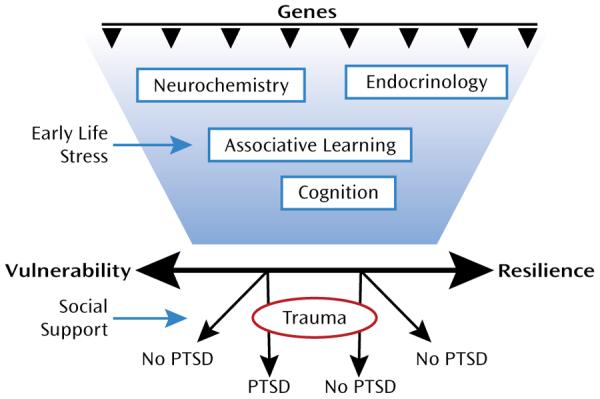
Schematic Diagram of Genetic, Neurobiological, and Environmental Interactions That Contribute to Vulnerability or Resilience in Relation to PTSD
Vulnerability to the development of PTSD after trauma exposure may be associated with an exaggerated fear response or an inability to control fear responses, which could either be a risk factor for the disorder (7) or an acquired trait of the illness (8). The DSM-IV (9) diagnosis of PTSD requires exposure to a traumatic event and a cluster of symptoms associated with that event (e.g., psychological and physiological reactions to trauma reminders and avoidance of such reminders). Consequently, several theorists (see reference 10, for example) have proposed that conditioning processes are involved in the etiology and maintenance of PTSD. Especially pertinent to this view is the idea that through the processes of Pavlovian conditioning, a neutral (conditioned) stimulus that occurs in temporal contiguity with an aversive (unconditioned) stimulus that innately elicits pain and fear acquires the ability to elicit a fear response in the absence of the unconditioned stimulus. Thus, neutral stimuli (the conditioned stimuli) present at the time of the trauma (the unconditioned stimulus) acquire the ability to elicit a conditioned fear reaction that can be triggered when the person subsequently encounters these or similar stimuli during the course of normal life. Consistent with this hypothesis, emotional and physiological reactivity to stimuli resembling the original traumatic event even years after the event's occurrence is a prominent characteristic of PTSD and has been reliably replicated in the laboratory (see references 11–13, for example). While PTSD is a complex disorder that includes the dysregulation of other emotions besides fear, such as anger or guilt, and is highly comorbid with depression, the study of fear lends itself best to translational approaches (14–16). In this review we first focus on the neural circuits that are involved in inhibition of fear responses and then discuss recent genetic findings in the area. We conclude with a discussion of how these results may be combined in a neurogenetic model that incorporates risk and resilience to trauma-related disorders and indicates prevention and treatment targets in the time course of development of the disorder.
Fear Inhibition as an Intermediate Phenotype
Fear inhibition involves learning of safety signals— that is, the ability to discriminate between danger and safety cues and to suppress fear responses in the presence of safety cues. In the laboratory fear inhibition can be measured by first using a fear conditioning paradigm for fear acquisition, which is then followed by the training of fear inhibition. Fear conditioning is based on a simple Pavlovian conditioning model, in which a neutral conditioned stimulus (CS; for example, a light) is paired with an aversive unconditioned stimulus (US; for example, an electric shock). After a number of pairings, the association is formed so that the CS alone elicits the conditioned response (CR; for example, a fear response). This basic model is used in animal as well as human research to investigate mechanisms of fear acquisition.
Two major laboratory models have been used for behavioral testing of fear inhibition in animals and humans: extinction and conditioned inhibition. While fear acquisition refers to learning that something is dangerous, extinction is a mechanism by which an individual learns that something that previously elicited fear is no longer dangerous—that is, that it is safe. In fear extinction paradigms, a stimulus that was previously paired with an aversive stimulus (the CS+) is then repeatedly presented without the US, so that it no longer elicits a fear response (17, 18). In a basic conditioned inhibition paradigm, the above CS+ pairing is intermingled at the time of training with a separate stimulus (CS−). In other words, the CS− does not co-occur with an aversive stimulus and thus represents safety or inhibition of fear. In another standard conditioned inhibition paradigm, one cue is paired with the aversive stimulus when presented alone (CS+, also referred to as A+) but not when presented in compound with a second cue (CS−, represented as AX−, indicating that the combination of A and X is not reinforced). In this model X should become a safety signal because it signals the absence of the aversive stimulus (19).
In humans, two physiological responses have been used as behavioral outcome measures for fear conditioning: acoustic startle response and skin conductance response. The acoustic startle response is characterized by an integrative reflex contraction of the skeletal musculature in response to a strong stimulus. It provides an excellent model to study emotional processing since the amygdala is directly connected with the startle circuit (16, 20–22). Fear-potentiated startle is the relative increase in the acoustic startle response elicited in the presence of a conditioned stimulus (CS+) that was previously paired with an aversive stimulus (US). The skin conductance response is an index of sympathetic nervous system activity that is frequently used in measuring fear acquisition and extinction in tandem with brain imaging studies using positron emission tomography (PET) or functional MRI (fMRI) (8, 23–26).
Unfortunately, traditional conditioned inhibition paradigms have a number of confounding issues, such as second-order conditioning, external inhibition, and configural learning, that make it difficult to discretely separate excitatory fear learning from inhibition of fear in neural circuits. Myers and Davis (27) developed an animal model using a conditional discrimination procedure that allows for the independent evaluation of excitation and inhibition of fear conditioning. In collaboration with this group, we developed a conditioned inhibition paradigm for use in humans (28) that contains a danger signal (AX+), a safety signal (BX−), and a safety transfer test (combination of A and B, where B reduces fear to A) (Figure 2). The procedure, referred to as a conditional discrimination (abbreviated as AX+/BX−), is based on a paradigm used in earlier learning theory experiments (29, 30). In this experiment, the response to stimulus X is conditional on the presence of either A or B. The A stimulus elicits fear potentiation of startle with training as the subject learns that A and X presented together predicts the US. Stimulus B elicits reduced startle compared to A (i.e., becomes inhibitory) in that B presented with X predicts safety from the US. The presentation of AB results in a reduced startle response compared to the response to A presented with a neutral stimulus because B has become inhibitory.
FIGURE 2. Conditional Discrimination Paradigm to Measure Inhibition of Learned Feara.
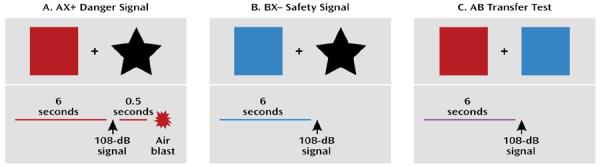
aWhile participating in physiological or neural imaging experiments, the subject observes a computer screen with different colored shapes. In the example in panel A, the danger signal (AX+) is represented by the red square and black star. In panel B, the safety signal (BX−) is represented by the blue square and black star. In panel C, the safety transfer test (combination of A and B, where B reduces fear to A) has both A (red square) and B (blue square) presented simultaneously. The aversive unconditioned stimulus (US) is the air blast that occurs only at the end of the AX+ danger signal. In all three conditions, fear-potentiated startle to the conditioned stimulus (CS) is elicited by a 108-dB startle signal, eliciting an eye blink reflex, which is measured with electromyography.
We translated this paradigm to use in clinical settings and have now demonstrated conditioned inhibition in healthy individuals (28) and in combat veterans with low levels of current PTSD symptoms (31). On the other hand, study subjects with high levels of PTSD symptoms were unable to reduce startle to AB trials (i.e., were unable to transfer fear inhibition). We have also replicated these findings in a sample of veterans with PTSD from the University Hospital Dubrava in Zagreb, Croatia (32), and in a civilian population in Atlanta with high levels of urban trauma (32a). Together these data suggest that PTSD is at least in part a disorder in which inhibition of fear is deficient, even when learned fear in the laboratory is separate from the index trauma(s). An alternative explanation is that fear excitation to the A stimulus in the AB compound is so exaggerated in PTSD that it overwhelms the inhibition from B. This would be consistent with our findings in Vietnam veterans, in that those with the most severe symptoms also had significantly more fear potentiation to the AX+ cue compared to healthy comparison subjects (31). However, our replication samples of combat-related PTSD in Croatia and civilian PTSD in Atlanta did not have increased potentiation to AX+; the group differences were limited to AB and BX− trials, which suggests a selective deficit in fear inhibition.
Both extinction tests and conditioned inhibition focus on active suppression of fear responses through learned safety signals; while fear itself may involve only subcortical areas of the brain located primarily in the limbic circuitry, safety signals may require a cognitive, cortical component (26, 33). This premise is supported by data from our laboratory showing that awareness of the association between the CS and the US is necessary for inhibiting fear responses on the AX+/BX− paradigm (34). Furthermore, a recent study by Weike and colleagues (33) examined the temporal domain of fear conditioning with a danger and safety signal and found that safety signal processing was slower than danger processing. The authors argued that top-down cognitive processes are involved in responses to safety signals, which accounts for the latency in response.
A recent meta-analysis of 15 studies using fear conditioning found that patients with anxiety disorders showed greater levels of fear responses compared to healthy comparison subjects (35). These data suggest that the fear response is overactive or that the inhibition of fear is deficient in PTSD, which has led researchers to use fear conditioning models to examine some of the core PTSD symptoms. One study (36) used a fear-potentiated startle paradigm with veterans diagnosed with PTSD and found equivalent levels of fear potentiation to the danger signal in the PTSD and comparison groups. However, participants with PTSD also potentiated to the safety cue, whereas the comparison subjects did not. Our recent data also show that increased fear responses to safety cues are related to the severity of current PTSD symptoms (31). A recent study of patients with panic disorder (37) found that these patients also had increased fear-potentiated startle responses to the safety cue; this finding may have been related to the patients' increased expectancy of the US during the safety cue. In that study the impaired discrimination between danger and safety appeared to involve both cognitive and physiological deficits. In our study of veterans with PTSD (31), we observed a dissociation between participants' cognitive awareness (they reported that they did not expect to receive an air blast US during the CS-− trial) and startle response, which was potentiated in response to the nonreinforced stimulus. On the other hand, a study by Orr and colleagues (38) that used skin conductance to examine fear conditioning in PTSD patients found that patients discriminated between the danger and safety cues better than did comparison subjects. In another study (39), similarly enhanced conditionability in PTSD patients was found when trauma-related cues were used as the US in fear conditioning; the enhanced fear conditioning was also related to slower extinction. Deficient fear extinction in PTSD has been found in several studies that used skin conductance as the physiological measure (26, 39, 40). A recent study of combat-exposed Vietnam veterans and their non-combat-exposed twins (41) found that combat-exposed veterans with PTSD did not have impaired extinction learning but rather had less extinction retention on the day after acquisition and extinction compared to exposed veterans without PTSD. Furthermore, impaired retention of extinction appeared to be an acquired trait related to the disorder since the twins of the veterans with PTSD did not show the same impairment.
While some data with combat veterans suggest that impaired fear inhibition may be an acquired trait (41) associated with current symptom severity (31), other studies have reported that heightened fear responses and decreased inhibition of fear may be predictors of the disorder. A prospective study of police academy cadets (42) found that greater skin conductance responses to threatening stimuli and slower habituation prior to trauma exposure were predictive of PTSD symptom severity after trauma exposure. A similar prospective study with firefighters (7) found that reduced extinction of fear-conditioned responses before the index trauma explained almost one-third of the variance in PTSD symptom severity in later traumatized individuals. It is possible that a decreased ability to inhibit fear is a risk factor for developing PTSD and contributes to the maintenance of the disorder, while decreased extinction retention is a state resulting from the disorder—given that these fear-inhibition phenotypes may have different neural underpinnings, this would explain the above studies.
For the purposes of this review, we will not define fear inhibition as either a vulnerability or an acquired trait of the disorder; more research is needed before such a determination can be made. However, the issue of whether it is a predisposition or a part of the PTSD syndrome itself does not dismiss the utility of impaired fear inhibition as a phenotype. With the development of new techniques for studying fear acquisition and fear inhibition in animal and human subjects, we can begin to understand how the neurobiology of fear is altered in PTSD. Below we review the animal and human data for some of the primary structures involved in fear conditioning and fear inhibition.
Neurocircuitry of Fear Inhibition
The Amygdala
The amygdala, part of the limbic system located in the temporal lobe of the brain, is an integral part of the fear circuitry (43–45). The amygdala comprises several nuclei, which can be roughly divided into the central nucleus and the basolateral nucleus, among several others (Figure 3). Animal studies have shown that the different nuclei function in different ways. For instance, the central nucleus regulates many aspects of the fear response, including the release of cortisol through the paraventricular nucleus of the hypothalamus, increase in startle response via the pons in the midbrain, and modulation of the autonomic nervous system through the lateral hypothalamus (46). Lesions of the central nucleus eliminate fear-conditioned responses, such as fear-potentiated startle (47) and freezing (43), in rodents. The basolateral nucleus projects to the central nucleus and appears to be the locus for associations between the CS and US that result in the acquisition of fear (48).
FIGURE 3. Inhibitory Control of Amygdala Regulation of Fear.
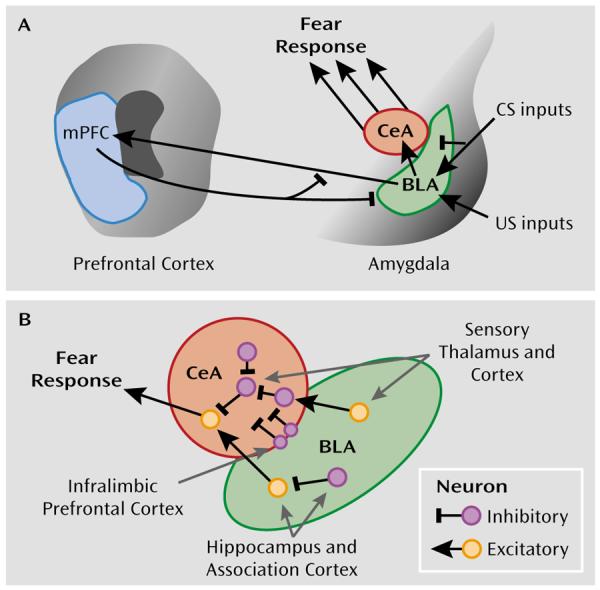
aPanel A is a schematic diagram illustrating the interaction of the basolateral nucleus (BLA) and central nucleus (CeA) of the amygdala with modulatory regions such as the medial prefrontal cortex (mPFC). The basolateral nucleus is thought to compare conditioned stimulus (CS) inputs and unconditioned stimulus (US) inputs regulating central nucleus activation of the hardwired fear and stress circuitry, leading to inhibition or activation of the fear response. Panel B illustrates recent research that has begun to determine the role of inhibitory neural circuitry in modulating the fear response at the cellular level (49, 50, 53, 54). Sensory inputs as well as associative inputs from the hippocampus and cortex project directly and indirectly to the central nucleus. “On” and “off” inhibitory circuits within the central nucleus are thought to differentially modulate fear output and extinction of fear. Additionally, direct projections from the infralimbic region of the medial prefrontal cortex activate inhibitory neurons in the intercalated region between the basolateral and central nuclei, serving to inhibit, in a top-down manner, the fear output of the central nucleus.
More recent work has begun to outline the cellular microcircuitry that underlies fear expression and inhibition (49, 50). As illustrated in Figure 3B, sensory and associative information projects directly and indirectly to the lateral and basolateral amygdala. Excitatory information from the basolateral nucleus is thought to be gated via inhibitory inputs at the level of the intercalated nuclei situated between the basolateral and central nuclei (51). These inputs are regulated via the medial prefrontal cortex and are thought to be required for extinction of fear (52, 53). Within the central nucleus, information flow is gated by “off” and “on” inhibitory neural networks that are thought to differentially regulate fear expression or inhibition (50, 54). Together, these data suggest that complex inhibitory neural circuitry controls fear behavior and its inhibition, which is dysregulated in pathological states that are marked by amygdala dysfunction.
The bed nucleus of the stria terminalis is a related part of the “extended amygdala” that appears to be associated with nonspecific fear, such as anxiety, which is unrelated to a predictable danger cue used in fear conditioning. Lesions of this structure eliminate anxiogenic effects of bright lights and corticotropin-releasing factor infusions in rodents (55). This region is hypothesized to be more involved in general, nonspecific anxiety and depression symptoms, whereas the central nucleus is thought to be more involved in fear, panic, and cue-specific stress responses.
In accord with animal research, brain imaging studies with humans have found that the amygdala modulates the fear response: left hemisphere damage in temporal lobectomy patients results in loss of fear-conditioned startle (56). In healthy intact humans, several studies using PET and fMRI have shown that presentation of fearful stimuli results in amygdala activation. The stimuli include fearful faces (57, 58) and conditioned fear cues (23, 25, 26). In one study (59), participants were instructed to expect a shock when the “threat” cue was on; that study also found amygdala activation in the threat compared to the safe condition. In a review of 55 imaging studies of the functional neuroanatomy of emotion (60), 25 studies related amygdala activation to fearful stimuli while four studies found activation to positive stimuli. These findings indicate that the amygdala plays an extensive role in regulating the fear response in humans as well as animals.
A preponderance of neuroimaging data from the past decade demonstrate that PTSD patients appear to have greater amygdala activation relative to comparison subjects (see reference 61 for a recent review). PET studies using combat scripts (62) and images (63, 64) and single photon emission computed tomography studies comparing responses to combat sounds and to white noise (65) found greater levels of amygdala activation in PTSD patients. Furthermore, recent fMRI studies have found that trauma-relevant words increase amygdala activation (66). This increased fear response extends beyond trauma-specific imagery: fearful faces also activate the amygdala in PTSD patients more than in comparison subjects (67, 68).
The Prefrontal Cortex
The prefrontal cortex has long been thought to play a role in behavioral inhibition. Nearly two decades ago, animal studies showed that lesions of the medial prefrontal cortex prior to original fear conditioning retard extinction to a tone (69). More recent studies have demonstrated that neurons in the prefrontal cortex may have inhibitory action on the amygdala (24, 70). Just as the amygdala has many subparts, so the prefrontal cortex can be subdivided into the medial and orbitofrontal prefrontal cortex. The anterior cingulate cortex, which is also part of the prefrontal cortex, has both ventromedial and dorsolateral components, which may play different roles in the expression and inhibition of fear, as will be discussed in greater detail below.
A study by Milad and Quirk (71) suggests that the rodent neuroanatomical analogue to the medial prefrontal cortex, the infralimbic prefrontal cortex, has enhanced activity following learning of an extinguished CS. Enhanced infralimbic prefrontal cortex activity was also shown to inhibit the fear response. These authors hypothesize that during the consolidation of extinction, a circuit running from the basolateral nucleus to the prefrontal cortex and back to inhibitory neurons within the amygdala may be strengthened such that when the extinguished CS is reexperienced, the infralimbic prefrontal cortex will represent a feed-forward inhibitory projection that will compete with the fear pathway represented within the basolateral-to-central nucleus projection (Figure 3) (72, 73). The preponderance of evidence indicates that neural plasticity within the amygdala and possibly within the medial prefrontal cortex occurs during the consolidation of extinction learning. As we have seen with studies of consolidation of fear conditioning (74–76), other molecular systems and brain regions, including sensory areas and associative cortical and subcortical areas, are undoubtedly also involved.
Neuroimaging studies in humans have used several paradigms that activate the prefrontal cortex, ranging from simple inhibition of a motor response, such as pressing a button, to more complex tasks in which the subject is required to suppress a response on a cognitive interference task. A simple task may involve responding to a letter when presented alone and withholding a response when the letter is paired with another letter or is shown with a colored background. This type of task is often referred to as a go/no-go task (77). A well-known and frequently used example of a complex task is the Stroop effect, where the meaning of a word (such as the word “red”) is in conflict with the color in which it is shown (for example, in blue ink). A novel example of this type of task is the multisource interference task, developed by Bush and Shin (78); in this task the number presented is in conflict with the position in which it is presented so that the subject is required to ignore the interfering information in order to correctly complete the task. When used in an fMRI procedure, this task reliably activates the dorsal anterior cingulate cortex (79).
While the above-mentioned tasks require cognitive inhibition, they do not necessarily involve suppression of emotion and thus may not necessarily map onto the fear inhibition circuitry. A more appropriate task to assess fear inhibition is the emotional Stroop test, in which the emotional content of a word competes with the cognitive content and must be ignored. This task also activates the anterior cingulate, but in a different area than the strictly cognitive interference tasks (80, 81). Emotionally relevant stimuli appear to be processed by the rostral or subgenual area of the anterior cingulate (68), which is anterior to the genu of the corpus callosum, while the dorsal region of the anterior cingulate appears to be more relevant for cognitive tasks (79).
Neuroimaging studies using fear conditioning paradigms demonstrate that fear acquisition and fear extinction also activate the prefrontal cortex, specifically the ventromedial prefrontal cortex (24). Recent developments in the spatial resolution of neuroimaging techniques have resulted in more fine-tuned examinations of this area of the brain. As mentioned above, the rostral or subgenual regions of the anterior cingulate are activated during the presentation of emotional stimuli; these areas are also activated during the regulation of fear (24, 82). Several lines of evidence suggest that this region of the ventromedial prefrontal cortex is associated with inhibition of fear: fMRI data indicate increased activation during extinction recall after extinction learning (24, 83). The ventromedial prefrontal cortex is also activated during fear reversal tasks in which the CS contingencies are switched after acquisition so that a previously conditioned danger cue (CS+) becomes the new safety cue (CS−) (82). Morphometric data show that the thickness of this cortical tissue is correlated with extinction retention (84). Furthermore, the blood-oxygen-level-dependent signal measured with fMRI is greater in the prefrontal cortex when subjects are instructed to “reappraise” a fearful cue—that is, when they actively suppress negative thoughts (85). While functional and morphometric data point to the rostral anterior cingulate during fear inhibition, such data on the dorsal region of the anterior cingulate suggest that this area is associated with fear acquisition (8). Given that this area is also implicated in cognitive tasks (79), it may be activated during the learning procedure of acquisition rather than the fear itself.
Exaggerated fear responses observed in PTSD and the impaired inhibition on conditioned inhibition tasks may be due to a weakened inhibitory control of the amygdala by the prefrontal cortex. PET studies of patients with PTSD show lower activation of the anterior cingulate cortex in response to the emotional Stroop task (86); however, PTSD patients have normal prefrontal cortex activation to nonemotional interference tasks (79, 86). On the other hand, a recent fMRI study of PTSD patients during acquisition, extinction learning, and extinction recall 24 hours later found increased amygdala activation in PTSD patients relative to comparison subjects during extinction learning, and decreased hippocampus and ventromedial prefrontal cortex activation during extinction recall (86a). Weakened prefrontal cortex control of the amygdala may be a risk factor for psychopathology; a recent study of children with depressed parents found a lack of anterior cingulate cortex activation to the emotional Stroop (87).
Interestingly, while all studies found amygdala activation in their study samples as a whole, there were also several instances of individual variability. For instance, LaBar et al. (23) found increased amygdala activation in seven of 10 subjects during early acquisition; Knight et al. (88) found increased amygdala activity only in those individuals who also showed an increased skin conductance response to the danger cue. These individual differences may be due to genotypes that would increase vulnerability to fearful stimuli (89). A recent study found greater fear-potentiated startle in individuals with the short allele of the serotonin transporter gene (90). A gene-by-environment interaction whereby a vulnerable individual is exposed to extreme trauma could lead to the development of PTSD.
Molecular and Genetic Mechanisms of Fear Inhibition
On a molecular level, fear conditioning involves new learning mediated by synaptic plasticity in the amygdala. Both associative fear conditioning and extinction of conditioned fear, a learning process by which a CS is no longer associated with the US, are dependent on activation of glutamate N-methyl-d-aspartate (NMDA) receptors. Administration of NMDA receptor antagonists either systemically (91, 92) or by direct infusion into the basolateral nucleus (93, 94) prior to extinction training blocks the extinction of fear memories. In addition, blockade of NMDA receptors after extinction training also impairs extinction, which suggests that NMDA receptors participate in the consolidation of extinction memories (95). In addition to these data, there is evidence that voltage-gated calcium channels are involved in mediating calcium-dependent synaptic plasticity, which may underlie extinction (96, 97). Additionally, a significant amount of data implicate brain-derived neurotrophic factor (BDNF) in the plasticity underlying fear and extinction learning through its TrkB receptor (74, 98).
There are also substantial data indicating that regulation of the inhibitory neurotransmitter γ-aminobutyric acid (GABA) is altered differentially in the acquisition of fear versus extinction. Injection of an inverse agonist, FG7142, which blocks GABA function, was shown to block the context-specific effects of extinction learning (99). Gephyrin, a scaffolding protein involved in GABA insertion into the surface membrane, is decreased at the protein and mRNA level in the amygdala following fear learning and is increased in the amygdala with extinction learning (100, 101). These data are consistent with enhanced amygdala excitability with fear learning and an increase in amygdala inhibitory tone following extinction. One study (102) demonstrated that blockade of GABA insertion within the amygdala impairs extinction of conditioned fear. Overall these data suggest that modulation of GABA-ergic microcircuitry within the central and basolateral nuclei is critically involved in the regulation of fear and its inhibition with extinction learning.
These molecular data suggest that expression of genes associated with neural plasticity (e.g., BDNF and glutamate receptors), neural inhibition (e.g., GABA receptors and cannabinoid receptors), and stress responsiveness (e.g., glucocorticoid receptors and corticotropin receptor) may be associated with the learning of extinction or impaired fear inhibition. Abnormal fear acquisition or inhibition, as described above, appears to be associated with PTSD, either as vulnerability factors preceding trauma exposure or as a consequence of trauma-related fear conditioning.
To date, much of the research on the genetic basis for PTSD has been gathered via twin studies. Data from these studies indicate that heritability accounts for 30%–40% of the variance in risk for PTSD (103–105). Despite known genetic contributions to risk for PTSD, there have been no linkage studies and only a handful of candidate gene association studies examining genetic main effects to date. In general, these studies have revealed that there are complex interactions between genetic and environmental factors and that many of the identified genetic polymorphisms are in the regulatory promoter regions and not necessarily in the coding regions (106, 107).
Several recent reviews have examined the genetics of PTSD (108, 109, 110), so we will not go into detail on this issue here. No genes have yet been reported that appear to have large main effects across the expected several replications in association with PTSD. Among the more replicated findings to date showing an effect are genes encoding the dopamine receptor 2 (111, 112) and the serotonin transporter (113, 114). However, these findings have not been uniformly replicated (115). As for the genes described above, although two studies have reported no associations with BDNF (116), there have been no reported studies, to our knowledge, of glutamatergic plasticity-related genes, and only one study has sought genetic links between GABA and PTSD (117). In sum, little is known about the genetic mechanisms of PTSD, including the potential genetic role of the glutamatergic and GABA-ergic systems.
Gene-by-Environment Interactions in PTSD
Although a small but growing body of psychiatric research has identified gene-by-environment interactions predicting other mental disorders or associated symptoms, to date only a handful of published studies have presented data on a gene-by-environment interaction predicting PTSD. The first focused on the serotonin promoter length polymorphism (5-HTTLPR), which was originally described as interacting with level of prior stress to predict adult depression (118). Kilpatrick and colleagues (119) identified a gene-by-environment interaction predictive of PTSD in an analysis of individuals exposed to hurricanes in south Florida. The study also classified PTSD patients according to the degree of social support available following trauma exposure. As part of the same hurricane study, Koenen and colleagues (120) reported that the “s” allele of the 5-HTTLPR polymorphism was associated with a lower risk of PTSD in low-risk environments (low crime and unemployment rates) but a higher risk of PTSD in high-risk environments. These results suggest that social environment modifies the effect of 5-HTTLPR genotype on PTSD risk.
The other primary series of studies exploring gene-by-environment effects examined FKBP5, a protein that modulates the glucocorticoid receptor (GR). FKBP5 is a co-chaperone that regulates GR sensitivity (121). Its binding to GR decreases nuclear translocation and decreases GR sensitivity. FKBP5 mRNA and protein expression are induced by GR activation, providing an ultrashort feedback loop for GR sensitivity. Polymorphisms in FKBP5 have been shown to associate with differential up-regulation of FKBP5 following GR activation and with differences in GR sensitivity and stress hormone system regulation. Because of the known role of abnormal GR sensitivity in PTSD (122, 123), FKBP5 appeared to be a good candidate gene that may underlie this component of PTSD and perhaps risk for PTSD. Additionally, FKBP5 polymorphisms had been reported to associate with peritraumatic dissociation (a known risk factor for PTSD) in medically injured children (124). Finally, FKBP5 blood mRNA levels have been found to differentially associate with PTSD in two separate studies (125, 126). In the largest candidate gene study of PTSD to date (5), our group showed a gene-by-environment effect of the polymorphisms in FKBP5 with a history of childhood maltreatment to predict level of adult PTSD symptoms in a traumatized civilian sample. Notably, no main effects were found for the FKBP5 genotype directly associating with PTSD symptoms, nor was there an effect of the gene interacting with adult trauma levels. These data suggest that the interaction of trauma, perhaps during a developmentally critical period, with the hypothalamic-pituitary-adrenal (HPA) axis stress-related genes alters amygdala regulation of fear and its inhibition later in life. These early developmental alterations would then increase risk or enhance resiliency in relation to the development of PTSD in adults following an index trauma.
Temporal Model of Pathophysiology and Treatment Options for PTSD
Conceptualizing PTSD as a disorder of fear conditioning (10, 127–129) leads to the use of fear inhibition experiments to identify vulnerable and resilient individuals and can be used for testing treatments that bolster resilience. Figure 4 shows a putative timeline for the development of fear-related disorders, including PTSD. It starts with factors that increase vulnerability, such as genotype (for example, AA genotype of the FKBP5 rs9296158 gene) and early life stress (such as childhood abuse). Exposure to trauma results in an association of all the external and internal stimuli present at the time of the event and the emotions of fear, horror, and helplessness that define the criteria of trauma. In an individual who will develop PTSD, these stimuli may later serve as reminders and cause symptoms of reexperiencing and hyperarousal. It is possible that intervention at this time, immediately following the event, can prevent the formation of a strong fear association and thus promote recovery. Several studies are under way in which victims of assault are treated in a hospital emergency department within hours after trauma exposure (130).
FIGURE 4. The Developmental Progression of PTSD.
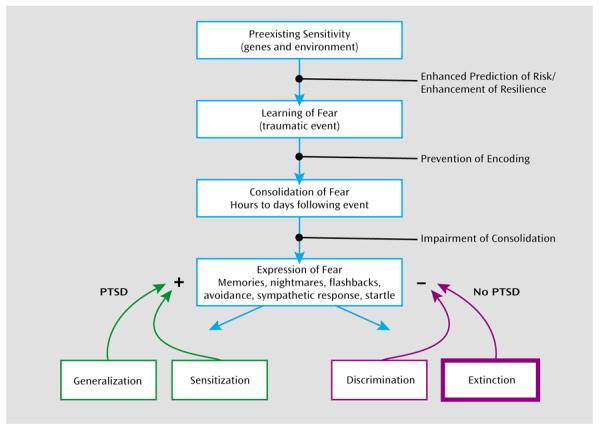
aThe strength and regulation of fearful memories is affected by numerous factors both before and after the traumatic or fearful event occurs. Genetic heritability comprises up to ~40% of the risk for both depression and PTSD, and early childhood abuse is a strong risk factor for all mood and anxiety disorders. Further understanding of the roles of genes and environment may allow enhanced prediction of risk and enhancement of resilience in vulnerable populations. Memories are not permanent at the time of the trauma, and psychological and pharmacological approaches to prevent the initial encoding of the trauma are under study. Memories then undergo a period of consolidation in which they shift from a labile state to a more permanent state. Impairing the consolidation (or even reconsolidation) would be an alternative way to prevent the sequelae of long-term trauma memories. The expression of traumatic memories, which can be the source of symptoms in fear-related disorders, is diminished by the process of extinction when repeated therapeutic exposures to the fear-related cues reduce or inhibit the fear memories over time. In contrast, there is some evidence that in individuals who develop PTSD and other pathology, a combination of avoidance of sufficient exposure with intrusive and uncontrollable memories leads to sensitization of the fear response. Enhancing discrimination and extinction of fear memories is a key aspect of recovery in the psychotherapeutic approaches to treating PTSD.
Once the fear memory is formed, it can still be modified by methods that interfere with fear memory consolidation and its potential reconsolidation (131). Consolidation and reconsolidation refer to the phenomenon in which a memory is repeatedly strengthened when trauma reminders become associated with hyperarousal symptoms; this results in a vicious circle by which fear memories lead to anxiety disorders. At this time point, it may be possible to intervene and modify the memory by associating it with safety rather than danger cues (132, 133). If the fear memory is consolidated, trauma reminders will elicit expression of the fear response—that is, amygdala hyperactivity (61). Exaggerated amygdala activity will be evident in the form of symptoms such as intrusive memories, nightmares, exaggerated startle responses (9), and sympathetic nervous system activation, which increases heart rate, respiration rate, and sweating (134). At this point in the course of the development of the disorder, individuals with healthy fear inhibition neurocircuitry might engage the prefrontal cortex to dampen amygdala activity. In fact, a new study (134a) using an extinction paradigm that combined a single reactivation trial 10 minutes prior to extinction found that the fear memory was significantly reduced. More importantly, even a year after extinction, the fear memory was still reduced, indicating resistance to spontaneous recovery of fear. The authors argued that the fear memory, once reactivated, did not have an opportunity to be reconsolidated because of extinction. Treatments that employ mechanisms of fear inhibition, such as extinction, can potentially strengthen the inhibitory controls of the prefrontal cortex on the amygdala, thereby promoting recovery from PTSD.
Although most of this review has focused on mechanisms of fear inhibition and extinction, the failure to extinguish occurs when the excitatory memory of the trauma/CS+ outcompetes the inhibitory memory. Resistance to extinguishing in PTSD may therefore be due either to abnormally strong excitatory conditioning during acquisition or to impaired inhibitory conditioning during extinction. In sum, the confounding contribution of inhibitory and excitatory processes to abnormalities in conditioned inhibition and extinction in PTSD is critical. Figure 4 illustrates how differential memory processes can both inhibit and excite fear memory expression, with PTSD as a potential pathological outcome.
A type of treatment for PTSD that has its basis in extinction is exposure therapy. The term exposure therapy refers to several behavioral and cognitive-behavioral treatment programs that involve confronting feared but safe thoughts, images, objects, situations, or activities in order to reduce pathological (unrealistic) fear, anxiety, and anxiety disorder symptoms. In the treatment of PTSD, exposure therapy usually involves prolonged imaginal exposure to the patient's memory of the trauma and in vivo exposure to various reminders of the trauma. This basic prolonged exposure protocol has been found to be highly effective in the treatment of women with PTSD following physical and sexual assault compared to waiting list or minimal attention control conditions (135–138). Similar exposure therapy programs have been successful with different trauma populations (139–141). Note that there are caveats to data on the prolonged exposure approaches to psychotherapy, however. Studies have often used limiting exclusion criteria and failed to address polysymptomatic presentations, which may render generalizability to a broader population of PTSD patients difficult to determine (142).
Foa and Kozak (143) suggested that two conditions are necessary for emotional processing to occur: activation of the fear memory and the incorporation of corrective information (e.g., that the feared consequence does not occur). These two conditions are met in exposure therapy when the patient confronts actual fear-related stimuli (in vivo exposure), intentionally creates an image of the feared situation, or intentionally retrieves a memory of the traumatic experience (imaginal exposure) and experiences the associated fear reactions (indicated by self-reports of distress or physiological signs of arousal) but in the absence of the feared consequence (e.g., being assaulted). These processes are basically the same as those that occur in extinction, which can be studied directly in animals. Thus, repeated presentations of the CS (in vivo exposure—extinction training) typically elicits the fear response (activation), which then diminishes over the course of repeated trials within an extinction session as well as over the course of successive extinction sessions. Another similarity across exposure therapy and extinction training is a partial return of the conditioned response following exposure/extinction training at the beginning of the next session, referred to as the return of fear (144) in the clinical literature and spontaneous recovery in the extinction literature. In addition, fear often returns in patients who undergo a subsequent major life stress (reinstatement) or even a change in context (renewal).
Pharmacology of Fear Inhibition
As the time course model for PTSD indicates (Figure 4), there may be several points at which therapeutic interventions can be done to prevent the development of PTSD. In addition to psychotherapy, several pharmacological approaches have been used at these time points. For instance, some data suggest that the administration of propranolol in the immediate aftermath of trauma may prevent fear consolidation (145). Studies with animal models have shown that propranolol interferes with the formation of emotional memories (146). Similarly, an early-intervention study of patients in an emergency department suggested that propranolol reduced the development of PTSD symptoms (145). If replicated, these results may provide an exciting approach to preventing PTSD if early intervention is possible, although the time window for response may be limited. However, in most cases, too much time may have passed between the trauma and the treatment, particularly in cases where the trauma occurred in childhood or in combat, when immediate treatment was not available. In such cases, the fear memory is fully consolidated and administration of propranolol would no longer be effective. The most appropriate treatment in these cases would involve exposure therapy, which strengthens fear inhibition through extinction. Pharmacological agents that enhance safety learning would have an important application in this treatment approach.
A recent study showed that d-cycloserine, a partial NMDA agonist, facilitated extinction when rats were tested drug free the next day (147). These results have been fully replicated using freezing as the measure of conditioned fear, even when d-cycloserine is administered up to 4 hours after extinction training (148). This finding led to the first successful clinical test of combining d-cycloserine with exposure-based psychotherapy (149), a result that has since been replicated in several other studies (150–153). Remarkably, the rodent studies have shown that d-cycloserine also seems to block later reinstatement (154). In addition, d-cycloserine leads to generalized extinction (155), where extinction training to one cue in the presence of d-cycloserine leads to a reduction of fear to another CS previously paired with the same US. This could be significant clinically because combining d-cycloserine with exposure-based psychotherapy to specific cues associated with the original trauma might generalize to other cues associated with that traumatic event, even though these are not dealt with explicitly during therapy. A recent meta-analysis of more than 40 animal and human trials examining d-cycloserine and extinction or exposure therapy concluded that d-cycloserine “is a useful target for translational research on augmenting exposure-based treatment via compounds that impact neuroplasticity” (156). Although no studies of the effects of d-cycloserine in exposure therapy for PTSD have yet been completed, several are under way.
The pharmacological enhancement of extinction or, more specifically, the pharmacological enhancement of emotional learning that takes place during psychotherapy, is an increasingly interesting avenue of research. Notably, neuroimaging studies suggest that the prefrontal cortex regions engaged by top-down emotion regulation strategies may inhibit the amygdala (24, 85). These connections may diminish fear through similar connections to the ventromedial prefrontal cortex that are thought to inhibit the amygdala during extinction. Also of note, one study suggested that orally administered d-cycloserine may lead to inhibited amygdala activity during repeated presentation of faces (157). A number of other avenues for enhancing extinction of fear are now being explored at the preclinical level. These include modulation of the cannabinoid system, which is known to influence local inhibitory circuits (158–160); modulation of GABA-ergic circuits directly (161); modulation of BDNF-dependent neural plasticity (74, 162); and enhancement of extinction through HPA axis modulation of cortisol (163, 164).
Conclusions
Significant progress has been made in recent years in understanding the neurobiology of conditioned fear and its inhibition. PTSD stands out among the leading psychiatric disorders in that the critical neural circuitry that may underlie the disorder is well understood. Thus the understanding of both the pathophysiology and novel treatment approaches for this devastating disorder may be particularly suited for bench-to-bedside research in mental health. Conceptualizing PTSD as a disorder of fear allows the use of tools that enhance fear inhibition to aid in the development of better treatments. While this approach may be phenomenologically narrow, it is clinically pragmatic: progress can be made through research on animal models and preclinical studies that can be translated to the clinical domain. Furthermore, it also provides a neurobiological intermediate phenotype to examine the relationship between genetic variation and a mental disorder. Such integrative, multidimensional analyses are of utmost importance to this field.
Acknowledgments
Dr. Ressler has received awards and/or funding support from the Burroughs Wellcome Foundation, NARSAD, NIMH, the National Institute on Drug Abuse, and Lundbeck, was a co-founder of SyneuRX/Therapade, and has a patent pending with Extinction Pharmaceuticals for d-cycloserine-based therapeutics. Dr. Jovanovic reports no financial relationships with commercial interests.
Supported in part by NIMH grant MH071537, the Emory and Grady Memorial Hospital General Clinical Research Center, NIH National Centers for Research Resources grant M01 RR00039, and the Burroughs Wellcome Fund.
References
- 1.Kessler RC, Sonnega A, Bromet E, Hughes M, Nelson CB. Posttraumatic stress disorder in the National Comorbidity Survey. Arch Gen Psychiatry. 1995;52:1048–1060. doi: 10.1001/archpsyc.1995.03950240066012. [DOI] [PubMed] [Google Scholar]
- 2.Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62:593–602. doi: 10.1001/archpsyc.62.6.593. [DOI] [PubMed] [Google Scholar]
- 3.Charney DS. Psychobiological mechanisms of resilience and vulnerability: implications for successful adaptation to extreme stress. Am J Psychiatry. 2004;161:195–216. doi: 10.1176/appi.ajp.161.2.195. [DOI] [PubMed] [Google Scholar]
- 4.Sterling P, Eyer J. Allostasis: a new paradigm to explain arousal pathology. In: Fischer S, Reason J, editors. Handbook of Life Stress, Cognition, and Health. John Wiley & Sons; Hoboken, NJ: 1988. pp. 629–649. [Google Scholar]
- 5.Binder EB, Bradley RG, Liu W, Epstein MP, Deveau TC, Mercer KB, Tang Y, Gillespie CF, Heim CM, Nemeroff CB, Schwartz AC, Cubells JF, Ressler KJ. Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA. 2008;299:1291–1305. doi: 10.1001/jama.299.11.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bradley RG, Binder EB, Epstein MP, Tang Y, Nair HP, Liu W, Gillespie CF, Berg T, Evces M, Newport DJ, Stowe ZN, Heim CM, Nemeroff CB, Schwartz A, Cubells JF, Ressler KJ. Influence of child abuse on adult depression: moderation by the corticotropin-releasing hormone receptor gene. Arch Gen Psychiatry. 2008;65:190–200. doi: 10.1001/archgenpsychiatry.2007.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guthrie RM, Bryant RA. Extinction learning before trauma and subsequent posttraumatic stress. Psychosomatic Med. 2006;68:307–311. doi: 10.1097/01.psy.0000208629.67653.cc. [DOI] [PubMed] [Google Scholar]
- 8.Milad MR, Quirk GJ, Pitman RK, Orr SP, Fischl B, Rauch SL. A role for the human dorsal anterior cingulate cortex in fear expression. Biol Psychiatry. 2007;62:1191–1194. doi: 10.1016/j.biopsych.2007.04.032. [DOI] [PubMed] [Google Scholar]
- 9.American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders, 4th ed (DSM-IV) American Psychiatric Association; Washington, DC: 1994. [Google Scholar]
- 10.Keane TM, Zimering RT, Caddell JM. A behavioral formulation of posttraumatic stress disorder in Vietnam veterans. Behav Ther. 1985;8:9–12. [Google Scholar]
- 11.Blanchard EB, Kolb LC, Gerardi RJ, Ryan DH, Pallmeyer TP. Cardiac response to relevant stimuli as an adjunctive tool for diagnosing post-traumatic stress disorder in Vietnam veterans. Behav Ther. 1986;17:592–606. [Google Scholar]
- 12.Pitman RK, Orr SP, Forgue DF, de Jong JB, Claiborn JM. Psychophysiologic assessment of posttraumatic stress disorder imagery in Vietnam combat veterans. Arch Gen Psychiatry. 1987;44:970–975. doi: 10.1001/archpsyc.1987.01800230050009. [DOI] [PubMed] [Google Scholar]
- 13.Orr SP, Metzger LJ, Lasko NB, Macklin ML, Hu FB, Shalev AY, Pitman RK. Physiologic responses to sudden, loud tones in monozygotic twins discordant for combat exposure: association with posttraumatic stress disorder. Arch Gen Psychiatry. 2003;60:283–288. doi: 10.1001/archpsyc.60.3.283. [DOI] [PubMed] [Google Scholar]
- 14.Mineka S, Oehlberg K. The relevance of recent developments in classical conditioning to understanding the etiology and maintenance of anxiety disorders. Acta Psychol (Amst) 2008;127:567–580. doi: 10.1016/j.actpsy.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 15.Myers KM, Toufexis DJ, Winslow JT, Jovanovic T, Norrholm SD, Duncan E, Davis M. Measurement of fear inhibition in rats, monkeys, and humans with and without posttraumatic stress disorder, using the AX+, BX− paradigm. In: Whalen PJ, Phelps EA, editors. The Human Amygdala. Guilford Press; New York: 2009. pp. 61–81. [Google Scholar]
- 16.Grillon C. Models and mechanisms of anxiety: evidence from startle studies. Psychopharmacology. 2008;199:421–437. doi: 10.1007/s00213-007-1019-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Myers KM, Ressler KJ, Davis M. Different mechanisms of fear extinction dependent on length of time since fear acquisition. Learn Mem. 2006;13:216–223. doi: 10.1101/lm.119806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Norrholm SD, Jovanovic T, Vervliet B, Myers KM, Davis M, Rothbaum BO, Duncan EJ. Conditioned fear extinction and reinstatement in a human fear-potentiated startle paradigm. Learn Mem. 2006;13:681–685. doi: 10.1101/lm.393906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grillon C, Ameli R. Conditioned inhibition of fear-potentiated startle and skin conductance in humans. Psychophysiology. 2001;38:807–815. [PubMed] [Google Scholar]
- 20.Davis M. The role of the amygdala in fear-potentiated startle: implications for animal models of anxiety. Trends Pharmacol Sci. 1992;13:35–41. doi: 10.1016/0165-6147(92)90014-w. [DOI] [PubMed] [Google Scholar]
- 21.LeDoux JE, Iwata J, Cicchetti P, Reis DJ. Different projections of the central amygdaloid nucleus mediate autonomic and behavioral correlates of conditioned fear. J Neurosci. 1988;8:2517–2529. doi: 10.1523/JNEUROSCI.08-07-02517.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.LeDoux JE. The Emotional Brain: The Mysterious Underpinnings of Emotional Life. Simon & Schuster; New York: 1996. [Google Scholar]
- 23.LaBar KS, Gatenby JC, Gore JC, LeDoux JE, Phelps EA. Human amygdala activation during conditioned fear acquisition and extinction: a mixed-trial fMRI study. Neuron. 1998;20:937–945. doi: 10.1016/s0896-6273(00)80475-4. [DOI] [PubMed] [Google Scholar]
- 24.Phelps EA, Delgado MR, Nearing KI, LeDoux JE. Extinction learning in humans: role of the amygdala and vmPFC. Neuron. 2004;43:897–905. doi: 10.1016/j.neuron.2004.08.042. [DOI] [PubMed] [Google Scholar]
- 25.Knight D. The role of the human amygdala in the production of conditioned fear responses. Neuroimage. 2005;26:1193–1200. doi: 10.1016/j.neuroimage.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 26.Bremner JD, Vermetten E, Schmahl C, Vaccarino V, Vythilingam M, Afzal N, Grillon C, Charney DS. Positron emission tomographic imaging of neural correlates of a fear acquisition and extinction paradigm in women with childhood sexual-abuse-related post-traumatic stress disorder. Psychol Med. 2005;35:791–806. doi: 10.1017/s0033291704003290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Myers KM, Davis M. AX+, BX− discrimination learning in the fear-potentiated startle paradigm: possible relevance to inhibitory fear learning in extinction. Learn Mem. 2004;11:464–475. doi: 10.1101/lm.74704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jovanovic T, Norrholm SD, Fiallos A, Keyes M, Myers K, Davis M, Duncan EJ. Fear potentiation and fear inhibition in PTSD. International Society for Traumatic Stress Studies, 20th Annual Meeting, Final Program and Proceedings; New Orleans. Nov 14–18, 2004; p. 67. poster presentation M-87. [Google Scholar]
- 29.Rescorla RA, Wagner AR. A theory of Pavlovian conditioning: variations in the effectiveness of reinforcement and nonreinforcement. In: Black AH, Prokasy WF, editors. Classical Conditioning II: Current Research and Theory. Appleton-Century-Crofts; New York: 1972. pp. 64–99. [Google Scholar]
- 30.Bouton MD, Swartzentruber D. Slow reacquisition following extinction: context, encoding, and retrieval mechanisms. J Exp Psychol Anim Behav Process. 1989;15:43–53. [Google Scholar]
- 31.Jovanovic T, Norrholm SD, Fennell JE, Keyes M, Fiallos A, Myers KM, Davis M, Duncan EJ. Posttraumatic stress disorder may be associated with impaired fear inhibition: relation to symptom severity. Psychiatry Res. 2009;157:151–160. doi: 10.1016/j.psychres.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jovanovic T, Norrholm SD, Jambrosic-Sakoman A, Esterajher S, Kozaric-Kovacic D, Myers KM, Davis M, Duncan E. Conditioned and external fear inhibition in combat-related PTSD in Croatian war veterans. Society for Neuroscience, 37th annual meeting; San Diego, Calif. Nov 3–7, 2007; Neuroscience Meeting Planner (online), abstract 639.6/FFF22 ( http://www. sfn.org/index.aspx?pagename=abstracts_ampublications) [Google Scholar]
- 32a.Jovanovic T, Norrholm SD, Blanding NQ, Davis M, Duncan E, Bradley B, Ressler KJ. Impaired fear inhibition is a biomarker of PTSD but not depression. Depress Anxiety. 2010;27:244–251. doi: 10.1002/da.20663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weike AI, Schupp HT, Hamm AO. In dubio pro defensio: initial activation of conditioned fear is not cue specific. Behav Neurosci. 2008;122:685–696. doi: 10.1037/0735-7044.122.3.685. [DOI] [PubMed] [Google Scholar]
- 34.Jovanovic T, Norrholm SD, Keyes M, Fiallos A, Jovanovic S, Myers KM, Davis M, Duncan EJ. Contingency awareness and fear inhibition in a human fear-potentiated startle paradigm. Behav Neurosci. 2006;120:995–1004. doi: 10.1037/0735-7044.120.5.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lissek S, Powers AS, McClure EB, Phelps EA, Woldehawariat G, Grillon C, Pine DS. Classical fear conditioning in the anxiety disorders: a meta-analysis. Behav Res Ther. 2005;43:1391–1424. doi: 10.1016/j.brat.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 36.Grillon C, Morgan CA., 3rd Fear-potentiated startle conditioning to explicit and contextual cues in Gulf War veterans with posttraumatic stress disorder. J Abnorm Psychol. 1999;108:134–142. doi: 10.1037//0021-843x.108.1.134. [DOI] [PubMed] [Google Scholar]
- 37.Lissek S, Rabin SJ, McDowell DJ, Dvir S, Bradford DE, Geraci M, Pine DS, Grillon C. Impaired discriminative fear-conditioning resulting from elevated fear responding to learned safety cues among individuals with panic disorder. Behav Res Ther. 2009;47:111–118. doi: 10.1016/j.brat.2008.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Orr SP, Metzger LJ, Lasko NB, Macklin ML, Peri T, Pitman RK. De novo conditioning in trauma-exposed individuals with and without posttraumatic stress disorder. J Abnorm Psychol. 2000;109:290–298. [PubMed] [Google Scholar]
- 39.Wessa M, Flor H. Failure of extinction of fear responses in posttraumatic stress disorder: evidence from second-order conditioning. Am J Psychiatry. 2007;164:1684–1692. doi: 10.1176/appi.ajp.2007.07030525. [DOI] [PubMed] [Google Scholar]
- 40.Peri T, Ben-Shakhar G, Orr SP, Shalev AY. Psychophysiologic assessment of aversive conditioning in posttraumatic stress disorder. Biol Psychiatry. 2000;47:512–519. doi: 10.1016/s0006-3223(99)00144-4. [DOI] [PubMed] [Google Scholar]
- 41.Milad MR, Orr SP, Lasko NB, Chang Y, Rauch SL, Pitman RK. Presence and acquired origin of reduced recall for fear extinction in PTSD: results of a twin study. J Psychiatr Res. 2008;42:515–520. doi: 10.1016/j.jpsychires.2008.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pole N, Neylan TC, Otte C, Henn-Hasse C, Metzler TJ, Marmar CR. Prospective prediction of posttraumatic stress disorder symptoms using fear potentiated auditory startle responses. Biol Psychiatry. 2009;65:235–240. doi: 10.1016/j.biopsych.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.LeDoux J. Brain mechanisms of emotion and emotional learning. Curr Opin Neurobiol. 1992;2:191–197. doi: 10.1016/0959-4388(92)90011-9. [DOI] [PubMed] [Google Scholar]
- 44.Davis M, Falls WA, Campeau S, Kim M. Fear-potentiated startle: a neural and pharmacological analysis. Behav Brain Res. 1993;58:175–198. doi: 10.1016/0166-4328(93)90102-v. [DOI] [PubMed] [Google Scholar]
- 45.Fanselow MS. Neural organization of the defensive behavior system responsible for fear. Psychon Bull Rev. 1994;1:429–438. doi: 10.3758/BF03210947. [DOI] [PubMed] [Google Scholar]
- 46.Davis M. The role of the amygdala in conditioned fear. In: Aggleton J, editor. The Amygdala: Neurobiological Aspects of Emotion, Memory and Mental Dysfunction. John Wiley & Sons; New York: 1992. pp. 255–305. [Google Scholar]
- 47.Davis M, Gendelman D, Tischler M, Gendelman P. A primary acoustic startle circuit: lesion and stimulation studies. J Neurosci. 1982;2:791–805. doi: 10.1523/JNEUROSCI.02-06-00791.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fanselow M, LeDoux J. Why we think plasticity underlying Pavlovian fear conditioning occurs in the basolateral amygdala. Neuron. 1999;23:229–232. doi: 10.1016/s0896-6273(00)80775-8. [DOI] [PubMed] [Google Scholar]
- 49.Quirk GJ, Gehlert DR. Inhibition of the amygdala: key to pathological states? Ann NY Acad Sci. 2003;985:263–272. doi: 10.1111/j.1749-6632.2003.tb07087.x. [DOI] [PubMed] [Google Scholar]
- 50.Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, Luthi A. Amygdala inhibitory circuits and the control of fear memory. Neuron. 2009;62:757–771. doi: 10.1016/j.neuron.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 51.Pare D, Quirk GJ, Ledoux JE. New vistas on amygdala networks in conditioned fear. J Neurophysiol. 2004;92:1–9. doi: 10.1152/jn.00153.2004. [DOI] [PubMed] [Google Scholar]
- 52.Likhtik E, Pelletier JG, Paz R, Paré D. Prefrontal control of the amygdala. J Neurosci. 2005;25:7429–7437. doi: 10.1523/JNEUROSCI.2314-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Likhtik E, Popa D, Apergis-Schoute J, Fidacaro GA, Paré D. Amygdala intercalated neurons are required for expression of fear extinction. Nature. 2008;454:642–645. doi: 10.1038/nature07167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Herry C, Ciocchi S, Senn V, Demmou L, Muller C, Luthi A. Switching on and off fear by distinct neuronal circuits. Nature. 2008;454:600–606. doi: 10.1038/nature07166. [DOI] [PubMed] [Google Scholar]
- 55.Davis M, Walker DL, Lee Y. Amygdala and bed nucleus of the stria terminalis: differential roles in fear and anxiety measured with the acoustic startle reflex. Philos Trans R Soc Lond B Biol Sci. 1997;352:1675–1687. doi: 10.1098/rstb.1997.0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Funayama ES, Grillon C, Davis M, Phelps EA. A double dissociation in the affective modulation of startle in humans: effects of unilateral temporal lobectomy. J Cogn Neurosci. 2001;13:721–729. doi: 10.1162/08989290152541395. [DOI] [PubMed] [Google Scholar]
- 57.Whalen PJ, Shin LM, McInerney SC, Fischer H, Wright CI, Rauch SL. A functional MRI study of human amygdala responses to facial expressions of fear versus anger. Emotion. 2001;1:70–83. doi: 10.1037/1528-3542.1.1.70. [DOI] [PubMed] [Google Scholar]
- 58.Pine DS, Grun J, Zarahn E, Fyer A, Koda V, Li W, Szeszko PR, Ardekani B, Bilder RM. Cortical brain regions engaged by masked emotional faces in adolescents and adults: an fMRI study. Emotion. 2001;1:137–147. doi: 10.1037/1528-3542.1.2.137. [DOI] [PubMed] [Google Scholar]
- 59.Phelps EA, O'Connor KJ, Gatenby JC, Gore JC, Grillon C, Davis M. Activation of the left amygdala to a cognitive representation of fear. Nat Neurosci. 2001;4:437–441. doi: 10.1038/86110. [DOI] [PubMed] [Google Scholar]
- 60.Phan KL, Wager T, Taylor SF, Liberzon I. Functional neuroanatomy of emotion: a meta-analysis of emotion activation studies in PET and fMRI. Neuroimage. 2002;16:331–348. doi: 10.1006/nimg.2002.1087. [DOI] [PubMed] [Google Scholar]
- 61.Liberzon I, Sripada CS. The functional neuroanatomy of PTSD: a critical review. Prog Brain Res. 2008;167:151–169. doi: 10.1016/S0079-6123(07)67011-3. [DOI] [PubMed] [Google Scholar]
- 62.Rauch SL, van der Kolk BA, Fisler RE, Alpert NM, Orr SP, Savage CR, Fischman AJ, Jenike MA, Pitman RK. A symptom provocation study of posttraumatic stress disorder using positron emission tomography and script driven imagery. Arch Gen Psychiatry. 1996;53:380–387. doi: 10.1001/archpsyc.1996.01830050014003. [DOI] [PubMed] [Google Scholar]
- 63.Shin LM, McNally RJ, Kosslyn SM, Thompson WL, Rauch SL, Alpert NM, Metzger LJ, Lasko NB, Orr SP, Pitman RK. Regional cerebral blood flow during script-driven imagery in childhood sexual abuse-related PTSD: a PET investigation. Am J Psychiatry. 1999;156:575–584. doi: 10.1176/ajp.156.4.575. [DOI] [PubMed] [Google Scholar]
- 64.Bremner J, Staib L, Kaloupek D, Southwick SW, Soufer R, Charney DS. Neural correlates of exposure to traumatic pictures and sound in Vietnam combat veterans with and without posttraumatic stress disorder: a positron emission tomography study. Biol Psychiatry. 1999;45:806–816. doi: 10.1016/s0006-3223(98)00297-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liberzon I, Taylor SF, Amdur R, Jung TD, Chamberlain KR, Minoshima S, Koeppe RA, Fig LM. Brain activation in PTSD in response to trauma-related stimuli. Biol Psychiatry. 1999;45:817–826. doi: 10.1016/s0006-3223(98)00246-7. [DOI] [PubMed] [Google Scholar]
- 66.Protopopescu X, Pan H, Tuescher O, Cloitre M, Goldstein M, Engelien W, Epstein J, Yang Y, Gorman J, LeDoux J, Silbersweig D, Stern E. Differential time courses and specificity of amygdala activity in posttraumatic stress disorder subjects and normal control subjects. Biol Psychiatry. 2005;57:464–473. doi: 10.1016/j.biopsych.2004.12.026. [DOI] [PubMed] [Google Scholar]
- 67.Rauch SL, Whalen PJ, Shin LM, McInerney SC, Macklin ML, Lasko NB, Orr SP, Pitman RK. Exaggerated amygdala response to masked facial stimuli in posttraumatic stress disorder: a fMRI study. Biol Psychiatry. 2000;47:769–776. doi: 10.1016/s0006-3223(00)00828-3. [DOI] [PubMed] [Google Scholar]
- 68.Shin LM, Wright CI, Cannistraro PA, Wedig MM, McMullin K, Martis B, Macklin ML, Lasko NB, Cavanagh SR, Krangel TS, Orr SP, Pitman RK, Whalen PJ, Rauch SL. A functional magnetic resonance imaging study of amygdala and medial prefrontal cortex responses to overtly presented fearful faces in posttraumatic stress disorder. Arch Gen Psychiatry. 2005;62:273–281. doi: 10.1001/archpsyc.62.3.273. [DOI] [PubMed] [Google Scholar]
- 69.Morgan MA, Romanski LM, LeDoux JE. Extinction of emotional learning: contribution of medial prefrontal cortex. Neurosci Lett. 1993;163:109–113. doi: 10.1016/0304-3940(93)90241-c. [DOI] [PubMed] [Google Scholar]
- 70.Grace AA, Rosenkranz JA. Regulation of conditioned responses of basolateral amygdala neurons. Physiol Behav. 2002;77:489–493. doi: 10.1016/s0031-9384(02)00909-5. [DOI] [PubMed] [Google Scholar]
- 71.Milad MR, Quirk GJ. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature. 2002;420:70–74. doi: 10.1038/nature01138. [DOI] [PubMed] [Google Scholar]
- 72.Peters J, Kalivas PW, Quirk GJ. Extinction circuits for fear and addiction overlap in prefrontal cortex. Learn Mem. 2009;16:279–288. doi: 10.1101/lm.1041309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vidal-Gonzalez I, Vidal-Gonzalez B, Rauch SL, Quirk GJ. Micro-stimulation reveals opposing influences of prelimbic and infralimbic cortex on the expression of conditioned fear. Learn Mem. 2006;13:728–733. doi: 10.1101/lm.306106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chhatwal JP, Stanek-Rattiner L, Davis M, Ressler KJ. Amygdala BDNF signaling is required for consolidation but not encoding of extinction. Nat Neurosci. 2006;9:870–872. doi: 10.1038/nn1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Maguschak KA, Ressler KJ. Beta-catenin is required for memory consolidation. Nat Neurosci. 2008;11:1319–1326. doi: 10.1038/nn.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ressler KJ, Paschall G, Zhou XL, Davis M. Regulation of synaptic plasticity genes during consolidation of fear conditioning. J Neurosci. 2002;22:7892–7902. doi: 10.1523/JNEUROSCI.22-18-07892.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Eagle D, Bari A, Robbins T. The neuropsychopharmacology of action inhibition: cross-species translation of the stop-signal and go/no-go tasks. Psychopharmacology (Berl) 2008;199:439–456. doi: 10.1007/s00213-008-1127-6. [DOI] [PubMed] [Google Scholar]
- 78.Bush G, Shin LM. The Multi-Source Interference Task: an fMRI task that reliably activates the cingulo-frontal-parietal cognitive/attention network. Nat Protoc. 2006;1:308–313. doi: 10.1038/nprot.2006.48. [DOI] [PubMed] [Google Scholar]
- 79.Shin LM, Bush G, Whalen PJ, Handwerger K, Cannistraro PA, Wright CI, Martis B, Macklin ML, Lasko NB, Orr SP, Pitman RK, Rauch SL. Dorsal anterior cingulate function in posttraumatic stress disorder. J Trauma Stress. 2007;20:701–712. doi: 10.1002/jts.20231. [DOI] [PubMed] [Google Scholar]
- 80.Whalen PJ, Bush G, McNally RJ, Wilhelm S, McInerney SC, Jenike MA, Rauch SL. The emotional counting Stroop paradigm: a functional magnetic resonance imaging probe of the anterior cingulate affective division. Biol Psychiatry. 1998;44:1219–1228. doi: 10.1016/s0006-3223(98)00251-0. [DOI] [PubMed] [Google Scholar]
- 81.Whalen PJ, Bush G, Shin LM, Rauch SL. The emotional counting Stroop: a task for assessing emotional interference during brain imaging. Nat Protocols. 2006;1:293–296. doi: 10.1038/nprot.2006.45. [DOI] [PubMed] [Google Scholar]
- 82.Schiller D, Levy I, Niv Y, LeDoux JE, Phelps EA. From fear to safety and back: reversal of fear in the human brain. J Neurosci. 2008;28:11517–11525. doi: 10.1523/JNEUROSCI.2265-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Milad MR, Wright CI, Orr SP, Pitman RK, Quirk GJ, Rauch SL. Recall of fear extinction in humans activates the ventromedial prefrontal cortex and hippocampus in concert. Biol Psychiatry. 2007;62:446–454. doi: 10.1016/j.biopsych.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 84.Milad MR, Quinn BT, Pitman RK, Orr SP, Fischl B, Rauch SL. Thickness of ventromedial prefrontal cortex in humans is correlated with extinction memory. Proc Natl Acad Sci USA. 2005;102:10706–10711. doi: 10.1073/pnas.0502441102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Delgado MR, Nearing KI, LeDoux JE, Phelps EA. Neural circuitry underlying the regulation of conditioned fear and its relation to extinction. Neuron. 2008;59:829–838. doi: 10.1016/j.neuron.2008.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bremner JD, Vermetten E, Vythilingam M, Afzal N, Schmahl C, Elzinga B, Charney DS. Neural correlates of the classic color and emotional Stroop in women with abuse-related posttraumatic stress disorder. Biol Psychiatry. 2004;55:612–620. doi: 10.1016/j.biopsych.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 86a.Milad MR, Pitman RK, Ellis CB, Gold AL, Shin LM, Lasko NB, Zeidan MA, Handwerger K, Orr SP, Rauch SL. Neurobiological basis of failure to recall extinction memory in posttraumatic stress disorder. Biol Psychiatry. 2009;66:1075–1082. doi: 10.1016/j.biopsych.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mannie ZN, Norbury R, Murphy SE, Inkster B, Harmer CJ, Cowen PJ. Affective modulation of anterior cingulate cortex in young people at increased familial risk of depression. Br J Psychiatry. 2008;192:356–361. doi: 10.1192/bjp.bp.107.043398. [DOI] [PubMed] [Google Scholar]
- 88.Knight DC, Waters NS, Bandettini PA. Neural substrates of explicit and implicit fear memory. Neuroimage. 2009;45:208–214. doi: 10.1016/j.neuroimage.2008.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Canli T, Lesch K. Long story short: the serotonin transporter in emotion regulation and social cognition. Nat Neurosci. 2007;10:1103–1109. doi: 10.1038/nn1964. [DOI] [PubMed] [Google Scholar]
- 90.Lonsdorf TB, Weike AI, Nikamo P, Schalling M, Hamm AO, Öhman A. Genetic gating of human fear learning and extinction: possible implications for gene-environment interaction in anxiety disorder. Psychol Sci. 2009;20:198–206. doi: 10.1111/j.1467-9280.2009.02280.x. [DOI] [PubMed] [Google Scholar]
- 91.Baker JD, Azorlosa JL. The NMDA antagonist MK-801 blocks the extinction of Pavlovian fear conditioning. Behav Neurosci. 1996;110:618–620. doi: 10.1037//0735-7044.110.3.618. [DOI] [PubMed] [Google Scholar]
- 92.Cox J, Westbrook RF. The NMDA receptor antagonist MK-801 blocks acquisition and extinction of conditioned hypoalgesia responses in the rat. Q J Exp Psychol B. 1994;47:187–210. [PubMed] [Google Scholar]
- 93.Falls WA, Miserendino MJ, Davis M. Extinction of fear-potentiated startle: blockade by infusion of an NMDA antagonist into the amygdala. J Neurosci. 1992;12:854–863. doi: 10.1523/JNEUROSCI.12-03-00854.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lee H, Kim JJ. Amygdalar NMDA receptors are critical for new fear learning in previously fear-conditioned rats. J Neurosci. 1998;18:8444–8454. doi: 10.1523/JNEUROSCI.18-20-08444.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Santini E, Muller RU, Quirk GJ. Consolidation of extinction learning involves transfer from NMDA-independent to NMDA-dependent memory. J Neurosci. 2001;21:9009–9017. doi: 10.1523/JNEUROSCI.21-22-09009.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cain CK, Godsil BP, Jami S, Barad M. The L-type calcium channel blocker nifedipine impairs extinction, but not reduced contingency effects, in mice. Learn Mem. 2005;12:277–284. doi: 10.1101/lm.88805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cain CK, Blouin AM, Barad M. L-type voltage gated calcium channels are required for extinction, but not for acquisition or expression of conditional fear in mice. J Neurosci. 2001;22:9113–9121. doi: 10.1523/JNEUROSCI.22-20-09113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rattiner LM, Davis M, French CT, Ressler KJ. Brain-derived neurotrophic factor and tyrosine kinase receptor B involvement in amygdala-dependent fear conditioning. J Neurosci. 2004;24:4796–4806. doi: 10.1523/JNEUROSCI.5654-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Harris JA, Westbrook RF. Evidence that GABA transmission mediates context-specific extinction of learned fear. Psychopharmacology. 1998;140:105–115. doi: 10.1007/s002130050745. [DOI] [PubMed] [Google Scholar]
- 100.Chhatwal JP, Myers KM, Ressler KJ, Davis M. Regulation of gephyrin and GABAA receptor binding within the amygdala after fear acquisition and extinction. J Neurosci. 2005;25:502–506. doi: 10.1523/JNEUROSCI.3301-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Heldt SA, Ressler KJ. Training-induced changes in the expression of GABAA-associated genes in the amygdala after the acquisition and extinction of Pavlovian fear. Eur J Neurosci. 2007;26:3631–3644. doi: 10.1111/j.1460-9568.2007.05970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lin HC, Mao SC, Gean PW. Block of gamma-aminobutyric acid-A receptor insertion in the amygdala impairs extinction of conditioned fear. Biol Psychiatry. 2009;66:665–673. doi: 10.1016/j.biopsych.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 103.True WR, Rice J, Eisen SA, Heath AC, Goldberg J, Lyons MJ, Nowak J. A twin study of genetic and environmental contributions to liability for posttraumatic stress symptoms. Arch Gen Psychiatry. 1993;50:257–264. doi: 10.1001/archpsyc.1993.01820160019002. [DOI] [PubMed] [Google Scholar]
- 104.Xian H, Chantarujikapong SI, Scherrer JF, Eisen SA, Lyons MJ, Goldberg J, Tsuang M, True WR. Genetic and environmental influences on posttraumatic stress disorder, alcohol and drug dependence in twin pairs. Drug Alcohol Depend. 2000;61:95–102. doi: 10.1016/s0376-8716(00)00127-7. [DOI] [PubMed] [Google Scholar]
- 105.Stein MB, Jang KL, Taylor S, Vernon PA, Livesley WJ. Genetic and environmental influences on trauma exposure and post-traumatic stress disorder symptoms: a twin study. Am J Psychiatry. 2002;159:1675–1681. doi: 10.1176/appi.ajp.159.10.1675. [DOI] [PubMed] [Google Scholar]
- 106.Smoller JW, Gardner-Schuster E, Misiaszek M. Genetics of anxiety: would the genome recognize the DSM? Depress Anxiety. 2008;25:368–377. doi: 10.1002/da.20492. [DOI] [PubMed] [Google Scholar]
- 107.Poulton R, Andrews G, Millichamp J. Gene-environment interaction and the anxiety disorders. Eur Arch Psychiatry Clin Neurosci. 2008;258:65–68. doi: 10.1007/s00406-007-0784-5. [DOI] [PubMed] [Google Scholar]
- 108.Broekman BF, Olff M, Boer F. The genetic background to PTSD. Neurosci Biobehav Rev. 2007;31:348–362. doi: 10.1016/j.neubiorev.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 109.Nugent NR, Amstadter AB, Koenen KC. Genetics of post-traumatic stress disorder: informing clinical conceptualizations and promoting future research. Am J Med Genet C Semin Med Genet. 2008;148C:127–132. doi: 10.1002/ajmg.c.30169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Norrholm SD, Ressler KJ. Genetics of anxiety and trauma-related disorders. Neuroscience. 2009;164:272–287. doi: 10.1016/j.neuroscience.2009.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Comings DE, Muhleman D, Gysin R. Dopamine D2 receptor (DRD2) gene and susceptibility to posttraumatic stress disorder: a study and replication. Biol Psychiatry. 1996;40:368–372. doi: 10.1016/0006-3223(95)00519-6. [DOI] [PubMed] [Google Scholar]
- 112.Voisey J, Swagell CD, Hughes IP, Morris CP, van Daal A, Noble EP, Kann B, Heslop KA, Young RM, Lawford BR. The DRD2 gene 957C>T polymorphism is associated with posttraumatic stress disorder in war veterans. Depress Anxiety. 2009;26:28–33. doi: 10.1002/da.20517. [DOI] [PubMed] [Google Scholar]
- 113.Lee HJ, Lee MS, Kang RH, Kim H, Kim SD, Kee BS, Kim YH, Kim YK, Kim JB, Yeon BK, Ohio KS, Ohio BH, Yoon JS, Lee C, Jung HY, Chee IS, Paik IH. Influence of the serotonin transporter promoter gene polymorphism on susceptibility to posttraumatic stress disorder. Depress Anxiety. 2005;21:135–139. doi: 10.1002/da.20064. [DOI] [PubMed] [Google Scholar]
- 114.Grabe HJ, Spitzer C, Schwahn C, Marcinek A, Frahnow A, Barnow S, Lucht M, Freyberger HJ, John U, Wallaschofski H, Volzke H, Rosskopf D. Serotonin transporter gene (SLC6A4) promoter polymorphisms and the susceptibility to posttraumatic stress disorder in the general population. Am J Psychiatry. 2009;166:926–933. doi: 10.1176/appi.ajp.2009.08101542. [DOI] [PubMed] [Google Scholar]
- 115.Gelernter J, Southwick S, Goodson S, Morgan A, Nagy L, Charney DS. No association between D2 dopamine receptor (DRD2) “A” system alleles, or DRD2 haplotypes, and posttraumatic stress disorder. Biol Psychiatry. 1999;45:620–625. doi: 10.1016/s0006-3223(98)00087-0. [DOI] [PubMed] [Google Scholar]
- 116.Zhang H, Ozbay F, Lappalainen J, Kranzler HR, van Dyck CH, Charney DS, Price LH, Southwick S, Yang BZ, Rasmussen A, Gelernter J. Brain derived neurotrophic factor (BDNF) gene variants and Alzheimer's disease, affective disorders, post-traumatic stress disorder, schizophrenia, and substance dependence. Am J Med Genet B Neuropsychiatr Genet. 2006;141B:387–393. doi: 10.1002/ajmg.b.30332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Feusner J, Ritchie T, Lawford B, Young RM, Kann B, Noble EP. GABA(A) receptor beta 3 subunit gene and psychiatric morbidity in a post-traumatic stress disorder population. Psychiatry Res. 2001;104:109–117. doi: 10.1016/s0165-1781(01)00296-7. [DOI] [PubMed] [Google Scholar]
- 118.Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, McClay J, Mill J, Martin J, Braithwaite A, Poulton R. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301:386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- 119.Kilpatrick DG, Koenen KC, Ruggiero KJ, Acierno R, Galea S, Resnick HS, Roitzsch J, Boyle J, Gelernter J. The serotonin transporter genotype and social support and moderation of posttraumatic stress disorder and depression in hurricane-exposed adults. Am J Psychiatry. 2007;164:1693–1699. doi: 10.1176/appi.ajp.2007.06122007. [DOI] [PubMed] [Google Scholar]
- 120.Koenen KC, Aiello AE, Bakshis E, Amstadter AB, Ruggiero KJ, Acierno R, Kilpatrick DG, Gelernter J, Galea S. Modification of the association between serotonin transporter genotype and risk of posttraumatic stress disorder in adults by county-level social environment. Am J Epidemiol. 2009;169:704–711. doi: 10.1093/aje/kwn397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Binder EB. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology. 2009;34(suppl 1):S186–S195. doi: 10.1016/j.psyneuen.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 122.Yehuda R, Yang R-K, Buchsbaum MS, Golier JA. Alterations in cortisol negative feedback inhibition as examined using the ACTH response to cortisol administration in PTSD. Psychoneuroendocrinology. 2006;31:447–451. doi: 10.1016/j.psyneuen.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 123.Yehuda R, Southwick SM, Krystal JH, Bremner D, Charney DS, Mason JW. Enhanced suppression of cortisol following dexamethasone administration in posttraumatic stress disorder. Am J Psychiatry. 1993;150:83–86. doi: 10.1176/ajp.150.1.83. [DOI] [PubMed] [Google Scholar]
- 124.Koenen KC, Saxe G, Purcell S, Smoller JW, Bartholomew D, Miller A, Hall E, Kaplow J, Bosquet M, Moulton S, Baldwin C. Polymorphisms in FKBP5 are associated with peritraumatic dissociation in medically injured children. Mol Psychiatry. 2005;10:1058–1059. doi: 10.1038/sj.mp.4001727. [DOI] [PubMed] [Google Scholar]
- 125.Segman RH, Shefi N, Goltser-Dubner T, Friedman N, Kaminski N, Shalev AY. Peripheral blood mononuclear cell gene expression profiles identify emergent post-traumatic stress disorder among trauma survivors. Mol Psychiatry. 2005;10:500–513. doi: 10.1038/sj.mp.4001636. [DOI] [PubMed] [Google Scholar]
- 126.Yehuda R, Cai G, Golier JA, Sarapas C, Galea S, Ising M, Rein T, Schmeidler J, Muller-Myhsok B, Holsboer F, Buxbaum JD. Gene expression patterns associated with posttraumatic stress disorder following exposure to the World Trade Center attacks. Biol Psychiatry. 2009;66:708–711. doi: 10.1016/j.biopsych.2009.02.034. [DOI] [PubMed] [Google Scholar]
- 127.Foa EB, Steketee G, Rothbaum BO. Behavioral/cognitive conceptualizations of post-traumatic stress disorder. Behav Ther. 1989;20:155–176. [Google Scholar]
- 128.Falls WA, Davis M. Behavioral and physiological analysis of fear inhibition. In: Friedman MJ, Charney DS, Deutch AY, editors. Neurobiological and Clinical Consequences of Stress: From Normal Adaptation to PTSD. Lippincott-Raven; Philadelphia: 1995. pp. 177–202. [Google Scholar]
- 129.Rothbaum BO, Davis M. Applying learning principles to the treatment of post-trauma reactions. Ann NY Acad Sci. 2003;1008:112–121. doi: 10.1196/annals.1301.012. [DOI] [PubMed] [Google Scholar]
- 130.Rothbaum BO, Houry D, Heekin M, Leiner AS, Daugherty J, Smith LS, Gerardi M. A pilot study of an exposure-based intervention in the ED designed to prevent posttraumatic stress disorder. Am J Emerg Med. 2008;26:326–330. doi: 10.1016/j.ajem.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 131.Nader K, Schafe GE, LeDoux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406:722–726. doi: 10.1038/35021052. [DOI] [PubMed] [Google Scholar]
- 132.Monfils M-H, Cowansage KK, Klann E, LeDoux JE. Extinction-reconsolidation boundaries: key to persistent attenuation of fear memories. Science. 2009;324:951–955. doi: 10.1126/science.1167975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kindt M, Soeter M, Vervliet B. Beyond extinction: erasing human fear responses and preventing the return of fear. Nat Neurosci. 2009;12:256–258. doi: 10.1038/nn.2271. [DOI] [PubMed] [Google Scholar]
- 134.Lang PJ, Davis M, Ohman A. Fear and anxiety: animal models and human cognitive psychophysiology. J Affect Disord. 2000;61:137–159. doi: 10.1016/s0165-0327(00)00343-8. [DOI] [PubMed] [Google Scholar]
- 134a.Schiller D, Monfils MH, Raio CM, Johnson DC, Ledoux JE, Phelps EA. Preventing the return of fear in humans using reconsolidation update mechanisms. Nature. 2010;463:49–53. doi: 10.1038/nature08637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Foa EB, Rothbaum BO, Riggs DS, Murdock TB. Treatment of posttraumatic stress disorder in rape victims: a comparison between cognitive-behavioral procedures and counseling. J Consult Clin Psychol. 1991;59:715–723. doi: 10.1037//0022-006x.59.5.715. [DOI] [PubMed] [Google Scholar]
- 136.Foa EB, Dancu CV, Hembree EA, Jaycox LH, Meadows EA, Street GP. A comparison of exposure therapy, stress inoculation training, and their combination for reducing posttraumatic stress disorder in female assault victims. J Consult Clin Psychol. 1999;67:194–200. doi: 10.1037//0022-006x.67.2.194. [DOI] [PubMed] [Google Scholar]
- 137.Resick PA, Nishith P, Weaver TL, Astin MC, Feuer CA. A comparison of cognitive-processing therapy with prolonged exposure and a waiting condition for the treatment of chronic post-traumatic stress disorder in female rape victims. J Consult Clin Psychol. 2002;70:867–879. doi: 10.1037//0022-006x.70.4.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rothbaum BO, Astin MC, Marsteller F. Prolonged exposure versus eye movement desensitization and reprocessing (EMDR) for PTSD rape victims. J Trauma Stress. 2005;18:607–616. doi: 10.1002/jts.20069. [DOI] [PubMed] [Google Scholar]
- 139.Marks I, Lovell K, Noshirvani H, Livanou M, Thrasher S. Treatment of posttraumatic stress disorder by exposure and/or cognitive restructuring: a controlled study. Arch Gen Psychiatry. 1998;55:317–325. doi: 10.1001/archpsyc.55.4.317. [DOI] [PubMed] [Google Scholar]
- 140.Paunovic N, Ost LG. Cognitive-behavior therapy vs exposure therapy in the treatment of PTSD in refugees. Behav Res Ther. 2001;39:1183–1197. doi: 10.1016/s0005-7967(00)00093-0. [DOI] [PubMed] [Google Scholar]
- 141.Taylor S, Thordarson DS, Maxfield L, Fedoroff IC, Lovell K, Ogrodniczuk J. Comparative efficacy, speed, and adverse effects of three PTSD treatments: exposure therapy, EMDR, and relaxation training. J Consult Clin Psychol. 2003;71:330–338. doi: 10.1037/0022-006x.71.2.330. [DOI] [PubMed] [Google Scholar]
- 142.Bradley R, Greene J, Russ E, Dutra L, Westen D. A multidimensional meta-analysis of psychotherapy for PTSD. Am J Psychiatry. 2005;162:214–227. doi: 10.1176/appi.ajp.162.2.214. [DOI] [PubMed] [Google Scholar]
- 143.Foa EB, Kozak MJ. Emotional processing of fear: exposure to corrective information. Psychol Bull. 1986;99:20–35. [PubMed] [Google Scholar]
- 144.Rachman S, Whittal M. The effect of an aversive event on the return of fear. Behav Res Ther. 1989;27:513–520. doi: 10.1016/0005-7967(89)90085-5. [DOI] [PubMed] [Google Scholar]
- 145.Pitman RK, Sanders KM, Zusman RM, Healy AR, Cheema F, Lasko NB, Cahill L, Orr SP. Pilot study of secondary prevention of posttraumatic stress disorder with propranolol. Biol Psychiatry. 2002;51:189–192. doi: 10.1016/s0006-3223(01)01279-3. [DOI] [PubMed] [Google Scholar]
- 146.Cahill L, Pham CA, Setlow B. Impaired memory consolidation in rats produced with beta-adrenergic blockade. Neurobiol Learn Mem. 2000;74:259–266. doi: 10.1006/nlme.1999.3950. [DOI] [PubMed] [Google Scholar]
- 147.Walker DL, Ressler KJ, Lu KT, Davis M. Facilitation of conditioned fear extinction by systemic administration or intraamygdala infusions of d-cycloserine as assessed with fear-potentiated startle in rats. J Neurosci. 2002;22:2343–2351. doi: 10.1523/JNEUROSCI.22-06-02343.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ledgerwood L, Richardson R, Cranney J. Effects of d-cycloserine on extinction of conditioned freezing. Behav Neurosci. 2003;117:341–349. doi: 10.1037/0735-7044.117.2.341. [DOI] [PubMed] [Google Scholar]
- 149.Ressler KJ, Rothbaum BO, Tannenbaum L, Anderson P, Graap K, Zimand E, Hodges L, Davis M. Cognitive enhancers as adjuncts to psychotherapy: use of d-cycloserine in phobic individuals to facilitate extinction of fear. Arch Gen Psychiatry. 2004;61:1136–1144. doi: 10.1001/archpsyc.61.11.1136. [DOI] [PubMed] [Google Scholar]
- 150.Guastella AJ, Richardson R, Lovibond PF, Rapee RM, Gaston JE, Mitchell P, Dadds MR. A randomized controlled trial of d-cycloserine enhancement of exposure therapy for social anxiety disorder. Biol Psychiatry. 2008;63:544–549. doi: 10.1016/j.biopsych.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 151.Kushner MG, Kim SW, Donahue C, Thuras P, Adson D, Kotlyar M, McCabe J, Peterson J, Foa EB. d-Cycloserine augmented exposure therapy for obsessive-compulsive disorder. Biol Psychiatry. 2007;62:835–838. doi: 10.1016/j.biopsych.2006.12.020. [DOI] [PubMed] [Google Scholar]
- 152.Hofmann SG, Meuret AE, Smits JAJ, Simon NM, Pollack MH, Eisenmenger K, Shiekh M, Otto MW. Augmentation of exposure therapy with d-cycloserine for social anxiety disorder. Arch Gen Psychiatry. 2006;63:298–304. doi: 10.1001/archpsyc.63.3.298. [DOI] [PubMed] [Google Scholar]
- 153.Wilhelm S, Buhlmann U, Tolin DF, Meunier SA, Pearlson GD, Reese HE, Cannistraro P, Jenike MA, Rauch SL. Augmentation of behavior therapy with d-cycloserine for obsessive-compulsive disorder. Am J Psychiatry. 2008;165:335–341. doi: 10.1176/appi.ajp.2007.07050776. [DOI] [PubMed] [Google Scholar]
- 154.Ledgerwood L, Richardson R, Cranney J. d-Cycloserine and the facilitation of extinction of conditioned fear: consequences for reinstatement. Behav Neurosci. 2004;118:505–513. doi: 10.1037/0735-7044.118.3.505. [DOI] [PubMed] [Google Scholar]
- 155.Ledgerwood L, Richardson R, Cranney J. d-Cycloserine facilitates extinction of learned fear: effects on reacquisition and generalized extinction. Biol Psychiatry. 2005;57:841–847. doi: 10.1016/j.biopsych.2005.01.023. [DOI] [PubMed] [Google Scholar]
- 156.Norberg MM, Krystal JH, Tolin DF. A meta-analysis of d-cycloserine and the facilitation of fear extinction and exposure therapy. Biol Psychiatry. 2008;63:1118–1126. doi: 10.1016/j.biopsych.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 157.Britton JC, Gold AL, Feczko EJ, Rauch SL, Williams D, Wright CI. d-Cycloserine inhibits amygdala responses during repeated presentations of faces. CNS Spectr. 2007;12:600–605. doi: 10.1017/s1092852900021398. [DOI] [PubMed] [Google Scholar]
- 158.Kamprath K, Marsicano G, Tang J, Monory K, Bisogno T, Di Marzo V, Lutz B, Wotjak CT. Cannabinoid CB1 receptor mediates fear extinction via habituation-like processes. J Neurosci. 2006;26:6677–6686. doi: 10.1523/JNEUROSCI.0153-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Chhatwal JP, Davis M, Maguschak KA, Ressler KJ. Enhancing cannabinoid neurotransmission augments the extinction of conditioned fear. Neuropsychopharmacology. 2005;30:516–524. doi: 10.1038/sj.npp.1300655. [DOI] [PubMed] [Google Scholar]
- 160.Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgansberger W, Di Marzo V, Lutz B. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- 161.Domschke K, Zwanzger P. GABAergic and endocannabinoid dysfunction in anxiety: future therapeutic targets? Curr Pharm Des. 2008;14:3508–3517. doi: 10.2174/138161208786848784. [DOI] [PubMed] [Google Scholar]
- 162.Heldt SA, Stanek L, Chhatwal JP, Ressler KJ. Hippocampus-specific deletion of BDNF in adult mice impairs spatial memory and extinction of aversive memories. Mol Psychiatry. 2007;12:656–670. doi: 10.1038/sj.mp.4001957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.de Quervain DJ, Margraf J. Glucocorticoids for the treatment of post-traumatic stress disorder and phobias: a novel therapeutic approach. Eur J Pharmacol. 2008;583:365–371. doi: 10.1016/j.ejphar.2007.11.068. [DOI] [PubMed] [Google Scholar]
- 164.Cai WH, Blundell J, Han J, Greene RW, Powell CM. Postreactivation glucocorticoids impair recall of established fear memory. J Neurosci. 2006;26:9560–9566. doi: 10.1523/JNEUROSCI.2397-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]