

Abstract
Y chromosomes have long been dismissed as “graveyards of genes,” but there is still much to be learned from the genetic relics of genes that were once functional on the human Y. We identified human X-linked genes whose gametologs have been pseudogenized or completely lost from the Y chromosome and inferred which evolutionary forces may be acting to retain genes on the Y. Although gene loss appears to be largely correlated with the suppression of recombination, we observe that X-linked genes with functional Y homologs evolve under stronger purifying selection and are expressed at higher levels than X-linked genes with nonfunctional Y homologs. Additionally, we support and expand upon the hypothesis that X inactivation is primarily driven by gene loss on the Y. Using linear discriminant analysis, we show that X-inactivation status can successfully classify 90% of X-linked genes into those with functional or nonfunctional Y homologs.
Keywords: Y degeneration, human sex chromosomes, X-chromosome inactivation, evolution, dosage compensation
Human sex chromosomes, X and Y, evolved from a pair of homologous autosomes approximately 180 million years ago (Mikkelsen et al. 2007; Rens et al. 2007). The human Y, being only 59 Megabases (Mb) long (Skaletsky et al. 2003), is dramatically smaller and has 10 times fewer genes than the 155-Mb human X chromosome (Ross et al. 2005). The human X chromosome includes regions of shared ancestry with the Y and X-specific regions not homologous with the Y (most such X-specific duplications and additions involve small-scale events, and they are distributed across the chromosome [Whibley et al. 2010]). Regions of shared ancestry between the X and Y include the X-conserved region (XCR, X-linked in eutherian and marsupial mammals), the X-added region (XAR, added to the sex chromosomes in eutherian mammals but autosomal in marsupials), and the X-transposed region (XTR, transposed from the human X to the human Y after human–chimpanzee divergence) (Ross et al. 2005). Although Y-specific amplification and limited gene acquisition from the autosomes onto the human Y have previously been described (Hughes et al. 2010), Y-specific gene loss from the ancestral X–Y pair has not been characterized in detail. Gene loss on the human Y chromosome may be mostly stochastic, driven by the accumulation of deleterious alleles hitchhiking along in the absence of homologous recombination (Charlesworth 1996; Charlesworth and Charlesworth 2000) or affected by selection acting on genes with male functions or male-limited expression patterns (as shown for the completely nonrecombining neo-Y chromosome in Drosophila miranda [Kaiser et al. 2011]) and thus preventing or slowing down their pseudogenization. Here, focusing on XCR and XAR, we aim to study whether features of X-linked genes inform us about evolutionary forces driving the evolution of Y-linked gene content.
Using comparative genomics and X–Y sequence comparisons, we assessed the status of the 723 consensus CDS genes listed for the human X chromosome, a set of consistently annotated and high-quality genes (supplementary fig. S1, Supplementary Material online). We first excluded the 17 pseudoautosomal (PAR) region genes, which still undergo X–Y recombination. Of the 706 non-PAR genes, 600 genes were classified as “ancestral X” due to the existence of sequence in homologous XAR or XCR regions in at least four out of eight assembled nonprimate genomes (mouse, rat, rabbit, cow, horse, or dog, opossum, or chicken; supplementary table S1 and fig. S1, Supplementary Material online), and 106 are classified as “notAncestral.” Of 600 ancestral X-linked genes, 19 have functional Y homologs (two of these are recently X transposed, XTR, and so excluded from further analysis, so we are left with 17), 266 have evidence of a pseudogenized Y homolog (one is in the XTR, and so is excluded, so we are left with 265), and 315 have no evidence of a functional or pseudogenized Y homolog, so are classified as “lost” on the Y chromosome (supplementary table S1 and fig. S1, Supplementary Material online). Of the 106 genes classified as “notAncestral,” many are members of multigene families, or genes with a single exon, suggesting that they have been independently added (duplicated or retrotransposed) onto the X, or onto the X and Y chromosomes; 31 have evidence of homologous Y sequence, and 75 have no evidence of a Y homolog (supplementary table S1, Supplementary Material online). Only one degraded Y exon was recovered for 29 of the 31 notAncestral X-linked genes with evidence of homologous Y sequence, and only two exons were recovered for the remaining two genes. Because these 31 genes do not appear to be conserved from the ancestral X chromosome, are nearly all part of multigene families, and only one exon was recovered on the Y, the similarity search likely found degraded duplicated or retrotransposed copies added to the Y that should not be included in further analyses. Further, among the 75 genes that did not pass our comparative genomics analysis, nearly all are part of multigene (often tandem) families or single exon genes suggesting they were duplicated or retrotransposed after the eutherian common ancestor. We conservatively exclude all “notAncestral” genes from the downstream analysis. To summarize, the 597 non-XTR X-linked genes that do, or likely once did, have homologous Y-linked sequence (gametologous) were divided into three sets: those with 1) 17 previously identified X-linked genes with functional Y gametologs [Ross et al. 2005]), 2) 265 X-linked genes with pseudogenized Y gametologs, and 3) 315 X-linked genes whose Y gametologs have either been completely lost from the Y chromosome or have diverged beyond our ability to detect them using presently available search algorithms (supplementary table S1 and fig. S1, Supplementary Material online).
The set of 265 X-degenerate Y-linked pseudogenes we identified is a large increase over the 27 previously described X-degenerate Y-linked pseudogenes ([Skaletsky et al. 2003; Hughes et al. 2012]; see Materials and Methods in supplementary note S1, Supplementary Material online). This increase may be due to our acceptance of many degraded pseudogenes (we accepted pseudogenes even if only one X-degenerate homologous exon is identified, so long as it had a sufficiently high score; supplementary note S1, Supplementary Material online). We can be confident that these are nonfunctional sequences because, for many genes, only a fraction of the gametologous Y-linked exons were found, many identified sequences contained multiple frameshift mutations and premature stop codons, and when multiple exons were recovered, they were often rearranged (supplementary table S1, Supplementary Material online). Furthermore, none of these correspond to previously identified Y-linked pseudogenes of autosomal origin (Skaletsky et al. 2003), and all of them have homologs on the ancestral X regions in at least four nonprimate species (supplementary note S1, Supplementary Material online), supporting the view that this set, even with the inclusion of highly degraded pseudogenes, represents only relics of the ancestral X–Y pair, and not recent duplications or retrotranspositions.
Finally, we identify a set of 315 genes proposed to have been lost from the ancestral Y chromosome, because they are found in homologous X regions in at least four nonprimate species but have no homologous Y sequence identifiable in our search (supplementary note S1, Supplementary Material online). It is possible that some of the genes in this set have pseudogene sequences that have diverged beyond our current ability to detect them. Our estimates of the numbers of genes lost and pseudogenized in each stratum are consistent with recent models describing the exponential decay, then leveling out, of Y chromosome gene loss (supplementary tables S1 and S2, Supplementary Material online [Hughes et al. 2012]).
Suppression of recombination between the X and Y is hypothesized to have occurred through a series of inversion events, leading to the formation of strata of varying X–Y divergence levels (Lahn and Page 1999; Ross et al. 2005; Lemaitre et al. 2009; Wilson and Makova 2009). If recombination helps to maintain the functional integrity of genes by preventing the accumulation of deleterious mutations (Charlesworth and Charlesworth 2000; Bachtrog 2008), then there should be more X-linked genes with functional Y homologs in the youngest strata (on the short arm of the X chromosome), and more X-linked genes with nonfunctional Y homologs near the tip of the long arm of the X chromosome. It was previously observed that X-linked genes with functional Y gametologs are qualitatively more abundant in younger versus older strata (Lahn and Page 1999), but comparisons between X-linked genes with pseudogenized versus lost Y homologs were not conducted. We tested this observation statistically and found that the distribution of X-linked genes with functional Y gametologs is significantly skewed to the short arm of the human X chromosome (where recombination was more recently suppressed, i.e., younger strata) versus X-linked genes with nonfunctional Y gametologs (one-sided ranked Wilcoxon test comparing the means of the two distributions, P = 0.00449; supplementary table S1, Supplementary Material online). Additionally, with our partitioning, we were able to look more closely at the set of X-linked genes with nonfunctional Y homologs and found that the distribution of X-linked genes with pseudogenized Y gametologs is significantly skewed toward the short arm of the X (in regions that more recently lost X–Y recombination) than the distribution of X-linked genes with lost Y gametologs (P = 0.04483; supplementary table S1, Supplementary Material online), showing that the absence of recombination not only leads to an accumulation of deleterious mutations (causing nonfunctionalization) but also to a higher likelihood of a gene being either deleted or mutated beyond homology recognition with its former X counterpart.
Evolutionary strata were first identified by observations of higher values of synonymous site divergence between X and Y gametologs, reflecting unique mutations accumulated on the X- and Y alleles of genes in older strata, and lower values of synonymous divergence in younger strata (Lahn and Page 1999). We tested whether there is a statistically significant correlation between synonymous divergence and position along the X chromosome using linear regression models of Xgene–Ygene pairs and Xgene–Ypseudogene pairs separately. We confirm the previous trends, and further observe a statistically significant relationship between increasing pairwise synonymous X–Y divergence and increasing physical distance from the Xpter for both Xgene–Ygene pairs and Xgene–Ypseudogene pairs across the whole X chromosome (P = 0.0001 and 0.0121, respectively; supplementary table S3, Supplementary Material online). However, despite the significant positive correlations between pairwise synonymous divergence and increasing distance from the Xpter, we did not observe strict stratum boundaries when we plotted pairwise synonymous divergence in windows along the length of the X chromosome (fig. 1). Limiting this analysis to genes with more than one exon, and Y-pseudogenes with evidence for more than one exon, to reduce the possibility of analyzing retrotransposed genes did not change this pattern (supplementary fig. S2, Supplementary Material online). Although these results suggest that gene decay on the Y chromosome might simply reflect evolutionary time that has passed since each recombination suppression event, we asked whether additional factors might be at play. Therefore, we next tested whether differences in X-linked genes’ 1) level of selective constraint, 2) functional importance, or 3) expression level might predict the evolutionary fate of their Y-linked gametologs.
Fig. 1.

Pairwise synonymous substitution rate across chromosome X. The pairwise Xgene–Ygene and Xgene–Ypseudogene synonymous substitution rates (dS) are plotted across the entire X chromosome in 10 Mb sliding windows, sliding by 1 Mb. We observe a trend from smaller dS values to larger dS values as we move from the short arm of the X chromosome (where the newest strata are located) to the end of the long arm (where the oldest strata are located) but do not identify strict boundaries using this method. For reference, the dS of X-linked genes with functional Y homologs (excluding the XTR) are plotted separately in red.
First, to test whether differences in the selective pressure acting on X-linked genes might indicate whether their Y gametologs will be retained or lost, we compared dN/dS ratios (the ratio of nonsynonymous to synonymous divergence) between the three classes of X-linked genes (those with Y-linked genes, pseudogenes, or lost Y gametologs). We compared dN/dS ratios along the human X-specific branch from three-way alignments of X homologs (human–chimpanzee–dog, human–dog–opossum, or human–opossum-platypus; supplementary note S1, Supplementary Material online). For all comparisons, we observed that X-linked genes with functional Y homologs have a lower dN/dS ratio than X-linked genes with pseudogenized or lost Y homologs (significant for the human–chimpanzee–dog comparison, Pgene-pseudogene = 0.0077; supplementary table S4, Supplementary Material online), suggesting stronger purifying selection acting on the former group. Thus, the strength of selective pressures acting on X-linked genes may be an important factor in determining whether gametologous Y-linked genes will be retained or lost.
Second, because X-linked genes without gametologous Y sequence are always hemizygous in males, we expected that such genes will be associated with human diseases more often than X-linked genes with functional Y gametologs (which might provide some redundancy in functionality). In contrast, we observed that X-linked genes with functional (4 of 17), pseudogenized (69 of 265), or lost (80 of 315) Y gametologs were all similarly associated with known human diseases (using Fisher’s exact tests, P = 1, 1, and 0.9267 for Ygene–Ypseudogene, Ygene–Ylost, and Ypseudogene–Ylost comparisons, respectively; supplementary note S1, Supplementary Material online). The results were similar when considering only the XAR (P = 0.7307, 1, and 1 for Ygene–Ypseudogene, Ygene–Ylost, and Ypseudogene–Ylost, respectively) or only the XCR (P = 1, 1, and 1 for Ygene–Ypseudogene, Ygene–Ylost, and Ypseudogene–Ylost, respectively). Thus, an X-linked gene’s association with human disease does not predict whether its Y-linked gametolog will be retained or lost.
Third, similar to Drosophila miranda neo-Y genes (Kaiser et al. 2011), we expected human X-linked genes with functional Y gametologs to be expressed at higher levels than X-linked genes without functional Y gametologs. Given the rapid evolution and importance of sex-biased genes (Ellegren and Parsch 2007), especially the high expression divergence of male-biased genes between species (Zhang et al. 2007), we also wondered whether X-linked genes expressed at high levels in the testes might be more likely to retain their Y homologs. Although previous comparisons showed that X-linked genes are more broadly expressed than their functional Y homologs (Wilson and Makova 2009), it was unclear whether, among X-linked genes, those with functional Y homologs show different expression patterns than those without functional Y homologs. Using RNAseq expression data (Brawand et al. 2011), we observed that X-linked genes with functional Y homologs are expressed at higher levels (at least 2-fold higher in the XAR) than X-linked genes with pseudogenized or lost Y homologs (table 1). In the younger XAR, where expression might be more similar to the ancestral state, these differences are significant in the brain and cerebellum of both male and female samples, but surprisingly, not in testis (table 1).
Table 1.
Expression of X-Linked Genes Classified by Y Functional Class.
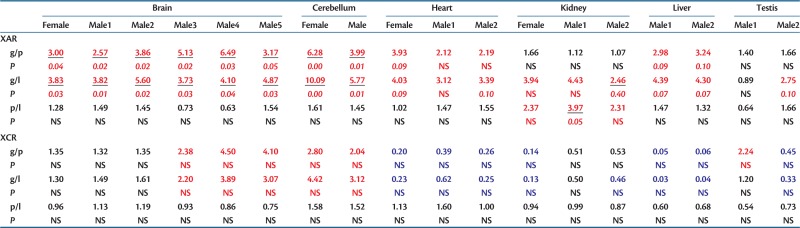 |
Note.—NS, not significant. Pairwise ratios of the median expression of X-linked genes with functional Y-linked gametologs (g), pseudogenized Y gametologs (p), or lost Y gametologs (l) in the XAR and the XCR. Cases where the ratio in expression abundance was >2, indicating that the class of X-linked genes in the numerator is expressed at least 2-fold greater than those in the denominator, are colored in red. Ratios lower than 0.5, indicating the X-linked genes in the numerator, are expressed at least 2-fold lower than those in the denominator, are colored in blue (supplementary note S1, Supplementary Material online). P values <0.1 from a permutation test with 10,000 replicates are shown below each comparison in italics, and those that are significant at a 0.05 level, before Bonferroni correction, are underlined.
So far, we have discussed factors that could influence whether an X-linked gene’s Y gametolog is retained over evolutionary time. A different perspective is to consider how gene loss on the Y might affect evolution of the X. Specifically, X chromosome inactivation (XCI) in female mammals is hypothesized to have evolved as a mechanism to achieve equal dosage of sex-linked genes between males and females, in response to the loss of expression and function of Y-linked gametologs in males (Charlesworth 1978; Carrel and Willard 2005; Carrel et al. 2006; Park et al. 2010). Thus, the proposed ancestral state, before the Y degenerated, is expression of all X-linked genes from both X chromosomes (fig. 2). For X-linked genes without functional Y homologs, the likely derived state is therefore inactivation of one copy in females, versus “escape from inactivation,” for X-linked genes with functional Y homologs (such genes are expressed in two copies—from both sex chromosomes—in males and females; fig. 2). If gene-specific X inactivation in mammals occurs in response to the loss of functional Y gametologs, X-linked genes with functional Y copies are expected to escape from inactivation in all females, X-linked genes with pseudogenized Y gametologs should escape in some, but not all individuals, and X-linked genes whose Y gametologs have been deleted should be silenced in nearly all individuals (Carrel and Willard 2005). Further, if X inactivation can only evolve after gene activity on the Y is reduced, then there should be a delay, such that X-linked dosage compensation lags behind Y-linked gene degeneration (Charlesworth 1978). Consistent with this, and previous experiments (Carrel and Willard 2005), we confirm that the average proportion of individuals (from nine cell lines assayed in [Carrel and Willard 2005]; supplementary note S1, Supplementary Material online) in which a gene escapes inactivation is significantly higher for X-linked genes with functional versus pseudogenized Y gametologs (fig. 3 and supplementary table S5, Supplementary Material online). We further found that X-linked genes with either functional or pseudogenized Y gametologs are significantly more likely to escape X-inactivation than X-linked genes that have lost their Y gametologs (fig. 3 and supplementary table S5, Supplementary Material online). This supports the hypothesis that there is an interplay between the X-linked gene’s inactivation status and the functionality of its Y gametolog. Additionally, the observation that some X-linked genes whose Y gametologs have been lost still escape inactivation (63 of 151 assayed genes escape XCI in at least one of nine cell lines; supplementary table S1, Supplementary Material online [Carrel and Willard 2005]) suggests that there is a lag between loss of function on the Y and evolution of inactivation on the X in females. Thus, we propose that the development of XCI is an active evolutionary process whereby genetic signals, resulting from the loss of functional X-degenerate genes on the Y, are still accumulating on the X in males to signal the inactivation of one of their X-linked gametologs in females.
Fig. 2.
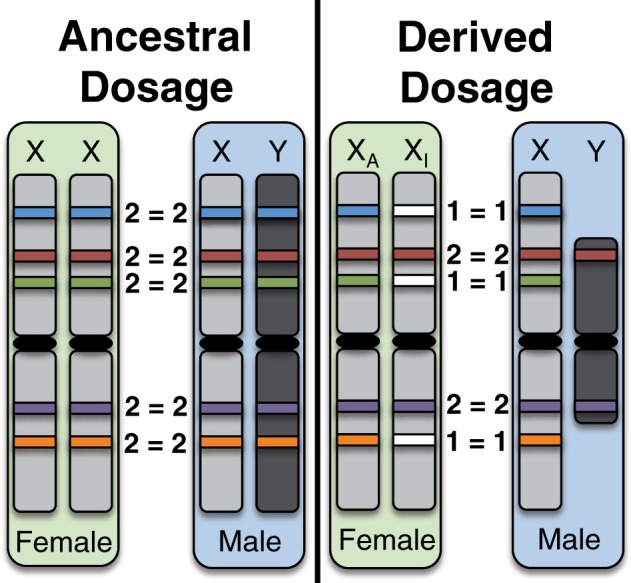
Schematic of ancestral versus derived dosage of sex-linked genes. Ancestrally, it is expected that all genes were expressed in two copies in females (from both X chromosomes) and in two copies in males (from the ancestral X and Y chromosomes). The derived condition, resulting from loss of gene content and expression in males on the Y chromosome, is expression of many sex-linked genes in a single copy in males, and so inactivation evolved to silence one copy of the gene in females (on the inactive X chromosome, XI) and resulted in expression of only one copy of the sex-linked gene in females (from the active X chromosome, XA). Filled rectangles represent expressed genes, whereas empty white rectangles represent silenced (inactivated) genes.
Fig. 3.
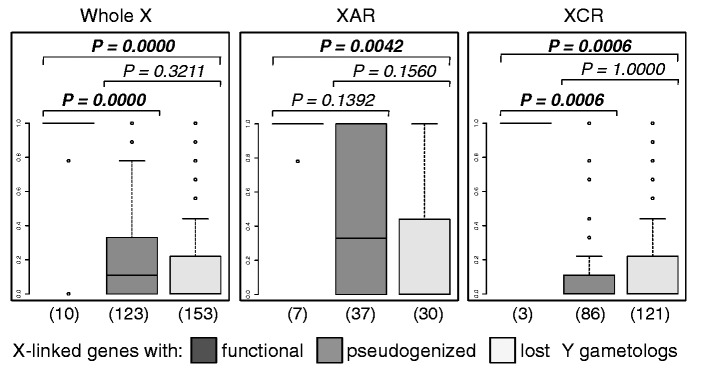
Average proportion of cell lines (out of nine assayed in [Carrel and Willard 2005]) for which an X-linked gene escapes inactivation. Boxplots representing the number of cell lines for which a gene escapes X-chromosome inactivation are shown for the sets X-linked genes with functional, pseudogenized, or absent (lost) Y-linked gametologs. P values testing for significant differences between each pairwise class of X-linked genes (with functional, pseudogenized, and lost Y gametologs) from permutation tests with 10,000 replicates are labeled at the top of the figures for the entire X (whole X), the XAR, and the XCR. P values that are significant after correction for multiple testing are shown in bold, and the number of genes assayed in each set is shown below each boxplot. The mean and median number of cell lines for which a gene escapes X-chromosome inactivation are reported in supplementary table S5, Supplementary Material online.
Because we find evidence that XCI evolves in response to gene loss on the Y chromosome, we wondered how well the current inactivation/escape status of X-linked genes on the inactive X discriminates between X-linked genes with functional or pseudogenized Y gametologs. We conducted linear discriminant analysis with jack-knifed leave-one-out cross-validation in R (lda(); [Team 2009]). The proportion of individuals in which an X-linked gene escapes inactivation (the XCI status) discriminates only moderately well between all three classes of genes (successfully classifying 51.76% of X-linked genes with functional, pseudogenized, or lost Y gametologs across the entire X), because of the challenges of discriminating between X-linked genes with pseudogenized versus lost Y gametologs. However, XCI status is highly predictive when discriminating whether its Y gametolog is functional or nonfunctional (pseudogenized or lost); this model successfully classifies genes in the XCR (96.19%) better than genes in the XAR (90.54%), as expected if X-inactivation might not yet be as well established in the younger XAR, with a success rate of 89.08% across the entire X chromosome.
It was previously observed that several X-linked genes without Y gametologs escape inactivation (Carrel and Willard 2005), but we observe that, on average, X-linked genes that have lost their Y gametologs (including pseudogenes) are subject to inactivation (supplementary table S5, Supplementary Material online). This difference results from our ability to classify X-linked genes lacking homologous Y sequence into those that once had Y gametologs, but lost them, from those that were likely added to the X chromosome independently (such “notAncestral” genes are excluded from the analyses above). The distribution of “notAncestral” X-linked genes was not significantly different from X-linked genes with pseudogenized or deleted Y homologs (two-sided Wilcoxon test, P = 0.0855, and P = 0.2239, respectively), and so differences in the time since recombination cessation should not affect comparisons. Curiously, we found that “notAncestral” genes escape XCI more often than X-linked genes with lost Y gametologs and showed patterns more similar to X-linked genes with pseudogenized Y gametologs (supplementary tables S1 and S5, Supplementary Material online). For example, one gene in this class, FAM9C, is thought to have arisen due to duplication on the X chromosome (Martinez-Garay et al. 2002), has no identifiable gametologous Y sequence, has no identifiable X-linked homologous sequence in the dog or opossum, and escapes XCI in seven out of the nine cell lines assayed (Carrel and Willard 2005). We therefore hypothesize that genes added independently to the X chromosome may not be under the same selective pressure to evolve dosage compensation between the sexes because they were not present on the ancestral X–Y chromosome pair. Alternatively, because these genes were added to the X only, after the cessation of X–Y recombination, immediately into an environment where they will be expressed in two copies in females and one copy in males, they may be sexually antagonistic (beneficial in females but detrimental in males) or may simply not be as sensitive to variations in dosage (Pessia et al. 2012). Finally, because silenced and escape regions tend to cluster and have distinct chromatin signatures (Carrel et al. 2006; Berletch et al. 2010), it is possible that genes that are added within or very near a segment of silenced or escape X-linked genes may be subject to the status of the region where they were added. Together these observations suggest that XCI largely evolves in response to the functional status of Y-linked gametologs.
In summary, we identified a significant skew of X-linked genes with functional, pseudogenized, and lost Y homologs on the X that suggests recombination suppression is a strong driver of gene loss on the Y chromosome. We further established that human X-linked genes that are highly expressed, especially in the brain, and X-linked genes with evidence of strong purifying selection, are more likely to retain functional Y homologs. Finally, we provided evidence supporting the hypothesis that X-inactivation evolves in response to gene loss on the Y chromosome and observed that there is likely some lag time between the loss of functionality on the Y and the inactivation of the X gametolog.
Supplementary Material
Supplementary note S1, data S1, tables S1–S5, and figures S1 and S2 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors are grateful to Laura Carrel and Steve Schaeffer for comments on the manuscript and to Lydia Krasilnikova for her assistance at the early stages of this project. This work was supported by NIH grants R01-GM072264–05S1 and R01-GM072264 to K.D.M. and R01-HD056452 to Laura Carrel and K.D.M., and an NSF Graduate Research Fellowship and fellowship from the Miller Institute for Basic Research to M.A.W.S.
References
- Bachtrog D. The temporal dynamics of processes underlying Y chromosome degeneration. Genetics. 2008;179:1513–1525. doi: 10.1534/genetics.107.084012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berletch JB, Yang F, Disteche CM. Escape from X inactivation in mice and humans. Genome Biol. Evol. 2010;11:213. doi: 10.1186/gb-2010-11-6-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brawand D, Soumillion M, Necsulea A, et al. (18 co-authors) The evolution of gene expression levels in mammalian organs. Nature. 2011;478:343–348. doi: 10.1038/nature10532. [DOI] [PubMed] [Google Scholar]
- Carrel L, Park C, Tyekucheva S, Dunn J, Chiaromonte F, Makova KD. Genomic environment predicts expression patterns on the human inactive X chromosome. PLoS Genet. 2006;2:1477–1486. doi: 10.1371/journal.pgen.0020151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434:400–404. doi: 10.1038/nature03479. [DOI] [PubMed] [Google Scholar]
- Charlesworth B. Model for evolution of Y chromsomes and dosage compensation. Proc Natl Acad Sci U S A. 1978;75:5618–5622. doi: 10.1073/pnas.75.11.5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B. Background selection and patterns of genetic diversity in Drosophila melanogaster. Genet Res. 1996;134:1289–1303. doi: 10.1017/s0016672300034029. [DOI] [PubMed] [Google Scholar]
- Charlesworth B, Charlesworth D. The degeneration of Y chromosomes. Philos Trans Royal Soc Biol Sci. 2000;355:1563–1572. doi: 10.1098/rstb.2000.0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren H, Parsch J. The evolution of sex-biased genes and sex-biased gene expression. Nat Rev Genet. 2007;8:689–698. doi: 10.1038/nrg2167. [DOI] [PubMed] [Google Scholar]
- Hughes JF, Skaletsky H, Brown LG, et al. (27 co-authors) Strict evolutionary conservation followed rapid gene loss on human and rhesus Y chromosomes. Nature. 2012;483:82–86. doi: 10.1038/nature10843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JF, Skaletsky H, Pyntikova T, et al. (17 co-authors) Chimpanzee and human Y chromosomes are remarkably divergent in structure and gene content. Nature. 2010;463:536–539. doi: 10.1038/nature08700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser VB, Zhou Q, Bachtrog D. Non-random gene loss from the Drosophila miranda neo-Y chromosome. Genome Biol Evol. 2011;3:1329–1337. doi: 10.1093/gbe/evr103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahn BT, Page DC. Four evolutionary strata on the human X chromosome. Science. 1999;286:964–967. doi: 10.1126/science.286.5441.964. [DOI] [PubMed] [Google Scholar]
- Lemaitre C, Braga MD, Gautier C, Sagot MF, Tannier E, Marais GA. Footprints of inversions at present and past pseudoautosomal boundaries in human sex chromosomes. Genome Biol Evol. 2009;1:56–66. doi: 10.1093/gbe/evp006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Garay I, Jablonka S, Sutajova M, Steuernagel P, Gal A, Kutsche K. A new gene family (FAM9) of low-copy repeats in Xp22.3 expressed exclusively in testis: implications for recombinations in this region. Genomics. 2002;80:259–267. doi: 10.1006/geno.2002.6834. [DOI] [PubMed] [Google Scholar]
- Mikkelsen TS, Wakefield MJ, Aken B, et al. (64 co-authors) Genome of the marsupial Monodelphis domestica reveals innovation in non-coding sequences. Nature. 2007;447:167–178. doi: 10.1038/nature05805. [DOI] [PubMed] [Google Scholar]
- Park C, Carrel L, Makova KD. Strong purifying selection at genes escaping X chromosome inactivation. Mol Biol Evol. 2010;27:2446–2450. doi: 10.1093/molbev/msq143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessia E, Makino T, Bailly-Bechet M, McLysaght A, Marais GA. Mammalian X chromosome inactivation evolved as a dosage-compensation mechanism for dosage-sensitive genes on the X chromosome. Proc Natl Acad Sci U S A. 2012;109:5346–5351. doi: 10.1073/pnas.1116763109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. Vienna (Austria): R Foundation for Statistical Computing; 2012. R: a language and environment for statistical computing. [Google Scholar]
- Rens W, O'Brien PC, Grützner F, et al. (13 co-authors) The multiple sex chromosomes of platypus and echidna are not completely identical and several share homology with the avian Z. Genome Biol. 2007;8:R243. doi: 10.1186/gb-2007-8-11-r243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross MT, Grafham DV, Coffey AJ, et al. (282 co-authors) The DNA sequence of the human X chromosome. Nature. 2005;434:325–337. doi: 10.1038/nature03440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaletsky H, Kudora-Kawaguchi T, Minx PJ, et al. (40 co-authors) The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature. 2003;423:825–837. doi: 10.1038/nature01722. [DOI] [PubMed] [Google Scholar]
- Whibley AC, Plagnol V, Tarpey PS, et al. (24 co-authors) Fine-scale survey of X chromosome copy number variants and indels underlying intellectual disability. Am J Hum Genet. 2010;87:173–188. doi: 10.1016/j.ajhg.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MA, Makova KD. Evolution and survival on eutherian sex chromosomes. PLoS Genet. 2009;5:e1000568. doi: 10.1371/journal.pgen.1000568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sturgill D, Parisi M, Kumar S, Oliver B. Constraint and turnover in sex-biased gene expression in the genus Drosophila. Nature. 2007;450:233–237. doi: 10.1038/nature06323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.