

Abstract
Rapid turnover of mobile elements drives the plasticity of bacterial genomes. Integrated bacteriophages (prophages) encode host-adaptive traits and represent a sizable fraction of bacterial chromosomes. We hypothesized that natural selection shapes prophage integration patterns relative to the host genome organization. We tested this idea by detecting and studying 500 prophages of 69 strains of Escherichia and Salmonella. Phage integrases often target not only conserved genes but also intergenic positions, suggesting purifying selection for integration sites. Furthermore, most integration hotspots are conserved between the two host genera. Integration sites seem also selected at the large chromosomal scale, as they are nonrandomly organized in terms of the origin–terminus axis and the macrodomain structure. The genes of lambdoid prophages are systematically co-oriented with the bacterial replication fork and display the host high frequency of polarized FtsK-orienting polar sequences motifs required for chromosome segregation. matS motifs are strongly avoided by prophages suggesting counter selection of motifs disrupting macrodomains. These results show how natural selection for seamless integration of prophages in the chromosome shapes the evolution of the bacterium and the phage. First, integration sites are highly conserved for many millions of years favoring lysogeny over the lytic cycle for temperate phages. Second, the global distribution of prophages is intimately associated with the chromosome structure and the patterns of gene expression. Third, the phage endures selection for DNA motifs that pertain exclusively to the biology of the prophage in the bacterial chromosome. Understanding prophage genetic adaptation sheds new lights on the coexistence of horizontal transfer and organized bacterial genomes.
Introduction
Bacterial viruses, commonly known as bacteriophages or phages, are numerous and have an important impact in the regulation of bacterial populations in the environment and in the human microbiome (Weinbauer 2004; Suttle 2005; Breitbart et al. 2008; Reyes et al. 2010). Bacteriophages are very abundant and very diverse. Their genomes can be single stranded or double stranded, made of DNA or RNA, in one or several linear or circular molecules (Abedon and Calendar 2005). The International Committee on Taxonomy of Viruses (ICTV) bases phage taxonomy on the shape of virion particle (King et al. 2011). However, distinct families can exchange large DNA fragments blurring classical taxonomical definitions (Hendrix et al. 1999). Exchange of functional modules between phages leads to reticulate evolution and may favor their evolvability (Botstein 1980). Modularity and genetic compaction lead to highly organized genomes of phages, where genes involved in related functions or expressed at the same moment in the phage infectious cycle are generally clustered together and expressed within the same operon (Ptashne 1992). A large group of otherwise unrelated phages (called "lambdoid" phages) share phage Lambda's genomic organization (Campbell and Botstein 1983). This is thought to facilitate viable genome assortment by recombination (Juhala et al. 2000). The rapid evolution of phages by mutation and recombination and their lack of universal genes (contrary to prokaryotes) render classical phylogenetic approaches of little use. Alternative methods based on gene repertoire relatedness have thus been proposed (Rohwer and Edwards 2002; Lima-Mendez et al. 2008b). Our understanding of phages is largely derived from the study of a few clades, most notably phages of enterobacteria. Accordingly, metagenomic studies find few sequences homologous to known phages (Edwards and Rohwer 2005; Angly et al. 2006; Reyes et al. 2010).
Phages are bacterial parasites whose transmission involves, with rare exceptions, the death of the host by completion of a lytic cycle. However, some phages, so-called temperate phages, have the ability to enter a lysogenic state and replicate vertically with the host (Kourilsky 1973; St-Pierre and Endy 2008). Most temperate phages integrate into the chromosome. Under specific physiological conditions, the prophage excises from the chromosome and enters the lytic cycle. Integration and excision are usually mediated by a site-specific tyrosine or serine recombinase (Nunes-Duby et al. 1998; Smith and Thorpe 2002). Some temperate phages remain in the cell under the extrachromosomal form, for example, phage N15 of Escherichia coli (Ravin 2011). Other prophages integrate and transpose randomly in genomes using DDE transposases, for example, Mu (Mizuuchi 1992). Satellite phages code for the information necessary to subvert virions from other phages but not for their own virion particle, for example, the P4 phage subverts virions from the P2 phage (Six and Klug 1973). Finally, Inoviridae are small single-stranded DNA (ssDNA) phages that integrate as prophages in the chromosome using the host recombinases (Huber and Waldor 2002). Thus, although the temperate Lambda phage model was instrumental in our understanding of phages (Ptashne 1992), the genetics of temperate phages is very diverse.
Prophages express very few genes. Among genes essential to their biology, they typically express a repressor of the lytic cycle (Ptashne 1992). Prophages and their bacterial hosts have aligned interests in avoiding further infection by mobile genetic elements. Hence, elements that are important in phage warfare are also useful to the host (Shinedling et al. 1987; Nechaev and Severinov 2008; Van Melderen and Saavedra De Bast 2009; Labrie et al. 2010). Some prophages carry cargo genes encoding traits adaptive to the host, among which are virulence factors in many bacterial pathogens (Ohnishi et al. 2001; Banks et al. 2002; Boyd and Brussow 2002; Brussow et al. 2004; Thomson et al. 2004; Abedon and Lejeune 2005; Winstanley et al. 2008). Not only do prophages encode traits that can increase the host fitness, they can also be used as biological weapons against other bacteria (Bossi et al. 2003; Brown et al. 2006). Several prophages have been shown to increase the growth rates of their hosts under particular conditions, even in the absence of competing mobile genetic elements (Edlin et al. 1977). These examples suggest a symbiotic association between phages and bacteria (Roossinck 2011). However, most intact prophages kill the bacterial cell upon induction of the lytic cycle. There is thus a delicate balance between lysogeny and induction of the lytic cycle, and this has important consequences in the interaction between phages and hosts. Understanding the way prophages integrate and remain in genomes is important to understand this balance and to quantify the contribution of prophages to bacterial fitness.
The integration of phages may affect a number of the organizational traits of the bacterial chromosome (Reyes-Lamothe et al. 2008; Rocha 2008). 1) Genes encoding functional neighbors or interacting proteins cluster in operons and superoperons (Lathe et al. 2000; Zaslaver et al. 2006). 2) The transcription of most genes, and especially essential genes, is co-oriented with the replication fork (Rocha and Danchin 2003). 3) Highly expressed genes concentrate near the origin of replication in fast growing bacteria to enjoy replication-associated gene dosage effects (Couturier and Rocha 2006). 4) Escherichia coli's chromosome is structured in four macrodomains and two nonstructured regions (Valens et al. 2004). Physical interactions are frequent within and rare between macrodomains. This chromosome structure has not yet been extensively investigated in other bacterial species. 5) The genome is packed with regulatory signals involved in cell processes such as translation, transcription, replication, chromosome structure, and segregation (Touzain et al. 2011). All these five organizational features are expected to constrain changes in bacterial genomes (Rocha 2004). Thus, large changes in chromosome structure are tolerated only when its organization is respected (Itaya et al. 2005; Cui et al. 2007; Esnault et al. 2007; Val et al. 2012). As a result, one would expect strong natural selection for phage integration in sites where it least affects the host fitness (Lawrence and Hendrickson 2003). Prophages are part of the chromosome. Thus, one would also expect selection for gene orientation and DNA motifs in the prophage matching the local and global chromosomal organization. Selection for such traits in phages is possible because most phages integrate at specific well-defined sites in the chromosome leaving reproducible prophage structures. Also, prophages and chromosomes have aligned interests whenever prophage organization within the genome improves, or at least does not negatively affect, the host fitness.
There have been indications that prophages are not randomly distributed in genomes. Notably, prophages encoding integrases of the tyrosine recombinase family tend to integrate at or close to the 3′ of transfer RNA (tRNA) or transfer-messenger RNA (tmRNA) genes possibly due to a preference for palindromic structures (Campbell 1992, 2003; Williams 2002, 2003). The current availability of very large data sets of complete genomes for Escherichia, Salmonella, and their phages opens up the possibility to study with a strong statistical basis the adaptation of prophages to the chromosome background. In this work, we focus on the patterns of phage integration and how these relate with local and global organizational features of the bacterial chromosome.
Results and Discussion
Identification of Prophages
We analyzed 47 completely sequenced genomes of E. coli, one from E. fergusonii, 20 from Salmonella enterica, and 1 from S. bongori (for details see supplementary table S1, Supplementary Material online). We identified prophages using Phage Finder (Fouts 2006), Prophinder (Lima-Mendez et al. 2008a), and PHAST (Zhou et al. 2011). We compared these independent predictions in the light of published information (Ohnishi et al. 2001; Casjens 2003; Canchaya et al. 2004; Thomson et al. 2004; Asadulghani et al. 2009). We precised prophage boundaries using sequence similarity to phages and the patterns of presence and absence of genes in the bacterial strains of the same species (see Materials and Methods). The few tandem prophages were curated manually. Smaller prophage remnants (putative defectives) are often very difficult to distinguish from other integrative elements. Therefore, we removed prophages smaller than 10 kb, as in Canchaya et al. (2003) and Casjens (2003). We removed 49 prophages with more than 25% of transposases in their gene repertoires. These elements are degraded and thus difficult to distinguish from other mobile elements. This resulted in the main data set of 500 prophages.
Prophages tend to be recently integrated in bacterial genomes and thus strain specific (Canchaya et al. 2003). Nevertheless, our data set includes some very closely related bacterial strains (fig. 1A and B), and some of the prophages may have arisen from the same integration event in an ancestral genome (henceforth named orthologous prophages). To control for pseudoreplication in the statistical analyses, we identified these prophages using similarity and positional scores (see Materials and Methods). This nonredundant data set (NRall) includes 418 prophages that have similarity scores lower than 90%. We also created an even smaller data set including 301 prophages in NRall that are larger than 30 kb (NRlong). These prophages are nonredundant and less affected by accumulation of mutations and pseudogenization events. By default, we present the statistics obtained using the main data set. Other data sets are mentioned only when relevant, for example, when leading to different conclusions. Comparison of the size of the main and the NRall data set suggests that most prophages are not orthologous.
Fig. 1.
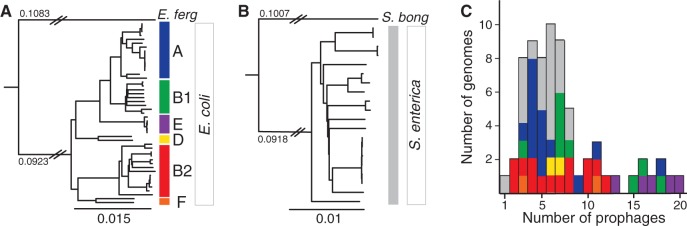
Core genome phylogenies and prophage content of Escherichia and Salmonella. (A) Maximum likelihood phylogenetic tree of the 47 Escherichia coli strains. (B) Maximum likelihood phylogenetic tree of the 20 Salmonella enterica strains. Escherichia fergusonii and S. bongori were used to root the trees of each species. The branch length separating E. fergusonii from the E. coli strains is not to scale (same for S. bongori); the numbers above the branch indicate the respective substitution rates per site. All nodes of the trees were supported with high bootstrap values (>97%), the few exceptions correspond to some terminal branches connecting very closely related strains. Phylogenetic groups of the strains are indicated with colors on the right part of each panel. (C) Distribution of the number of prophages per genome. Colors correspond to the phylogenetic groups of panels A and B.
The number of prophages in genomes is highly variable regardless of their phylogenetic group (fig. 1C). It ranges from 2 to 20 in Escherichia (up to 13.5% of the genome of O157:H7 str. EC4115) and from 1 to 8 in Salmonella (up to 4.9% of the genome of Newport str. SL254) (see supplementary table S1, Supplementary Material online). On average, Escherichia genomes have more prophage genes than Salmonella's (5.6% vs. 3.5%; Student’s test, P < 0.0005). Independent of this effect, larger genomes have more prophages (fig. 2A; Spearman's ρ = 0.52, P < 0.0001). To investigate how prophages contribute to the diversity of the repertoire of gene families in both E. coli and S. enterica, we computed the pan genomes of these species (see Materials and Methods). In both species, we found approximately 3,000 genes present in more than 90% of the strains (persistent genes), although the fraction of core genes (present in 100% of the strains) is smaller in E. coli (1,983 genes vs. 2,628 in S. enterica) (fig. 2B). The accessory genome, consisting of the genes present in less than 90% of the strains, is much larger in E. coli (∼18,100 genes) than in S. enterica (∼6,800 genes). Importantly, E. coli pan genomes remain larger when analyzing the same number of genomes of the two species (fig. 2C). The larger E. coli accessory genome is consistent with the high abundance of prophages in this species. Indeed, prophages account for 41% and 31% of the accessory genes in E. coli and S. enterica, respectively. A total of 75% of prophage genes are present in less than two strains in E. coli (80% in S. enterica), suggesting that upon acquisition, they tend to be rapidly lost, contributing to the open pan genome of these two species (fig. 2C). Prophages are important contributors to genome plasticity (Ohnishi et al. 2001; Banks et al. 2002; Casjens 2003; Canchaya et al. 2004). In these clades, they account for a large fraction of the accessory genome determining variations in genome size.
Fig. 2.
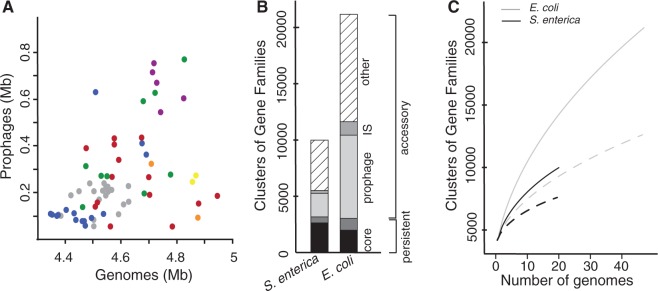
Contribution of prophages to chromosome plasticity. (A) Scatter plot of cumulative size of resident prophages against the size of the host genome (Spearman's ρ = 0.52, P < 0.0001). Colors correspond to the phylogenetic groups as in figure 1. (B) Fraction of the core, persistent, and accessory genes in the pan genome of Salmonella enterica (left) and Escherichia coli (right). The core genome corresponds to the genes present in all strains, the persistent genome to the genes present in more than 90% of the strains. The accessory genome is split in three categories: the prophages, the insertion, sequences (IS), and the other genes. (C) Escherichia coli (in gray) and S. enterica (in black) pan genomes according to the number of sequenced genomes. The dotted lines correspond to pan genomes after removing prophage elements.
The Diversity of Prophages
We made sequence similarity analyses between the proteomes of all phages of enterobacteria and all detected prophages of Escherichia and Salmonella. With these results, we built phage classification schemes based on trees and on graphs (see Materials and Methods). In the following, we use the tree representation because it is easier to compare with classical protein phylogenies and does not involve the choice of clustering parameters. Prophages were classified by comparing their position in the cladogram with those of a set of 147 phages and 50 prophages classified in GenBank or in the literature (Casjens 2003) (see Materials and Methods and supplementary fig. S1, Supplementary Material online). Six different features were thus attributed to each prophage, when possible: 1) the nucleic acid type (double stranded DNA [dsDNA] or ssDNA), 2) the life style (temperate or virulent), 3) the type lambdoid or nonlambdoid, 4) the order, 5) the viral family (based on the particle structure), and 6) the genus (see supplementary table S2, Supplementary Material online). The nucleic acid type and the life style were confidently determined for all the prophages. The taxonomic order, a family, and a genus were attributed to 75% of the prophages (supplementary table S2, Supplementary Material online). The remaining 25% prophages are on average much smaller (median size of 19 kb vs. 40 kb for classed prophages, P < 0.0001, Wilcoxon test). Almost one third of unclassified prophages lack an integrase (vs. 12% in the NRlong data set, see later). These traits suggest that many unclassified elements are prophage relics, which might justify their unreliable classification. Some of the few large unclassified prophages may be previously nondescribed classes or chimeras. Indeed, the Stx-like group of prophages is related to both Lambda-like (Siphoviridae) and P22-like (Podoviridae) phages (Garcia-Aljaro et al. 2009) and was classed apart from both. A second group of prophages was classed independently of the genera defined by the ICTV: the "SfV-like" phages. Such elements display unique features as they are lambdoid and have a Myoviridae tail structure (Allison et al. 2002; Mmolawa et al. 2003). Importantly, our method of classification can be sensitive to the inclusion of small genomes in the data set (Wolf et al. 2002; Snel et al. 2005). To test the robustness of the classification tree, we applied the same procedure to the 301 NRlong prophages. We found identical classifications for 90% of the prophages. Hence, small phage genomes may affect the topology of the cladogram but do not introduce major changes in the classification. In the following analyses, we use the classification based on the entire data set as this allows classing all prophages.
Temperate and virulent phages form clearly distinct clades in our classification. Accordingly, no single prophage was positioned among virulent phages in the tree (fig. 3A). The majority of prophages are from the Myoviridae, Siphoviridae, and Podoviridae families (126, 223 and 30 prophages, respectively), with only three occurrences of Inoviridae. Two thirds of the prophages are lambdoid. Escherichia coli and S. enterica have significantly different distributions of phage genera (P < 0.0001, χ2 test), with the latter lacking Inoviruses, Epsilon15-like, Mu-like, and phiC31-like prophages. However, a wide diversity of viruses, including filamentous phages, were previously observed in Salmonella (Ackermann 2007), suggesting that a larger sampling will partially correct for this effect. The most noticeable difference between the species is the very high fraction of Lambda-like prophages in E. coli (50%) relative to S. enterica (23%) (P < 10−6, χ2 test). Interestingly, within a few groups (Lambda, SfV, P22, and P2), the phages of E. coli and S. enterica are well separated in the classification (fig. 3B). This suggests that host switching happens rarely and/or that it is accompanied with rapid evolution of specific gene repertoires.
Fig. 3.

Classification of prophages. (A) Phylogenetic tree of phages and prophages based on gene repertoire relatedness (see Materials and Methods). Phage/prophage families are colored according to the color key. The phage/prophage genus is indicated in the inner circle. The members of the “lambdoid” group are indicated in the second circle. The classification of phages/prophages into temperate and virulent is indicated in the third circle. White clusters correspond to unclassified clades. (B) Phylogenetic tree as in (A) but restricted to temperate phages/prophages. Red branches correspond to Salmonella phages/prophages and black branches to Escherichia phages/prophages. Labels indicate some types of phages/prophages of interest and mentioned in the text.
Integration Hotspots
Comparative analyses of prophage locations are complicated by the high plasticity of the genomes of Escherichia and Salmonella (Vernikos et al. 2007; Touchon et al. 2009). To facilitate this analysis, we localized prophages relative to the closest flanking core genes. Escherichia coli and S. enterica genomes are mostly collinear (see supplementary table S1, Supplementary Material online), and only 4% of prophages are within a rearrangement breakpoint region. These few elements were removed from the analysis of integration loci. The remaining 369 E. coli prophages were found in 58 distinct integrative loci and the 102 S. enterica prophages in 24 distinct integrative loci (fig. 4). Loci are shared by an average of 6.4 and 4.2 prophages within E. coli and within S. enterica genomes, respectively. Importantly, similar trends are found with the NRlong data set (5.4 and 3 in E. coli and S. enterica, respectively). We simulated 1,000 times the expected number of integration locations if they took place at random. In this case, one would expect to find 336.2 (95% interval of confidence [CI]: ±0.3) distinct loci in E. coli (1.1 prophage per locus) and 99.8 (95% CI: ±0.1) in S. enterica (1 prophage per locus). Hence, prophages have significant integration hotspots in the genomes. A total of 19 of the 24 integrative loci of S. enterica (80%) are also integration loci in E. coli (fig. 4). Hence, the turnover of prophages is very high but restricted to a few sites in the bacterial chromosome that are often conserved for many millions of generations.
Fig. 4.

Distribution of prophages at integration hotspots. The x axis indicates the position of the hotspots of phage integration in the genomes of Escherichia coli (top) and Salmonella enterica (bottom). The positions of the “integrative loci” (on top for E. coli and bottom for S. enterica) are indicated as positions in the core genome. For example, position 634 in E. coli refers to prophages integrated 3′ of the 634th core gene in the reference genome of E. coli (MG1655 see Materials and Methods). The bars indicate the number of genomes with at least one prophage integrated among E. coli (top) and S. enterica (bottom). Colors in the bars correspond to the phylogenetic group of the genomes as in figure 1. The presence of prophages in E. fergusonii and in S. bongori is represented by a black rectangle above (respectively below) the bars of E. coli (respectively S. enterica). The 19 integrative loci conserved between E. coli and S. enterica genomes are connected in the middle of the figure. “Putative targets” of integration are also indicated in the middle part of the figure (details in the keys). The identification of tRNA (amino acid), sRNA, and protein coding genes are reported at the top and the bottom of the graphs, next to the indication of the flanking core gene (details in supplementary table S3, Supplementary Material online).
Hotspots flanking tRNA or tmRNA genes have often been described and could result from integrases targeting conserved palindromic sequences (Williams 2002). However, these genes flank only 15% of E. coli and 37% of S. enterica integration sites (fig. 4 and supplementary table S3, Supplementary Material online) and only 8 of the 19 conserved hotspots between the two species. The tRNA gene pool is highly variable in these two species (Withers et al. 2006), but the tRNA genes flanking these integration loci are present in a single copy in all strains of E. coli and S. enterica. These tRNAs are not a random sample of the tRNAs of E. coli and S. enterica: They are present in all genomes in one single copy and they decode the least used anticodon of 4- or 6-codon amino acids (supplementary table S4, Supplementary Material online). This might represent selection for elements that are lowly expressed (the case of rarely used tRNAs [Dong et al. 1996]), highly conserved in genomes (core genes), and present in unique positions (allowing coevolution between the temperate phage and the host).
Many recently identified small RNA (sRNA) genes also include palindromes forming hairpins (Waters and Storz 2009). Hence, we analyzed the colocalization of prophages with 441 sRNAs identified in recent large-scale studies of Escherichia and Salmonella (Huang et al. 2009; Raghavan et al. 2011; Shinhara et al. 2011; Kroger et al. 2012) (see Materials and Methods). A total of 11 (19%) and 4 (17%) additional integration sites (after removing the overlap with tRNA genes) are located close (<1 kb) to conserved sRNA genes in E. coli and S. enterica, respectively (fig. 4 and supplementary table S3, Supplementary Material online). No further sRNAs were identified when the detection window was extended to 5 kb. We found that eight sRNA genes form stable secondary structures (i.e., more stable than 90% of random sequences with same size and composition, see Materials and Methods). Two of these genes (ryeB in Salmonella and ryeE in E. coli) were previously known to be targeted by phages (Wassarman et al. 2001; Balbontin et al. 2008). Therefore, sRNAs might also be important integration sites.
We investigated the specific features of the 64% (E. coli) and 46% (S. enterica) of integration loci that are not associated with tRNAs, tmRNAs, ribosomal RNAs (rRNAs), or sRNAs (henceforth named noncoding RNA [ncRNAs]). Integration into protein coding sequences has been described within icd (Wang et al. 1997) and ompW in E. coli (Creuzburg et al. 2011) and lepA in S. enterica (Hermans et al. 2006). Indeed, we find these three loci among the most occupied hotspots (fig. 4). Integration leads to duplication of the 3′-end without affecting the length of the ORF in the first case, whereas the gene is disrupted in the second case (supplementary table S3, Supplementary Material online). We identified 15 additional protein encoding genes disrupted due to phage integration (ssuA, yneJ, wrbA, intQ, tqsA, intR, mlrA yecE, yfaT eutB, yfhL, prfC, yjbN, SPC_4453, and Z5614) (fig. 4 and supplementary table S3, Supplementary Material online). The intQ and intR genes encode integrases and might correspond to pseudogenes of previous prophages. Surprisingly, the other genes are well conserved within E. coli, and eight of them (ssuA, wrbA, prfC, yecE, yneJ, tqsA, yfaT, and eutB) would be part of the E. coli core genome if they had not been disrupted by phage integration. These cases correspond to sites less frequently occupied by prophages (3.5 prophages per site on average). Two of them were disrupted by Mu-like prophages that integrate randomly in the host genome (Bukhari and Metlay 1973). Thus, some protein encoding genes are hotspots even though this leads to their disruption. However, most of these integration loci are poorly populated suggesting that these are secondary integration sites.
Strikingly, 50% of E. coli and 25% of S. enterica integrative loci are neither next to ncRNA genes nor within protein coding genes. Many of these loci have few or even one single prophage and may represent secondary integration sites. However, five of these loci are occupied at higher frequencies than the average loci (11.8 prophages, P < 0.02, Wilcoxon test). This is the case of the integration site of phage Lambda (Otsuka et al. 1988). Contrary to ncRNA genes, intergenic regions are under few constraints, and integration sites in these regions are expected to evolve fast. Nevertheless, we observe four such hotspots shared by E. coli and S. enterica (i.e., 21% of all conserved loci). Conservation of intergenic sequences at such large evolutionary distances requires strong purifying selection. This may result from selection for lysogeny, which is adaptive for the host, and for constancy of integration sites, which favors coevolution of phage and bacterial genome structures.
Tropism of Phage Integration
We also studied the tropism of phage integration from the point of view of the phage. In E. coli, Inovirus, Epsilon15-like, and phiC31-like phages integrate each at one single site (supplementary table S5, Supplementary Material online). Stx-like, P4-like, P22-like, and SfV-like phages integrate at a small number of different sites (2, 3, 5, and 5 sites, respectively). On the other hand, P2-like and lambda-like phages integrate into many sites (13 and 21 sites, respectively). Expectedly, we found Mu-like phages integrated randomly in the chromosome. Integration loci tend to be genus specific because few sites (8/4 in E. coli/S. enterica) include more than one phage genus. Of these, two sites show an extreme prophage diversity including almost all genera of prophages and even other mobile genetic elements such as integrative conjugative elements and pathogenicity islands (i.e., sites flanking tRNAThr and tmRNA, supplementary fig. S2, Supplementary Material online). We found no obvious association between phage genus and target type (i.e., tRNA, tmRNA, sRNA, or protein coding gene) (supplementary table S5, Supplementary Material online). We found 15 integrative sites containing only unclassified prophages in E. coli (4 in S. enterica) (supplementary table S5, Supplementary Material online), which typically correspond to small elements ongoing genetic degradation. This suggests that some integration sites provide a more favorable genetic background than others.
We then tested whether integration tropisms were associated with the phylogeny of the phage integrases. We found that 413 of the 500 prophages (83%) contained an integrase, all tyrosine recombinases. This percentage rose to 89% among NRlong prophages. Phages lacking integrases may have lost them after integration or use other means to integrate. Accordingly, Mu-like prophages and Inoviruses lacked such integrases (1% of the NRlong prophages). We constructed a phylogenetic tree of the integrases to associate integrase similarity with integration tropism. The deeper nodes of the tree are poorly supported limiting the conclusions that can be taken from ancient evolutionary events (fig. 5). The more recent nodes show clusters of phages of the same genus. This includes P2-like, P4-like, and Epsilon15-like prophages. Lambdoid prophages are intermingled in the tree as expected because they showed no commonalities in terms of integration sites. Importantly, integrases from elements integrated at the same locus form terminal clades in the tree, that is, closely related integrases tend to integrate at the same sites. The few apparent exceptions were all examined in detail and concern loci with multiple close integrations where one element is correctly grouped in the tree and the other is inserted in a nearby sequence and clusters elsewhere in the tree (supplementary fig. S3, Supplementary Material online).
Fig. 5.

Phylogeny of the integrases. The maximum likelihood tree was made from a trimmed alignment of 332 tyrosine recombinases and rooted using the midpoint root. Bootstrap values (out of 1,000 replicates) are given in percents in the tree and are shown when exceeding 50%. Prophage types are indicated in the first column. The species hosting the prophage is shown in the second column. The third column shows that blocks of closely related integrases correspond to phages integrated at the same loci. One given block puts together a given number of integrases that are together in the phylogenetic tree and are associated with a single locus.
Distribution of Prophages in the Chromosome
The propensity for integration by site-specific recombination varies with genomic regions in S. enterica (Garcia-Russell et al. 2004). Unfortunately, there are no data available on the large-scale structure of the chromosome of Salmonella. In E. coli, the chromosome is structured in domains and macrodomains that are associated with specific local properties, such as DNA compactedness (Wiggins et al. 2010). This might affect patterns of prophage integration or excision. Accordingly, prophages and their integration loci are not randomly distributed among the four macrodomains and the two nonstructured (NS-left and NS-right) domains of the E. coli chromosome (both P < 0.0005, χ2 test). The latter have the lowest number of prophage loci (3 in NS-right and 0 in NS-left), followed by the origin of replication (Ori) macrodomain (nine loci) (fig. 6B). Contrary to the four macrodomains, NS regions show high intracellular mobility and interact with their surrounding domains (Valens et al. 2004). This should not disfavor integration events and indeed we find that the frequency of transposases in this region is not significantly different from the rest of the genome (P = 0.77, χ2 test). Furthermore, NS regions integrate some well-known pathogenicity islands encoding tyrosine recombinases (Napolitano et al. 2011), for example, PAI-LEE, PAI-ICFT073, and PAI-IIIEDL933 (Blum et al. 1994; McDaniel et al. 1995; Dobrindt et al. 2002). Core genes in these regions have sequence compositions similar to the rest of the genome (51% in GC content, P = 0.2, Wilcoxon test) suggesting this is not the cause of a putative integration bias. The frequency of tRNA or sRNA genes in these regions is also not different from expected (P > 0.05, χ2 test). Essential genes are 50% more abundant than expected in NS regions (P < 10−7, χ2 test), but their density (10% vs. 6% in the entire genome) seems too low to lead to a general avoidance of prophages in these regions because of the over-representation of genes for which inactivation is lethal. Interestingly, the average Codon Adaptation Index of genes in the NS regions and the Ori macrodomain is higher than in the rest of the genome (0.414 vs. 0.396, P < 10−6, Wilcoxon test). High expression of neighboring genes might render prophages less stable. On the other hand, macrodomains are located in different regions of the cell. Notably, the NS-right region is closer to the cell center, followed by the Ori, the Right, and the terminus of replication (Ter) macrodomain that is the closest to the cell poles (NS-left and left were not tested) (Meile et al. 2011). This might render the Ter and the nearby macrodomains more susceptible to integration by phages, especially because phage infection might preferentially take place at cell poles (Edgar et al. 2008; Guerrero-Ferreira et al. 2011).
Fig. 6.
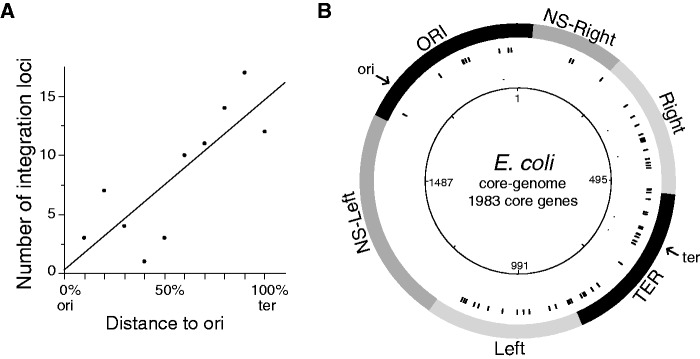
Distribution of prophages in the chromosome. (A) Number of loci with prophages in function of the distance to the origin of replication. Distribution of integration loci in function of the distance to the origin of replication (ori: origin and ter: terminus). (B) Circular representation of the distribution of the prophages in function of the macrodomains of Escherichia coli. Circles represent the following (from the inside out): 1, position in the core genome; 2, location of the integration locus; and 3, location of the four macrodomains and the two nonstructured (NS-right and NS-left) domains of the E. coli chromosome.
The frequency of prophages (and integration loci) increases with the distance to the origin of replication both in Escherichia and Salmonella (fig. 6A, respectively, Spearman's ρ = 0.79, P < 0.006 and ρ = 0.82, P < 0.005). The frequency of ncRNA genes is not higher in this region (P > 0.6, χ2 test) and cannot justify the observed pattern. We then tested whether macrodomain structure was sufficient to explain these patterns. For this, we analyzed the abundance of prophages within each macrodomain. We divided each macrodomain in equally sized terminus-proximal and terminus-distal regions. The intra-macrodomain regions nearer the terminus have 24% more prophages and 24% more integration loci than the intra-macrodomain regions nearer the origin of replication (respectively, P < 10−6 and P = 0.055, χ2 tests). Hence, prophages are more abundant in certain macrodomains, and within the macrodomains, they are more abundant in regions closer to the terminus of replication.
Prophage Polarization
The genes of lambdoid prophages show a preference for co-orientation with the bacterial replication fork, and this is not explained by their tropism toward some tRNAs (Campbell 2002). Indeed, we found no loci specificity toward lambdoid phages after accounting for phage genus. Bacterial genes, and especially essential genes, are also predominantly co-orientated with the replication fork, presumably to minimize effects of the collisions between the replication fork and the RNA polymerase (Rocha and Danchin 2003). To study these patterns, we defined prophage transcription polarity as the fraction of the prophage coding sequences in the most gene-rich strand of the prophage. We analyzed two subsets: the lambdoids (330 prophages) and the nonlambdoid Myoviridae (104 prophages), which are the largest clade of the remaining prophages. Together these groups make 87% of our data set. Most prophages were highly polarized with an average of 77% of the coding nucleotides in the most gene-rich strand (76% in lambdoids and 79% in nonlambdoid Myoviridae).
The co-orientation of a large fraction of the prophage genome does not necessarily entail co-orientation of prophage genes with the bacterial replication fork. We defined prophage replication polarization as the predominant orientation of genes relative to the direction of the bacterial replication fork. We found that 85% of prophages are predominantly co-oriented with the bacterial replication fork (P < 10−15 in the three data sets: all, NRall, and NRlong, χ2 test) (fig. 7). The effect is much stronger in lambdoid prophages (98% of prophages, P < 10−15, χ2 test) than for the average host gene (∼57% both in E. coli and S. enterica) and for the E. coli essential genes (71%). Replication polarization of nonlambdoid Myoviridae is not significant (56%, P > 0.05, χ2 test). Hence, replication polarity, contrary to transcription polarity, is specific to lambdoids. Interestingly, among lambdoid phages, the smaller and presumably more degraded prophages are less often co-oriented with the replication fork than the NRlong prophages (88% vs. 100%, P < 10−6, χ2 test). Lambdoid prophages might thus degrade faster when antioriented with the replication fork.
Fig. 7.
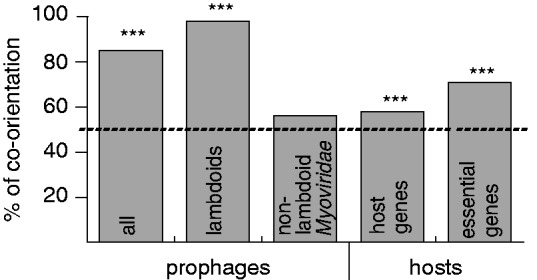
Percentage of prophages and host genes co-oriented with the replication fork. The dotted line shows the polarization under random expectation (50%). P < 0.05 (*); P < 0.01 (**); P < 0.001 (***).
If the replication polarity of lambdoids is caused by collisions between the bacterial RNA polymerases and replication forks, as proposed for bacteria, then the transcription of genes expressed in the prophage should be preferentially co-oriented with the replication fork. Most genes are silent in the prophage state, with the notable exception of the repressor of the lytic cycle. We thus identified a total of 115 cI repressors of the lytic cycle among the 330 lambdoid prophages (see Materials and Methods). A majority of these (90%) were found antioriented with the replication fork. This result is in stark contradiction with the hypothesis that collisions between RNA polymerase and the replication fork cause co-orientation of prophage genes with the bacterial replication fork. Inversion of Lambda prophages in E. coli lacks strong phenotypes in terms of bacterial growth or genetic instability (Campbell 2002). This suggests that prophage polarization does not have a strong impact on the cell's physiology. Co-orientation of lambdoids with the replication fork might thus be associated with their particular genetic organization and how it accommodates in the bacterial chromosome, for example, in terms of DNA motifs (see later). Alternatively, this might be due to some association between the mechanism of phage integration and the bacterial replication fork. This association was found in several DDE recombinases (Peters and Craig 2001) but to the best of our knowledge not in integrases using tyrosine recombinase activity.
Distribution of DNA Motifs in Prophages
The genomes of Escherichia and Salmonella are packed with signals that regulate cellular processes affecting the chromosome at large scales such as macrodomain formation (matS) and chromosome segregation (FtsK-orienting polar sequences [KOPS]) (Touzain et al. 2011). The MatP protein interacts with the 13 bp matS sites to organize the terminus of replication of the chromosome into the Ter macrodomain (Mercier et al. 2008). The motif matS is thus concentrated in the Ter macrodomain and absent from the rest of the chromosome. We found no single matS motif in any of the prophages. This is statistically unexpected given the motif size and composition (see Materials and Methods, P < 0.004, χ2 test). The absence of matS in the prophages of the Ter macrodomain is not statistically significant but might simply result from the lack of statistical power (P = 0.1, χ2 test). Indeed, prophages of the Ter macrodomain of E. coli display a strong underrepresentation of matS motifs when compared with the host Ter macrodomain (P < 10−15, χ2 test). The density of matS in the Ter macrodomain of E. coli K12 MG1655 is low (1 every 49 kb). The average size of the NRlong prophages is 44 kb. Therefore, integration of a prophage lacking matS probably has no disruptive effect in the formation of the macrodomain. However, this does not explain the significant avoidance of matS in prophages. The matS motif defines the Ter macrodomain and is absent from the rest of the chromosome (Mercier et al. 2008). Avoidance of matS in prophages outside the Ter macrodomain might be caused by its potential disruptive effect. Phages recombine frequently to produce mosaic structures. Hence, lack of matS in phages integrating at the Ter macrodomain could increase the probability of producing viable recombinant genomes with phages integrating at other chromosomal sites. These results suggest that motifs can be strongly counter selected in prophages when they disrupt chromosomal structure.
KOPS motifs are octamers that orient the transport of DNA by FtsK at the last stages of chromosome segregation (Bigot et al. 2005; Levy et al. 2005). KOPS are more frequent in the ter-proximal regions and in co-orientation with the replication fork (Bigot et al. 2005). KOPS are more abundant than expected in the chromosome (9.6 × 10−5 KOPS/nt) and in lambdoid prophages (9.5 × 10−5 KOPS/nt, both P < 0.01, χ2 test). They are also strongly co-oriented with the replication fork (respectively, 90% and 86%). We observed lower density of KOPS in nonlambdoid Myoviridae prophages (5.1 × 10−5 KOPS/nt, P > 0.1, χ2 test) and even lower in virulent phages (3.1 × 10−5 KOPS/nt, P > 0.7, χ2 test). Interestingly, the density of KOPS in lambdoids mirrors the trends of the rest of the genome: KOPS density is lower in prophages in the Ori-proximal half of the chromosome than in Ter-proximal half (7.2 × 10−5 vs. 1.0 × 10−4 KOPS/nt, P < 10−5, χ2 test). Furthermore, the density of KOPS in the Ter-proximal half and in its lambdoid prophages is very similar (9.6 × 10−5 vs. 1.0 × 10−4 KOPS/nt, P > 0.4, χ2 test). This suggests selection for the over-representation of polarized KOPS in lambdoids to match the chromosomal organization.
Conclusion
Our study shows that phages integrate in ways that minimize their negative effects on the chromosome organization. This coevolution of phages and bacteria involves selection for integration sites, gene order, and DNA motifs that affect the biology of the bacterial chromosome. Phage integration is restricted to a few sites that are conserved over very long evolutionary periods. Targeting slow evolving sequences (especially RNA genes) is adaptive for phages. However, many prophages integrate at sites in intergenic regions that are conserved between E. coli and S. enterica. This suggests selection for the conservation of integration sites as a means of promoting lysogeny over lysis and facilitating long-term coevolution of temperate phages and bacteria. Prophage organization is also important at the chromosome scale because prophage density increases along the replichores and differs markedly among macrodomains. This might result from integration biases caused by different accessibility of chromosomal regions to prophages. It might also result from selection for regions of low gene expression. Accordingly, phage abundance increases along the ori–ter axis. The expression of the tmRNA gene, an important integration hotspot, is important for the function of the neighboring P22-like phages and pathogenicity islands (Julio et al. 2000). This suggests that integration sites might provide other functions besides a site-specific recombination point, for example, regulation of gene expression. Accordingly, we find that prophages avoid integration in the most expressed tRNA genes and in the chromosomal regions with the highest fraction of highly expressed genes. This suggests that they avoid proximity to regions highly transcribed. Transcriptional spillover from nearby genes could lead to expression of phage genes and destabilization of the lysogen. Importantly, temperate phages show avoidance and over-representation of DNA motifs that are relevant only at the prophage state in the context of the biology of the host. This adds a constraint to the evolution of temperate phages that is absent from virulent phages. Learning the way prophages minimize their impact on genome organization might provide key information on how to modify genomes with minimal impact on bacterial fitness.
Materials and Methods
Data
A data set of 69 complete genomes of Escherichia and Salmonella was downloaded from the NCBI RefSeq (ftp://ftp.ncbi.nih.gov/genomes/Bacteria/, last accessed January 2012). It consists of 20 S. enterica, 1 S. bongori, 47 E. coli, and 1 E. fergusonii genomes. A total of 299 complete genomes of phages infecting enterobacterial hosts were also downloaded from the NCBI RefSeq (ftp://ftp.ncbi.nih.gov/genomes/Viruses/, last accessed December 2011).
Identification of Prophages
Prophages were detected using Phage Finder (Fouts 2006), PHAST (Zhou et al. 2011), and Prophinder (Lima-Mendez et al. 2008a). These three phage-finding programs combine sequence comparisons to known phage or prophage genes, comparisons to known bacterial genes, tRNA genes, dinucleotide analysis, and identification of integration sites. Phages infecting the enteric bacteria E. coli and S. enterica are the most intensively studied, many sequences are available, and it is therefore less probable to miss prophages due to a gap of knowledge on phages for these genera. We removed small prophages (<10 kb) and elements with a large number of insertion sequences (IS; >25% of the predicted ORFs). IS elements were detected as in Touchon and Rocha (2007). Prophage borders and the few prophages coded in tandem were manually validated using different types of information: gene annotation, PFAM protein functions, and core/pan genome definition in bacterial genomes (see later). Prophage genes integrate together and are thus expected to share similar patterns of presence/absence in bacterial genomes. The frequency of gene families in pan genomes (see later) follows a U-distribution, where most families are present in either very few or many genomes (Touchon et al. 2009). Families of genes in prophages, because they tend to be strain specific, are among the low-frequency genes. On the other hand, genes involved in the core functions of the bacterial cell tend to be among high-frequency genes. Hence, when a bordering gene corresponds to a persistent gene (present in at least 90% of strains), it was removed of the predicted prophage. A blastp (with an e value < 0.001) of the detected prophages was performed against the rest of the bacterial hosts to check for the presence of further undetected elements. Any cluster of 10 or more genes (with a maximal distance of 3 kb between two consecutive genes) was further inspected. Because of their small sizes (typically <10 kb), Inoviruses were detected using a dedicated procedure. They were searched by similarity to known phages by blastp (with an e value < 0.001). When at least four proteins of the reference genomes (GenBank IDs NC_001332, NC_001954, NC_002014 and NC_003287) were detected in a 10 kb window, the putative prophage was checked with GenBank annotations and its borders were manually confirmed as described earlier.
Classification of Phages
Prophages were classed by comparison to previously classed phages by building a common gene content matrix. First, homologous proteins were identified as unique reciprocal best hits with >40% similarity in amino acid sequence and <20% of difference in protein length as in Touchon et al. (2009). The similarity score was determined with the BLOSUM60 matrix and the Needleman–Wunsch end gap free alignment algorithm. We measured gene repertoire relatedness between pairs of (pro)phages as:  with S(Ai,Bi) the similarity score of the pair i of homologous proteins shared by (pro)phage A and (pro)phage B (varying from 0.4 to 1), M the total number of homologs between (pro)phages A and B and nA and nB the total number of proteins of (pro)phage A and B, respectively.
with S(Ai,Bi) the similarity score of the pair i of homologous proteins shared by (pro)phage A and (pro)phage B (varying from 0.4 to 1), M the total number of homologs between (pro)phages A and B and nA and nB the total number of proteins of (pro)phage A and B, respectively.
The gene repertoire relatedness matrix between all pairs of phage/prophages was used to calculate a tree using BioNJ (Gascuel 1997). We then classed phages/prophages using the gene repertoire similarity tree. Prophages were classed according to the phages/prophages with known classification with which they branched together (forming a monophyletic subtree with the classified (pro)phages branching basally, see supplementary fig. S1, Supplementary Material online). For many prophages, we consistently inferred different features: 1) the nucleic acid type: dsDNA/ssDNA/ssRNA; 2) the ICTV taxonomic order: Caudovirales/non-Caudovirales; 3) the life style: temperate/virulent; 4) the type: lambdoid/nonlambdoid; 5) the ICTV family: Siphoviridae/Podoviridae/Myoviridae; and 6) the ICTV genus: Lambda-like/P22-like/P2-like/Epsilon15-like/PhiC31-like/Mu-like/P4-like/Inovirus. Temperate/virulent life styles and the lambdoid membership could be determined from literature data for most phages of the databank. In addition to the genera defined by the ICTV, two supplementary groups were considered as a genus due to their unique features: "SfV-like" phages that can be defined as lambdoid Myoviridae (Allison et al. 2002; Mmolawa et al. 2003) and considered as an independent Myoviridae group, albeit not officially elevated to the rank of genus (Lavigne et al. 2009) and "Stx-like" phages as they constitute a group of very closely related lambdoid phages carrying the Stx toxin and displaying Siphoviridae/Podoviridae hybrid structures (Garcia-Aljaro et al. 2009). The identification of P4 prophages is more complicated because these satellite phages lack structural genes, and there is only one reference sequence in GenBank. P4 encodes one characteristic protein, Sid, which is responsible for its parasitic behavior. Sid functions as a head size determination of phage P2, preventing P2 to integrate its genome within its own capsids (Dearborn et al. 2012). Prophages were classed as P4-like when branching next to P4 (GenBank ID NC_001609) in the tree and if they contained the Sid protein (blastp e value < 0.001). Sid is a good marker of P4-like phages because it was not found in prophages distant from P4 in the tree.
Identification of Core and Pan Genomes
A preliminary set of orthologs was defined by identifying unique pairwise reciprocal best hits, with at least 60% similarity in amino acid sequence and less than 20% of difference in protein length. The list was then refined using information on the distribution of similarity of these putative orthologs and data on gene order conservation (as in Touchon et al. [2009]). The analysis of orthology was made for every pair of genomes of each clade (E. coli and S. enterica). The core genome consists of genes found in all strains of a clade and was defined as the intersection of pairwise lists of positional orthologs.
Definition of Integration Loci
The E. coli and S. enterica core genomes were used to define the integration loci of the detected prophages. Each prophage was localized relative to the two closest flanking core genes of the species. By convention, an integration locus was defined by the relative position of the left core gene among the core genome of the species. For example, the locus 135 in E. coli corresponds to a prophage located between the 135th and the 136th core genes of the E. coli core genome. The relative positions of the loci were defined by the order of the core genes in E. coli K12 MG1655 strain and S. enterica LT2 strain. These strains were used as references for E. coli and S. enterica gene orders, respectively, because they represent the most likely configuration of the chromosome in the ancestor of each species. Few rearrangements were observed (respectively, 2.3 and 2 in average for E. coli and S. enterica genomes) compared with the two reference genomes. Integration loci located between two nonsuccessive core genes, that is, with rearrangements in between them were removed.
Clades Phylogenetic Trees
We extended the species core genomes by adding genomes of the two earliest diverging available species, E. fergusonii and S. bongori. We made multiple alignments of each family of core proteins using muscle v3.6 (Edgar 2004) with default parameters and back-translated these alignments to DNA. The concatenated alignments of core genes were given to Tree-puzzle 5.2 (Schmidt et al. 2002) to compute the distance matrix between genomes using maximum likelihood under the Hasegawa–Kishino–Yano + G(8) + I model. The tree of the core genome was built from the distance matrix using BioNJ (Gascuel 1997). We made 1,000 bootstrap experiments on the concatenated sequences to assess the robustness of the topology. The topology of these trees is congruent with previous whole-genome phylogenetic analyses of E. coli and S. enterica (Touchon et al. 2009; Touchon and Rocha 2010). Groups’ terminology is based on the latest update of E. coli strains classification (Tenaillon et al. 2010).
Identification of Integrases, cI Repressors, and Phylogenetic Analysis
Integrase and cI repressor proteins were searched using PFAM protein profiles for tyrosine recombinase (PF00589), serine recombinase (PF07508 and PF00239), and cI repressor (PF07022) obtained from the PFAM database, version 26.0 (http://pfam.sanger.ac.uk/, last accessed January 2012). Prophages were searched with these profiles using hmmpfam (e value < 0.001, coverage of >50% of the profile) (Eddy 2011). The multiple alignment of the 413 tyrosine recombinase proteins was made with muscle v3.6 (Edgar 2004). Informative regions were selected using BMGE with the BLOSUM30 matrix (Criscuolo and Gribaldo 2010). Poorly aligned sequences were manually removed from the alignment. The final alignment of 332 sequences was used to reconstruct the phylogenetic tree using the maximum likelihood method implemented in TREEFINDER (Jobb et al. 2004) under a mixed + G(5) model, which was estimated as the best-fit model with the Akaike information criterion. The tree topology was assessed with 1,000 bootstrap replicates using the same model.
Identification of ncRNA
The tRNA genes were identified using tRNAscan-SE 1.23 (Lowe and Eddy 1997). The tmRNA genes were detected by sequence similarity search using blastn, having at least 90% of identity sequence and less than 20% of difference in sequence length with the original sequence identified in E. coli (Lee et al. 1978). A single tmRNA gene was thus identified in each genome of Escherichia and Salmonella. Other sRNA genes were identified using two recent published data sets from E. coli (Raghavan et al. 2011; Shinhara et al. 2011) and one from Salmonella (Kroger et al. 2012). The 328 sRNA sequences reported in E. coli K12 MG1655 strain and the 113 sRNA sequences identified in S. enterica SL1344 strain were then blasted against all genome sequences analyzed in this study. For each sRNA, only the best match within each host genome with at least 80% of identity sequence and length coverage of 50% was considered. We found 326 and 195 sRNAs in Escherichia and Salmonella genomes, respectively, with 153 nonredundant sRNA genes shared by all Escherichia strains, 123 shared by all Salmonella genomes, and 73 shared between all Escherichia and all Salmonella genomes. RNA genes were considered as putative integration targets of a prophage when found at less than 1 kb of prophages borders. A sRNA was not considered as a putative integration target if a core gene of the host was found between the sRNA and the prophage. RNA genes located within a prophage (or a neighboring prophage) were not considered as potential integration targets. sRNA secondary structures were predicted with RNAfold (Gruber et al. 2008). Each sequence was shuffled 1,000 times keeping nucleotide composition constant, and the distribution of minimum free energies was computed with the 1,000 randomized sequences. For each sRNA, the predicted structure was considered as reliable when its minimum free energy was found among the 10% most stable structures of the distribution of minimum energy for the random sequences.
Identification of Targeted CDS
The identification of putative integration targets within protein coding genes was made by searching for homologies between the sequences flanking the prophage and proteins in the pan genome using tblastn (Altschul et al. 1997) (e value < 0.001). We took 1 kb sequences around each prophage limit. When both prophage flanking regions matched the same protein, we aligned them independently to the corresponding gene with needle (Rice et al. 2000) using the end gap free option. Two cases were then considered: 1) phage integration led to the duplication of one end of the CDS and 2) the CDS was disrupted due to phage integration. The first situation was identified when one hit corresponded to the entire CDS and the other hit to a smaller fragment. The second case was recognized when none of the hits corresponded to the entire query CDS and when they were found aligned to complementary parts of the query CDS (i.e., nonoverlapping but converging at the same position).
Identification of Macrodomains, Essential Genes, Origin and Terminus of Replication, KOPS, and matS Motifs
Macrodomain borders were delineated as in Scolari et al. (2011). Essential genes were defined as in Baba et al. (2006). We used the sequences patterns reviewed by Touzain et al. (2011) to identify KOPS (GGG[ATGC]AGGG) (Bigot et al. 2005) and matS (GTGAC[AG][AGTC][TC]GTCAC) (Mercier et al. 2008) sequences in the 69 Escherichia and Salmonella complete genomes using Fuzznuc (http://emboss.bioinformatics.nl/cgi-bin/emboss/fuzznuc, last accessed January 3, 2013). To identify the origin of replication, we searched using blastn, the best hit with the known oriC sequence of E. coli K12 MG1655 of 378 bp in the other genomes. This sequence is well conserved in Salmonella (>86% of identity sequence in all length) and Escherichia (98.7% of identity sequence) replicons. To identify the terminus of replication, we searched using Fuzznuc the known dif site sequence (GGTGCGCATAATGTATATTATGTTAAAT) (Hendrickson and Lawrence 2007) and also the terC sequence of E. coli K12 MG1655 (GGATGTTGTAACTA) in all the genomes analyzed (Duggin and Bell 2009). Both sequences are well conserved between the two species and are close to each other along the chromosome (<20 kb). Cumulative GC and AT skews analysis in 10 kb sliding windows (Greub et al. 2003), the switch of KOPS orientation (Bigot et al. 2005), and the identification of the dnaA gene (Mackiewicz et al. 2004) close to the origin were used to confirm/support the predictions. We then classed all prophage genes and KOPS motifs according to their orientation relative to the replication fork movement.
Statistics on Oligonucleotide Usage
Over-representation of KOPS and matS motifs was determined by comparison to the expected frequencies of these motifs in the different genomes. The expected frequencies of KOPS were calculated using a Markov maximal order model as in Schbath (1997). As KOPS motifs display a degenerate nucleotide at position 4, random expectation was calculated for each one of the four possible KOPS motifs independently. The degenerate matS motifs are longer (13 nucleotides), and their random frequencies cannot be estimated confidently with the Markov maximum order model because such long motifs are expected at very low frequencies. Random expectation of these motifs was then estimated using the hosts' or (pro)phages' nucleotide content:
F(matS) = f(G)3 · f(C)3 · f(A)2 · f(T)2 · f(A/G) · f(T/C), with f(X) the frequency of nucleotide X in the genome.
Supplementary Material
Supplementary tables S1–S5 and figures S1–S3 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
This work was supported by a European Research Council starting grant (EVOMOBILOME no. 281605) and a grant from the Ministère de l'enseignement supérieur et de la recherche to L.-M.B.
References
- Abedon ST, Calendar RL. The bacteriophages. New York: Oxford University Press; 2005. [Google Scholar]
- Abedon ST, Lejeune JT. Why bacteriophage encode exotoxins and other virulence factors. Evol Bioinform Online. 2005;1:97–110. [PMC free article] [PubMed] [Google Scholar]
- Ackermann HW. Salmonella phages examined in the electron microscope. Methods Mol Biol. 2007;394:213–234. doi: 10.1007/978-1-59745-512-1_11. [DOI] [PubMed] [Google Scholar]
- Allison GE, Angeles D, Tran-Dinh N, Verma NK. Complete genomic sequence of SfV, a serotype-converting temperate bacteriophage of Shigella flexneri. J Bacteriol. 2002;184:1974–1987. doi: 10.1128/JB.184.7.1974-1987.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angly FE, Felts B, Breitbart M, et al. (18 co-authors) The marine viromes of four oceanic regions. PLoS Biol. 2006;4:e368. doi: 10.1371/journal.pbio.0040368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asadulghani M, Ogura Y, Ooka T, Itoh T, Sawaguchi A, Iguchi A, Nakayama K, Hayashi T. The defective prophage pool of Escherichia coli O157: prophage-prophage interactions potentiate horizontal transfer of virulence determinants. PLoS Pathog. 2009;5:e1000408. doi: 10.1371/journal.ppat.1000408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2 doi: 10.1038/msb4100050. 2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbontin R, Figueroa-Bossi N, Casadesus J, Bossi L. Insertion hot spot for horizontally acquired DNA within a bidirectional small-RNA locus in Salmonella enterica. J Bacteriol. 2008;190:4075–4078. doi: 10.1128/JB.00220-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks DJ, Beres SB, Musser JM. The fundamental contribution of phages to GAS evolution, genome diversification and strain emergence. Trends Microbiol. 2002;10:515–521. doi: 10.1016/s0966-842x(02)02461-7. [DOI] [PubMed] [Google Scholar]
- Bigot S, Saleh OA, Lesterlin C, Pages C, El Karoui M, Dennis C, Grigoriev M, Allemand JF, Barre FX, Cornet F. KOPS: DNA motifs that control E. coli chromosome segregation by orienting the FtsK translocase. EMBO J. 2005;24:3770–3780. doi: 10.1038/sj.emboj.7600835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum G, Ott M, Lischewski A, Ritter A, Imrich H, Tschape H, Hacker J. Excision of large DNA regions termed pathogenicity islands from tRNA-specific loci in the chromosome of an Escherichia coli wild-type pathogen. Infect Immun. 1994;62:606–614. doi: 10.1128/iai.62.2.606-614.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossi L, Fuentes JA, Mora G, Figueroa-Bossi N. Prophage contribution to bacterial population dynamics. J Bacteriol. 2003;185:6467–6471. doi: 10.1128/JB.185.21.6467-6471.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botstein D. A theory of modular evolution for bacteriophages. Ann N Y Acad Sci. 1980;354:484–490. doi: 10.1111/j.1749-6632.1980.tb27987.x. [DOI] [PubMed] [Google Scholar]
- Boyd EF, Brussow H. Common themes among bacteriophage-encoded virulence factors and diversity among the bacteriophages involved. Trends Microbiol. 2002;10:521–529. doi: 10.1016/s0966-842x(02)02459-9. [DOI] [PubMed] [Google Scholar]
- Breitbart M, Haynes M, Kelley S, et al. (13 co-authors) Viral diversity and dynamics in an infant gut. Res Microbiol. 2008;159:367–373. doi: 10.1016/j.resmic.2008.04.006. [DOI] [PubMed] [Google Scholar]
- Brown SP, Le Chat L, De Paepe M, Taddei F. Ecology of microbial invasions: amplification allows virus carriers to invade more rapidly when rare. Curr Biol. 2006;16:2048–2052. doi: 10.1016/j.cub.2006.08.089. [DOI] [PubMed] [Google Scholar]
- Brussow H, Canchaya C, Hardt WD. Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol Mol Biol Rev. 2004;68:560–602. doi: 10.1128/MMBR.68.3.560-602.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukhari AI, Metlay M. Genetic mapping of prophage Mu. Virology. 1973;54:109–116. doi: 10.1016/0042-6822(73)90120-7. [DOI] [PubMed] [Google Scholar]
- Campbell A. Prophage insertion sites. Res Microbiol. 2003;154:277–282. doi: 10.1016/S0923-2508(03)00071-8. [DOI] [PubMed] [Google Scholar]
- Campbell A, Botstein D. Evolution of the lambdoid phages. In: Hendrix RW, Roberts JW, Stahl FW, Weisberg RA, editors. Lambda II. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory; 1983. pp. 365–380. [Google Scholar]
- Campbell AM. Chromosomal insertion sites for phages and plasmids. J Bacteriol. 1992;174:7495–7499. doi: 10.1128/jb.174.23.7495-7499.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell AM. Preferential orientation of natural lambdoid prophages and bacterial chromosome organization. Theor Popul Biol. 2002;61:503–507. doi: 10.1006/tpbi.2002.1604. [DOI] [PubMed] [Google Scholar]
- Canchaya C, Fournous G, Brussow H. The impact of prophages on bacterial chromosomes. Mol Microbiol. 2004;53:9–18. doi: 10.1111/j.1365-2958.2004.04113.x. [DOI] [PubMed] [Google Scholar]
- Canchaya C, Proux C, Fournoux G, Bruttin A, Brussow H. Prophage genomics. Microbiol Mol Biol Rev. 2003;67:238–276. doi: 10.1128/MMBR.67.2.238-276.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casjens S. Prophages and bacterial genomics: what have we learned so far? Mol Microbiol. 2003;49:277–300. doi: 10.1046/j.1365-2958.2003.03580.x. [DOI] [PubMed] [Google Scholar]
- Couturier E, Rocha EPC. Replication-associated gene dosage effects shape the genomes of fast-growing bacteria but only for transcription and translation genes. Mol Microbiol. 2006;59:1506–1518. doi: 10.1111/j.1365-2958.2006.05046.x. [DOI] [PubMed] [Google Scholar]
- Creuzburg K, Heeren S, Lis CM, Kranz M, Hensel M, Schmidt H. Genetic background and mobility of variants of the gene nleA in attaching and effacing Escherichia coli. Appl Environ Microbiol. 2011;77:8705–8713. doi: 10.1128/AEM.06492-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criscuolo A, Gribaldo S. BMGE (Block Mapping and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol Biol. 2010;10:210. doi: 10.1186/1471-2148-10-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui T, Moro-oka N, Ohsumi K, Kodama K, Ohshima T, Ogasawara N, Mori H, Wanner B, Niki H, Horiuchi T. Escherichia coli with a linear genome. EMBO Rep. 2007;8:181–187. doi: 10.1038/sj.embor.7400880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dearborn AD, Laurinmaki P, Chandramouli P, Rodenburg CM, Wang S, Butcher SJ, Dokland T. Structure and size determination of bacteriophage P2 and P4 procapsids: function of size responsiveness mutations. J Struct Biol. 2012;178:215–224. doi: 10.1016/j.jsb.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrindt U, Blum-Oehler G, Nagy G, Schneider G, Johann A, Gottschalk G, Hacker J. Genetic structure and distribution of four pathogenicity islands (PAI I(536) to PAI IV(536)) of uropathogenic Escherichia coli strain 536. Infect Immun. 2002;70:6365–6372. doi: 10.1128/IAI.70.11.6365-6372.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Nilsson L, Kurland CG. Co-variation of tRNA abundance and codon usage in Escherichia coli at different growth rates. J Mol Biol. 1996;260:649–663. doi: 10.1006/jmbi.1996.0428. [DOI] [PubMed] [Google Scholar]
- Duggin IG, Bell SD. Termination structures in the Escherichia coli chromosome replication fork trap. J Mol Biol. 2009;387:532–539. doi: 10.1016/j.jmb.2009.02.027. [DOI] [PubMed] [Google Scholar]
- Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol. 2011;7:e1002195. doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Rokney A, Feeney M, Semsey S, Kessel M, Goldberg MB, Adhya S, Oppenheim AB. Bacteriophage infection is targeted to cellular poles. Mol Microbiol. 2008;68:1107–1116. doi: 10.1111/j.1365-2958.2008.06205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edlin G, Lin L, Bitner R. Reproductive fitness of P1, P2, and Mu lysogens of Escherichia coli. J Virol. 1977;21:560–564. doi: 10.1128/jvi.21.2.560-564.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards RA, Rohwer F. Viral metagenomics. Nat Rev Microbiol. 2005;3:504–510. doi: 10.1038/nrmicro1163. [DOI] [PubMed] [Google Scholar]
- Esnault E, Valens M, Espeli O, Boccard F. Chromosome structuring limits genome plasticity in Escherichia coli. PLoS Genet. 2007;3:e226. doi: 10.1371/journal.pgen.0030226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouts DE. Phage_Finder: automated identification and classification of prophage regions in complete bacterial genome sequences. Nucleic Acids Res. 2006;34:5839–5851. doi: 10.1093/nar/gkl732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Aljaro C, Muniesa M, Jofre J, Blanch AR. Genotypic and phenotypic diversity among induced, stx2-carrying bacteriophages from environmental Escherichia coli strains. Appl Environ Microbiol. 2009;75:329–336. doi: 10.1128/AEM.01367-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Russell N, Harmon TG, Le TQ, Amaladas NH, Mathewson RD, Segall AM. Unequal access of chromosomal regions to each other in Salmonella: probing chromosome structure with phage lambda integrase-mediated long-range rearrangements. Mol Microbiol. 2004;52:329–344. doi: 10.1111/j.1365-2958.2004.03976.x. [DOI] [PubMed] [Google Scholar]
- Gascuel O. BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol Biol Evol. 1997;14:685–695. doi: 10.1093/oxfordjournals.molbev.a025808. [DOI] [PubMed] [Google Scholar]
- Greub G, Mege JL, Raoult D. Parachlamydia acanthamoebae enters and multiplies within human macrophages and induces their apoptosis. Infect Immun. 2003;71:5979–5985. doi: 10.1128/IAI.71.10.5979-5985.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber AR, Lorenz R, Bernhart SH, Neubock R, Hofacker IL. The Vienna RNA websuite. Nucleic Acids Res. 2008;36:W70–W74. doi: 10.1093/nar/gkn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Ferreira RC, Viollier PH, Ely B, Poindexter JS, Georgieva M, Jensen GJ, Wright ER. Alternative mechanism for bacteriophage adsorption to the motile bacterium Caulobacter crescentus. Proc Natl Acad Sci U S A. 2011;108:9963–9968. doi: 10.1073/pnas.1012388108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson H, Lawrence JG. Mutational bias suggests that replication termination occurs near the dif site, not at Ter sites. Mol Microbiol. 2007;64:42–56. doi: 10.1111/j.1365-2958.2007.05596.x. [DOI] [PubMed] [Google Scholar]
- Hendrix RW, Smith MCM, Burns RN, Ford ME, Hatfull GF. Evolutionary relationships among diverse bacteriophages and prophages: all the world's a phage. Proc Natl Acad Sci U S A. 1999;96:2192–2197. doi: 10.1073/pnas.96.5.2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermans AP, Beuling AM, van Hoek AH, Aarts HJ, Abee T, Zwietering MH. Distribution of prophages and SGI-1 antibiotic-resistance genes among different Salmonella enterica serovar Typhimurium isolates. Microbiology. 2006;152:2137–2147. doi: 10.1099/mic.0.28850-0. [DOI] [PubMed] [Google Scholar]
- Huang HY, Chang HY, Chou CH, Tseng CP, Ho SY, Yang CD, Ju YW, Huang HD. sRNAMap: genomic maps for small non-coding RNAs, their regulators and their targets in microbial genomes. Nucleic Acids Res. 2009;37:D150–D154. doi: 10.1093/nar/gkn852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KE, Waldor MK. Filamentous phage integration requires the host recombinases XerC and XerD. Nature. 2002;417:656–659. doi: 10.1038/nature00782. [DOI] [PubMed] [Google Scholar]
- Itaya M, Tsuge K, Koizumi M, Fujita K. Combining two genomes in one cell: stable cloning of the Synechocystis PCC6803 genome in the Bacillus subtilis 168 genome. Proc Natl Acad Sci U S A. 2005;102:15971–15976. doi: 10.1073/pnas.0503868102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobb G, von Haeseler A, Strimmer K. TREEFINDER: a powerful graphical analysis environment for molecular phylogenetics. BMC Evol Biol. 2004;4:18. doi: 10.1186/1471-2148-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Juhala RJ, Ford ME, Duda RL, Youlton A, Hatfull GF, Hendrix RW. Genomic sequences of bacteriophages HK97 and HK022: pervasive genetic mosaicism in the lambdoid bacteriophages. J Mol Biol. 2000;299:27–51. doi: 10.1006/jmbi.2000.3729. [DOI] [PubMed] [Google Scholar]
- Julio SM, Heithoff DM, Mahan MJ. ssrA (tmRNA) plays a role in Salmonella enterica serovar Typhimurium pathogenesis. J Bacteriol. 2000;182:1558–1563. doi: 10.1128/jb.182.6.1558-1563.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King AMQ, Lefkowitz E, Adams MJ, Carstens EB. Virus taxonomy: ninth report of the International Committee on Taxonomy of Viruses. Waltham (MA): Elsevier; 2011. [Google Scholar]
- Kourilsky P. Lysogenization by bacteriophage lambda. I. Multiple infection and the lysogenic response. Mol Gen Genet. 1973;122:183–195. doi: 10.1007/BF00435190. [DOI] [PubMed] [Google Scholar]
- Kroger C, Dillon SC, Cameron AD, et al. (21 co-authors) The transcriptional landscape and small RNAs of Salmonella enterica serovar Typhimurium. Proc Natl Acad Sci U S A. 2012;109:E1277–E1286. doi: 10.1073/pnas.1201061109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrie SJ, Samson JE, Moineau S. Bacteriophage resistance mechanisms. Nat Rev Microbiol. 2010;8:317–327. doi: 10.1038/nrmicro2315. [DOI] [PubMed] [Google Scholar]
- Lathe WC, Snel B, Bork P. Gene context conservation of a higher order than operons. Trends Biochem Sci. 2000;25:474–479. doi: 10.1016/s0968-0004(00)01663-7. [DOI] [PubMed] [Google Scholar]
- Lavigne R, Darius P, Summer EJ, Seto D, Mahadevan P, Nilsson AS, Ackermann HW, Kropinski AM. Classification of Myoviridae bacteriophages using protein sequence similarity. BMC Microbiol. 2009;9:224. doi: 10.1186/1471-2180-9-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence JG, Hendrickson H. Lateral gene transfer: when will adolescence end? Mol Microbiol. 2003;50:739–749. doi: 10.1046/j.1365-2958.2003.03778.x. [DOI] [PubMed] [Google Scholar]
- Lee SY, Bailey SC, Apirion D. Small stable RNAs from Escherichia coli: evidence for the existence of new molecules and for a new ribonucleoprotein particle containing 6S RNA. J Bacteriol. 1978;133:1015–1023. doi: 10.1128/jb.133.2.1015-1023.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy O, Ptacin JL, Pease PJ, Gore J, Eisen MB, Bustamante C, Cozzarelli NR. Identification of oligonucleotide sequences that direct the movement of the Escherichia coli FtsK translocase. Proc Natl Acad Sci U S A. 2005;102:17618–17623. doi: 10.1073/pnas.0508932102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima-Mendez G, Van Helden J, Toussaint A, Leplae R. Prophinder: a computational tool for prophage prediction in prokaryotic genomes. Bioinformatics. 2008a;24:863–865. doi: 10.1093/bioinformatics/btn043. [DOI] [PubMed] [Google Scholar]
- Lima-Mendez G, Van Helden J, Toussaint A, Leplae R. Reticulate representation of evolutionary and functional relationships between phage genomes. Mol Biol Evol. 2008b;25:762–777. doi: 10.1093/molbev/msn023. [DOI] [PubMed] [Google Scholar]
- Lowe T, Eddy S. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackiewicz P, Zakrzewska-Czerwinska J, Zawilak A, Dudek MR, Cebrat S. Where does bacterial replication start? Rules for predicting the oriC region. Nucleic Acids Res. 2004;32:3781–3791. doi: 10.1093/nar/gkh699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel TK, Jarvis KG, Donnenberg MS, Kaper JB. A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc Natl Acad Sci U S A. 1995;92:1664–1668. doi: 10.1073/pnas.92.5.1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meile JC, Mercier R, Stouf M, Pages C, Bouet JY, Cornet F. The terminal region of the E. coli chromosome localises at the periphery of the nucleoid. BMC Microbiol. 2011;11:28. doi: 10.1186/1471-2180-11-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercier R, Petit MA, Schbath S, Robin S, El Karoui M, Boccard F, Espeli O. The MatP/matS site-specific system organizes the terminus region of the E. coli chromosome into a macrodomain. Cell. 2008;135:475–485. doi: 10.1016/j.cell.2008.08.031. [DOI] [PubMed] [Google Scholar]
- Mizuuchi K. Transpositional recombination: mechanistic insights from studies of Mu and other elements. Annu Rev Biochem. 1992;61:1011–1051. doi: 10.1146/annurev.bi.61.070192.005051. [DOI] [PubMed] [Google Scholar]
- Mmolawa PT, Schmieger H, Heuzenroeder MW. Bacteriophage ST64B, a genetic mosaic of genes from diverse sources isolated from Salmonella enterica serovar typhimurium DT 64. J Bacteriol. 2003;185:6481–6485. doi: 10.1128/JB.185.21.6481-6485.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano MG, Almagro-Moreno S, Boyd EF. Dichotomy in the evolution of pathogenicity island and bacteriophage encoded integrases from pathogenic Escherichia coli strains. Infect Genet Evol. 2011;11:423–436. doi: 10.1016/j.meegid.2010.12.003. [DOI] [PubMed] [Google Scholar]
- Nechaev S, Severinov K. The elusive object of desire—interactions of bacteriophages and their hosts. Curr Opin Microbiol. 2008;11:186–193. doi: 10.1016/j.mib.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes-Duby SE, Kwon HJ, Tirumalai RS, Ellenberger T, Landy A. Similarities and differences among 105 members of the Int family of site-specific recombinases. Nucleic Acids Res. 1998;26:391–406. doi: 10.1093/nar/26.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi M, Kurokawa K, Hayashi T. Diversification of Escherichia coli genomes: are bacteriophages the major contributors? Trends Microbiol. 2001;9:481–485. doi: 10.1016/s0966-842x(01)02173-4. [DOI] [PubMed] [Google Scholar]
- Otsuka AJ, Buoncristiani MR, Howard PK, Flamm J, Johnson C, Yamamoto R, Uchida K, Cook C, Ruppert J, Matsuzaki J. The Escherichia coli biotin biosynthetic enzyme sequences predicted from the nucleotide sequence of the bio operon. J Biol Chem. 1988;263:19577–19585. [PubMed] [Google Scholar]
- Peters JE, Craig NL. Tn7 recognizes transposition target structures associated with DNA replication using the DNA-binding protein TnsE. Genes Dev. 2001;15:737–747. doi: 10.1101/gad.870201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptashne M. Genetic switch: phage lambda and higher organisms. Cambridge (MA): Blackwell; 1992. [Google Scholar]
- Raghavan R, Groisman EA, Ochman H. Genome-wide detection of novel regulatory RNAs in E. coli. Genome Res. 2011;21:1487–1497. doi: 10.1101/gr.119370.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravin NV. N15: the linear phage-plasmid. Plasmid. 2011;65:102–109. doi: 10.1016/j.plasmid.2010.12.004. [DOI] [PubMed] [Google Scholar]
- Reyes A, Haynes M, Hanson N, Angly FE, Heath AC, Rohwer F, Gordon JI. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature. 2010;466:334–338. doi: 10.1038/nature09199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Lamothe R, Wang X, Sherratt D. Escherichia coli and its chromosome. Trends Microbiol. 2008;16:238–245. doi: 10.1016/j.tim.2008.02.003. [DOI] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Rocha EPC. Order and disorder in bacterial genomes. Curr Opin Microbiol. 2004;7:519–527. doi: 10.1016/j.mib.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Rocha EPC. The organisation of the bacterial genome. Annu Rev Genet. 2008;42:211–233. doi: 10.1146/annurev.genet.42.110807.091653. [DOI] [PubMed] [Google Scholar]
- Rocha EPC, Danchin A. Essentiality, not expressiveness, drives gene strand bias in bacteria. Nat Genet. 2003;34:377–378. doi: 10.1038/ng1209. [DOI] [PubMed] [Google Scholar]
- Rohwer F, Edwards R. The phage proteomic tree: a genome-based taxonomy for phage. J Bacteriol. 2002;184:4529–4535. doi: 10.1128/JB.184.16.4529-4535.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roossinck MJ. The good viruses: viral mutualistic symbioses. Nat Rev Microbiol. 2011;9:99–108. doi: 10.1038/nrmicro2491. [DOI] [PubMed] [Google Scholar]
- Schbath S. An efficient statistic to detect over- and under-represented words in DNA sequences. J Comput Biol. 1997;4:189–192. doi: 10.1089/cmb.1997.4.189. [DOI] [PubMed] [Google Scholar]
- Schmidt HA, Strimmer K, Vingron M, von Haeseler A. TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics. 2002;18:502–504. doi: 10.1093/bioinformatics/18.3.502. [DOI] [PubMed] [Google Scholar]
- Scolari VF, Bassetti B, Sclavi B, Lagomarsino MC. Gene clusters reflecting macrodomain structure respond to nucleoid perturbations. Mol Biosyst. 2011;7:878–888. doi: 10.1039/c0mb00213e. [DOI] [PubMed] [Google Scholar]
- Shinedling S, Parma D, Gold L. Wild-type bacteriophage T4 is restricted by the lambda rex genes. J Virol. 1987;61:3790–3794. doi: 10.1128/jvi.61.12.3790-3794.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinhara A, Matsui M, Hiraoka K, Nomura W, Hirano R, Nakahigashi K, Tomita M, Mori H, Kanai A. Deep sequencing reveals as-yet-undiscovered small RNAs in Escherichia coli. BMC Genomics. 2011;12:428. doi: 10.1186/1471-2164-12-428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Six EW, Klug CA. Bacteriophage P4: a satellite virus depending on a helper such as prophage P2. Virology. 1973;51:327–344. doi: 10.1016/0042-6822(73)90432-7. [DOI] [PubMed] [Google Scholar]
- Smith MC, Thorpe HM. Diversity in the serine recombinases. Mol Microbiol. 2002;44:299–307. doi: 10.1046/j.1365-2958.2002.02891.x. [DOI] [PubMed] [Google Scholar]
- Snel B, Huynen MA, Dutilh BE. Genome trees and the nature of genome evolution. Ann Rev Microbiol. 2005;59:191–209. doi: 10.1146/annurev.micro.59.030804.121233. [DOI] [PubMed] [Google Scholar]
- St-Pierre F, Endy D. Determination of cell fate selection during phage lambda infection. Proc Natl Acad Sci U S A. 2008;105:20705–20710. doi: 10.1073/pnas.0808831105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suttle CA. Viruses in the sea. Nature. 2005;437:356–361. doi: 10.1038/nature04160. [DOI] [PubMed] [Google Scholar]
- Tenaillon O, Skurnik D, Picard B, Denamur E. The population genetics of commensal Escherichia coli. Nat Rev Microbiol. 2010;8:207–17. doi: 10.1038/nrmicro2298. [DOI] [PubMed] [Google Scholar]
- Thomson N, Baker S, Pickard D, et al. (15 co-authors) The role of prophage-like elements in the diversity of Salmonella enterica serovars. J Mol Biol. 2004;339:279–300. doi: 10.1016/j.jmb.2004.03.058. [DOI] [PubMed] [Google Scholar]
- Touchon M, Hoede C, Tenaillon O, et al. (41 co-authors) Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. PLoS Genet. 2009;5:e1000344. doi: 10.1371/journal.pgen.1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touchon M, Rocha EP. Causes of insertion sequences abundance in prokaryotic genomes. Mol Biol Evol. 2007;24:969–981. doi: 10.1093/molbev/msm014. [DOI] [PubMed] [Google Scholar]
- Touchon M, Rocha EP. The small, slow and specialized CRISPR and anti-CRISPR of Escherichia and Salmonella. PLoS One. 2010;5:e11126. doi: 10.1371/journal.pone.0011126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touzain F, Petit MA, Schbath S, El Karoui M. DNA motifs that sculpt the bacterial chromosome. Nat Rev Microbiol. 2011;9:15–26. doi: 10.1038/nrmicro2477. [DOI] [PubMed] [Google Scholar]
- Val ME, Skovgaard O, Ducos-Galand M, Bland MJ, Mazel D. Genome engineering in Vibrio cholerae: a feasible approach to address biological issues. PLoS Genet. 2012;8:e1002472. doi: 10.1371/journal.pgen.1002472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valens M, Penaud S, Rossignol M, Cornet F, Boccard F. Macrodomain organization of the Escherichia coli chromosome. EMBO J. 2004;23:4330–4341. doi: 10.1038/sj.emboj.7600434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Melderen L, Saavedra De Bast M. Bacterial toxin-antitoxin systems: more than selfish entities? PLoS Genet. 2009;5:e1000437. doi: 10.1371/journal.pgen.1000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernikos GS, Thomson NR, Parkhill J. Genetic flux over time in the Salmonella lineage. Genome Biol. 2007;8:R100. doi: 10.1186/gb-2007-8-6-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yang CH, Lee G, Chang F, Wilson H, del Campillo-Campbell A, Campbell A. Integration specificities of two lambdoid phages (21 and e14) that insert at the same attB site. J Bacteriol. 1997;179:5705–5711. doi: 10.1128/jb.179.18.5705-5711.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassarman KM, Repoila F, Rosenow C, Storz G, Gottesman S. Identification of novel small RNAs using comparative genomics and microarrays. Genes Dev. 2001;15:1637–1651. doi: 10.1101/gad.901001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters LS, Storz G. Regulatory RNAs in bacteria. Cell. 2009;136:615–628. doi: 10.1016/j.cell.2009.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinbauer MG. Ecology of prokaryotic viruses. FEMS Microbiol Rev. 2004;28:127–181. doi: 10.1016/j.femsre.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Wiggins PA, Cheveralls KC, Martin JS, Lintner R, Kondev J. Strong intranucleoid interactions organize the Escherichia coli chromosome into a nucleoid filament. Proc Natl Acad Sci U S A. 2010;107:4991–4995. doi: 10.1073/pnas.0912062107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KP. Integration sites for genetic elements in prokaryotic tRNA and tmRNA genes: sublocation preference of integrase subfamilies. Nucleic Acids Res. 2002;30:866–875. doi: 10.1093/nar/30.4.866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KP. Traffic at the tmRNA gene. J Bacteriol. 2003;185:1059–1070. doi: 10.1128/JB.185.3.1059-1070.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winstanley C, Langille MGI, Fothergill JL, et al. (19 co-authors) Newly introduced genomic prophage islands are critical determinants of in vivo competitiveness in the Liverpool Epidemic Strain of Pseudomonas aeruginosa. Genome Res. 2008;19:12–23. doi: 10.1101/gr.086082.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Withers M, Wernisch L, dos Reis M. Archaeology and evolution of transfer RNA genes in the Escherichia coli genome. RNA. 2006;12:933–942. doi: 10.1261/rna.2272306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf YI, Rogozin IB, Grishin NV, Koonin EV. Genome trees and the tree of life. Trends Genet. 2002;18:472–479. doi: 10.1016/s0168-9525(02)02744-0. [DOI] [PubMed] [Google Scholar]
- Zaslaver A, Mayo A, Ronen M, Alon U. Optimal gene partition into operons correlates with gene functional order. Phys Biol. 2006;3:183–189. doi: 10.1088/1478-3975/3/3/003. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;39:W347–W352. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.