

Abstract
We report a major update of the MAFFT multiple sequence alignment program. This version has several new features, including options for adding unaligned sequences into an existing alignment, adjustment of direction in nucleotide alignment, constrained alignment and parallel processing, which were implemented after the previous major update. This report shows actual examples to explain how these features work, alone and in combination. Some examples incorrectly aligned by MAFFT are also shown to clarify its limitations. We discuss how to avoid misalignments, and our ongoing efforts to overcome such limitations.
Keywords: multiple sequence alignment, metagemone, protein structure, progressive alignment, parallel processing
Introduction
Multiple sequence alignment (MSA) plays an important role in evolutionary analyses of biological sequences. MAFFT is an MSA program, first released in 2002 (Katoh et al. 2002). Because of its high performance (Nuin et al. 2006; Golubchik et al. 2007; Dessimoz and Gil 2010; Letsch et al. 2010; Sahraeian and Yoon 2011; Sievers et al. 2011), MAFFT is becoming popular in recent years. After reviewing the previous version (version 6) in Katoh and Toh (2008b), we have been continuously improving its accuracy, speed, and utility in practical situations. These improvements and techniques were mostly reported in individual papers (Katoh et al. 2009; Katoh and Toh 2010; Katoh and Frith 2012; Katoh and Standley 2013). In this report, we demonstrate the different kinds of analyses that can be achieved with the new features, alone and in combination, using realistic examples. We also discuss limitations of current version by giving examples of sequences incorrectly aligned by MAFFT, and describe our ongoing efforts to overcome these limitations.
Basic Concepts and Usage
As listed in table 1, MAFFT version 7 has options for various alignment strategies, including progressive methods (PartTree, FFT-NS-1, and L-INS-1) (Feng and Doolittle 1987; Higgins and Sharp 1988; Katoh and Toh 2007), iterative refinement methods (FFT-NS-i, L-INS-i, E-INS-i, and G-INS-i) (Barton and Sternberg 1987; Berger and Munson 1991; Gotoh 1993; Katoh et al. 2005), and structural alignment methods for RNAs (Q-INS-i and X-INS-i; Katoh and Toh 2008a). See Katoh and Toh (2008b) for details of these strategies. According to a recent comparative study based on the MetAl metric (Blackburne and Whelan 2012a, 2012b), there are two significantly different classes of MSA methods, similarity-based methods and evolution-based methods. MAFFT is classified as a similarity-based method. However, evolutionary information is useful even for similarity-based methods, because the sequences to be aligned are generated from a common ancestor in the course of evolution. In this respect, MAFFT takes evolutionary information into account.
Table 1.
Options of MAFFT Version 7.
| Option name | Command | |
|---|---|---|
| For a large-scale alignment: progressive methods with the PartTree algorithm | ||
| NW-NS-PartTree1 | mafft ––parttree ––retree 1 input | Distance is by the 6mer method. |
| NW-NS-PartTree2 | mafft ––parttree ––retree 2 input | Distance is by the 6mer method. Guide tree is re-built. |
| NW-NS-DPPartTree1 | mafft ––dpparttree ––retree 1 input | Distance is estimated based on DP. |
| NW-NS-DPPartTree2 | mafft ––dpparttree ––retree 2 input | Distance is estimated based on DP. Guide tree is re-built. |
| For a medium-scale alignment: progressive methods | ||
| FFT-NS-1 | mafft ––retree 1 input | Approximately two times faster than the default. |
| FFT-NS-2 | mafft input | Default. |
| For a small-scale alignment: iterative refinement methods | ||
| FFT-NS-i | mafft ––maxiterate 16 input | Fastest of the four in this category. Uses WSP score (Gotoh 1995) only. |
| G-INS-i | mafft ––maxiterate 16 ––globalpair input | Uses WSP score and consistency (Notredame et al. 1998) score from global alignments. |
| L-INS-i | mafft ––maxiterate 16 ––localpair input | Uses WSP score and consistency score from local alignments. |
| E-INS-i | mafft ––maxiterate 16 ––genafpair input | Uses WSP score and consistency score from local alignments with a generalized affine gap cost (Altschul 1998). |
| If not sure which option to use | ||
| Automatic | mafft ––auto | Selects an appropriate option from FFT-NS-2, FFT-NS-i and L-INS-i, according to the size of input data. |
| For a small-scale RNA alignment: structural alignment methods | ||
| Q-INS-i | mafft-qinsi input | Structure information is included in iterative refinement step. |
| X-INS-i-scarnapair | mafft-xinsi ––scarnapair input | Uses pairwise structural alignment by MXSCARNA (Tabei et al. 2008). |
| To add new sequences into an existing MSA | ||
| Add | mafft ––add mew msa | The simplest option for alignment extension. |
| Addprofile | mafft ––addprofile msa1 msa2 | msa1 must form a monophyletic cluster. |
| Addfragments | mafft ––addfragments new msa | Suitable for short new sequences. |
| Addfragments, LAST | mafft ––addfragments new ––lastmultipair msa | Faster option, LAST (Kiełbasa et al. 2011) is required. |
| Addfragments, 6mer | mafft ––addfragments new ––6merpair msa | Faster option for conserved data. |
| Parameters | ||
| ––bl #, ––jtt #, ––tm # | Score matrices for protein alignment. | |
| ––kimura # | Score matrix for nucleotide alignment. | |
| Utility options | ||
| ––anysymbol | See main text. | |
| ––reorder | ||
| ––clustalout | ||
| ––phylipout | ||
| ––namelength # | ||
| ––adjustdirection | ||
| ––adjustdirectionaccurately | ||
| ––seed msa1 –seed msa2 … | ||
| ––treein treefile | ||
| ––treeout | ||
| ––thread # | ||
Note.—N, the number of sequences; L, the sequence length; input, new, unaligned sequences in the multi-fasta format; msa, msa1, msa2, aligned sequences in the multi-fasta format; treefile, input guide tree file.
All the options of MAFFT assume that the input sequences are all homologous, that is, descended from a common ancestor. Thus, all the letters in the input data are aligned. Genomic rearrangement or domain shuffling is not assumed, and thus the order of the letters in each sequence is always preserved, although the sequences can be reordered according to similarity. Most options in MAFFT assume that almost all the pairs in the input sequences can be aligned, locally or globally. In such a situation, there is a tradeoff between accuracy and speed. For example, the PartTree option (Katoh and Toh 2007) is a fast and rough method, whereas L-INS-i and G-INS-i are slower and more accurate. RNA structural alignment methods are generally more accurate and computationally more expensive because they need additional calculations (Katoh and Toh 2008a). However, this tradeoff does not always hold. In particular, the new options to add sequences into an existing alignment (Katoh and Frith 2012), requires careful consideration of this tradeoff, as discussed later.
Profile Alignments
MAFFT has a subprogram, mafft-profile, to align two existing alignments.
mafft-profile alignment1 alignment2 > output
This method separately converts alignment1 and alignment2 to profiles and then aligns the two profiles. It means that the two input alignments are assumed to be phylogenetically isolated from each other, like figure 1A. Careless application of this method results in serious misalignments, as discussed in later section.
Fig. 1.

Assumptions on the phylogenetic relationship in different options of MAFFT. (A) mafft-profile, (B) ––addprofile, (C), misuse of mafft-profile, and (D) ––add or ––addprofile.
MAFFT version 7 has an alternative option, ––addprofile, which is safer against misuses.
mafft --addprofile alignment1 alignment2 > output
This option accepts two existing alignments, alignment1 and alignment2, and assumes a phylogenetic relationship shown in figure 1B. That is, alignment1 is assumed to form a monophyletic cluster, but alignment2 is not assumed to form a monophyletic cluster. The cluster of alignment1 can be placed in any phylogenetic position in the tree of alignment2. Moreover, this option checks whether alignment1 forms a monophyletic cluster. If not, it returns an error message and asks user to use the ––add option (see the following section).
Adding Unaligned Sequences into an MSA
As a result of advances in sequencing technologies, we increasingly need MSAs consisting of a larger number of sequences. There are several different approaches to enable construction of large MSAs, such as rapid algorithms and parallelization. Here, we describe an alternate approach: use of an existing alignment. There already exist databases of carefully aligned and annotated sequences (Cole et al. 2009; Sigrist et al. 2010; Punta et al. 2012), in which each MSA consists of a small number (typically up to ∼1,000) of sequences. We can use such MSAs as a backbone to build a larger MSA containing newly sequenced data. This is more efficient than rebuilding the entire MSA from a set of ungapped sequences. Moreover, this approach is relatively robust to low-quality sequences resulting from sequencing errors, misassemblies, and other factors. Such noise usually has a negative effect on the quality of an MSA, but there are situations where biologically important information is contained in low-quality sequences. In such a case, we first select highly reliable sequences to build a backbone MSA, and then add the other sequences, including low-quality ones, into the MSA. As a result, the quality of the final MSA is less affected by the low-quality sequences.
Inappropriate Applications of Profile Alignment
The mafft-profile program is not useful for this purpose. There are two types of misapplications. One is as follows: 1) convert an existing alignment to a profile, 2) align new sequences and convert them to a profile, and 3) align the two profiles. This procedure is inappropriate for adding new sequences because it assumes a phylogenetic relationship as illustrated in figure 1A.
Another misapplication is as follows: 1) convert the existing alignment to a profile, 2) separately align each new sequence to the profile of the existing alignment, and 3) construct a full alignment from the individual alignments computed in the previous step. This approach is more reasonable than the first one but still problematic, because the phylogenetic positions of new sequences are assumed at the root of the tree, as illustrated in figure 1C. Results of this procedure for two cases are shown in table 2 and figure 2.
Table 2.
Comparison of Different Options Using the 16S.B.ALL Data Set (Mirarab et al. 2012).
| Data | Method | Accuracy | CPU Time | Actual Timea |
|---|---|---|---|---|
| Case 1 | mafft ––multipair ––addfragments frags existingmsa | 0.9969 | 6.67 days | 18.3 h |
| mafft ––6merpair ––addfragments frags existingmsa | 0.9949 | 3.76 h | 36.2 min | |
| mafft ––localpair ––add frags existingmsa | 0.9707 | 23.4 daysb | 2.43 daysb | |
| mafft ––6merpair ––add frags existingmsa | 0.9604 | 1.32 h | 1.44 h | |
| profile alignment | 0.2779 | 15.5 h | 1.60 h | |
| Case 2 | mafft ––6merpair ––addfragments frags existingmsa | 0.9969 | 4.54 h | 33.8 min |
| Case 3 | mafft ––6merpair ––addfragments frags existingmsa | 0.9949 | 1.79 days | 5.91 h |
Note.—The estimated alignments were compared with the CRW alignment to measure the accuracy (the number of correctly aligned letters/the number of aligned letters in the CRW alignment). Calculations were performed on a Linux PC with 2.67 GHz Intel Xeon E7-8837/256 GB RAM (for the case marked with superscript alphabet “b”), or on a Linux PC with 3.47 GHz Intel Xeon X5690/48 GB RAM (for the other cases). Case 1: 13,822 sequences in the existing alignment × 13,821 fragments; Case 2: 1,000 sequences in the existing alignment × 138,210 fragments; Case 3: 13,822 sequences in the existing alignment × 138,210 fragments.
aWall-clock time with 10 cores. Command-line argument for parallel processing is ––thread 10.
bFull command-line options are as follows: mafft ––localpair ––weighti 0 ––add frags existingmsa.
Fig. 2.

ITS alignments by different options of MAFFT, displayed on Jalview (Waterhouse et al. 2009). (A, B) Incorrect alignments by the FFT-NS-2 and L-INS-i algorithms, respectively. (C) An incorrect alignment by mafft-profile. The full-length sequences were aligned with the L-INS-i algorithm and then each new sequence was separately added to the full-length alignment, using mafft-profile. (D) Reasonable alignment by a two-step strategy. The ––6merpair ––addfragments option was used at the second step. (E) Reordered version of D; sequences are ordered such that similar sequences are placed closely. All calculations were performed using 16 cores on a Linux PC with 2.67 GHz Intel Xeon E7-8837/256 GB RAM.
The ––add and ––addfragments Options
To overcome this limitation of profile alignment, in 2010, we implemented an option, ––add, to add unaligned sequences to an existing MSA. This option assumes that each new sequence was derived from a branch in the tree of an existing alignment, as illustrated in figure 1D. This option works almost identically to the standard progressive method, except that the alignment calculation is skipped at the nodes whose children are all in the existing alignment.
Along with popularization of second-generation sequencers, we sometimes need to align short reads to an existing alignment. Several tools (Berger and Stamatakis 2011; Löytynoja et al. 2012; Sun and Buhler 2012) for this purpose were developed between 2011 and 2012. A limitation of the ––add option in MAFFT for this purpose was pointed out in Löytynoja et al. (2012). Thus, we implemented a new option, ––addfragments, which does not consider the relationship among the sequences to be added. Details of the ––add and ––addfragments options are described in Katoh and Frith (2012).
Test Case 1: Fungal Internal Transcribed Spacers Sequences
Here, we discuss how the ––addfragments option works, using an actual case. Internal transcribed spacers (ITSs) are spacer regions located between structural ribosomal RNAs. The structure of the rDNA region in a eukaryotic genome is 18S − ITS1 − 5.8S − ITS2 − 28S. Here, we use a data set consisting of ITS1 and ITS2 sequences obtained from environmental samples (Chen W, personal communication). Each sequence has either ITS1 or ITS2 region only, extracted from 454 pyrosequencing data using FungalITSextractor (Nilsson et al. 2010). In addition, several fungal genomic sequences that fully cover ITS1 + 5.8S rRNA + ITS2 are available from public databases.
Suppose a situation where we need an MSA of approximately 300 full-length sequences and approximately 5,000 ITS1 or ITS2 sequences. One possible solution is to build an entire MSA at once. The result of the default option (FFT-NS-2) of MAFFT is obviously incorrect, as shown in figure 2A. ITS1 and ITS2 regions are forced to be aligned to each other. Even if a more computationally expensive (and usually more accurate) method, L-INS-i, is applied (CPU time = 98 h), the alignment is still obviously incorrect (fig. 2B).
Two-step strategies can solve this type of problem. That is, a set of full-length sequences taken from databases are first aligned to build a backbone MSA, and then the new ITS1 and ITS2 sequences are added into this backbone MSA, using the ––addfragments option.
Step 1: mafft --auto full_length_sequences >\ backbone_msa
Step 2: mafft --addfragments \ new_sequences backbone_msa > output
The second command is equivalent to
mafft --multipair --addfragments \ new_sequences backbone_msa > output
in which Dynamic Programming (DP) is used to compare the distances between every new sequence and every sequence in the backbone MSA (––multipair is selected by default).
mafft --6merpair --addfragments \ new_sequences backbone_msa > output
where distances are rapidly estimated using the number of shared 6mers, instead of DP.
The result of the latter option (––6merpair ––addfragments) is shown in fig. 2D and E. The difference between D and E is just in the order of sequences; the sequences were reordered according to similarity using the ––reorder option in E. In this alignment, ITS1 and ITS2 are clearly separated and aligned to appropriate positions in the full-length alignment. Moreover, this strategy is computationally much less expensive (CPU time = 15 min [first step] + 1.5 min [second step]) than the full application of L-INS-i (CPU time = 98 h). The former option (––multipair ––addfragments) also returns a similar result to the latter (––6merpair) but is slower (CPU time = 48.6 min [second step]).
This case suggests that it is crucial to select a strategy appropriate to the problem of interest. The most time-consuming method, L-INS-i, is not always the most accurate one. The difficulty of this problem for standard approaches comes from the fact that ITS1 sequences and ITS2 sequences are not homologous to each other and most pairwise alignments are impossible. Because of these nonhomologous pairs, the distance matrix used for the guide tree calculation is not additive; the distances between ITS1 and full-length sequences and those between ITS2 and full-length sequences are close to zero, whereas the distances between ITS1 and ITS2 are quite large. In this situation, it is difficult for normal distance-based tree-building methods to give a reasonable tree. Moreover, in the alignment step, the objective function of the L-INS-i is affected by inappropriate pairwise alignment scores between ITS1 and ITS2. Such problems can be avoided by just ignoring the relationship between ITS1 and ITS2, as done in the ––addfragments option.
In addition, a result of the second type of misuse of mafft-profile (discussed earlier) is shown in figure 2C. Some new sequences are correctly aligned but others are obviously incorrectly aligned (note that the order of sequences in fig. 2C is identical that in fig. 2D). These misalignments are due to an incorrect assumption on phylogenetic placement of new sequences shown in figure 1C.
Test Case 2: Bacterial SSU rRNA
Another case is the 16S.B.ALL data set by Mirarab et al. (2012). It consists of an MSA of 13,822 bacterial SSU rRNA sequences, taken from the Gutell Comparative RNA Website (CRW) (Cannone et al. 2002) and 138,210 fragmentary sequences, which are originally included in the CRW alignment but ungapped and artificially truncated. In Katoh and Standley (2013), we used a subset (13,821 fragmentary sequences) prepared by Mirarab et al. (2012). In addition to this subset, here we use the full data set (138,210 fragmentary sequences), to examine the scalability. Suppose a situation where we already have a manually curated (or backbone) MSA and a newly determined set of many fragmentary sequences in a metagenomics project, and we need an entire MSA of them.
The first four lines in table 2 (case 1) show the performances of various options for such an analysis, with a relatively small data set (13,822 sequences in the existing alignment × 13,821 fragments). The accuracy of each resulting MSA was evaluated by comparing the MSA with the original CRW alignment. CPU time and wall-clock time for each method are also listed. As the sequences in this data set are highly conserved, the difference in accuracy between the default (––multipair ––addfragments) and the faster option (––6merpair ––addfragments) is small.
Again, the tradeoff between accuracy and speed does not hold. The application of a computationally expensive method based on L-INS-1 (––localpair ––add) has no advantage, because the extra computational time is spent on the comparison of nonoverlapping fragmentary sequences, which have no reasonable solutions.
The “profile alignment” line in table 2 shows results of the second type of misuse of profile alignment (discussed earlier), in which the given alignment is converted to a profile and each new sequence is separately aligned to the profile. This result clearly indicates that the application of profile alignment must be avoided in this case, too. Users do not need to be too worried about this misuse, because this calculation is disabled in MAFFT unless the user modifies the code or writes a wrapper script.
The last two lines in table 2 (Cases 2 and 3) show the performance of the fast option (––6merpair ––addfragments) for a larger number (138,210) of fragmentary sequences. The number of sequences in the existing alignment is 1,000 and 13,822 in cases 2 and 3, respectively. This fast option gives a reasonable quality of result in a reasonable computing time. At present, the default option (––multipair ––addfragments) cannot handle cases 2 and 3. Simulation-based benchmarks in Katoh and Frith (2012) suggested that, for cases with more divergent sequences, the accuracy of the default option is higher than that of the fast option. We are now trying to improve the scalability of the default option.
Parallelization
MAFFT version 7 has an option for parallel processing, ––thread (Katoh and Toh 2010). This feature is currently supported on Mac OS X in addition to Linux, but not yet supported on Windows for technical reasons. With the ––thread n option, it runs in parallel with n threads. The number of threads can be automatically determined by ––thread –1. This option sets the number of threads as the number of physical cores, not the number of logical cores in Intel’s hyperthreaded CPUs.
For progressive methods, the result with the multithread version is identical to that of the serial processing version. However, for iterative refinement methods, the results are not always identical. We confirmed that the accuracy of the parallel version in this case is comparable with that of the serial version (Katoh and Toh 2010). The efficiency of parallelization depends on the alignment strategy. In the case of the ––addfragments option, the efficiency is acceptably high as shown in table 2.
Utility Options
MAFFT version 7 also has several enhanced options for peripheral functions.
Estimating the Direction of DNA Sequences
In the case of nucleotide alignments, if some of input sequences have an incorrect direction relative to the other sequences, the directions can be automatically adjusted by the ––adjustdirection option. We use an algorithm with a time complexity of 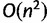 , where n is the number of sequences (Katoh and Standley 2013). It is slow when the distances are calculated with DP. However, when the distance is rapidly calculated based on the number of shared 6mers, the speed is reasonable. This option is also available on the web version, with the “Adjust direction” button.
, where n is the number of sequences (Katoh and Standley 2013). It is slow when the distances are calculated with DP. However, when the distance is rapidly calculated based on the number of shared 6mers, the speed is reasonable. This option is also available on the web version, with the “Adjust direction” button.
MAFFT cannot handle more complicated sequences with genomic rearrangements (translocations, duplications, or inversions). The web version of MAFFT displays dot plots between the first sequence and the remaining sequences, using the LAST local alignment program (Kiełbasa et al. 2011), for every nucleotide alignment run. By viewing the dot plots, a user can easily check for genomic rearrangements and the directions of input sequences. See Katoh and Standley (2013) for details and an example.
Input/Output
MAFFT version 7 has several enhancements in the flexibility of input/output. The following options related to input/output are available and can be combined with other options.
––anysymbol If the input data include unusual letters, like U, J, etc., (in the case of protein data), MAFFT stops by default. The ––anysymbol option allows these letters and nonalphabetical letters.
––preservecase By default, amino acid sequences are converted to upper case and nucleotide sequences are converted to lower case. This behavior can be changed by using the ––preservecase option.
––reorder The order of sequences is the same as the input sequences by default, but the sequences can be sorted according to similarity to each other by the ––reorder option.
––phylipout and ––clustalout The output format is multi-fasta by default, but the phylip (interleaved) format and the clustal format can be selected.
Guide Tree and Phylogenetic Positions of New Sequences
Users can check the guide tree by using the ––treeout option. In the case of ––addfragments, the estimated phylogenetic positions of new sequences are shown together with the estimated tree of the existing alignment. The alignment calculation is performed based on this phylogenetic estimation. It is also possible to compute such phylogenetic information only, without alignment, by the ––retree 0 option. An example of output is shown in Figure 3A.
Fig. 3.

(A) A part of output of the ––treeout option showing the phylogenetic positions of new sequences (new#) in the tree of the existing alignment (backbone#), estimated before the alignment calculation. This file also shows a Newick format tree of the existing alignment (not shown in this figure). For each new sequence, the nearest sequence in the existing alignment (nearest sequence), approximate distance to the nearest sequence (approximate distance), and the members of the sister group (sister group) are shown. (B) Graphical representation of (A).
Note that this phylogenetic information is roughly estimated before the MSA calculation, not based on the MSA. Especially, with the fast option, ––6merpair, the estimation is very rough. With the ––multipair option (default), the estimation is expected to be better, but it needs a relatively long computational time. For more rigorous estimation of phylogenetic positions of new sequences, specially designed tools, such as pplacer (Matsen et al. 2010), PaPaRa (Berger and Stamatakis 2011), PAGAN (Löytynoja et al. 2012), SEPP (Mirarab et al. 2012), or combinations of them including MAFFT, should be tried.
Parameters
For amino acid alignment, MAFFT uses the BLOSUM62 matrix by default. For nucleotide alignment, a 200PAM log-odds scoring matrix is generated assuming that the transition rate is twice the transversion rate. These matrices are suitable for aligning distantly related sequences. We selected these default parameters based on an expectation that, if the program works well for difficult (distantly related) cases, it should also work well for easy cases.
It is unclear whether this expectation is always correct. For example, in a benchmark using simulated protein sequences (Löytynoja et al. 2012) generated by INDELiBLE (Fletcher and Yang 2009), when we tested a more stringent scoring matrix, JTT 1PAM (Jones et al. 1992) with weaker gap penalties than the default, the benchmark scores were considerably improved. Despite this observation, we consistently used the default parameters in the benchmark in Katoh and Frith (2012), because it does not make sense to arbitrarily adjust parameters to a simulation setting. This observation suggests that the current default parameters of MAFFT might not be very suitable for aligning closely related sequences. However, this idea must be checked using actual biological sequences.
User can select different scoring matrices other than the default. For amino acid alignment, ––bl 45, ––bl 62, ––bl 80, ––jtt N, and ––tm N are accepted, where N is an expected evolutionary distance among input sequences. The ––bl, ––jtt, and ––tm options mean BLOSUM (Henikoff S and Henikoff JG 1992), JTT (Jones et al. 1992), and a transmembrane model (Jones et al. 1994), respectively. A user-defined scoring matrix can also be accepted, by ––aamatrix. For nucleotide alignments, ––kimura N is accepted, where N is an expected evolutionary distance among input sequences. Gap penalties can be adjusted by ––op, ––exp, ––lop, and ––lexp options.
One possible extension is to use different scoring matrices and gap penalties for different sequence pairs according to the divergence level, like ClustalW (Thompson et al. 1994). More studies using actual sequence data will be necessary before implementing this extension. It will also be necessary to adjust gap penalties, preferably based on a realistic evolutionary model of insertions and deletions.
Use of Structural Information
We have discussed possible improvements in MSAs of closely related sequences in the previous section. MSA of distantly related sequences is still a challenging problem.
Test Case 3: PIN Domain
Figure 4 shows a typical limitation of sequence level alignment for a highly divergent set of three PIN-domain containing proteins: human regnase-1, VPA0982 from Vibrio parahaemolyticus, nuclease domain of taq polymerase from Thermus aquaticus. These three proteins share a magnesium-binding site composed of three conserved aspartic acids. Figure 4A shows a superposition of the three structures (Protein Databank identifiers 3v33, 2qip, and 1taq, respectively). The middle aspartic acid is indicated by sphere-representation, colored red. In Figure 4B, a typical MSA (by MAFFT-L-INS-i) is shown wherein the middle aspartic acid position is misaligned. In Figure 4C, a structure-informed MSA (described below), with the middle aspartic acid correctly aligned, is shown.
Fig. 4.

(A) Superposition of 3v33, 2qip, and 1taq structures visualized by PyMOL (Schrödinger LLC 2010). (B) MAFFT-L-INS-i sequence alignment displayed on jalview (Waterhouse et al. 2009). Misaligned Ds are highlighed in red. (C) Structure-informed MSA with correctly aligned Ds; Alpha helices and beta sheets are shown in blue and yellow, respectively, in (A–C).
Strategy for Integrating Structural Alignments and MAFFT
It has long been known that structural information can be used to improve MSA calculations. This was the basis of the 3D Coffee program (O’Sullivan et al. 2004), and later the PROMALS3D package (Pei et al. 2008). Here, we address incorporation of protein structural information in MAFFT-based MSA construction. There are both conceptual issues and technical issues that complicate the process. Conceptually, we have to define structural similarity in such a way that it can easily be used in sequence alignments. We discuss our approach to this problem below in the context of integrating MAFFT with the structural alignment program ASH (Standley et al. 2004, 2007). On the technical level, structural information complicates matters simply because protein structures contain more information and more noise than sequence information.
Here, we focus on one essential feature of ASH: the equivalence score that is used to define structural similarity. A particular element in the structural similarity matrix takes the form of a Gaussian-shaped function of the inter-residue distance
where dij is the distance between two alpha carbons i and j in the two input structures and d0 is a parameter that defines tolerance in the score. The default behavior is to set d0 to 4 Å. The goal of ASH is to maximize the sum of eij over aligned residues. The residue-level equivalences, which form the basis of all ASH alignments, provide a convenient route for combining MAFFT and ASH. We can, for example, set a threshold value of eij and incorporate highly confident parts of the alignment into MAFFT to “seed” the MSA calculation. If we consider the case of the three PIN domain-containing structures in Figure 4, we can first compute structural alignments for the three unique pairs using ASH (ash_3v33A-2qipA, ash_3v33A-1taqA, and ash_2qipA-1taqA). If we set a threshold for residue equivalence at  0.5, we can then combine the equivalence-filtered alignments into MAFFT using the seed option (Katoh et al. 2009):
0.5, we can then combine the equivalence-filtered alignments into MAFFT using the seed option (Katoh et al. 2009):
mafft-linsi --seed ash_3v33A-2qipA \ --seed ash_3v33A-1taqA\
--seed ash_2qipA-1taqA \ sequences > output
Because the sequence identities between the aligned structures are low, we see an improvement in the resulting MSA relative to conventional MAFFT (Fig. 4). Based on this approach, we are developing an integrative service for protein structure-informed MSA construction.
Acknowledgments
The authors thank Drs. Wen Chen, C. André Lévesque, and Christopher Lewis, Agriculture and Agri-Food Canada, for permitting the use of the ITS data in this article and providing other challenging problems. This work was supported by Platform for Drug Discovery, Informatics, and Structural Life Science from the Ministry of Education, Culture, Sports, Science and Technology, Japan, and the Computational Biology Research Center, National Institute of Advanced Industrial Science and Technology (AIST), Japan.
References
- Altschul SF. Generalized affine gap costs for protein sequence alignment. Proteins. 1998;32:88–96. [PubMed] [Google Scholar]
- Barton GJ, Sternberg MJ. A strategy for the rapid multiple alignment of protein sequences. Confidence levels from tertiary structure comparisons. J Mol Biol. 1987;198:327–337. doi: 10.1016/0022-2836(87)90316-0. [DOI] [PubMed] [Google Scholar]
- Berger MP, Munson PJ. A novel randomized iterative strategy for aligning multiple protein sequences. Comput Appl Biosci. 1991;7:479–484. doi: 10.1093/bioinformatics/7.4.479. [DOI] [PubMed] [Google Scholar]
- Berger SA, Stamatakis A. Aligning short reads to reference alignments and trees. Bioinformatics. 2011;27:2068–2075. doi: 10.1093/bioinformatics/btr320. [DOI] [PubMed] [Google Scholar]
- Blackburne BP, Whelan S. Class of multiple sequence alignment algorithm affects genomic analysis. Mol Biol Evol. 2012a doi: 10.1093/molbev/mss256. Advance access published December 4, 2012, doi:10.1093/molbev/mss256. [DOI] [PubMed] [Google Scholar]
- Blackburne BP, Whelan S. Measuring the distance between multiple sequence alignments. Bioinformatics. 2012b;28:495–502. doi: 10.1093/bioinformatics/btr701. [DOI] [PubMed] [Google Scholar]
- Cannone JJ, Subramanian S, Schnare MN, et al. (14 co-authors) The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics. 2002;3:2. doi: 10.1186/1471-2105-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JR, Wang Q, Cardenas E, et al. (11 co-authors) The ribosomal database project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37:D141–D145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessimoz C, Gil M. Phylogenetic assessment of alignments reveals neglected tree signal in gaps. Genome Biol. 2010;11:R37. doi: 10.1186/gb-2010-11-4-r37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng DF, Doolittle RF. Progressive sequence alignment as a prerequisite to correct phylogenetic trees. J Mol Evol. 1987;25:351–360. doi: 10.1007/BF02603120. [DOI] [PubMed] [Google Scholar]
- Fletcher W, Yang Z. INDELible: a flexible simulator of biological sequence evolution. Mol Biol Evol. 2009;26:1879–1888. doi: 10.1093/molbev/msp098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golubchik T, Wise MJ, Easteal S, Jermiin LS. Mind the gaps: evidence of bias in estimates of multiple sequence alignments. Mol Biol Evol. 2007;24:2433–2442. doi: 10.1093/molbev/msm176. [DOI] [PubMed] [Google Scholar]
- Gotoh O. Optimal alignment between groups of sequences and its application to multiple sequence alignment. Comput Appl Biosci. 1993;9:361–370. doi: 10.1093/bioinformatics/9.3.361. [DOI] [PubMed] [Google Scholar]
- Gotoh O. A weighting system and algorithm for aligning many phylogenetically related sequences. Comput Appl Biosci. 1995;11:543–551. doi: 10.1093/bioinformatics/11.5.543. [DOI] [PubMed] [Google Scholar]
- Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins DG, Sharp PM. CLUSTAL: a package for performing multiple sequence alignment on a microcomputer. Gene. 1988;73:237–244. doi: 10.1016/0378-1119(88)90330-7. [DOI] [PubMed] [Google Scholar]
- Jones DT, Taylor WR, Thornton JM. The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci. 1992;8:275–282. doi: 10.1093/bioinformatics/8.3.275. [DOI] [PubMed] [Google Scholar]
- Jones DT, Taylor WR, Thornton JM. A mutation data matrix for transmembrane proteins. FEBS Lett. 1994;339:269–275. doi: 10.1016/0014-5793(94)80429-x. [DOI] [PubMed] [Google Scholar]
- Katoh K, Asimenos G, Toh H. Multiple alignment of DNA sequences with MAFFT. Methods Mol Biol. 2009;537:39–64. doi: 10.1007/978-1-59745-251-9_3. [DOI] [PubMed] [Google Scholar]
- Katoh K, Frith MC. Adding unaligned sequences into an existing alignment using MAFFT and LAST. Bioinformatics. 2012;28:3144–3146. doi: 10.1093/bioinformatics/bts578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Kuma K, Toh H, Miyata T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005;33:511–518. doi: 10.1093/nar/gki198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM. MAFFT: iterative refinement and additional methods. Methods Mol Biol. Forthcoming 2013 doi: 10.1007/978-1-62703-646-7_8. [DOI] [PubMed] [Google Scholar]
- Katoh K, Toh H. PartTree: An algorithm to build an approximate tree from a large number of unaligned sequences. Bioinformatics. 2007;23:372–374. doi: 10.1093/bioinformatics/btl592. [DOI] [PubMed] [Google Scholar]
- Katoh K, Toh H. Improved accuracy of multiple ncRNA alignment by incorporating structural information into a MAFFT-based framework. BMC Bioinformatics. 2008a;9:212. doi: 10.1186/1471-2105-9-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Toh H. Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform. 2008b;9:286–298. doi: 10.1093/bib/bbn013. [DOI] [PubMed] [Google Scholar]
- Katoh K, Toh H. Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics. 2010;26:1899–1900. doi: 10.1093/bioinformatics/btq224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiełbasa SM, Wan R, Sato K, Horton P, Frith MC. Adaptive seeds tame genomic sequence comparison. Genome Res. 2011;21:487–493. doi: 10.1101/gr.113985.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letsch HO, Kuck P, Stocsits RR, Misof B. The impact of rRNA secondary structure consideration in alignment and tree reconstruction: simulated data and a case study on the phylogeny of hexapods. Mol Biol Evol. 2010;27:2507–2521. doi: 10.1093/molbev/msq140. [DOI] [PubMed] [Google Scholar]
- Löytynoja A, Vilella AJ, Goldman N. Accurate extension of multiple sequence alignments using a phylogeny-aware graph algorithm. Bioinformatics. 2012;28:1684–1691. doi: 10.1093/bioinformatics/bts198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsen FA, Kodner RB, Armbrust EV. pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics. 2010;11:538. doi: 10.1186/1471-2105-11-538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirarab S, Nguyen N, Warnow T. SEPP: SATé-enabled phylogenetic placement. Pac Symp Biocomput. 2012;17:247–258. doi: 10.1142/9789814366496_0024. [DOI] [PubMed] [Google Scholar]
- Nilsson RH, Veldre V, Hartmann M, Unterseher M, Amend A, Bergsten J, Kristiansson E, Ryberg M, Jumpponen A, Abarenkov K. An open source software package for automated extraction of ITS1 and ITS2 from fungal ITS sequences for use in high-throughput community assays and molecular ecology. Fungal Ecology. 2010;3:284–287. [Google Scholar]
- Notredame C, Holm L, Higgins DG. COFFEE: an objective function for multiple sequence alignments. Bioinformatics. 1998;14:407–422. doi: 10.1093/bioinformatics/14.5.407. [DOI] [PubMed] [Google Scholar]
- Nuin PA, Wang Z, Tillier ER. The accuracy of several multiple sequence alignment programs for proteins. BMC Bioinformatics. 2006;7:471. doi: 10.1186/1471-2105-7-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan O, Suhre K, Abergel C, Higgins DG, Notredame C. 3DCoffee: combining protein sequences and structures within multiple sequence alignments. J Mol Biol. 2004;340:385–395. doi: 10.1016/j.jmb.2004.04.058. [DOI] [PubMed] [Google Scholar]
- Pei J, Kim BH, Grishin NV. PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 2008;36:2295–2300. doi: 10.1093/nar/gkn072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punta M, Coggill PC, Eberhardt RY, et al. (16 co-authors) The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahraeian SM, Yoon BJ. PicXAA-R: efficient structural alignment of multiple RNA sequences using a greedy approach. BMC Bioinformatics. 2011;12:S38. doi: 10.1186/1471-2105-12-S1-S38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger LLC. The PyMOL Molecular Graphics System, Version 1.3r1. Portland. 2010 Oregon: Schrödinger, LLC. [Google Scholar]
- Sievers F, Wilm A, Dineen D, et al. (12 co-authors) Fast scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigrist CJ, Cerutti L, de Castro E, Langendijk-Genevaux PS, Bulliard V, Bairoch A, Hulo N. PROSITE, a protein domain database for functional characterization and annotation. Nucleic Acids Res. 2010;38:D161–D166. doi: 10.1093/nar/gkp885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standley D, Toh H, Nakamura H. Ash structure alignment package: sensitivity and selectivity in domain classification. BMC Bioinformatics. 2007;8:116. doi: 10.1186/1471-2105-8-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standley DM, Toh H, Nakamura H. Detecting local structural similarity in proteins by maximizing number of equivalent residues. Proteins. 2004;57:381–391. doi: 10.1002/prot.20211. [DOI] [PubMed] [Google Scholar]
- Sun H, Buhler JD. PhyLAT: a phylogenetic local alignment tool. Bioinformatics. 2012;28:1336–1344. doi: 10.1093/bioinformatics/bts158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabei Y, Kiryu H, Kin T, Asai K. A fast structural multiple alignment method for long RNA sequences. BMC Bioinformatics. 2008;9:33. doi: 10.1186/1471-2105-9-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]