

Abstract
The strongest mucosal immune responses are induced following mucosal Ag delivery and processing in the mucosal lymphoid tissues, and much is known regarding the immunological parameters which regulate immune induction via this pathway. Recently, experimental systems have been identified in which mucosal immune responses are induced following nonmucosal Ag delivery. One such system, footpad delivery of Venezuelan equine encephalitis virus replicon particles (VRP), led to the local production of IgA Abs directed against both expressed and codelivered Ags at multiple mucosal surfaces in mice. In contrast to the mucosal delivery pathway, little is known regarding the lymphoid structures and immunological components that are responsible for mucosal immune induction following nonmucosal delivery. In this study, we have used footpad delivery of VRP to probe the constituents of this alternative pathway for mucosal immune induction. Following nonmucosal VRP delivery, J chain-containing, polymeric IgA Abs were detected in the peripheral draining lymph node (DLN), at a time before IgA detection at mucosal surfaces. Further analysis of the VRP DLN revealed up-regulated α4β7 integrin expression on DLN B cells, expression of mucosal addressin cell adhesion molecule 1 on the DLN high endothelia venules, and production of IL-6 and CC chemokines, all characteristics of mucosal lymphoid tissues. Taken together, these results implicate the peripheral DLN as an integral component of an alternative pathway for mucosal immune induction. A further understanding of the critical immunological and viral components of this pathway may significantly improve both our knowledge of viral-induced immunity and the efficacy of viral-based vaccines.
Because the vast majority of harmful pathogens rely on penetration of a mucosal barrier to initiate infection, innate and adaptive immune mechanisms which uphold the integrity of the mucosal surface are paramount for mediating protection from invading microbes. To date, vaccination has proven to be one of the most effective strategies of prophylactic immuno-modulation against mucosal pathogens; however, mucosal infections with numerous viral, bacterial, and fungal agents still pose a significant threat to human health. Therefore, the development of vaccine regimens capable of stimulating protective mucosal immunity represent a powerful opportunity to intercede in the disease course of many infectious organisms (1).
It is well established that, compared with nonmucosal delivery, the strongest mucosal immune responses are induced following mucosal Ag delivery (1–3). Stimulation of this “natural pathway” occurs following nasal or oral Ag exposure and results in Ag capture and processing in the MALT, found in close proximity to each mucosal surface (2, 4). As a consequence, B cells enter the systemic circulation, up-regulate homing receptors, such as the α4β7 integrin, and migrate back to the mucosal surface where they were activated (5, 6). The α4β7 integrin (also termed the mucosal homing receptor) is critical for lymphocyte homing to the small intestine where it binds to mucosal addressin cell adhesion molecule 1 (MAdCAM-1)5 on mucosal lymph node high endothelial venules (HEVs) (7, 8). Mucosal B cells next differentiate into IgA-secreting plasma cells where they produce large amounts of dimeric/polymeric IgA Abs by inclusion of the joining chain, or J chain, during Ab secretion (9, 10). Indeed, J chain-containing IgA molecules are specifically transported onto mucosal surfaces via the pIgR (11).
Although mucosal Ag delivery is the most efficient method of inducing local mucosal Ab responses, a growing body of evidence supports the existence of an additional pathway capable of stimulating mucosal Ab synthesis and/or protection from mucosal challenge following nonmucosal Ag delivery (reviewed in Refs. 12 and 13). Peripheral, or nonmucosal delivery of viral (14–21) and bacterial (22, 23) agents, as well as immunomodulatory factors (24, 25) significantly augment mucosal Ab and/or T cell responses. Likewise, several specialized nonmucosal delivery approaches have been developed which promote mucosal immune induction such as parenteral targeting of mucosal lymphoid tissues using anti-MAdCAM-1 Abs (26), Ag delivery through the skin in the presence of known mucosal adjuvants (transcutaneous immunization) (27–29), and direct Ag inoculation into a peripheral lymph node (targeted lymph node immunization) (30, 31).
Our laboratory recently demonstrated that nonmucosal delivery of modified virus particles derived from the alphavirus, Venezuelan equine encephalitis virus (VEE), stimulates IgG and IgA Ab production at multiple mucosal surfaces to both expressed and codelivered Ags (18–20). The particles used in these studies, termed VEE replicon particles (VRP), express only the viral nonstructural or replicase components responsible for replication of the genomic RNA and can be engineered to express high levels of a heterologous protein (32). VRP efficiently infect DCs following footpad inoculation in mice and replicate the modified viral genome to high levels in the draining lymph node (DLN) (33). However, VRP fail to propagate beyond the first infected cell, since progeny virions are not produced following infection (34). In this study, we have used nonmucosal delivery of VRP as a model system to dissect the individual components of a peripheral mucosal immune induction pathway. VRP were used as an adjuvant in two separate test Ag systems: OVA and inactivated influenza virus. In this report, we demonstrate that several markers of mucosal lymphoid tissues are present in the lymph node draining the VRP inoculation site, such as mucosal IgA Abs, mucosal cytokines (IL-6, TNF-α), and a mucosal homing profile. These observations are consistent with a model in which, following nonmucosal VRP delivery, the DLN is converted into the functional equivalent of a mucosal inductive site and serves as a component of an alternative pathway for mucosal immune induction. These studies provide a framework for the identification of the critical components of an alternative pathway for mucosal immune induction and the potential to improve vaccines targeting mucosal pathogens.
Materials and Methods
VEE replicon constructs
The construction and packaging of VRP was performed as previously described (32, 35). Briefly, confluent monolayers of BHK-21 cells were co-electroporated with three in vitro-transcribed RNAs, the replicon RNA, and two defective helper RNAs encoding the viral structural genes. Only the replicon RNA is packaged into VRP, because the helper RNAs lack the viral packaging signal. In this study, three different replicon constructs were used: 1) replicons expressing GFP (GFP-VRP), 2) replicons expressing the hemagglutinin (HA) gene from influenza virus (HA-VRP), and 3) replicons which lack a functional transgene downstream of the 26S promoter (null VRP) (18). HA-VRP and null VRP were quantitated by immunocytochemistry of infected BHK cells with antisera against HA (32) and null VRP (18), respectively. GFP-VRP were quantitated by immunofluorescence of infected BHK cells. All replicon particles used in this study were packaged in the wild-type (V3000) envelope.
Animals and immunizations
Seven- to 10-wk-old female BALB/c mice were immunized with Ag and/or VRP in a 0.01-ml volume in the rear footpad(s) as previously described (18) according to protocols approved by the Institutional Animal Care and Use Committee. Briefly, animals were immunized at weeks 0 and 4 with Ag alone or Ag coinoculated with either VRP or CpG DNA as an adjuvant. Chicken egg albumin (OVA) was purchased from Sigma-Aldrich; inactivated influenza virus (I-Flu) was purchased from Charles River Laboratories and was dialyzed against PBS in a Slidalyzer cassette (Pierce) according to the manufacturer's guidelines before use. CpG DNA (oligodeoxynucleotide 1826) was purchased from InvivoGen. Diluent consisted of low endotoxin, filter-sterilized PBS, except for the lymphoid organ culture experiments (see below), in which 110 mM Ca2+, 50 mM Mg2+, and 0.1% (v/v) donor calf serum were included.
Lymphoid organ cultures
Lymphoid cultures were prepared as previously described (18, 19). Briefly, spleen, nasal tissue, and draining popliteal lymph nodes were harvested from immunized animals and placed in Eppendorf tubes containing 1 ml of wash buffer (HBSS containing 100 U/ml penicillin, 100 μg/ml streptomycin, 110 mM Ca2+, 50 mM Mg2+, and 15 mM HEPES) and washed three times by aspiration and resuspension. Spleen and nasal tissue were placed in individual wells of a 48-well tissue culture plate in 0.3 ml of medium (RPMI 1640 medium (Life Technologies) containing 15 mM HEPES, 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, 50 μg/ml gentamicin, 2 mM l-glutamine (Life Technologies), and 0.25 μg/ml amphotericin B), and DLNs were placed in individual wells in a 96-well tissue culture plate in 0.1 ml of medium. Plates were incubated at 37°C for 7 days to allow Ab secretion from tissue-resident B cells into the supernatant. Following incubation, supernatants were collected, clarified by centrifugation at 4°C, and analyzed for the presence of Ag-specific Abs by ELISA and/or large molecular mass IgA Abs by nonreducing Western blot. A limited set of tissue samples from an additional time point (day 21 after boost) was previously published in Thompson et al. (18) (Fig. 1) as evidence of VRP-induced mucosal immune induction.
FIGURE 1.

The DLN is an early site of IgA production following VRP infection. Groups of BALB/c mice were immunized in the rear footpad with HA-VRP (1 × 105 IU), I-Flu alone (10 μg), or I-Flu (10 μg) plus GFP-VRP (1 × 105 IU) as an adjuvant at weeks 0 and 4. At the indicated time points, lymphoid organ cultures were established from the spleen (A and B), nasal epithelium (C and D), and DLN (E and F) and evaluated for the presence of flu-specific IgG (A, C, and E) and IgA Abs (B, D, and F) by ELISA. Values represent mean ± SEM. *, p < 0.05; **, p < 0.01; and ***, p < 0.001 compared with I-Flu alone, as determined by ANOVA.
Preparation of DLN extracts
Draining popliteal lymph nodes were dissected from immunized animals, and each lymph node was placed in a 1.5- ml tube (Kontes) with 0.1 ml of PBS containing protease inhibitors (complete mini protease inhibitor mixture tablet (Roche)). DLNs were physically homogenized with a plastic pestle (Kontes) with the aid of a hand-held motor and were frozen at −20°C. Following thaw, debris were pelleted by centrifugation at 4°C, and supernatants were analyzed for IgA Abs by ELISA or for cytokine production by Beadlyte multiplex LUMINEX custom analysis performed by Millipore/Upstate Biotechnology (see below). Extracts were compared from individual lymph nodes across the various immunization groups. As a control, lymph node extracts were prepared from individual Peyer's patches (PPs) exactly as described for the popliteal lymph nodes (homogenized in 0.1 ml of PBS).
Sera, fecal extracts, and vaginal washes
All sample collection was performed as previously described (18). Blood was harvested from either the tail vein, following cardiac puncture, or from the submandibular plexus from individual animals, and sera were collected following centrifugation in microtainer serum separator tubes (BD Biosciences). For fecal extracts, fresh fecal pellets (5–8, ≈100–150 mg) were isolated from individual animals and placed in a 1.5-ml Eppendorf tube containing 1 ml of fecal extract buffer (PBS containing 10% (v/v) normal goat serum and 0.1% (v/v) Kathon CG/ICP (Supeleco)). Samples were vortexed for at least 10 min until all pellets were disrupted into a homogenous mixture. Samples were clarified by centrifugation at 4°C, and supernatants were transferred to fresh tubes and stored at −20°C before analysis by ELISA (see below). Vaginal washes were performed by lavage of the exterior vaginal opening with 0.07 ml of PBS 8–10 times. Lavage samples were stored at −20°C and clarified at 4°C before ELISA analysis (see below).
Flow cytometric analysis
DLNs were harvested from immunized animals, and the overall mass of the lymph nodes was determined by weighing individual lymph nodes on an analytical balance. Each lymph node was next disrupted with a razor blade and a hemostat, and single-cell suspensions were created by agitating each lymph node in complete RPMI medium (RPMI 1640 medium containing 10% (v/v) FBS, 2 mM l-glutamine, 50 μg/ml gentamicin, 100 U/ml penicillin, 100 μg/ml streptomycin, and 15 mM HEPES) containing 2.5 mg/ml collagenase A (Roche Applied Science) and 17 μg/ml DNase I (Roche Applied Science) for 30 min at 37°C. Single-cell suspensions were then stained with Abs directed against CD3, CD19, CD45 (B220), CD11c, CD11b (all purchased from eBioscience) as well as α4β7 integrin (LPAM-1; clone DATK32) purchased from BD Pharmingen and examined on a BD Biosciences FACSCalibur flow cytometer. Stained cells were analyzed using CellQuest software (BD Biosciences).
ELISA
ELISAs for influenza- and OVA-specific Abs were performed on serum, fecal extracts, vaginal washes, and lymphoid culture supernatants as previously described (18). Briefly, Ag solutions (either 250 ng/ml influenza virus in carbonate buffer or 1 mg/ml OVA in PBS) were incubated in 96-well plates overnight at 4°C to allow Ags to bind to the plate. Excess Ag was removed, and blocking solution (PBS containing 5% milk for flu or 1×Sigmablock (Sigma-Aldrich) for OVA) was added for 2 h for flu or overnight for OVA at room temperature. Following removal of blocking solution, plates were incubated at room temperature for 2 h (flu) or overnight (OVA) with serial dilutions of individual samples diluted in the appropriate blocking buffer. Plates were washed with a multichannel plate washer (Nunc) and incubated for 1 h with HRP-conjugated secondary goat anti-mouse γ- or α-chain-specific Abs (Southern Biotechnology Associates or Sigma). Finally, plates were again washed, o-phenylenediamine dihydrochloride substrate was added for 30 min, and the reaction was stopped with the addition of 0.1 M NaF. Ab end point titers are reported as the reciprocal of the highest dilution that resulted in an OD450 ≥ 0.2. In lymphoid culture supernatants, end point titers for flu-specific IgA are reported as the reciprocal of the highest dilution that results in an OD450 reading at least 2 SDs greater than values obtained from mock-vaccinated animals (Fig. 1). Data are presented as the geometric mean ± SEM.
Analysis of polymeric IgA
Lymph node (popliteal and PP) extracts were assayed for the presence of polymeric IgA Abs by nonreducing Western blot analysis. Proteins were separated in Laemmli buffer in the absence of reducing agent (no 2-ME) by 6% SDS-PAGE and transferred to polyvinylidene difluoride membranes (Bio-Rad) in transfer buffer (48 mM Tris, 39 mM glycine, and 10% methanol) at 12 V for 1 h. Membranes were subsequently blocked in PBS with 5% dry milk/0.1% Tween 20 (Sigma-Aldrich) at room temperature overnight. Blocked membranes were next washed in PBS with 1% dry milk/ 0.1% Tween 20 and incubated with a goat anti-IgA Ab (Sigma-Aldrich and/or Southern Biotechnology Associates) at room temperature for 2 h. Membranes were again washed and then incubated with a rabbit anti-goat HRP-conjugated secondary Ab (Sigma-Aldrich) at room temperature for 1 h. Membranes were washed again and HRP-conjugated Abs were detected via chemiluminescence with ECL detection reagents (Amersham Pharmacia). For analysis of influenza-specific, polymeric IgA, lymphoid culture supernatants were mixed with either influenza virus or an irrelevant virus (Girdwood virus) for 2 h at 4°C, and virus-Ab complexes were cen-trifuged at 60,000 × g for 30 min through a sucrose cushion. Pelleted virus-Ab complexes were resuspended in nonreducing sample buffer and analyzed by IgA Western blotting as described above.
Detection of J chain
The presence of J chain was evaluated in a Western blot assay on DLN PBS extracts and vaginal wash samples before and after immunoprecipitation with anti-IgA-coupled agarose beads (Open Biosystems). Pre- and post-IgA precipitates derived from DLN extracts were separated in Laemmli buffer by 15% SDS-PAGE and probed with anti-IgA-HRP (Southern Biotechnology Associates), or anti-IgM-HRP (Southern Biotechnology Associates) as described above, or with a biotinylated anti-mouse J chain mAb (36) (provided by T. Leanderson, Lund University, Lund, Sweden), followed by incubation with streptavidin-HRP and detection via ECL as described above. The mouse J chain protein is detected as an ∼25-kDa band in SDS-PAGE with the mAb used here (T. Leanderson, personal communication) (36).
Statistical analysis
Ab titers and cytokine values were evaluated for statistically significant differences by either the ANOVA or Mann-Whitney U test (GraphPad INSTAT). A p ≤0.05 was considered significant.
Immunofluoresent staining of DLNs
To examine the addressin profile present in the DLN of immunized animals, DLNs were harvested, snap frozen in liquid nitrogen, and sectioned. Frozen sections were stained with Abs against PNAd and MAdCAM-1 as previously described (37, 38) and analyzed by confocal microscopy (Zeiss LSM510).
Cytokine/chemokine analysis
PBS homogenates of lymph nodes (see above) were analyzed for the presence of IL-1β, TNF-α, IL-5, IL-6, IFN-γ, RANTES, GM-CSF, MIP-1β, TGF-β1, TGF-β2, and TGF-β3 on a Luminex machine. Samples were analyzed by the Upstate Biotechnology/Millipore Custom Multiplex cytokine analysis service. DLN samples were diluted 1/10 in PBS plus protease inhibitors (see above) and were analyzed by Upstate Biotechnology. At least four individual lymph nodes for each inoculum and time point were analyzed. For statistical purposes, any analyte with a value below the assay limit of detection was assigned a value of the limit of detection −1 pg/ml.
Results
The DLN is an early site of IgA production following VRP infection
To further characterize mucosal immune induction in the VRP system, the kinetics and anatomical localization of IgA production following VRP delivery were determined. Groups of female BALB/c mice were immunized in a rear footpad at weeks 0 and 4 with either diluent, HA-VRP (1 × 105 infectious units (IU)), or formalin-inactivated I-Flu (10 μg), which serves as a non-VRP-vectored Ag control. To evaluate the ability of VRP to induce mucosal immunity to a codelivered Ag, an additional group of animals received I-Flu (10 μg) mixed with GFP-VRP (1 × 105 IU) as an adjuvant. Memory IgA responses were evaluated following secondary inoculation, as our preliminary studies failed to detect IgA responses after a primary immunization (data not shown). Groups of three animals were sacrificed on days 3, 7, 14, and 28 after boost, and lymphoid organ cultures (18, 19) were generated from the spleen (a characteristic systemic lymphoid tissue), the nasal epithelium (a characteristic mucosal surface), and the draining popliteal lymph node (a candidate component of an alternative mucosal immune induction pathway). Organ culture supernatants were evaluated for the presence of influenza (flu)-specific IgG and IgA Abs by ELISA. In general, the kinetics of flu-specific IgG and IgA Ab synthesis was similar in a given tissue (Fig. 1). The dose of I-Flu used in this study (10 μg) was chosen because preliminary studies showed that it induced an Ag-specific systemic IgG response similar to that of HA-VRP, allowing assessment of the role of VRP in IgA production under conditions of equivalent overall immune stimulation. Following delivery of I-Flu alone, flu-specific IgG (Fig. 1A) and IgA (Fig. 1B) Abs were detectable in spleen organ culture supernatants; however, flu-specific IgA Abs were significantly increased following either delivery of HA-VRP or when GFP-VRP were coadministered with I-Flu. Likewise, significantly increased flu-specific IgG (Fig. 1C) and IgA (Fig. 1D) Abs were detected in the nasal epithelium of animals immunized with VRP-containing inocula. Flu-specific IgG (Fig. 1 E) and IgA (Fig. 1F) Abs also were detected in the supernatants from the DLNs following delivery of Ag alone. These values were again significantly increased in animals immunized with VRP as expression vectors or as adjuvants. Taken together, these results further validate the utility of VRP expression vectors and VRP adjuvants.
This study was designed also to determine the anatomical location in which IgA Abs were first produced in VRP-immunized animals. Flu-specific IgA Ab responses peaked in the DLNs at day 3 after boost (Fig. 1F), a time at which such Abs were essentially undetectable at the mucosal surface (Fig. 1D). In general, the responses induced by HA-VRP and I-Flu plus VRP were not statistically different from one another at most time points. The exceptions to this statement are that I-Flu plus VRP induced statistically stronger responses compared with HA-VRP at: 1) day 3 DLN IgG (p < 0.01), 2) day 14 DLN IgG (p < 0.001), and 3) day 3 DLN IgA (p < 0.01). These results suggest that VRP-induced IgA production occurs in the DLN before production at the mucosal surface and provide a foundation for the further study of VRP-stimulated IgA synthesis in the DLN in the context of mucosal immune induction.
The data presented in Fig. 1 suggested that IgA Abs were produced in the DLN following VRP delivery and that such production was dependent upon signals provided by the VRP and/or VRP infection, since equivalent levels of Ag-specific IgA Abs were not produced in the DLN following delivery of Ag alone. This study was extended to determine whether IgA production was occurring in vivo and was not an artifact of ex vivo incubation. Therefore, groups of female BALB/c mice were immunized in a rear footpad at weeks 0 and 4 with I-Flu alone (1 μg), or I-Flu (1 μg) code-livered with null VRP (1 × 105 IU), and DLNs were harvested at days 1, 3, 7, and 14 after boost and homogenized in 0.1 ml of PBS. Supernatants were evaluated for the presence of flu-specific IgA Abs by ELISA. Fecal extracts also were prepared from immunized animals as a measure of mucosal immune induction (18, 20). Ag-specific IgA Abs were detectable in DLN homogenates and the levels peaked at day 3 after boost, similar to the kinetics of IgA production in the DLN in the lymphoid culture system (Fig. 2A). Conversely, Ag-specific IgA Abs peaked in fecal extracts at day 7 after boost, a time after peak production in the DLN (Fig. 2B). I-Flu alone did not induce significant IgA production in either tissue.
FIGURE 2.

VRP induce IgA Ab production in the DLN in vivo. Groups of BALB/c mice were immunized in the rear footpad with I-Flu alone (1 μg), or I-Flu (1 μg) plus null VRP (1 × 105 IU) at weeks 0 and 4. At the indicated time points, flu-specific IgA Abs were evaluated in DLN homogenates (A), fecal extracts (B), and PP homogenates (C) by ELISA. Values represent mean ± SEM. *, p < 0.05; **, p < 0.02; and ***, p < 0.003 compared with I-Flu alone, as determined by the Mann-Whitney U test.
We next sought to evaluate IgA production in the mucosa-draining lymphoid tissues in the VRP system. As a first step, a single PP from each of six individual animals was harvested and homogenized as described for DLNs. PP homogenates were evaluated for the presence of flu-specific IgA Abs by ELISA; however, the mean values produced in PP homogenates at all four time points tested were below the limits of detection of the ELISA and were not statistically distinct from the assay background (Fig. 2C). Taken together, these results suggest that, following nonmucosal VRP infection, Ag-specific IgA Abs are produced in vivo in the DLN before production at mucosal surfaces.
VRP stimulate high molecular mass IgA Ab production in the DLN
Although delivery of VRP-containing inocula clearly increased the levels of Ag-specific IgA Abs produced in the DLNs, Ag-specific IgA Abs also were produced at detectable levels following delivery of I-Flu alone. Based on the idea that mucosal IgA Abs are polymeric, the molecular mass of the IgA species produced in the DLN was determined following delivery of both VRP-containing and non-VRP-containing inocula. Supernatants from day 3 DLN cultures were analyzed by SDS-PAGE under nonreducing conditions for the presence of IgA Abs. To compare the ratio of monomeric to polymeric IgA on a per lymph node basis, an equal amount of DLN supernatants were evaluated in the Western blot assay. The IgA Abs in the DLN following delivery of I-Flu alone were predominantly monomeric, with an apparent molecular mass of ∼160 kDa (Fig. 3A). Interestingly, the delivery of either HA-VRP or I-Flu plus a VRP adjuvant resulted in the production of monomeric forms of DLN IgA as well as large molecular (>250 kDa) forms not present at significant levels following delivery of Ag alone (Fig. 3A). IgA Abs were not detected in the contralateral popliteal lymph node under these conditions.
FIGURE 3.
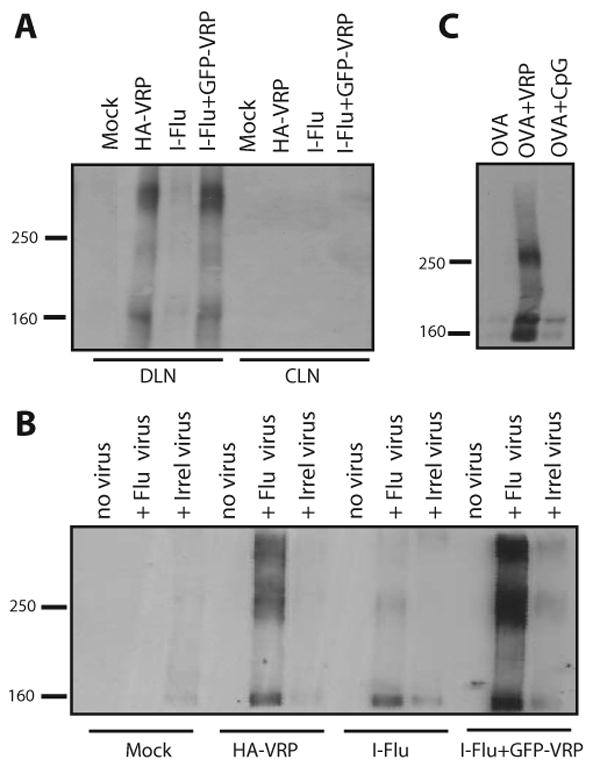
VRP induce the production of large molecular mass IgA Abs in the DLN. Groups of BALB/c mice were immunized in the rear footpad with HA-VRP (1 × 105 IU), I-Flu alone (10 μg), or I-Flu (10 μg) plus GFP-VRP (1 × 105 IU) at weeks 0 and 4. At day 3 after boost, DLN and contralateral lymph node (CLN) culture supernatants were evaluated for IgA Abs by nonreducing Western blot analysis (A). Day 3 DLN supernatants were then mixed with influenza virus to form virus-Ab complexes and complexes were purified via ultracentrifugation before nonreducing Western blot analysis for IgA (B). Groups of BALB/c mice were immunized in the rear footpads with OVA alone (10 μg), OVA (10 μg) plus null VRP (1 × 105 IU), or OVA (10 μg) plus CpG DNA (1 μg) at weeks 0 and 4 and DLNs were harvested at day 3 and DLN PBS homogenates were evaluated for IgA Abs by nonreducing Western blot analysis as in A (C). The gel lanes in A have been rearranged from the original order for esthetic purposes; however, the data are all from the same gel from a single experiment.
To determine whether VRP-induced high-molecular mass IgA Abs were specific for the influenza Ag, day 3 DLN supernatants were incubated with influenza virus or an irrelevant virus (Girdwood virus) and were pelleted through a sucrose cushion by ultracentrifugation. This procedure utilized cosedimentation of influenza virions as a means to affinity purify virus-specific Abs from non-flu-specific Abs before Western blot analysis. The flu-specific IgA Abs present in the DLN following delivery of I-Flu were predominantly monomeric. Immunization with either HA-VRP or I-Flu plus GFP-VRP resulted in the production of both a monomeric species of flu-specific IgA, as well as two high-molecular mass forms not present at appreciable levels following delivery of I-Flu alone (Fig. 3B). Taken together, these results suggest that VRP infection provides a signal which promotes the production of high-molecular mass IgA molecules in the DLN.
To evaluate the presence of polymeric IgA Abs in the DLN in vivo, groups of female BALB/c mice were immunized at weeks 0 and 4 with either OVA alone (10 μg) or OVA mixed with null VRP (1 × 105 IU). An additional group of animals was immunized with OVA (10 μg) mixed with CpG DNA (1 μg) as a control adjuvant known to induce mucosal immunity following nonmucosal delivery, albeit at lower levels compared with VRP (18). At day 3 after boost, DLN homogenates were prepared and evaluated for the presence of IgA Abs by nonreducing Western blot analysis as described above. As shown in Fig. 3C, IgA Abs were not detectable in the DLN following delivery of OVA alone or following codelivery of OVA with CpG DNA. In contrast, both monomeric and dimeric forms of IgA were produced in the VRP DLNs, suggesting that high-molecular mass IgA Abs are produced in the DLN in vivo.
Role of exogenous Ag in DLN polymeric IgA production
An additional experiment was performed to determine whether the production of polymeric IgA Abs in the DLN was dependent upon exogenous Ag stimulation, or whether Ags naturally present in the DLN have the capacity to promote IgA production during a concomitant VRP infection. Groups of female BALB/c mice were immunized at weeks 0 and 4 with either I-Flu alone (1 μg), I-Flu (1 μg) mixed with null VRP (1 × 105 IU), or null VRP alone (1 × 105 IU), and DLN homogenates in PBS were prepared at day 3 after boost and analyzed for the presence of IgA by Western blot under nonreducing conditions. As shown in Fig. 4A, VRP induced the production of both monomeric and polymeric forms of IgA following codelivery with Ag. In contrast, IgA Abs were not detected following delivery of either Ag alone or null VRP alone when comparing equal proportions of DLN lysate, suggesting that antigenic stimulation, in addition to VRP infection, is required to promote polymeric IgA synthesis in the DLN.
FIGURE 4.

Role of exogenous Ag in DLN polymeric IgA production. Groups of BALB/c mice were immunized in the rear footpads at weeks 0 and 4 with I-Flu alone (1 μg), I-Flu (1 μg) coinoculated with null VRP (1 × 105 IU), or null VRP alone (1 × 105 IU). At day 3 after boost, DLNs were harvested and PBS extracts were prepared and equal proportions of lysates were analyzed for the presence of IgA Abs by Western blot under nonreducing conditions (A). DLN Abs were evaluated for the presence of the J chain by reducing Western blot analysis. The mouse J chain protein is detected as an ∼25-kDa band in SDS-PAGE with the mAb used here (T. Leanderson, personal communication) (36) (B). Vaginal wash (VW) fluids and DLN PBS extracts were probed with an anti-mouse J chain mAb before and following IgA IP (B). IgA Abs were also evaluated in serum and VW fluids by nonreducing Western blot analysis (C). Additionally, levels of IgA Abs were evaluated in increasing amounts of I-Flu DLN lysates (1×, 4×, 8×, 12×) compared with 1× of I-Flu plus VRP lysates (C).
At mucosal sites, the formation of dimeric and polymeric IgA is dependent upon incorporation of the J chain during IgA secretion (10). Therefore, to further characterize the mucosal nature of DLN IgA Abs, we next evaluated day 3 DLN PBS homogenates (described above) for the presence of J chain in a Western blot assay. Our analysis of DLN homogenates revealed the presence of IgM Abs in addition to IgA Abs (data not shown). Therefore, DLN homogenates were subjected to immunoprecipitation (IP) using anti-IgA-agarose beads to analyze J chain content specifically in the IgA fractions in the DLN, as opposed to J chain present in pentameric IgM complexes. IgM Abs were no longer detected following IgA IP (data not shown). As a control, J chain content was also analyzed in vaginal wash fluids from immunized mice by SDS-PAGE under reducing conditions. An ∼24-kDa band, corresponding to the J chain (36) (T. Leanderson, personal communication), was present in vaginal washes from animals immunized with I-Flu alone and I-Flu plus VRP (Fig. 4B). Comparing equal proportions of the DLN lysates, a J chain band of weak intensity was present in post-IgA IP lysates prepared from animals immunized with I-Flu alone. In contrast, the J chain was readily detectable in both pre- and post-IgA IP DLN lysates following immunization with I-Flu plus VRP (Fig. 4B), indicating that the J chain is incorporated into VRP-induced polymeric DLN IgA.
Although the data presented in Figs. 3 and 4A suggested that delivery of both VRP and Ag were required for DLN IgA production, we considered the possibility that IgA Abs may, in fact, be produced in the DLN following delivery of Ag alone at levels not detectable when comparing equal proportions of DLN lysates. Therefore, day 3 DLN lysates were subjected to IgA Western blot analysis as described above; however, increasing amounts of I-Flu DLN lysates (1×, 4×, 8×, and 12×) were electrophoresed and compared with a 1× amount of I-Flu plus VRP lysate (Fig. 4C). As additional controls, monomeric and polymeric IgA Abs were also analyzed in serum and vaginal wash fluids. Consistent with previous reports (39, 40), both monomeric and polymeric species of IgA were present in the serum of immunized animals, regardless of the inoculum (Fig. 4C). Additionally, IgA Abs present in vaginal lavage fluids were almost exclusively polymeric (Fig. 4C). Although inclusion of VRP in the inoculum clearly increased IgA production in the DLN, analysis of increasing amounts of DLN lysate from I-Flu-inoculated animals revealed both monomeric and polymeric IgA species present in the DLNs when 8- and 12-fold more lysate was analyzed compared with I-Flu plus VRP (Fig. 4C). Furthermore, polymeric IgA Abs were present in day 3 DLN lysates from unmanipulated animals as well as PBS-immunized mice, albeit at lower levels (data not shown). Taken together, these results demonstrate that peripheral delivery of VRP significantly increased monomeric and polymeric IgA production in the draining peripheral lymph node.
Characterization of DLN cells following VRP delivery
The observation that the DLN appeared to serve as the earliest site of IgA production, and high-molecular mass Ag-specific IgA following VRP delivery, led us to further characterize the general characteristics of the DLN under these conditions. The overall mass of the DLN, a general marker of inflammation, as well as the general cellularity of the DLN were determined. Groups of animals were immunized with either I-Flu alone (1 μg), I-Flu (1 μg) mixed with null VRP (1 × 105 IU), or null VRP alone (1 × 105 IU), and DLNs were harvested at days 0, 1, and 3 after boost. DLNs were carefully weighed on an analytical balance to determine overall mass, and single-cell suspensions were created by collagenase digestion. Total cell counts were performed by trypan blue exclusion, and cells were analyzed by flow cytometry for the presence of B cells, T cells, and dendritic cells (DCs) following staining with Abs directed against the appropriate cell surface markers (CD19, CD3, and CDllc, respectively). As shown in Fig. 5A, VRP delivery in the presence or absence of codelivered Ag resulted in a 2- to 4-fold increase in the overall mass of the DLN, depending upon the time point examined. Moreover, the total cellularity of the VRP DLN was increased by 4- to 6-fold (Fig. 5B). VRP induced a proportional 3- to 10-fold increase in B cells, T cells, and CD11b− DCs in the DLN at days 1 and 3 after boost (Fig. 5, C–E). Also, VRP infection led to an ∼20-to 40-fold increase in the number of CD11b+ DCs in the DLN at days 1 and 3 after boost, both in the presence and absence of exogenous Ag (Fig. 5F). These results suggest that VRP delivery results in significantly increased cellularity in the inflamed DLN independent of exogenous Ag delivery, with a preferential recruitment of a CD11b+ DCs.
FIGURE 5.

Characterization of VRP DLN cells. Groups of BALB/c mice were immunized in the rear footpads at weeks 0 and 4 with I-Flu alone (1 μg), I-Flu (1 μg) coinoculated with null VRP (1 × 105 IU), or null VRP alone (1 × 105 IU). At days 0, 1, and 3 after boost, DLNs were harvested and weighed on an analytical balance and single-cell suspensions were prepared by collagenase digestion. The mass of each lymph node was determined (A) and total number of cells was determined by trypan blue exclusion (B). The number of B cells (C), T cells (D), CD11b− DCs (E), and CD11b+ DCs (F) was evaluated by flow cytometry. Values are presented as geometric mean ± SEM. *, p < 0.05; **, p < 0.01; and ***, p < 0.001 compared with I-Flu alone, as determined by ANOVA.
Presence of α4β7 integrin-positive B cells in the VRP DLN
Because VRP infection resulted in recruitment of B cells to the DLN, we next sought to evaluate such cells for characteristics of mucosal B cells, such as expression of the mucosal homing receptor. Groups of female BALB/c mice were immunized in a rear footpad at weeks 0 and 4 with either OVA alone (10 μg) or OVA (10 μg) coinoculated with null VRP (1 × 105 IU), and DLNs were harvested at days 1 and 3 after boost. Single-cell suspensions were generated by collagenase digestion, and DLN B cells were analyzed for expression of CD19, B220, and the α4β7 integrin by flow cytometry. Additionally, single-cell suspensions prepared from the mesenteric lymph node (MLN) were also analyzed as a representative mucosal lymphoid tissue. As shown in Fig. 6A, the α4β7 integrin was expressed to similar levels on B cells isolated from the MLN at both days 1 and 3, regardless of the immunizing inoculum. Analysis of DLN B cells revealed two populations of B cells according to B220 expression levels; a B220highCD19high population and a B220lowCD19high population (Fig. 6). Expression levels of the α4β7 integrin were analyzed on both populations at days 1 and 3 after boost. As shown in Fig. 6B, a small B220low population of B cells was present in the DLN of animals inoculated with OVA alone at day 1 after boost. Increased α4β7 integrin expression was observed in a subset of these B220low B cells (Fig. 6B, see inset). Delivery of OVA in the presence of VRP resulted in a significantly increased population of B220low B cells with up-regulated α4β7 integrin expression at day 1 after boost (Fig. 6B). The α4β7 integrin levels were comparable to those of MLN B cells (Fig. 6A). Interestingly, although a B220low population was still present in the DLN following delivery of OVA in the presence or absence of VRP at day 3 after boost, this population of cells no longer expressed increased levels of the α4β7 integrin (Fig. 6B). Increased α4β7 integrin expression was detected on a subset of B220high B cells at day 3 following delivery of OVA alone (Fig. 6B).
FIGURE 6.
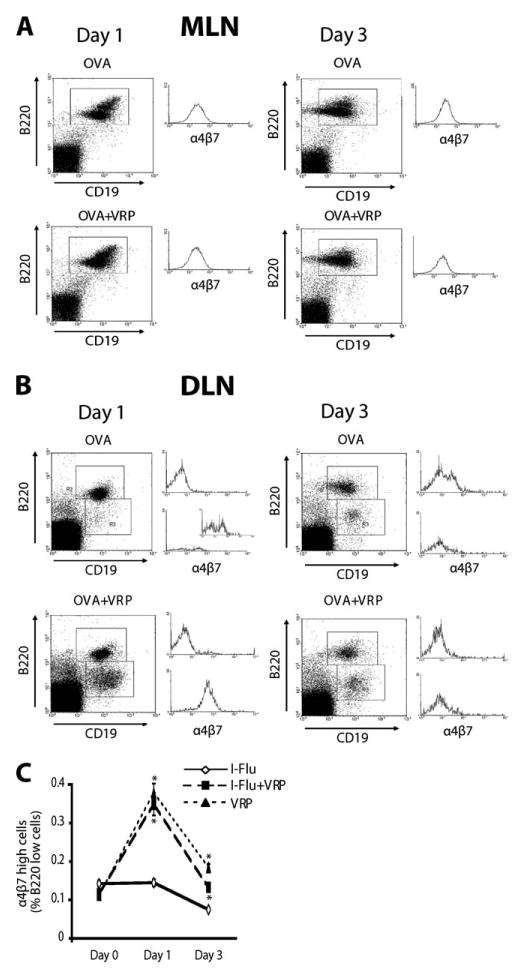
VRP induce the expression of the mucosal homing receptor on DLN B cells. Groups of animals were immunized in the rear footpad at weeks 0 and 4 with OVA alone (10 μg) or OVA (10 μg) coinoculated with null VRP (1 × 105 IU). At days 1 and 3 after boost, DLN cells were stained with Abs directed against CD19, B220, and the α4β7 integrin and analyzed by flow cytometry. As a positive control, α4β7 integrin expression was examined on MLN cells (A). α4β7 integrin expression in both B220high and B220low DLN cells is shown in B. The histograms in A and B are all on the same scale, except the expanded inset in B. To confirm the up-regulation of α4β7 integrin expression specifically in the B220 low population following delivery of a viral Ag, α4β7 integrin expression was analyzed on DLN cells at days 0, 1, and 3 after boost following delivery of I-Flu alone (1 μg), I-Flu (1 μg) plus null VRP (1 × 105 IU), or null VRP alone (1 × 105 IU) (C). Values are presented as geometric mean ± SEM. *, p < 0.001 compared with I-Flu alone, as determined by ANOVA.
To determine the proportion of B220low DLN B cells with up-regulated α4β7 integrin expression, as well as to evaluate the role of exogenous Ag delivery in mucosal homing receptor expression following delivery of a viral Ag, an additional experiment was performed. Groups of mice were immunized at weeks 0 and 4 with either I-Flu alone (1 μg), I-Flu (1 μg) plus null VRP (1 × 105 IU), or null VRP alone (1 × 105 IU), and α4β7 integrin expression was evaluated on DLN B cells at day 0 (before boost), day 1, and day 3 after boost. As shown in Fig. 6C, α4β7 integrin expression was induced in the B220low population at day 1 after boost and was again undetectable at day 3. Moreover, VRP induced increased α4β7 integrin expression either when coinoculated with a viral Ag or when delivered in the absence of any exogenous Ag, suggesting that the signals which catalyze increased mucosal homing receptor expression are active during VRP infection. Taken together, these data suggest that peripheral inoculation of VRP results in a mucosal homing profile of DLN B cells that is similar to the homing profile of B cells isolated from mucosal lymphoid tissues (the MLN).
VRP induce MAdCAM-1 expression on the HEVs of the DLN
Although PNAd expression can be detected on the HEVs of both systemic and mucosal lymphoid tissues (7, 8), MAdCAM-1 expression is distinctive characteristic of mucosal lymphoid tissues (41). Therefore, next we evaluated MAdCAM-1 expression levels in the VRP DLN. Female BALB/c mice were immunized in both rear footpads with either I-Flu alone (1 μg), null VRP alone (1 × 105 IU), I-Flu (1 μg) plus null VRP (1 × 105 IU), or I-Flu (1 μg) plus CpG DNA (1 μg) at weeks 0 and 4. DLNs were harvested from immunized animals at days 1, 2, and 3 after boost, snap frozen in liquid nitrogen, sectioned, and analyzed for expression of MAdCAM-1 and PNAd by confocal microscopy. Sections derived from MLN served as a mucosal lymphoid tissue-positive control. As expected, expression of both PNAd (green) and MAdCAM-1 (red) were readily detectable on the HEV of the MLN, including double-positive cells (yellow, Fig. 7A). PNAd staining was abundant in the DLN following delivery of I-Flu alone at all time points examined, consistent with a systemic lymphoid tissue phenotype (Fig. 7B). Interestingly, MAdCAM-1 expression was detected in the DLN following delivery of I-Flu in the presence of VRP beginning at day 2 after boost, with increased expression at day 3 after boost. Expression of MAdCAM-1 was also noted on the DLN following null VRP injections, although expression was not as abundant (Fig. 7C). Analysis of MAdCAM-1 staining suggested that endothelial cells lining the HEV up-regulated MAdCAM-1, similar to expression in mucosal lymphoid tissues, such as the MLN (Fig. 7, C and D). Both “1st type” staining, in which a single cell inside the vessel is detected (these cells could be endothelial cells), as well as “2nd type,” in which MAdCAM-1 is clearly expressed by the endothelial cells were apparent in the VRP DLN (Fig. 7D). Additionally, positive staining was also detected in the DLN following delivery of I-Flu plus CpG DNA at day 3, however, to a lesser extent (Fig. 7D). As MAdCAM-1 expression was present following delivery of null VRP alone as well as Ag plus null VRP, these data suggest this “profile change” is Ag independent. These results suggest that stimulation of an alternative mucosal inductive pathway by VRP results in an HEV profile in the DLN which, distinct from traditional systemic lymphoid tissues, exhibits characteristics of mucosal lymph nodes.
FIGURE 7.
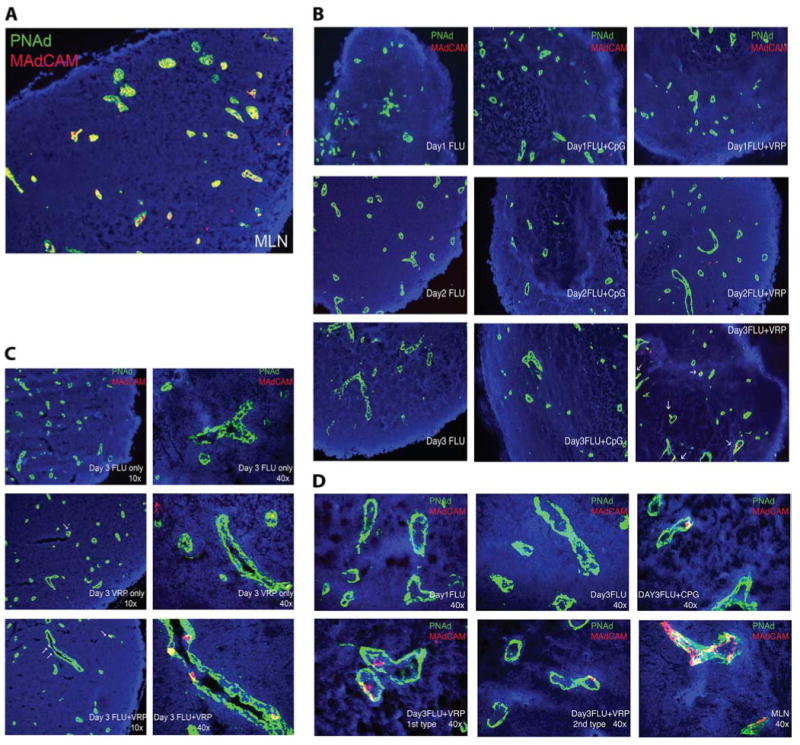
VRP up-regulate MAdCAM-1 in the DLN. Groups of female BALB/c mice were immunized with at weeks 0 and 4 with I-Flu alone (1 μg), I-Flu (1 μg) coinoculated with null VRP (1 × 105 IU), or I-Flu (1 μg) coinoculated with CpG DNA (1 μg) and DLNs were harvested and snap frozen in OCT in liquid nitrogen at days 1, 2, and 3 after boost. MLN (A) and DLN sections were stained with Abs directed against MAdCAM-1 (red) and PNAd (green) analyzed by confocal microscopy and visualized at ×10 (B) and ×40 (C).
VRP drive mucosal cytokine/chemokine production in the DLN
To further characterize the mucosal characteristics of the VRP-DLN, the expression levels of representative Th1/Th2 cytokines, inflammatory cytokines, and mucosally relevant chemokines were evaluated in the DLN in a Luminex assay. Groups of female BALB/c mice were immunized in the rear footpads at weeks 0 and 4 with either OVA alone (10 μg), OVA (10 μg) coinoculated with null VRP (1 × 105 IU), or OVA (10 μg) plus CpG DNA (1 μg) and DLNs were harvested at 6, 12, and 24 h after boost. DLN homogenates in PBS were prepared as described above. Homogenates were analyzed for expression of IL-1β, IL-5, IL-6, TNF-α, IFN-γ, MIP-1β, RANTES, TGF-β1, 2, and 3 in a LUMINEX assay by Upstate Biotechnology/Millipore (Fig. 8). Although generally VRP-induced cytokine/chemokine production peaked at 6 h after boost and decreased over the time course, subtle differences existed between the kinetics of expression of the individual proteins. The inclusion of VRP in the inoculum significantly increased expression of IL-1β (11-fold), IL-5 (3.5-fold), IL-6 (16.5-fold), TNF-α (7-fold), IFN-γ (5-fold), MIP-1β (27-fold), and RANTES (6.5-fold) at the 6- and/or 12-h time points compared with delivery of Ag alone. Additionally, preliminary experiments suggest that VRP likewise up-regulate IL-4 in the DLN (data not shown). Although significant levels of all three subtypes of TGF-β were present in the DLN at 6 h, VRP infection did not induce a statistically significant increase in these proteins and responses were quite variable (data not shown). As a control, we also evaluated the levels of the same eight cytokines in PP homogenates from the same animals; however, protein levels in this tissue were below the limits of the LUMINEX assay (data not shown). This analysis suggests that VRP significantly augment the production of several mucosally relevant cytokines and chemokines in the DLN which may affect the development of specific T cell subsets involved in the mucosal adjuvant effect.
FIGURE 8.

VRP up-regulate mucosal cytokine/chemokine production in the DLN. Groups of female BALB/c mice were immunized with at weeks 0 and 4 with OVA alone (10 μg), OVA (10 μg) coinoculated with null VRP (1 × 105 IU), or OVA (10 μg) inoculated with CpG DNA (1 μg), and PBS homogenates were created from the DLNs at 6, 12, and 24 h after boost. DLN homogenates were evaluated for the presence of IL-1β (A), IL-5 (B), IL-6 (C), TNF-α (D), IFN-γ (E), MIP-1α (F), and RANTES (G) via LUMINEX. Values are presented as geometric mean ± SEM. *, p < 0.03 compared with OVA alone, as determined by the Mann-Whitney U test.
Discussion
Mucosal Ag delivery in the presence of strong mucosal adjuvants, such as the bacterial enterotoxins, results in Ag processing in the mucosal lymphoid structures, stimulates potent mucosal IgA responses, and has revealed the components and immune inductive requirements of the natural pathway of mucosal immune induction (1, 42, 43). However, it has become increasingly clear that nonmucosal Ag delivery also can result in mucosal immune induction under specific conditions (12–14, 16, 17, 19–25, 28, 30, 31, 44). The molecular mechanisms which regulate peripherally induced mucosal immunity are poorly understood at present. The examples are varied and suggest either a consistent unifying mechanism is responsible for such induction under all circumstances or, conversely, that numerous diverse signals have the capacity to drive mucosal immunity following nonmucosal delivery.
It is possible that mucosal immune induction following nonmucosal delivery is merely a result of stimulating components of the natural pathway, as has been proposed by McKenzie et al. (26), following peripheral delivery of anti-MAdCAM-1 Abs. Conversely, mucosal immune induction following nonmucosal delivery may be a result of stimulating an alternative pathway for mucosal immune induction, a pathway with components distinct from those of the natural pathway. Another, nonmutually exclusive, possibility is that specific vaccine signals promote the generation of a “mucosal-like” site outside of the mucosal compartment. We hypothesize that this is the mechanism by which VRP promote mucosal immune induction following nonmucosal delivery. Several characteristics of mucosal lymphoid tissues are observed in the DLN of VRP-immunized mice, suggesting that under these conditions, the DLN is “converted” to the functional equivalent of a mucosal inductive site.
To test this “conversion” hypothesis in the VRP system, we first sought to determine the anatomical location in which IgA Abs are produced following VRP delivery. Such analysis revealed that Ag-specific IgA Abs were first detected in the popliteal lymph node draining the site of VRP inoculation, before Ab production at mucosal surfaces. Moreover, further characterization of DLN IgA Abs revealed the presence of J chain-containing, polymeric forms in the DLN, which resemble IgA Abs found at mucosal sites. This finding is consistent with the production of oligomeric IgA Abs in the peripheral DLN following delivery of CFA (45), and is supportive of a model in which the DLN serves as the primary inductive tissue for the activation of mucosal immune responses following footpad VRP delivery.
Interestingly, the peripheral DLN has been implicated in three other models of mucosal immune activation following nonmucosal delivery. First, Enioutina et al. (25) demonstrated that the active form of vitamin D3 augments mucosal IgA responses in a peripheral delivery model. As in the VRP system, IgA-producing cells, and a “mucosal-like” cytokine profile were present in the DLN following vitamin D delivery; however, the presence of J chain-positive, polymeric IgA was not evaluated in those studies (25, 46). Second, following i.m. rotavirus delivery, cells isolated from the peripheral DLN induced gut IgA and IgG responses following adoptive transfer into naive recipients (15). Third, in targeted lymph node immunization, Ags are inoculated directly into the lymph node itself, promoting Ag acquisition and processing by lymph node-resident cells. This inoculation regimen induces IgA Ab synthesis in the local inoculated lymph node as well as in mucosal secretions, in the absence of Ag processing in other peripheral lymphoid structures (30). Additionally, expression of RANTES and MIP-1β, both of which were highly up-regulated in the DLN by VRP, correlated with lymph node IgA production in the DLN in this model (47). Indeed, Lehner et al. (47) proposed that the affected lymph node serves as an inductive site for the generation of cells with protective properties at the local mucosal surface. These examples, along with the data presented here, suggest a critical role for the DLN in peripherally induced mucosal immune induction.
It is attractive to speculate that, as a consequence of B cell activation within the lymph node draining VRP inoculation, such cells undergo IgA class switch, initiate pIgA production, and migrate to the mucosal surface where they continue to secrete pIgA Abs some days later. Characterization of the VRP DLN revealed the presence of both α4β7 integrin-positive B cells and expression of MAdCAM-1 on the HEVs, potentially implicating mucosal homing receptor interactions in VRP-induced mucosal immunity. Interestingly, however, α4β7 integrin-positive B cells were present in the DLN at day 1 after boost, a time in which MAdCAM-1 expression was not detected. Instead, MAdCAM-1 expression kinetics correlated with the kinetics of IgA Ab production in the DLN, suggesting that the involvement of MAdCAM-1 may not be in the recruitment of IgA-producing cells to the DLN but rather in the retention of such cells in the DLN. Further experimentation will be required to define the precise role of the α4β7 integrin-MAdCAM-1 pathway in mucosal immune induction following nonmucosal VRP infection.
Identification of the essential cell type(s) in the DLN which promote a mucosal environment has yet to be performed; however, we considered the potential role of lymph node DCs in the VRP experimental system. A large body of evidence supports the notion that mucosal DCs are qualitatively distinct from systemic DCs in priming mucosally relevant B cell and T cell responses (Refs. 48–51 and reviewed in Refs. 52 and 53). For example, Mora et al. (54) recently demonstrated increased α4β7 integrin expression and mucosal homing of CD8+ T cells cocultured with mucosal, but not systemic DCs. Likewise, Sato et al. (55) demonstrated that mucosal CDllb+ DCs promote IgA synthesis from cocultured B cells by producing large amounts of IL-6, as mucosal DCs cultured in the presence of an anti-IL-6 Ab failed to stimulate IgA production. It is interesting in this regard that VRP infection stimulated a strong IL-6 response in the DLN.
Of note, the mucosal DCs responsible for driving IgA production in the experimental system described by Sato et al. were CDllb+, the same population of DCs that were dramatically increased in the DLN of VRP-immunized mice. Indeed, preliminary experiments suggest that the CDllb+ DC population represents the major target of VRP infection in the DLN (data not shown); however, whether these cells represent the cellular source of DLN IL-6 or the DLN/mucosal IgA response remains to be determined. It is possible that VRP not only rely upon the DLN for mucosal immune induction, but that VRP infection promotes a “mucosal DC-like” phenotype in DLN CD11b+ DCs, potentially through IL-6 production. Of note, Mora et al. (48) recently demonstrated the ability of mucosal DC-derived retinoic acid to promote IgA secretion and increased α4β7 integrin expression in vitro (48). It will be interesting to determine whether VRP-infected DCs possess the capacity to drive either increased α4β7 integrin expression or IgA production in vitro, and specifically whether soluble IL-6 and/or retinoic acid are implicated in the VRP system.
The data presented here implicate the DLN in mucosal immune induction in the VRP system; however, elements of the natural mucosal induction pathway also may be involved. It is possible that VRP-infected DCs migrate beyond the DLN to PPs and/or MLN following footpad delivery and initiate mucosal immune induction following migration to known mucosal inductive tissues as has been proposed following both transcutaneous immunization (56) and Ag delivery in the presence of vitamin D3 (57). However, a series of experiments have failed to experimentally support such an hypothesis. Analysis of PP, MLN, and nasal-associated lymphoid tissue following peripheral delivery of GFP-VRP has failed to reproducibly detect VRP-infected cells in these tissues (J. M. Thompson, E. M. Richmond, and R. E. Johnston, unpublished observations). Additionally, Ag-specific Abs were not detected in PP homogenates from VRP-immunized animals under conditions where Ag-specific polymeric IgA was readily detectable in the DLN. Thus, it is unlikely that infection of mucosal lymphoid tissues represents the predominant mechanism of immune induction in the VRP system.
In summary, nonmucosal delivery of alphavirus replicon particles has been used as a model system to dissect the organization of an alternative pathway for mucosal immune induction. The draining peripheral lymph node appears to be the central component of this pathway, as the VRP-infected DLN produced J chain-containing, polymeric forms of IgA, increased mucosal cytokines, a population of B cells harboring a mucosal homing phenotype, and mucosal-like HEVs. Identification of the essential viral and immunological factors which regulate mucosal immune induction following nonmucosal VRP delivery may shed new light on an alternative pathway for mucosal immune induction. Moreover, such insights may allow robust stimulation of this alternative pathway, resulting in vaccines with protective efficacy against mucosal pathogens and consequently reduced morbidity/mortality associated with such infections.
Acknowledgments
We thank Nancy Davis for critical review of this manuscript and members of the Carolina Vaccine Institute for helpful discussions. We acknowledge Martha Collier for preparation of VRP used in these studies. We are grateful to Tomas Leanderson (Lund University) for providing the anti-mouse J chain mAb used in this study.
Footnotes
This work was supported by grants from the National Institutes of Allergy and Infectious Diseases/National Institutes of Health: P01-AI046023 (to R.E.J.), R01-AI051990 (to R.E.J.), U01-AI070976 (to R.E.J.), and T32-AI007419 (to J.M.T.).
Abbreviations used in this paper: MAdCAM-1, mucosal addressin cell adhesion molecule 1; DC, dendritic cell; DLN, draining lymph node; HA, hemagglutinin; HEV, high endothelial venule; I-Flu, inactivated influenza virus; MLN, mesenteric lymph node; VEE, Venezuelan equine encephalitis virus; VRP, VEE replicon particles; PP, Peyer's patch; PNAd, peripheral lymph node addressin; IP, immunoprecipitation; IU, infectious unit.
Disclosures J.M.T. and R.E.J. are listed inventors on a patent application related to the subject matter of this article. J.M.T. and R.E.J. are consultants and R.E.J. is the Executive Director of Global Vaccines, Inc., a not-for-profit company that holds a license to the technology described in this article.
References
- 1.Neutra MR, Kozlowski PA. Mucosal vaccines: the promise and the challenge. Nat Rev Immunol. 2006;6:148–158. doi: 10.1038/nri1777. [DOI] [PubMed] [Google Scholar]
- 2.McGhee JR, Czerkinsky C, Mestecky J. Mucosal vaccines: an overview. In: Ogra PL, Mestecky J, Lamm ME, Strober W, Bienenstock J, McGhee J, editors. Mucosal Immunology. 2nd. Academic; London: 1999. pp. 741–758. [Google Scholar]
- 3.Brandtzaeg P. Role of secretory antibodies in the defence against infections. Int J Med Microbiol. 2003;293:3–15. doi: 10.1078/1438-4221-00241. [DOI] [PubMed] [Google Scholar]
- 4.McGhee JR, Lamm ME, Strober W. Mucosal immune responses: an overview. In: Ogra PL, Mestecky J, Lamm ME, Strober W, Bienstock J, McGhee J, editors. Mucosal Immunology. 2nd. Academic; London: 1999. pp. 485–506. [Google Scholar]
- 5.Brandtzaeg P, Farstad IN, Haraldsen G. Regional specialization in the mucosal immune system: primed cells do not always home along the same track. Immunol Today. 1999;20:267–277. doi: 10.1016/s0167-5699(99)01468-1. [DOI] [PubMed] [Google Scholar]
- 6.Brandtzaeg P, Johansen FE. Mucosal B cells: phenotypic characteristics, transcriptional regulation, and homing properties. Immunol Rev. 2005;206:32–63. doi: 10.1111/j.0105-2896.2005.00283.x. [DOI] [PubMed] [Google Scholar]
- 7.Butcher EC. Lymphocyte homing and intestinal immunity. In: Ogra PL, Mestecky J, Lamm ME, Strober W, Bienenstock J, McGhee JR, editors. Mucosal Immunology. 2nd. Academic; London: 1999. pp. 507–522. [Google Scholar]
- 8.Arbones ML, Ord DC, Ley K, Ratech H, Maynard-Curry C, Otten G, Capon DJ, Tedder TF. Lymphocyte homing and leukocyte rolling and migration are impaired in L-selectin-deficient mice. Immunity. 1994;1:247–260. doi: 10.1016/1074-7613(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 9.Mestecky J, Moro I, Underdown BJ. Mucosal immunoglobulins. In: Ogra PL, Mestecky J, Lamm ME, Strober W, Bienenstock J, McGhee J, editors. Mucosal Immunology. 2nd. Academic; London: 1999. pp. 133–152. [Google Scholar]
- 10.Johansen FE, Braathen R, Brandtzaeg P. Role of J chain in secretory immunoglobulin formation. Scand J Immunol. 2000;52:240–248. doi: 10.1046/j.1365-3083.2000.00790.x. [DOI] [PubMed] [Google Scholar]
- 11.Mestecky J, Lue C, Russell MW. Selective transport of IgA: cellular and molecular aspects. Gastroenterol Clin North Am. 1991;20:441–471. [PubMed] [Google Scholar]
- 12.Bouvet JP, Decroix N, Pamonsinlapatham P. Stimulation of local antibody production: parenteral or mucosal vaccination? Trends Immunol. 2002;23:209–213. doi: 10.1016/s1471-4906(02)02186-5. [DOI] [PubMed] [Google Scholar]
- 13.Underdown BJ, Plotkin SA. The induction of mucosal protection by parenteral immunization. In: Ogra PL, Mestecky J, Lamm ME, Strober W, Bienenstock J, McGhee JR, editors. Mucosal Immunology. 2nd. Academic; London: 1999. pp. 719–728. [Google Scholar]
- 14.Coffin SE, Klinek M, Offit PA. Induction of virus-specific antibody production by lamina propria lymphocytes following intramuscular inoculation with rotavirus. J Infect Dis. 1995;172:874–878. doi: 10.1093/infdis/172.3.874. [DOI] [PubMed] [Google Scholar]
- 15.Coffin SE, Clark SL, Bos NA, Brubaker JO, Offit PA. Migration of antigen-presenting B cells from peripheral to mucosal lymphoid tissues may induce intestinal antigen-specific IgA following parenteral immunization. J Immunol. 1999;163:3064–3070. [PubMed] [Google Scholar]
- 16.Musey L, Ding Y, Elizaga M, Ha R, Celum C, McElrath MJ. HIV-1 vaccination administered intramuscularly can induce both systemic and mucosal T cell immunity in HIV-1-uninfected individuals. J Immunol. 2003;171:1094–1101. doi: 10.4049/jimmunol.171.2.1094. [DOI] [PubMed] [Google Scholar]
- 17.Charles PC, Brown KW, Davis NL, Hart MK, Johnston RE. Mucosal immunity induced by parenteral immunization with a live attenuated Venezuelan equine encephalitis virus vaccine candidate. Virology. 1997;228:153–160. doi: 10.1006/viro.1996.8381. [DOI] [PubMed] [Google Scholar]
- 18.Thompson JM, Whitmore AC, Konopka JL, Collier ML, Richmond EMB, Davis NL, Staats HF, Johnston RE. Mucosal and systemic adjuvant activity of alphavirus replicon particles. Proc Natl Acad Sci USA. 2006;103:3722–3727. doi: 10.1073/pnas.0600287103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.LoBue AD, Lindesmith L, Yount B, Harrington PR, Thompson JM, Johnston RE, Moe CL, Baric RS. Multivalent norovirus vaccines induce strong mucosal and systemic blocking antibodies against multiple strains. Vaccine. 2006;24:5220–5234. doi: 10.1016/j.vaccine.2006.03.080. [DOI] [PubMed] [Google Scholar]
- 20.Harrington PR, Yount B, Johnston RE, Davis N, Moe C, Baric RS. Systemic, mucosal, and heterotypic immune induction in mice inoculated with Venezuelan equine encephalitis replicons expressing Norwalk virus-like particles. J Virol. 2002;76:730–742. doi: 10.1128/JVI.76.2.730-742.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ogra PL, Kerr-Grant D, Umana G, Dzierba J, Weintraub D. Antibody response in serum and nasopharynx after naturally acquired and vaccine-induced infection with rubella virus. N Engl J Med. 1971;285:1333–1339. doi: 10.1056/NEJM197112092852401. [DOI] [PubMed] [Google Scholar]
- 22.Peters C, Peng X, Douven D, Pan ZK, Paterson Y. The induction of HIV Gag-specific CD8+ T cells in the spleen and gut-associated lymphoid tissue by parenteral or mucosal immunization with recombinant Listeria monocytogenes HIV Gag. J Immunol. 2003;170:5176–5187. doi: 10.4049/jimmunol.170.10.5176. [DOI] [PubMed] [Google Scholar]
- 23.Kauppi M, Eskola J, Kayhty H. Anti-capsular polysaccharide antibody concentrations in saliva after immunization with Haemophilus influenzae type b conjugate vaccines. Pediatr Infect Dis J. 1995;14:286–294. doi: 10.1097/00006454-199504000-00008. [DOI] [PubMed] [Google Scholar]
- 24.Egan MA, Chong SY, Hagen M, et al. A comparative evaluation of nasal and parenteral vaccine adjuvants to elicit systemic and mucosal HIV-1 peptide-specific humoral immune responses in cynomolgus macaques. Vaccine. 2004;22:3774–3788. doi: 10.1016/j.vaccine.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 25.Enioutina EY, Visic D, McGee ZA, Daynes RA. The induction of systemic and mucosal immune responses following the subcutaneous immunization of mature adult mice: characterization of the antibodies in mucosal secretions of animals immunized with antigen formulations containing a vitamin D3 adjuvant. Vaccine. 1999;17:3050–3064. doi: 10.1016/s0264-410x(99)00147-4. [DOI] [PubMed] [Google Scholar]
- 26.McKenzie BS, Corbett AJ, Johnson S, Brady JL, Pleasance J, Kramer DR, Boyle JS, Jackson DC, Strugnell RA, Lew AM. Bypassing luminal barriers, delivery to a gut addressin by parenteral targeting elicits local IgA responses. Intl Immunol. 2004;16:1613–1622. doi: 10.1093/intimm/dxh163. [DOI] [PubMed] [Google Scholar]
- 27.Glenn GM, Kenney RT, Ellingsworth LR, Frech SA, Hammond SA, Zoeteweij JP. Transcutaneous immunization and immunostimulant strategies: capitalizing on the immunocompetence of the skin. Expert Rev Vaccines. 2003;2:253–267. doi: 10.1586/14760584.2.2.253. [DOI] [PubMed] [Google Scholar]
- 28.Glenn GM, Taylor DN, Li X, Frankel S, Montemarano A, Alving CR. Transcutaneous immunization: a human vaccine delivery strategy using a patch. Nat Med. 2000;6:1403–1406. doi: 10.1038/82225. [DOI] [PubMed] [Google Scholar]
- 29.Yu J, Cassels F, Scharton-Kersten T, Hammond SA, Hartman A, Angov E, Corthesy B, Alving C, Glenn G. Transcutaneous immunization using colonization factor and heat-labile enterotoxin induces correlates of protective immunity for enterotoxigenic Escherichia coli. Infect Immun. 2002;70:1056–1068. doi: 10.1128/IAI.70.3.1056-1068.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lehner T, Bergmeier LA, Tao L, Panagiotidi C, Klavinskis LS, Hussain L, Ward RG, Meyers N, Adams SE, Gearing AJ. Targeted lymph node immunization with simian immunodeficiency virus p27 antigen to elicit genital, rectal, and urinary immune responses in nonhuman primates. J Immunol. 1994;153:1858–1868. [PubMed] [Google Scholar]
- 31.Kawabata S, Miller CJ, Lehner T, Fujihashi K, Kubota M, McGhee JR, Imaoka K, Hiroi T, Kiyono H. Induction of Th2 cytokine expression for p27-specific IgA B cell responses after targeted lymph node immunization with simian immunodeficiency virus antigens in rhesus macaques. J Infect Dis. 1998;177:26–33. doi: 10.1086/513811. [DOI] [PubMed] [Google Scholar]
- 32.Pushko P, Parker M, Ludwig GV, Davis NL, Johnston RE, Smith JF. Replicon-helper systems from attenuated Venezuelan equine encephalitis virus: expression of heterologous genes in vitro and immunization against heterologous pathogens in vivo. Virology. 1997;239:389–401. doi: 10.1006/viro.1997.8878. [DOI] [PubMed] [Google Scholar]
- 33.MacDonald GH, Johnston RE. Role of dendritic cell targeting in Venezuelan equine encephalitis virus pathogenesis. J Virol. 2000;74:914–922. doi: 10.1128/jvi.74.2.914-922.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frolov I, Hoffman TA, Pragai BM, Dryga SA, Huang HV, Schlesinger S, Rice CM. Alphavirus-based expression vectors: strategies and applications. Proc Nat Acad Sci. 1996;93:11371–11377. doi: 10.1073/pnas.93.21.11371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davis NL, Caley IJ, Brown KW, et al. Vaccination of macaques against pathogenic simian immunodeficiency virus with Venezuelan equine encephalitis virus replicon particles. J Virol. 2000;74:371–378. doi: 10.1128/jvi.74.1.371-378.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kallberg E, Leanderson T. A subset of dendritic cells express joining chain (J-chain) protein. Immunology. 2008;123:590–599. doi: 10.1111/j.1365-2567.2007.02733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao X, Deak E, Soderberg K, Linehan M, Spezzano D, Zhu J, Knipe DM, Iwasaki A. Vaginal submucosal dendritic cells, but not Langerhans cells, induce protective Th1 responses to herpes simplex virus-2. J Exp Med. 2003;197:153–162. doi: 10.1084/jem.20021109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soderberg KA, Linehan MM, Ruddle NH, Iwasaki A. MAdCAM-1 expressing sacral lymph node in the lymphotoxin β-deficient mouse provides a site for immune generation following vaginal herpes simplex virus-2 infection. J Immunol. 2004;173:1908–1913. doi: 10.4049/jimmunol.173.3.1908. [DOI] [PubMed] [Google Scholar]
- 39.Kerr MA. The structure and function of human IgA. Biochem J. 1990;271:285–296. doi: 10.1042/bj2710285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Monteiro RC, Van De Winkel JG. IgA Fc receptors. Annu Rev Immunol. 2003;21:177–204. doi: 10.1146/annurev.immunol.21.120601.141011. [DOI] [PubMed] [Google Scholar]
- 41.Streeter PR, Berg EL, Rouse BTN, Bargatze RF, Butcher EC. A tissue-specific endothelial cell molecule involved in lymphocyte homing. Nature. 1988;331:41–46. doi: 10.1038/331041a0. [DOI] [PubMed] [Google Scholar]
- 42.Elson CO, Dertzbaugh MT. Mucosal adjuvants. In: Ogra PL, Mestecky J, Lamm ME, Strober W, Bienenstock J, McGhee JR, editors. In Mucosal Immunology. 2nd. Academic; London: 1999. pp. 817–838. [Google Scholar]
- 43.Rappuoli R, Pizza M, Douce G, Dougan G. Structure and mucosal adjuvanticity of cholera and Escherichia coli heat-labile enterotoxins. Immunol Today. 1999;20:493–500. doi: 10.1016/s0167-5699(99)01523-6. [DOI] [PubMed] [Google Scholar]
- 44.Decroix N, Quan CP, Pamonsinlapatham P, Bouvet JP. Mucosal immunity induced by intramuscular administration of free peptides in-line with PADRE: IgA antibodies to the ELDKWA epitope of HIV gp41. Scand J Immunol. 2002;56:59–65. doi: 10.1046/j.1365-3083.2002.01113.x. [DOI] [PubMed] [Google Scholar]
- 45.Kaartinen M, Hurme M, Makela O. Evidence for oligomeric IgA production by peripheral rat lymph nodes. Nature. 1974;252:329–330. doi: 10.1038/252329a0. [DOI] [PubMed] [Google Scholar]
- 46.Daynes RA, Enioutina EY, Butler S, Mu HH, McGee ZA, Araneo BA. Induction of common mucosal immunity by hormonally immunomodulated peripheral immunization. Infect Immun. 1996;64:1100–1109. doi: 10.1128/iai.64.4.1100-1109.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lehner T, Wang Y, Cranage M, et al. Protective mucosal immunity elicited by targeted iliac lymph node immunization with a subunit SIV envelope and core vaccine in macaques. Nat Med. 1996;2:767–775. doi: 10.1038/nm0796-767. [DOI] [PubMed] [Google Scholar]
- 48.Mora JR, Iwata M, Eksteen B, et al. Generation of gut-homing IgA-secreting B Cells by intestinal dendritic cells. Science. 2006;314:1157–1160. doi: 10.1126/science.1132742. [DOI] [PubMed] [Google Scholar]
- 49.Spalding DM, Williamson SI, Koopman WJ, McGhee JR. Preferential induction of polyclonal IgA secretion by murine Peyer's patch dendritic cell-T cell mixtures. J Exp Med. 1984;160:941–946. doi: 10.1084/jem.160.3.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dudda JC, Lembo A, Bachtanian E, Huehn J, Siewert C, Hamann A, Kremmer E, Förster R, Martin SF. Dendritic cells govern induction and reprogramming of polarized tissue-selective homing receptor patterns of T cells: important roles for soluble factors and tissue microenvironments. Eur J Immunol. 2005;35:1056–1065. doi: 10.1002/eji.200425817. [DOI] [PubMed] [Google Scholar]
- 51.Johansson-Lindbom B, Svensson M, Wurbel MA, Malissen B, Márquez G, Agace W. Selective generation of gut tropic T cells in gut-associated lymphoid tissue (GALT): requirement for GALT dendritic cells and adjuvant. J Exp Med. 2003;198:963–969. doi: 10.1084/jem.20031244. epub September 8, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sato A, Iwasaki A. Intestinal epithelial barrier and mucosal immunity. Cell Mol Life Sci. 2005;62:1333–1338. doi: 10.1007/s00018-005-5037-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Iwasaki A. Mucosal dendritic cells. Annu Rev Immunol. 2007;25:381–418. doi: 10.1146/annurev.immunol.25.022106.141634. [DOI] [PubMed] [Google Scholar]
- 54.Mora JR, Bono MR, Manjunath N, Weninger W, Cavanagh LL, Rosemblatt M, von Andrian UH. Selective imprinting of gut-homing T cells by Peyer's patch dendritic cells. Nature. 2003;424:88–93. doi: 10.1038/nature01726. [DOI] [PubMed] [Google Scholar]
- 55.Sato A, Hashiguchi M, Toda E, Iwasaki A, Hachimura S, Kaminogawa S. CD11b+ Peyer's patch dendritic cells secrete IL-6 and induce IgA secretion from naive B cells. J Immunol. 2003;171:3684–3690. doi: 10.4049/jimmunol.171.7.3684. [DOI] [PubMed] [Google Scholar]
- 56.Belyakov IM, Hammond SA, Ahlers JD, Glenn GM, Berzofsky JA. Transcutaneous immunization induces mucosal CTLs and protective immunity by migration of primed skin dendritic cells. J Clin Invest. 2004;113:998–1007. doi: 10.1172/JCI20261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Enioutina EY, Bareyan D, Daynes RA. Vitamin D3-mediated alterations to myeloid dendritic cell trafficking in vivo expand the scope of their antigen presenting properties. Vaccine. 2007;25:1236–1249. doi: 10.1016/j.vaccine.2006.10.008. [DOI] [PubMed] [Google Scholar]