

Abstract
Vitamin B12-dependent enzymes function in core biochemical pathways in Mycobacterium tuberculosis, an obligate pathogen whose metabolism in vivo is poorly understood. Although M. tuberculosis can access vitamin B12 in vitro, it is uncertain whether the organism is able to scavenge B12 during host infection. This question is crucial to predictions of metabolic function, but its resolution is complicated by the absence in the M. tuberculosis genome of a direct homologue of BtuFCD, the only bacterial B12 transport system described to date. We applied genome-wide transposon mutagenesis to identify M. tuberculosis mutants defective in their ability to use exogenous B12. A small proportion of these mapped to Rv1314c, identifying the putative PduO-type ATP : co(I)rrinoid adenosyltransferase as essential for B12 assimilation. Most notably, however, insertions in Rv1819c dominated the mutant pool, revealing an unexpected function in B12 acquisition for an ATP-binding cassette (ABC)-type protein previously investigated as the mycobacterial BacA homologue. Moreover, targeted deletion of Rv1819c eliminated the ability of M. tuberculosis to transport B12 and related corrinoids in vitro. Our results establish an alternative to the canonical BtuCD-type system for B12 uptake in M. tuberculosis, and elucidate a role in B12 metabolism for an ABC protein implicated in chronic mycobacterial infection.
Keywords: tuberculosis, vitamin B12, corrinoids, BtuFCD, BacA
2. Introduction
The genome of Mycobacterium tuberculosis, obligate human pathogen and causative agent of tuberculosis, encodes three B12-dependent enzymes. Previous work in our laboratory has established that both the methylmalonyl-coenzyme A (CoA) mutase, MutAB [1], and the metH-encoded methionine synthase [2] are functional, and require B12 for activity. Mycobacterium tuberculosis also possesses a predicted pathway for B12 biosynthesis [3], but appears not to produce the cofactor in vitro [1,2] or in macrophages [4]. Nevertheless, the bacillus can use exogenous vitamin B12 and encodes a B12-responsive riboswitch that suppresses transcription of the alternative, B12-independent methionine synthase, metE, in B12-replete conditions [2]. These observations imply a role for the cofactor in M. tuberculosis pathogenesis. However, it is uncertain whether B12 is available during infection, and which mycobacterial genes are required for its uptake and assimilation.
Vitamin B12 and B12 derivatives are members of the cobalamin group of corrinoid macrocycles [5]. Cobalamins are structurally complex, comprising a defining tetrapyrrole framework with a centrally chelated cobalt ion held in place by a lower axial base, dimethylbenzimidazole and an upper ligand that determines the cofactor form (figure 1). The cyano group in vitamin B12 (cyanocobalamin, CNCbl) must be replaced by deoxyadenosine and methyl ligands, respectively, during conversion to the biologically active cofactors: adenosylcobalamin (AdoCbl or coenzyme B12), which is required by methylmalonyl-CoA mutase, and methylcobalamin (MeCbl), which serves as an intermediary in the synthesis of methionine from homocysteine and methyltetrahydrofolate [6]. The reactivity of B12 cofactors derives from the cobalt-coordinated organic ligands [7] and, together with the size of the cobalamin core, underlies the need for multi-component systems to mediate controlled translocation and delivery of B12 across the cell membrane to its target enzyme [8].
Figure 1.
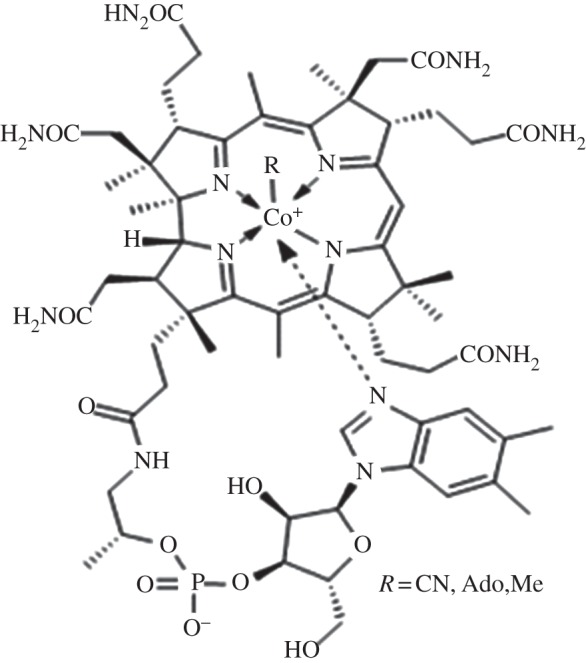
Structure of vitamin B12 and B12-derived cofactors.
Although bioinformatic analyses have predicted alternative vitamin transporters [9], BtuCD–BtuF remains the only confirmed bacterial B12 transport system identified to date [10]. The Escherichia coli model is the best characterized: a high-affinity corrinoid transporter, BtuB, operates with the TonB–ExbBD complex to traffic B12 across the outer membrane into the periplasm [11] where it is captured by the btuF-encoded B12-binding protein and delivered to the ATP-binding cassette (ABC) importer, BtuCD, which spans the cytoplasmic membrane [12]. Mycobacterium tuberculosis is characterized by a notoriously complex cell envelope comprising a cytoplasmic membrane and an external cell wall [13]. However, despite its demonstrated ability to use exogenous B12 [2,4], the proteins involved in mycobacterial B12 transport and assimilation are unknown: M. tuberculosis is included in the small number of B12-using bacteria that lack a candidate BtuFCD-type B12 transport system [3,9,14] as well as an identifiable homologue of TonB [15].
In this study, we used random mutagenesis to identify genes whose disruption abrogated the ability of M. tuberculosis to use exogenous vitamin B12 in vitro. Our results establish an essential role in B12 uptake for Rv1819c, a predicted ABC protein implicated in chronic infection in vivo [16], thereby revealing an alternative to the well-characterized BtuCD system for B12 transport.
3. Material and methods
3.1. Bacterial strains and growth conditions
Strains, plasmids and oligonucleotides are described in the electronic supplementary material, table S1. Mycobacterium tuberculosis was grown on Middlebrook 7H10 (Difco) supplemented with 0.5 per cent glycerol and Middlebrook OADC enrichment (Difco) or in Middlebrook 7H9 supplemented with 0.2 per cent glycerol, Middlebrook OADC and 0.05 per cent Tween 80 or 0.05 per cent tyloxapol, as required. For propionate utilization experiments, 7H9 broth was supplemented with 0.5 per cent bovine serum albumin fraction V (Sigma), 0.085 per cent NaCl and 0.1 per cent (w/v) sodium propionate, as described [1]. Hygromycin (hyg), kanamycin (kan) and gentamicin (gent) were used at 50, 25 and 2.5 μg ml−1, respectively, CNCbl and AdoCbl at 10 µg ml−1, (CN)2Cbi at 1 µM and 3-nitropropionate (3NP) at 0.1 mM.
3.2. Construction of transposon mutant library
A library of transposon (Tn) mutants was constructed in M. tuberculosis H37Rv ΔmetH, using the MycoMarT7 phage as described [17]. For the primary screen, transductants were plated across multiple 7H10 plates containing 20 µg ml−1 kan and 10 µg ml−1 CNCbl at a density of 20 000 colony forming units (CFU) per plate. The secondary screen was performed in duplicate in microtitre plate format and, for each Tn mutant, comprised four parallel wells containing 0.1 per cent propionate plus 20 µg ml−1 kan as base medium in each well: the first well constituted a growth control and contained only the base medium; in well 2, 10 µg ml−1 CNCbl was added to the base medium; in well 3, the base medium was supplemented with 0.1 mM 3NP; and in well 4, 0.1 mM 3NP and 10 µg ml−1 CNCbl were added.
3.3. Identification of transposon insertion sites
A combination of Tn-linker [18] and rescue cloning [19] strategies was applied to identify Tn insertion sites using the oligonucleotides in the electronic supplementary material, table S1.
3.4. Construction of mutant strains of Mycobacterium tuberculosis
Mycobacterium tuberculosis mutants were constructed using suicide plasmids described in electronic supplementary material, table S1. Genetic complementation used tweety-based vectors [20].
3.5. DNA sequencing
Mycobacterium tuberculosis genomic DNA was sequenced using an Illumina GenomeAnalyzer II, as described previously [21].
3.6. Homology modelling
The initial detection of crystal structures related to Rv1819c was performed using HHsearch [22] and COMA [23]. The Rv1819c model was then generated using a previously described iterative approach [24,25]. Briefly, both the set of structural templates and corresponding alignments were refined until the resulting model stopped improving and the visual inspection revealed no significant flaws.
4. Results
4.1. A forward genetic screen identifies B12 uptake mutants
We showed previously that deletion of the B12-dependent methionine synthase, MetH, renders M. tuberculosis sensitive to vitamin B12 during growth on solid medium [2]. This phenotype depends on the function of a B12 riboswitch that is located immediately upstream of metE, the gene encoding an alternative, B12-independent methionine synthase in M. tuberculosis. In wild-type M. tuberculosis, exogenous B12 suppresses transcription of metE by binding to the riboswitch [2], possibly ensuring efficient B12-dependent methionine synthesis by MetH. In the metH deletion mutant, however, riboswitch-mediated suppression of metE in response to B12 effectively results in the complete shutdown of methionine synthase activity, thereby eliminating production of an essential amino acid and so inhibiting bacillary growth [2]. This effect is most profoundly manifest on solid medium, where exposure to 10 µg ml−1 CNCbl results in a 3log10-fold reduction in viable CFU of ΔmetH knockout mutants [2]. Here, we exploited the observed B12 sensitivity of metH mutants in a genetic screen designed to elucidate a potential B12 transport system in M. tuberculosis (figure 2). To this end, we constructed an unmarked metH knockout of the laboratory strain, M. tuberculosis H37RvJO [21] (electronic supplementary material, figure S1a) and confirmed that it phenocopied the previously described hygromycin (hyg)-marked ΔmetH (BB) deletion mutant [2] during growth on B12-containing solid medium (see the electronic supplementary material, figure S1b). The unmarked ΔmetH knockout was used as background strain in which to generate a Tn mutant library using the MycoMarT7 phage [17] that carries a kan resistance marker and inserts randomly at TA dinucleotides [19]. In the primary screen, the library of insertion mutants was plated on solid medium containing kan and CNCbl to enable the identification of genes whose disruption alleviated the growth defect of the metH mutant (figure 2a). In total, 612 individual clones were isolated, each of which was picked and regrown in standard liquid medium; of these, 35 grew poorly or not at all and were eliminated, leaving 577 ‘B12-resistant’ insertion mutants for further analysis.
Figure 2.

Identification of genes required for B12 transport and assimilation. (a) Schematic of the screening cascade. The ΔmetH Tn library was plated on selective medium containing 10 µg ml−1 CNCbl. 612 ‘B12-resistant’ clones were isolated and regrown in standard liquid medium, eliminating 35 mutants owing to poor (n = 14) or absent (n = 21) growth. A secondary screen tested the B12 uptake ability of the remaining 577 insertion mutants in a four-well microtitre assay using 0.1% propionate (Prop) plus 20 µg ml−1 kanamycin as base medium (well 1) supplemented with 10 µg ml−1 CNCbl (well 2), 3NP (well 3) and 3NP plus 10 µg ml−1 CNCbl (well 4). A total of 84 mutants failed to grow in well 4, suggesting impaired B12 uptake. Each determination was performed in duplicate, and the results confirmed in batch culture. (b) Insertion mutants with disrupted B12 uptake ability.
Previously, in characterizing the ΔmetH (BB) mutant, we noted the high frequency at which suppressor mutants arose spontaneously on B12-containing solid medium, with single-nucleotide polymorphisms (SNPs) in the B12 riboswitch located upstream of metE accounting for approximately 10–20 per cent of these [2]. In the current screen, we used dual selection on kan and CNCbl in order to limit the potentially confounding effects of spontaneous riboswitch mutations: according to these criteria, growth on CNCbl plus kan would require successful transduction with the kan-resistant Tn as well as disruption—spontaneous or Tn-mediated—of B12-dependent growth inhibition. Nevertheless, we predicted that a significant proportion of B12-resistant mutants might contain Tn insertions in the riboswitch motif. So, in order to minimize the impact of disruptions to the B12 riboswitch, we applied a secondary screen (figure 2a) to determine the capacity of the insertion mutants to assimilate exogenous CNCbl for growth in liquid medium containing propionate in the presence of 3NP, an inhibitor of the key methylcitrate cycle enzyme, isocitrate lyase [26]. Two prior observations informed the design of this screen: (i) the inhibitory effect of genetic (ΔprpDC) or chemical (3NP) abrogation of methylcitrate cycle enzymes during growth in liquid medium containing propionate can be alleviated by supplementing the culture with CNCbl, thereby enabling M. tuberculosis to use propionate as a carbon source via the methylmalonyl pathway that includes the B12-dependent methylmalonyl-CoA mutase, MutAB [1]; (ii) for reasons that are not clear, B12-mediated growth inhibition is less effective in liquid versus solid medium—that is, the ΔmetH mutant can grow in B12-supplemented liquid medium (see the electronic supplementary material, figure S1c).
The secondary screen therefore assessed the ability of all 577 Tn mutants to use exogenous CNCbl for growth in liquid medium containing propionate in the presence of 3NP (figure 2a). The majority of Tn mutants (n = 493) phenocopied the parental ΔmetH strain in this assay, and were eliminated as candidate B12 uptake mutants. In contrast, the remaining 84 Tn mutants were unable to grow in well 4, suggesting impaired ability to use exogenous B12 for methylmalonyl pathway-dependent propionate catabolism. To verify these results, 43 of the 84 mutants were selected at random for phenotypic confirmation of disrupted B12 uptake in batch culture (data not shown) and on B12-containing solid medium (see the electronic supplementary material, figure S2a).
4.2. Disruption of Rv1819c eliminates the ability of Mycobacterium tuberculosis to use exogenous B12 in vitro
Insertions in Rv1819c accounted for 72 of the 84 Tn mutants (figure 2b; electronic supplementary material, figure S2a–c) and, moreover, mapped throughout the 1920 bp gene (electronic supplementary material, figure S2d). This result strongly suggested a role in B12 uptake for a predicted ABC transport protein previously identified as the putative M. tuberculosis homologue of BacA [16,27], a protein of cryptic function implicated in chronic infection in multiple host–pathogen models [25]. In their study of M. tuberculosis Rv1819c, Domenech et al. [16] constructed a deletion mutant by allelic exchange mutagenesis (see the electronic supplementary material, figure S2b). We assessed the ability of this mutant—referred to as ΔbacA::hyg by Domenech et al. [16]—to use exogenous B12 for MutAB-dependent growth in propionate (electronic supplementary material, figure S3a). Consistent with the inferred role of Rv1819c in B12 uptake, the ΔbacA::hyg strain grew very poorly in propionate plus 3NP supplemented with B12, reproducing the phenotype of the ΔmetH Rv1819c::Tn mutants (figure 3). By contrast, the complemented derivative carrying a full-length copy of Rv1819c at the attB site, referred to as ΔbacA::pKLMt5 in the original study [16], was able to use B12 for growth (see the electronic supplementary material, figure S3a). Similarly, integration of full-length Rv1819c at attB restored the B12-sensitive phenotype of a randomly selected ΔmetH Rv1819c::Tn mutant during growth on solid medium supplemented with CNCbl (see the electronic supplementary material, figure S3b), and reversed the inability of the same mutant to use B12 for growth in propionate-containing liquid medium supplemented with 3NP (figure 3), confirming the essentiality of Rv1819c in this assay.
Figure 3.
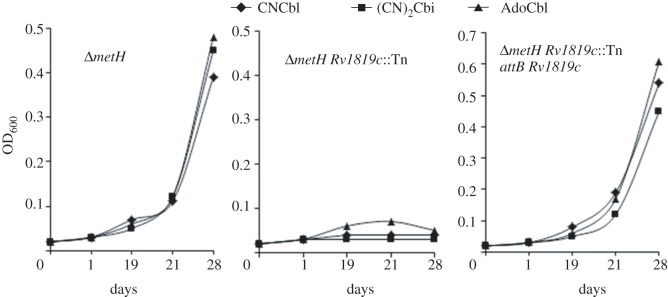
Disruption of Rv1819c eliminates the ability of M. tuberculosis ΔmetH to use corrinoids for growth in 0.1% propionate-containing 3NP. Data are from a representative experiment performed in duplicate.
It was noticeable in the propionate utilization experiment (see the electronic supplementary material, figure S3a) that the ΔbacA::hyg mutant started to replicate after two to three weeks of apparent growth arrest, possibly indicating the emergence of suppressor mutants. To circumvent this complication, we deleted the prpDC locus [28] in this strain, thereby negating the need to use 3NP to eliminate methylcitrate pathway function [1]. In contrast to the single prpDC deletion mutant, the double ΔbacA::hyg ΔprpDC knockout exhibited no growth at all in propionate over the 28-day time course (see the electronic supplementary material, figure S3c), even when supplemented with CNCbl, strongly suggesting that Rv1819c is required for the assimilation of exogenous B12 to enable methylmalonyl-CoA pathway function.
4.3. Spontaneous B12-resistant mutants carrying non-synonymous single-nucleotide polymorphisms in Rv1819c
We reported previously that SNPs in the metE-associated B12 riboswitch accounted for 10–20 per cent of all B12-resistant mutants isolated after plating the ΔmetH (BB) knockout on medium containing CNCbl, whereas the remaining B12-resistance mutations were unknown [2]. To investigate the possibility that mutations in Rv1819c might account for B12 resistance in those clones lacking riboswitch mutations, we plated the ΔmetH (BB) strain on medium containing CNCbl and sequenced the riboswitch region and Rv1819c locus in 10 spontaneous B12-resistant mutants. Consistent with previous results [2], two isolates carried independent mutations in the highly conserved B12-box motif within the metE riboswitch [29], namely C → T transversions at positions −155 and −163 relative to the metE start codon, respectively. Notably, four other B12-resistant mutants had wild-type riboswitch sequences, but contained non-synonymous SNPs in Rv1819c (see the electronic supplementary material, table S2), supporting the inferred role of Rv1819c in B12 uptake. To eliminate the possibility that an additional, unidentified mutation (or mutations) might account for the observed phenotype, we sequenced the genome of a representative Rv1819c point mutant, SP09 (see the electronic supplementary material, table S2). The parental, B12-sensitive strain, ΔmetH (BB), was differentiated from the laboratory strain, H37RvJO [21], only in the targeted deletion of metH sequence. Moreover, the Rv1819c mutation constituted the sole polymorphism separating SP09 from its ΔmetH (BB) parent and, importantly, complementation with wild-type Rv1819c at the attB locus restored B12 sensitivity to both SP09 and SP18 (see the electronic supplementary material, figure S4).
In the primary Tn screen (figure 2a), ‘B12-resistant’ mutants had been selected on kan and CNCbl in order to limit the potentially confounding effects of spontaneous riboswitch mutations. To verify the utility of this approach, we analysed the insertion sites in a random selection of 20 of the 493 ΔmetH Tn mutants subsequently eliminated in the secondary screen owing to their inability to use exogenous B12 for growth in propionate. All 20 mutants contained insertions in the B12 riboswitch region directly upstream of metE (data not shown), confirming that disrupted riboswitch function represents a major mechanism for loss of B12 regulation in strains which carry an intact Rv1819c gene.
4.4. Rv1819c is essential for corrinoid transport in Mycobacterium tuberculosis
Mycobacterium tuberculosis is predicted to encode a complete pathway for B12 biosynthesis, including enzymes required for the conversion of the B12 precursor, cobinamide, to AdoCbl through the addition of dimethylbenzimidazole and deoxyadenosine ligands [3]. The E. coli corrinoid transporter, BtuFCD, mediates uptake of cobinamide as well as CNCbl and AdoCbl [30], suggesting that Rv1819c might fulfil a corresponding role in M. tuberculosis. In support of this idea, cobinamide—provided as the dicyanide salt, (CN)2Cbi—was unable to complement the growth defect of ΔmetH Rv1819c::Tn mutants in propionate in the presence of 3NP, mimicking similar observations with AdoCbl and CNCbl (figure 3). Insertions in Rv1819c also alleviated the growth inhibitory effect of AdoCbl, CNCbl and (CN)2Cbi on the metH knockout mutant on solid medium, a phenotype that was reversed upon complementation with wild-type Rv1819c (see the electronic supplementary material, figure S5). In combination, these results confirmed the essentiality of Rv1819c for corrinoid transport in M. tuberculosis.
4.5. Impaired vitamin B12 uptake in spontaneous bleomycin-resistant Rv1819c mutants
Domenech et al. [16] showed that deletion of Rv1819c decreased the susceptibility of M. tuberculosis to the glycopeptide antibiotic, bleomycin, a phenotype commonly associated with BacA function [31–33]. We determined the minimum inhibitory concentration (MIC) of bleomycin against a selected Rv1819c::Tn mutant (electronic supplementary material, figure S6a) as well as the spontaneous B12-resistant mutants, SP09 and SP18 (see the electronic supplementary material, figure S6b), and observed values comparable to that reported for ΔbacA::hyg [16]. To explore further the overlap between B12 uptake and bleomycin susceptibility, we isolated spontaneous bleomycin-resistant (BleoR) mutants in two different genetic backgrounds, ΔprpDC and the unmarked metH knockout, by plating the strains on solid medium containing 3 µg ml−1 bleomycin (10 × MIC). Five BleoR mutants each of the ΔprpDC and ΔmetH knockouts were selected at random, and shown to be defective in their ability to use B12 for MutAB-dependent growth in propionate (see the electronic supplementary material, figure S7a). Moreover, the spontaneous BleoR mutants of ΔmetH were resistant to CNCbl during growth on solid medium (see the electronic supplementary material, figure S7b). All five BleoR mutants derived from the ΔprpDC strain carried nonsense mutations in Rv1819c, whereas missense mutations in Rv1819c were identified in four of five spontaneous BleoR ΔmetH mutants (see the electronic supplementary material, table S2). Moreover, complementation with full-length Rv1819c reversed the inability of the spontaneous Rv1819c point mutants of ΔprpDC to catabolize propionate in liquid medium supplemented with CNCbl (figure 4), and restored the bleomycin susceptibility of SP09 to wild-type levels (see the electronic supplementary material, figure S6c).
Figure 4.
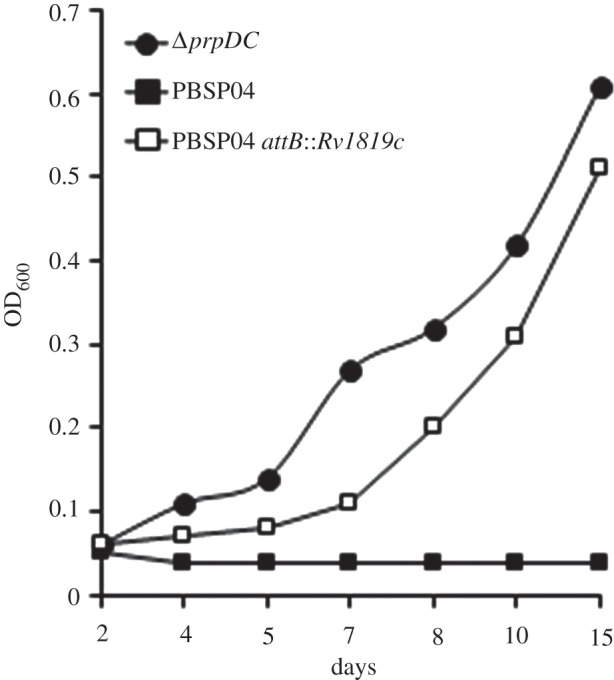
Impaired B12 uptake in a spontaneous bleomycin-resistant (BleoR) prpDC mutant, PBSP04, carrying a SNP in Rv1819c (see the electronic supplementary material, table S2). Strains were grown in 0.1% propionate supplemented with 3NP and CNCbl. Data are from a representative experiment performed in duplicate.
4.6. Rv1819c encodes an ATP-binding cassette-type transporter
Rv1819c was previously included in a group of ‘BacA-related’ proteins identified on the basis of their similarity to the highly conserved BacA and SbmA proteins of Sinorhizobium and E. coli, respectively [27]. Unlike BacA/SbmA orthologues, however, which are predicted to require an interaction with a separate cytoplasmic protein for function, Rv1819c encodes both transmembrane (TMD) and nucleotide-binding (NBD) domains of an ABC transport protein on a single polypeptide. Sequence similarity analyses using only the TMD located M. tuberculosis Rv1819c in a cluster distinct from BacA/SbmA (see the electronic supplementary material, figure S8a). Moreover, these analyses indicated that Rv1819c was more closely related to ABC proteins other than BacA/SbmA in both E. coli and Sinorhizobium, namely YddA [34] and ExsE [35], respectively. The Rv1819c NBD similarly identified YddA and ExsE as close homologues in an equivalent similarity search (see the electronic supplementary material, figure S8b), together with the recently described human ABC-type B12 transporter, ABCD4 [36].
We built a homology model of Rv1819c based on the crystal structures of two polyspecific ABC exporters, Staphylococcus aureus Sav1866 [37] and Salmonella typhimurium MsbA [38]. Consistent with known ABC protein architecture [39], Rv1819c is predicted to form a homodimer (figure 5), with each subunit comprising an N-terminal TMD fused to a highly conserved NBD that features all the motifs characteristic of functional ABC transporters (see the electronic supplementary material, figure S9a). Unlike Sav1866 and MsbA, though, the TMD domain of Rv1819c possesses an extra N-terminal region which is predicted to contain an additional transmembrane helix (see the electronic supplementary material, figure S9b). Proteomic analyses in the closely related M. bovis BCG suggest that this region is present in the mature protein [40], and therefore is not a signal peptide. However, in the absence of a close structural template containing seven transmembrane helices, we omitted the first 65 N-terminal residues in building the Rv1819c model. The predicted structure nevertheless provides a useful framework for the interpretation of experimental data. Notably, all three SNPs which resulted in substituted amino acids in the spontaneous B12-resistant and BleoR mutants (see the electronic supplementary material, table S2) affect residues located in conserved regions of Rv1819c (figure 5). While the structural consequences of the P349T and G411D mutations require further investigation, L442S affects a conserved position in the putative nucleotide-binding pocket formed by two interacting ABC domains. In Sav1866, the corresponding residue, Ile356, makes a van der Waals contact with the sugar moiety of the bound ADP [37], and so supports the inferred association between a distorted pocket and crippled protein function.
Figure 5.
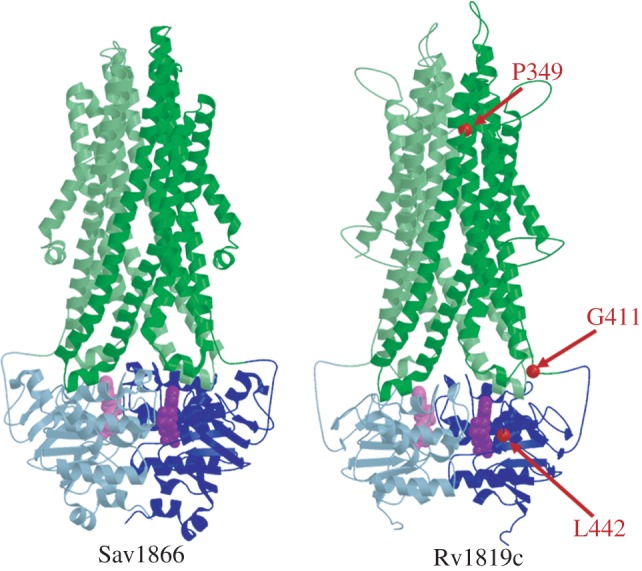
Rv1819c encodes an ABC-type transporter. Computational model of M. tuberculosis Rv1819c (amino acid residues 66–639) compared with the x-ray structure of Staphylococcus aureus Sav1866 (PDB code: 2HYD) [37]. The model is based on the crystal structures of Sav1866 and the ABC lipid flippase, MsbA, from Salmonella typhimurium (PDB code: 3B60) [38], both of which contain transmembrane- (green) and nucleotide-binding (blue) domains fused into a single polypeptide chain that interacts to form a homodimer in the active protein. The two subunits in both structures are denoted by the different colour intensities. ADP molecules bound to each subunit are shown in purple and light purple, respectively. Residues substituted in spontaneous Rv1819c mutants are indicated with red arrows.
4.7. A PduO-type adenosyltransferase is required for assimilation of vitamin B12
CNCbl must be adenosylated to generate the active cofactor, AdoCbl [41] (figure 6a). The genome of M. tuberculosis is predicted to encode both CobO (Rv2849c) and PduO (Rv1314c) ATP:co(I)rrinoid adenosyltransferases [3], non-homologous enzymes that catalyse this reaction in other bacteria [42,43]. It was notable, therefore, that six putative B12 uptake mutants (figure 2) contained Tn insertions in pduO (see the electronic supplementary material, figure S2d), because this suggested that an impaired ability to convert exogenous CNCbl to the cofactor form could confer B12 resistance in the primary screen, as well as eliminate the ability of M. tuberculosis to use B12 for growth in propionate. To confirm the role of PduO-dependent adenosylation in these phenotypes, we evaluated the abilities of the ΔmetH pduO::Tn mutants to assimilate different corrinoids for growth in propionate in the presence of 3NP (figure 6b). The mutants were unable to use either cyano form, CNCbl or (CN)2Cbi, both of which are adenosylated in the biosynthetic pathway to AdoCbl (figure 6a). In contrast, supplementation with AdoCbl itself restored growth in this assay (figure 6b), suggesting bypass of PduO function. In combination, these observations implicate PduO as sole active adenosyltransferase in M. tuberculosis during growth in vitro.
Figure 6.
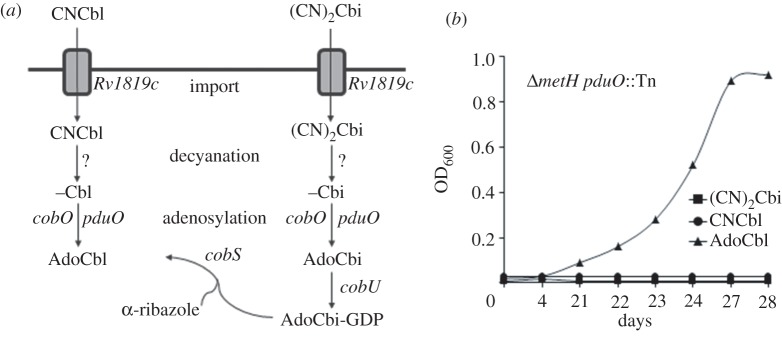
PduO is essential for B12 salvage and assimilation. (a) Predicted steps in late-stage AdoCbl biosynthesis and salvage in M. tuberculosis. Cobinamide is provided in vitro as a dicyanide salt, (CN)2Cbi. (b) The ΔmetH pduO::Tn mutant cannot use CNCbl or (CN)2Cbi for growth in 0.1% propionate-containing 3NP. Data are from a representative experiment performed in duplicate.
5. Discussion
Our results identify Rv1819c as sole corrinoid transporter in M. tuberculosis under standard in vitro conditions and, moreover, establish the capacity of the organism to scavenge corrinoids. The association of Rv1819c with B12 uptake is unexpected, particularly given previous studies suggesting that Rv1819c might function in ATP-dependent peptide transport [16,44]. Moreover, the properties enabling polyspecific translocation of compounds such as bleomycin, B12 and antimicrobial peptides which lack obvious structural similarity remain unclear [45]. In Gram-positive organisms, ABC-mediated importers function together with a high-affinity substrate-binding protein (SBP) that is anchored to the extracytoplasmic membrane [46]. Although M. tuberculosis possesses in excess of 30 ABC transporters, as well as 15 putative SBPs [47], we failed to identify a candidate B12-binding protein, raising the possibility that Rv1819c-mediated B12 import occurs in the absence of a specific SBP or that multiple proteins fulfil this role [48]. In most bacteria, the components of the ABC transporters involved in the uptake of ferric siderophores, haem and vitamin B12 are closely related [49]. However, our screen identified Rv1819c as sole transport candidate, excluding the possibility that other ABC proteins might perform overlapping functions in mycobacterial B12 transport, at least under in vitro conditions. Instead, in associating Rv1819c with B12 uptake, our results add to the expanding complement of atypical mycobacterial nutrient acquisition systems. For example, a novel pathway was recently elucidated that enables the scavenging of haem [50]—a tetrapyrrole which, like B12, is derived from δ-aminolaevulinic acid via a uroporphyrinogen III intermediate [5]. In that system, uptake is mediated by the combined activity of a haem-binding protein and the MmpL family members MmpL3 and MmpL11—predicted RND-type efflux pumps which have been associated with multiple cellular functions [51]. In addition to haem import, recent evidence suggests that MmpL3 fulfils an essential role in exporting trehalose monomycolate across the cell membrane for incorporation into cell wall mycolic acids [52,53], and it has also been implicated in the susceptibility of M. tuberculosis to diverse small molecules [54,55]. It is tempting, therefore, to consider the analogy with Rv1819c—itself a predicted export protein that has now been implicated in the uptake of antimicrobial peptides [16,44] and vitamin B12, and might also play a role in cell wall biogenesis [16].
Rv1819c has been extensively investigated as M. tuberculosis BacA [16,44]. Unlike BacA/SbmA orthologues, however, deletion of Rv1819c does not render M. tuberculosis hypersusceptible to other antimicrobial drugs and cell disrupting agents [16]. Moreover, our structural model of M. tuberculosis Rv1819c predicts an ABC transporter comprising both TMD and NBD within a single polypeptide. This distinguishes the mycobacterial protein from BacA proteins in Brucella and other intracellular pathogens [32] that contain the TMD only and, importantly, is supported by sequence analyses that situate Rv1819c in a separate cluster from the BacA subfamily even when based on TMD sequence alone. The M. tuberculosis protein also differs from BacA proteins in its potential role in pathogenesis. While the essentiality of the mycobacterial protein for the maintenance of chronic infection in vivo [16] is reminiscent of BacA-like phenotype, closer inspection of the comparative in vivo infection dynamics of different ‘bacA’ mutants suggests divergent function: for example, in contrast to the Brucella and Sinorhizobium deletion mutants [27,32], the M. tuberculosis Rv1819c knockout is not impaired in its ability to establish an infection [16]. It is tempting, therefore, to consider the virulence defect of the Rv1819c deletion mutant in the light of recent studies describing the accumulation during chronic infection of cholesterol-rich lipid bodies inside foamy macrophages and their infecting bacilli [56,57]. That is, Rv1819c might function to ensure adequate supply of host-derived corrinoids for the B12-dependent utilization of propionate derived from cholesterol catabolism, a possibility that requires further investigation.
Although designed to detect a putative vitamin B12 transporter, our screen also established the essentiality of the PduO-type adenosyltransferase for the assimilation of exogenous corrinoids. The M. tuberculosis genome contains both pduO and cobO adenosyltransferases; therefore, the inferred inactivity of the alternative enzyme in vitro might indicate functional adaptation of CobO to de novo B12 biosynthesis [3], or to specific environmental conditions, including anaerobiosis [41]. Intracellular trafficking of B12 in humans requires the sequential activity of multiple proteins which fulfil dual roles as molecular chaperones and in the enzymatic modification of the cofactor [58]. For example, MMACHC catalyses the reductive decyanation of CNCbl [59] while mediating LMBD1-dependent [60] transfer from the lysosome into the cytoplasm. Recent evidence further suggests that this process is facilitated by the interaction of LMBD1 with ABCD4 [36]—an ABC transporter and homologue of Rv1819c (see the electronic supplementary material, figure S8). In a subsequent step, the ATP:corrinoid adenosyltransferase attaches the axial ligand and ensures delivery of the resulting AdoCbl cofactor across the mitochondrial membrane to its target enzyme, methylmalonyl-CoA mutase [61]. Given that Tn-mediated disruption of pduO alleviated the B12 sensitivity of the metH mutant in the primary screen, it is tempting to speculate that PduO might function not merely in enzymatic conversion of exogenous corrinoids, but also in delivery of the active cofactor into the cytoplasm. Our current model for the translocation of B12 across the mycobacterial cell wall into the cytoplasm therefore proposes the sequential activity of the ABC transporter, Rv1819c and the PduO-type adenosyltransferase (figure 6a).
Our Tn screen also identified four low-frequency insertions associated with compromised B12 uptake (figure 2b). Targeted sequencing of Rv1819c and the metE riboswitch in these strains excluded spontaneous mutations as the underlying cause of the observed B12 phenotypes. A single mutant carried an insertion in rpfB, which encodes a resuscitation-promoting factor. To explore this result further, we retested ΔmetH rpfB::Tn in parallel with an rpfB deletion mutant of H37Rv, constructed previously [62]. Although the Tn mutant was not able to use exogenous CNCbl for growth in propionate-containing medium, the ΔrpfB knockout strain phenocopied wild-type H37Rv in this assay (data not shown), thereby excluding a role for RpfB in B12 uptake. It is possible that rpfB::Tn possesses an additional, unidentified polymorphism, affecting B12 assimilation; alternatively, polar effects on the downstream gene, ksgA, encoding dimethyladenosine transferase, might contribute to the observed phenotype [63], a possibility under investigation. Two additional Tn insertions mapped to mymA, encoding a putative flavin-dependent monooxygenase. The predicted role of MymA in the maintenance of cell wall ultrastructure [64] suggests that compromised B12 uptake in these mutants might be non-specific; however, this requires further investigation, and is complicated by the fact that mymA is the first gene in a seven-gene operon [65]. We also isolated a mutA::Tn mutant, whose inability to use propionate for growth in B12-containing medium is consistent with impaired methylmalonyl-CoA mutase function. The basis for the B12 resistance of this mutant in the primary screen is unclear, however, and probably also the result of an additional spontaneous mutation. The final Tn insertion mapped to Rv2927c, a gene which previous saturation mutagenesis studies have predicted as essential for growth of M. tuberculosis in vitro [66,67]. Although the function of Rv2927c is unknown, it has been proposed to operate as part of the cell division machinery [68]. It seems probable that, like the mymA::Tn mutants, the failure of Rv2927c::Tn to assimilate B12 is non-specific. However, given the inferred requirement for PduO-dependent adenosylation in the assimilation of exogenous B12, the prediction that Rv2927c might function in de novo adenosine nucleotide biosynthesis [69] is intriguing, and the subject of current investigation.
6. Acknowledgements
This work was financially supported by a Swiss–South African Joint Research Programme grant (to V.M. and J.D.M), and by grants from the South African Medical Research Council (to V.M.), the National Research Foundation (to V.M.) and the Howard Hughes Medical Institute (International Research Scholar's grants to V.M. and Č.V.). We thank Eric Rubin and Chris Sassetti for providing the MycoMar phage and for valuable advice, Clif Barry and Helena Boshoff for insightful discussions and for the ΔbacA::hyg knockout mutant, and Candice Soares De Melo for technical assistance.
Supplementary Material
References
- 1.Savvi S, Warner DF, Kana BD, McKinney JD, Mizrahi V, Dawes SS. 2008. Functional characterization of a vitamin B12-dependent methylmalonyl pathway in Mycobacterium tuberculosis: implications for propionate metabolism during growth on fatty acids. J. Bacteriol. 190, 3886–3895 10.1128/JB.01767-07 (doi:10.1128/JB.01767-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Warner DF, Savvi S, Mizrahi V, Dawes SS. 2007. A riboswitch regulates expression of the coenzyme B12-independent methionine synthase in Mycobacterium tuberculosis: implications for differential methionine synthase function in strains H37Rv and CDC1551. J. Bacteriol. 189, 3655–3659 10.1128/JB.00040-07 (doi:10.1128/JB.00040-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rodionov DA, Vitreschak AG, Mironov AA, Gelfand MS. 2003. Comparative genomics of the vitamin B12 metabolism and regulation in prokaryotes. J. Biol. Chem. 278, 41 148–41 159 10.1074/jbc.M305837200 (doi:10.1074/jbc.M305837200) [DOI] [PubMed] [Google Scholar]
- 4.Griffin JE, Pandey AK, Gilmore SA, Mizrahi V, McKinney JD, Bertozzi CR, Sassetti CM. 2012. Cholesterol catabolism by Mycobacterium tuberculosis requires transcriptional and metabolic adaptations. Chem. Biol. 19, 218–227 10.1016/j.chembiol.2011.12.016 (doi:10.1016/j.chembiol.2011.12.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Warren MJ, Raux E, Schubert HL, Escalante-Semerena JC. 2002. The biosynthesis of adenosylcobalamin (vitamin B12). Nat. Prod. Rep. 19, 390–412 10.1039/b108967f (doi:10.1039/b108967f) [DOI] [PubMed] [Google Scholar]
- 6.Banerjee R, Ragsdale SW. 2003. The many faces of vitamin B12: catalysis by cobalamin-dependent enzymes. Annu. Rev. Biochem. 72, 209–247 10.1146/annurev.biochem.72.121801.161828 (doi:10.1146/annurev.biochem.72.121801.161828) [DOI] [PubMed] [Google Scholar]
- 7.Krautler B. 2005. Vitamin B12: chemistry and biochemistry. Biochem. Soc. Trans. 33, 806–810 10.1042/BST0330806 (doi:10.1042/BST0330806) [DOI] [PubMed] [Google Scholar]
- 8.Nielsen MJ, Rasmussen MR, Andersen CB, Nexo E, Moestrup SK. 2012. Vitamin B12 transport from food to the body's cells—a sophisticated, multistep pathway. Nat. Rev. Gastroenterol. Hepatol. 9, 345–354 10.1038/nrgastro.2012.76 (doi:10.1038/nrgastro.2012.76) [DOI] [PubMed] [Google Scholar]
- 9.Rodionov DA, et al. 2009. A novel class of modular transporters for vitamins in prokaryotes. J. Bacteriol. 191, 42–51 10.1128/JB.01208-08 (doi:10.1128/JB.01208-08) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korkhov VM, Mireku SA, Locher KP. 2012. Structure of AMP-PNP-bound vitamin B12 transporter BtuCD–F. Nature 490, 367–372 10.1038/nature11442 (doi:10.1038/nature11442) [DOI] [PubMed] [Google Scholar]
- 11.Noinaj N, Guillier M, Barnard TJ, Buchanan SK. 2010. TonB-dependent transporters: regulation, structure, and function. Annu. Rev. Microbiol. 64, 43–60 10.1146/annurev.micro.112408.134247 (doi:10.1146/annurev.micro.112408.134247) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lewinson O, Lee AT, Locher KP, Rees DC. 2010. A distinct mechanism for the ABC transporter BtuCD–BtuF revealed by the dynamics of complex formation. Nat. Struct. Mol. Biol. 17, 332–338 10.1038/nsmb.1770 (doi:10.1038/nsmb.1770) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brennan PJ, Nikaido H. 1995. The envelope of mycobacteria. Annu. Rev. Biochem. 64, 29–63 10.1146/annurev.bi.64.070195.000333 (doi:10.1146/annurev.bi.64.070195.000333) [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y, Rodionov DA, Gelfand MS, Gladyshev VN. 2009. Comparative genomic analyses of nickel, cobalt and vitamin B12 utilization. BMC Genomics 10, 78. 10.1186/1471-2164-10-78 (doi:10.1186/1471-2164-10-78) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mirus O, Strauss S, Nicolaisen K, von Haeseler A, Schleiff E. 2009. TonB-dependent transporters and their occurrence in cyanobacteria. BMC Biol. 7, 68. 10.1186/1741-7007-7-68 (doi:10.1186/1741-7007-7-68) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Domenech P, Kobayashi H, LeVier K, Walker GC, Barry CE., 3rd 2009. BacA, an ABC transporter involved in maintenance of chronic murine infections with Mycobacterium tuberculosis. J. Bacteriol. 191, 477–485 10.1128/JB.01132-08 (doi:10.1128/JB.01132-08) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sassetti CM, Boyd DH, Rubin EJ. 2001. Comprehensive identification of conditionally essential genes in mycobacteria. Proc. Natl Acad. Sci. USA 98, 12 712–12 717 10.1073/pnas.231275498 (doi:10.1073/pnas.231275498) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kwon YM, Ricke SC. 2000. Efficient amplification of multiple transposon-flanking sequences. J. Microbiol. Methods 41, 195–199 10.1016/S0167-7012(00)00159-7 (doi:10.1016/S0167-7012(00)00159-7) [DOI] [PubMed] [Google Scholar]
- 19.Rubin EJ, Akerley BJ, Novik VN, Lampe DJ, Husson RN, Mekalanos JJ. 1999. In vivo transposition of mariner-based elements in enteric bacteria and mycobacteria. Proc. Natl Acad. Sci. USA 96, 1645–1650 10.1073/pnas.96.4.1645 (doi:10.1073/pnas.96.4.1645) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pham TT, Jacobs-Sera D, Pedulla ML, Hendrix RW, Hatfull GF. 2007. Comparative genomic analysis of mycobacteriophage Tweety: evolutionary insights and construction of compatible site-specific integration vectors for mycobacteria. Microbiology 153, 2711–2723 10.1099/mic.0.2007/008904-0 (doi:10.1099/mic.0.2007/008904-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ioerger TR, et al. 2010. Variation among genome sequences of H37Rv strains of Mycobacterium tuberculosis from multiple laboratories. J. Bacteriol. 192, 3645–3653 10.1128/JB.00166-10 (doi:10.1128/JB.00166-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Söding J. 2005. Protein homology detection by HMM–HMM comparison. Bioinformatics 21, 951–960 10.1093/bioinformatics/bti125 (doi:10.1093/bioinformatics/bti125) [DOI] [PubMed] [Google Scholar]
- 23.Margelevičius M, Venclovas Č. 2010. Detection of distant evolutionary relationships between protein families using theory of sequence profile–profile comparison. BMC Bioinform. 11, 89. 10.1186/1471-2105-11-89 (doi:10.1186/1471-2105-11-89) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Venclovas Č, Margelevičius M. 2009. The use of automatic tools and human expertise in template-based modeling of CASP8 target proteins. Proteins 77(Suppl 9), 81–88 10.1002/prot.22515 (doi:10.1002/prot.22515) [DOI] [PubMed] [Google Scholar]
- 25.Warner DF, Ndwandwe DE, Abrahams GL, Kana BD, Machowski EE, Venclovas C, Mizrahi V. 2010. Essential roles for imuA‘- and imuB-encoded accessory factors in DnaE2-dependent mutagenesis in Mycobacterium tuberculosis. Proc. Natl Acad. Sci USA 107, 13 093–13 098 10.1073/pnas.1002614107 (doi:10.1073/pnas.1002614107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Honer Zu, Bentrup K, Miczak A, Swenson DL, Russell DG. 1999. Characterization of activity and expression of isocitrate lyase in Mycobacterium avium and Mycobacterium tuberculosis. J. Bacteriol. 181, 7161–7167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.LeVier K, Walker GC. 2001. Genetic analysis of the Sinorhizobium meliloti BacA protein: differential effects of mutations on phenotypes. J. Bacteriol. 183, 6444–6453 10.1128/JB.183.21.6444-6453.2001 (doi:10.1128/JB.183.21.6444-6453.2001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muñoz-Elías EJ, Upton AM, Cherian J, McKinney JD. 2006. Role of the methylcitrate cycle in Mycobacterium tuberculosis metabolism, intracellular growth, and virulence. Mol. Microbiol. 60, 1109–1122 10.1111/j.1365-2958.2006.05155.x (doi:10.1111/j.1365-2958.2006.05155.x) [DOI] [PubMed] [Google Scholar]
- 29.Vitreschak AG, Rodionov DA, Mironov AA, Gelfand MS. 2003. Regulation of the vitamin B12 metabolism and transport in bacteria by a conserved RNA structural element. RNA 9, 1084–1097 10.1261/rna.5710303 (doi:10.1261/rna.5710303) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bradbeer C, Kenley JS, Di Masi DR, Leighton M. 1978. Transport of vitamin B12 in Escherichia coli. Corrinoid specificities of the periplasmic B12-binding protein and of energy-dependent B12 transport. J. Biol. Chem. 253, 1347–1352 [PubMed] [Google Scholar]
- 31.Ichige A, Walker GC. 1997. Genetic analysis of the Rhizobium meliloti bacA gene: functional interchangeability with the Escherichia coli sbmA gene and phenotypes of mutants. J. Bacteriol. 179, 209–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.LeVier K, Phillips RW, Grippe VK, Roop RM, 2nd, Walker GC. 2000. Similar requirements of a plant symbiont and a mammalian pathogen for prolonged intracellular survival. Science 287, 2492–2493 10.1126/science.287.5462.2492 (doi:10.1126/science.287.5462.2492) [DOI] [PubMed] [Google Scholar]
- 33.Yorgey P, Lee J, Kordel J, Vivas E, Warner P, Jebaratnam D, Kolter R. 1994. Posttranslational modifications in microcin B17 define an additional class of DNA gyrase inhibitor. Proc. Natl Acad. Sci. USA 91, 4519–4523 10.1073/pnas.91.10.4519 (doi:10.1073/pnas.91.10.4519) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Linton KJ, Higgins CF. 1998. The Escherichia coli ATP-binding cassette (ABC) proteins. Mol. Microbiol. 28, 5–13 10.1046/j.1365-2958.1998.00764.x (doi:10.1046/j.1365-2958.1998.00764.x) [DOI] [PubMed] [Google Scholar]
- 35.Gibson KE, Campbell GR, Lloret J, Walker GC. 2006. CbrA is a stationary-phase regulator of cell surface physiology and legume symbiosis in Sinorhizobium meliloti. J. Bacteriol. 188, 4508–4521 10.1128/JB.01923-05 (doi:10.1128/JB.01923-05) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coelho D, et al. 2012. Mutations in ABCD4 cause a new inborn error of vitamin B12 metabolism. Nat. Genet. 44, 1152–1155 10.1038/ng.2386 (doi:10.1038/ng.2386) [DOI] [PubMed] [Google Scholar]
- 37.Dawson RJ, Locher KP. 2006. Structure of a bacterial multidrug ABC transporter. Nature. 443, 180–185 10.1038/nature05155 (doi:10.1038/nature05155) [DOI] [PubMed] [Google Scholar]
- 38.Ward A, Reyes CL, Yu J, Roth CB, Chang G. 2007. Flexibility in the ABC transporter MsbA: alternating access with a twist. Proc. Natl Acad. Sci. USA 104, 19 005–19 010 10.1073/pnas.0709388104 (doi:10.1073/pnas.0709388104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rees DC, Johnson E, Lewinson O. 2009. ABC transporters: the power to change. Nat. Rev. Mol. Cell Biol. 10, 218–227 10.1038/nrm2646 (doi:10.1038/nrm2646) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zheng J, Liu L, Wei C, Leng W, Yang J, Li W, Wang J, Jin Q. 2012. A comprehensive proteomic analysis of Mycobacterium bovis bacillus Calmette-Guerin using high resolution Fourier transform mass spectrometry. J. Proteomics 77, 357–371 10.1016/j.jprot.2012.09.010 (doi:10.1016/j.jprot.2012.09.010) [DOI] [PubMed] [Google Scholar]
- 41.Sheppard DE, Penrod JT, Bobik T, Kofoid E, Roth JR. 2004. Evidence that a B12-adenosyl transferase is encoded within the ethanolamine operon of Salmonella enterica. J. Bacteriol. 186, 7635–7644 10.1128/JB.186.22.7635-7644.2004 (doi:10.1128/JB.186.22.7635-7644.2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Debussche L, Couder M, Thibaut D, Cameron B, Crouzet J, Blanche F. 1991. Purification and partial characterization of Cob(I)alamin adenosyltransferase from Pseudomonas denitrificans. J. Bacteriol. 173, 6300–6302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johnson CL, Pechonick E, Park SD, Havemann GD, Leal NA, Bobik TA. 2001. Functional genomic, biochemical, and genetic characterization of the Salmonella pduO gene, an ATP:cob(I)alamin adenosyltransferase gene. J. Bacteriol. 183, 1577–1584 10.1128/JB.183.5.1577-1584.2001 (doi:10.1128/JB.183.5.1577-1584.2001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arnold MF, et al. 2013. Partial complementation of Sinorhizobium meliloti bacA mutant phenotypes by the Mycobacterium tuberculosis BacA protein. J. Bacteriol. 195, 389–398 10.1128/JB.01445-12 (doi:10.1128/JB.01445-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mattiuzzo M, Bandiera A, Gennaro R, Benincasa M, Pacor S, Antcheva N, Scocchi M. 2007. Role of the Escherichia coli SbmA in the antimicrobial activity of proline-rich peptides. Mol. Microbiol. 66, 151–163 10.1111/j.1365-2958.2007.05903.x (doi:10.1111/j.1365-2958.2007.05903.x) [DOI] [PubMed] [Google Scholar]
- 46.Ames GF. 1986. Bacterial periplasmic transport systems: structure, mechanism, and evolution. Annu. Rev. Biochem. 55, 397–425 10.1146/annurev.bi.55.070186.002145 (doi:10.1146/annurev.bi.55.070186.002145) [DOI] [PubMed] [Google Scholar]
- 47.Braibant M, Gilot P, Content J. 2000. The ATP binding cassette (ABC) transport systems of Mycobacterium tuberculosis. FEMS Microbiol. Rev. 24, 449–467 10.1111/j.1574-6976.2000.tb00550.x (doi:10.1111/j.1574-6976.2000.tb00550.x) [DOI] [PubMed] [Google Scholar]
- 48.Berntsson RP, Ter Beek J, Majsnerowska M, Duurkens RH, Puri P, Poolman B, Slotboom DJ. 2012. Structural divergence of paralogous S components from ECF-type ABC transporters. Proc. Natl Acad. Sci. USA 109, 13 990–13 995 10.1073/pnas.1203219109 (doi:10.1073/pnas.1203219109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koster W. 2001. ABC transporter-mediated uptake of iron, siderophores, heme and vitamin B12. Res. Microbiol. 152, 291–301 10.1016/S0923-2508(01)01200-1 (doi:10.1016/S0923-2508(01)01200-1) [DOI] [PubMed] [Google Scholar]
- 50.Tullius MV, et al. 2011. Discovery and characterization of a unique mycobacterial heme acquisition system. Proc. Natl Acad. Sci. USA 108, 5051–5056 10.1073/pnas.1009516108 (doi:10.1073/pnas.1009516108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Domenech P, Reed MB, Barry CE., 3rd 2005. Contribution of the Mycobacterium tuberculosis MmpL protein family to virulence and drug resistance. Infect Immun. 73, 3492–3501 10.1128/IAI.73.6.3492-3501.2005 (doi:10.1128/IAI.73.6.3492-3501.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tahlan K, et al. 2012. SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 56, 1797–1809 10.1128/AAC.05708-11 (doi:10.1128/AAC.05708-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grzegorzewicz AE, et al. 2012. Inhibition of mycolic acid transport across the Mycobacterium tuberculosis plasma membrane. Nat. Chem. Biol. 8, 334–341 10.1038/nchembio.794 (doi:10.1038/nchembio.794) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stanley SA, et al. 2012. Identification of novel inhibitors of M. tuberculosis growth using whole cell based high-throughput screening. ACS Chem. Biol. 7, 1377–1384 10.1021/cb300151m (doi:10.1021/cb300151m) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.La Rosa V, et al. 2012. MmpL3 is the cellular target of the antitubercular pyrrole derivative BM212. Antimicrob. Agents Chemother. 56, 324–331 10.1128/AAC.05270-11 (doi:10.1128/AAC.05270-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cáceres N, Tapia G, Ojanguren I, Altare F, Gil O, Pinto S, Vilaplana C, Cardona PJ. 2009. Evolution of foamy macrophages in the pulmonary granulomas of experimental tuberculosis models. Tuberculosis (Edinb) 89, 175–182 10.1016/j.tube.2008.11.001 (doi:10.1016/j.tube.2008.11.001) [DOI] [PubMed] [Google Scholar]
- 57.Peyron P, et al. 2008. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog. 4, e1000204. 10.1371/journal.ppat.1000204 (doi:10.1371/journal.ppat.1000204) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Banerjee R, Gherasim C, Padovani D. 2009. The tinker, tailor, soldier in intracellular B12 trafficking. Curr. Opin. Chem. Biol. 13, 484–491 10.1016/j.cbpa.2009.07.007 (doi:10.1016/j.cbpa.2009.07.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim J, Gherasim C, Banerjee R. 2008. Decyanation of vitamin B12 by a trafficking chaperone. Proc. Natl Acad. Sci. USA 105, 14 551–14 554 10.1073/pnas.0805989105 (doi:10.1073/pnas.0805989105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rutsch F, et al. 2009. Identification of a putative lysosomal cobalamin exporter altered in the cblF defect of vitamin B12 metabolism. Nat. Genet. 41, 234–239 10.1038/ng.294 (doi:10.1038/ng.294) [DOI] [PubMed] [Google Scholar]
- 61.Padovani D, Labunska T, Palfey BA, Ballou DP, Banerjee R. 2008. Adenosyltransferase tailors and delivers coenzyme B12. Nat. Chem. Biol. 4, 194–196 10.1038/nchembio.67 (doi:10.1038/nchembio.67) [DOI] [PubMed] [Google Scholar]
- 62.Downing KJ, Betts JC, Young DI, McAdam RA, Kelly F, Young M, Mizrahi V. 2004. Global expression profiling of strains harbouring null mutations reveals that the five rpf-like genes of Mycobacterium tuberculosis show functional redundancy. Tuberculosis (Edinb) 84, 167–179 10.1016/j.tube.2003.12.004 (doi:10.1016/j.tube.2003.12.004) [DOI] [PubMed] [Google Scholar]
- 63.Tufariello JM, Jacobs WR, Jr, Chan J. 2004. Individual Mycobacterium tuberculosis resuscitation-promoting factor homologues are dispensable for growth in vitro and in vivo. Infect. Immun. 72, 515–526 10.1128/IAI.72.1.515-526.2004 (doi:10.1128/IAI.72.1.515-526.2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Singh A, Gupta R, Vishwakarma RA, Narayanan PR, Paramasivan CN, Ramanathan VD, Tyagi AK. 2005. Requirement of the mymA operon for appropriate cell wall ultrastructure and persistence of Mycobacterium tuberculosis in the spleens of guinea pigs. J. Bacteriol. 187, 4173–4186 10.1128/JB.187.12.4173-4186.2005 (doi:10.1128/JB.187.12.4173-4186.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Singh A, Jain S, Gupta S, Das T, Tyagi AK. 2003. mymA operon of Mycobacterium tuberculosis: its regulation and importance in the cell envelope. FEMS Microbiol. Lett. 227, 53–63 10.1016/S0378-1097(03)00648-7 (doi:10.1016/S0378-1097(03)00648-7) [DOI] [PubMed] [Google Scholar]
- 66.Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. 2011. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 7, e1002251. 10.1371/journal.ppat.1002251 (doi:10.1371/journal.ppat.1002251) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sassetti CM, Boyd DH, Rubin EJ. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48, 77–84 10.1046/j.1365-2958.2003.03425.x (doi:10.1046/j.1365-2958.2003.03425.x) [DOI] [PubMed] [Google Scholar]
- 68.Flores AR, Parsons LM, Pavelka MS., Jr 2005. Characterization of novel Mycobacterium tuberculosis and Mycobacterium smegmatis mutants hypersusceptible to beta-lactam antibiotics. J. Bacteriol. 187, 1892–1900 10.1128/JB.187.6.1892-1900.2005 (doi:10.1128/JB.187.6.1892-1900.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Galagan JE, et al. 2010. TB database 2010: overview and update. Tuberculosis (Edinb.) 90, 225–235 10.1016/j.tube.2010.03.010 (doi:10.1016/j.tube.2010.03.010) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.