

Abstract
Physiological changes in pregnancy, including changes in body composition and metabolic enzyme activity, can alter drug pharmacokinetics. A semi-mechanistic metabolism model was developed to describe the pharmacokinetics of two cytochrome P450 3A (CYP3A) substrates, midazolam and nifedipine, in obstetrics patients. The model parameters were optimized to fit the data of oral midazolam pharmacokinetics in pregnant women, by increasing CYP3A-induced hepatic metabolism 1.6-fold in the model with no change in gut wall metabolism. Fetal metabolism had a negligible effect on maternal plasma drug concentrations. Validation of the model was performed by applying changes in volume of distribution and metabolism, consistent with those observed for midazolam, to the pharmacokinetics parameters of immediate-release nifedipine in healthy volunteers. The predicted steady-state areas under the concentration–time curve (AUCs) for nifedipine were within 15% of the data observed in pregnant women undergoing treatment for preterm labor. This model predicts the pharmacokinetics of two CYP3A substrates in pregnancy, and may be applicable to other CYP3A substrates as well.
The results of database surveys in developed countries show that 44–93% of pregnant women take at least one prescription medication other than vitamins and iron.1 Pregnancy leads to a number of physiologic changes that alter drug metabolism and disposition. Despite the impact of pregnancy on pharmacokinetics, this population is underrepresented in and often excluded from clinical trials.2,3 Therefore, for many drugs, the pharmacokinetics and effects in pregnant women are largely unknown. One means of overcoming the ethical and practical constraints associated with studying the effects of individual drugs in pregnant women is to use pharmacometric models to predict drug disposition at various stages of pregnancy.
Traditionally, physiology-based pharmacokinetic (PBPK) models incorporate physiologic parameters (such as blood flow and tissue volumes) with drug-specific parameters (such as physicochemical properties and in vitro metabolism data) to predict the concentration of drugs in various tissues. Such PBPK models of pregnancy have historically been used by toxicologists to predict the concentration of toxicants, including drugs, in various tissues.4,5 More recently, pharmacokinetic modeling has come to be accepted by regulatory agencies and pharmaceutical scientists as an important component of clinical pharmacology studies that address drug efficacy and drug–drug interactions.6
We have proposed a semi-mechanistic pharmacokinetic model of drug metabolism, based on standard one- or two-compartment models and incorporating both hepatic and intestinal metabolism.7,8,9 This approach uses compartmental pharmacokinetic parameters available from the literature or derived from clinical study data. Metabolic enzyme activity can be estimated from in vitro rate constants or from clinical data. This model has been validated for the cytochrome P450 3A (CYP3A) probe substrate, midazolam, using data from drug–drug interaction studies with ketoconazole, clarithromycin, diltiazem, and erythromycin in healthy volunteers.7,8,9
Here, we adapt this semi-mechanistic model to describe drug disposition in pregnancy, incorporating placental and fetal compartments and physiologic changes associated with pregnancy (Figure 1 and Table 1). The parameters of the model are optimized using midazolam clinical pharmacokinetic data in pregnancy;10 sensitivity analyses are performed to illustrate the impact of fetal metabolism, changes in hepatic blood flow, plasma protein binding, and CYP3A activity on plasma midazolam concentrations. Based on the adjusted parameter estimates determined for midazolam, the model was used for predicting the concentration levels of another CYP3A substrate, nifedipine, in pregnant women.
Figure 1.

Obstetric semi-mechanistic metabolism model. Central and peripheral volumes of distribution are derived from empirical two-compartment pharmacokinetics analyses. Clearances in gut wall, liver, and fetal liver (CLGW, CLH, CLfetal) are estimated by the well-stirred model. CLR, renal clearance; ka, absorption rate constant; QPV, portal vein blood flow; QH, hepatic blood flow; QHA, hepatic artery blood flow; Qplac placental blood flow; Qfet, fetal (umbilical vein) blood flow.
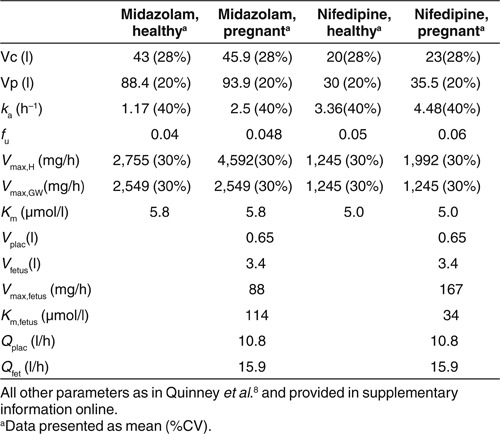
Table 1. Model parameter valuesa.
Results
Postpartum midazolam model
The healthy volunteer model for midazolam7,8,9 was used to simulate plasma midazolam concentrations after a 2-mg oral dose of the drug. Predicted plasma midazolam concentrations were similar to those observed in women 6–10 weeks postpartum (Figure 2a).10 Observed and predicted areas under the concentration–time curve (AUCs) from time 0 to ∞ 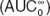 were 17.4 and 16.9 ng·h/ml, respectively (residual sum of squares, 20.8; percentage error, 3%).
were 17.4 and 16.9 ng·h/ml, respectively (residual sum of squares, 20.8; percentage error, 3%).
Figure 2.
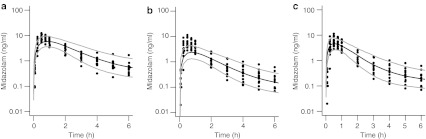
Predicted and observed midazolam concentrations. (a) Predictions of postpartum concentrations of midazolam by the healthy volunteer semi-mechanistic metabolism model. (b) Initial predictions by the obstetric model based on empiric changes assumed from the literature. (c) Visual predictive check of final obstetric midazolam model. Observed data are from women at 26–30 weeks of gestation.10 The participants received a single 2 mg dose of midazolam orally. Median and 5th and 95th prediction intervals are indicated by black and gray lines, respectively.
Midazolam obstetric model
The initial semi-mechanistic obstetrics model for midazolam metabolism was based on the model derived from data in healthy human volunteers, with the addition of placental and fetal compartments (Figure 1), a 20% increase in fraction unbound (fu),10 a twofold increase in intestinal and hepatic CYP3A clearance (Vmax),11 and an increased volume of distribution (Vd)12 based on reports in the literature. In comparison with observed midazolam data at 28–32 weeks' gestation,10 the model underestimated
(5.7 vs. 9.5 ng·h/ml) and maximal plasma concentration (Cmax) (2.1 vs. 6.4 ng/ml) (Figure 2b and Table 2).
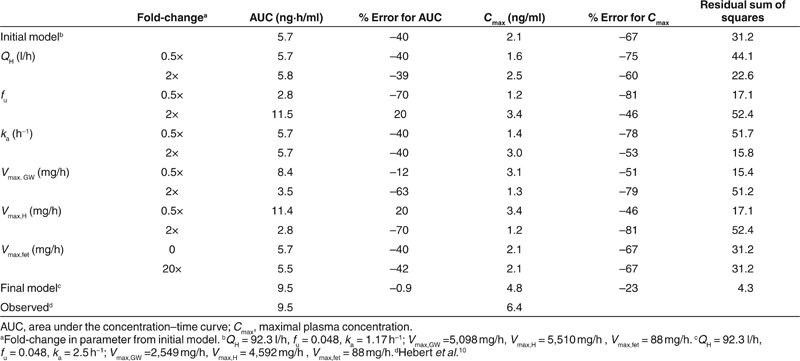
Table 2. Effect of varying model parameters on AUC, Cmax, and model fit (residual sum of squares).
Sensitivity analyses
In order to identify the impact of individual parameters on the midazolam concentration–time curve, select parameter values were varied 0.5- and 2-fold relative to the initial model (Figure 3 and Table 2). Changes in hepatic blood flow (QH) and the absorption rate constant (ka) had little effect on AUC, but a large effect on Cmax. A twofold change in fu had large effects on the plasma concentration–time curve. Similarly, reducing Vmax,GW or Vmax,H increased both the AUC and the Cmax of midazolam.
Figure 3.

Sensitivity analysis of obstetric semi-mechanistic metabolism model. The solid line indicates predicted maternal plasma drug concentration based on the initial obstetric semi-mechanistic metabolism model. Broken and dotted lines represent twofold decreases and increases, respectively, in (a) hepatic blood flow, QH; (b) absorption rate constant, ka; (c) fraction unbound, fu; (d) intestinal rate of metabolism, Vmax,GW; and (e) hepatic rate of metabolism, Vmax,H. (f) Effect of fetal metabolism on maternal plasma concentrations as predicted by the initial model (solid line), assuming no contribution from fetal metabolism (dotted line), and with a 20-fold increase in cytochrome P450 3A7 (CYP3A7) Vmax (dashed line).
The contribution of the fetus to maternal drug pharmacokinetics was also examined using sensitivity analysis. Fetal hepatic Vmax was estimated to be 88 mg/h, based on in vitro CYP3A7 metabolism of midazolam and estimated expression of CYP3A7 in the fetal liver.13,14 Completely excluding fetal clearance (Vmax,fet = 0) did not alter the maternal concentration–time course of midazolam. Even increasing the Vmax,fet by as much as 20-fold reduced midazolam AUC in maternal plasma only minimally (Table 1). Although fetal metabolism has little effect on maternal plasma drug concentration, it may impact fetal exposure. However, given the low intrinsic clearance of midazolam by the fetal liver, even a complete exclusion of fetal metabolism is predicted to increase fetal
of midazolam by only 2%. Additionally, changes in fetal blood flow (Quv and Qplac) did not significantly affect plasma concentrations (see Supplementary Figure S1 online). It therefore appears likely that the effect of fetal metabolism on maternal plasma midazolam concentration is minimal.
The volume of distribution of midazolam is expected to be higher in pregnancy because of increased plasma volume, increased extracellular volume, and decreased binding. We assumed that in pregnancy, the increase in the central volume of distribution is proportional to the change in plasma volume, and that peripheral volume of distribution accounts for additional increases in the total volume of distribution. Sensitivity analysis indicates that the maternal plasma drug concentration–time profile was not sensitive to changes in the volume of the peripheral compartment (see Supplementary Figure S2 online).
The original model, which anticipated that the effects of pregnancy on CYP3A Vmax would be similar in the gut wall and the liver, led to underestimation of Cmax. Based on results of the sensitivity analysis and iterative model fitting, the initial model of midazolam disposition in pregnancy was revised such that ka was increased to 2.5 h–1, Vmax,H was increased 1.6-fold, and Vmax,GW was left unchanged from the healthy volunteer model. This modified pregnancy model was able to accurately predict midazolam AUC within 1%, but continued to underestimate Cmax by 23% (Figure 2c).
Application to nifedipine
In order to validate the increased hepatic CYP3A activity associated with pregnancy, we applied our model to another CYP3A substrate, nifedipine. Using data from the literature, we constructed a model of nifedipine pharmacokinetics in healthy volunteers (Table 1).15,16 The two-compartment parameters and in vitro metabolism kinetics adequately predicted the pharmacokinetics of oral nifedipine (10 mg) in healthy volunteers (Figure 4a). The mean predicted AUC0–8 of nifedipine, 156 ng·h/ml (95% confidence interval of 149–163), was similar to the mean observed AUC0–8, 180 ng·h/ml (95% confidence interval of 150–210). Parameter adjustments were made as in the midazolam model (Table 1), and the steady-state concentration of nifedipine after oral administration of a 20 mg immediate-release capsule every 6 h was predicted in pregnant subjects (Figure 4b). As compared to observed data in subjects taking nifedipine for preterm labor,17 the median AUC0–6 was underpredicted by 11% (210 (121–299) ng·h/ml vs. 237 (224–253) ng·h/ml), and Cmax was underpredicted by 3% (178 (166–188) ng/ml vs. 184 (90–308) ng/ml).
Figure 4.

Model validation with nifedipine. (a) Plasma concentration–time curve for nifedipine in healthy volunteers after a single 10 mg oral dose. (b) Steady-state plasma concentration–time profile of nifedipine 10 mg given orally every 6 h to women at 24–34 weeks of gestation. Solid lines represent the 5th and 95th percentiles of the simulations (n = 1,000). The broken line represents the median of the simulations. Open and closed circles represent the observed nifedipine plasma concentrations of (a) nine studies in healthy volunteers, and (b) individual observations in pregnant women, respectively.
Discussion
Pharmacokinetic studies of the CYP3A substrates, midazolam and nifedipine, have recently been conducted in pregnant women (25–34 weeks of gestation).10,17 Both drugs are predominately metabolized by CYP3A, the intrinsic clearance (CLint) showing CYP3A4 > CYP3A5 > CYP3A7.13 Unlike many other CYP3A substrates, neither drug is significantly transported by p-glycoprotein.18,19 Clinical studies10,11 indicate that CYP3A activity increases in pregnancy. By incorporating various physiologic changes of pregnancy into a pharmacokinetic model, we attempted to isolate the effect of pregnancy on CYP3A activity.
PBPKs models have long been used to estimate fetal toxicity.5 However, relatively few models have been developed to estimate the pharmacokinetics of therapeutic drugs in pregnancy. The primary purpose of this model is to estimate changes in drug clearance and maternal pharmacokinetics of drugs. We have simplified the model to focus on important organs of drug metabolism, namely, intestine and liver. Although not impacting the pharmacokinetics of these CYP3A substrates, placental aromatase and (perhaps) fetal hepatic enzymes may play a role in the metabolism of other drugs (e.g., methadone as a substrate of CYP19). Therefore, we believe that separation of fetal and placental compartments may be relevant in the case of some drugs. This is similar to models proposed by Gabrielsson and Paalzow.5 We have additionally collapsed the PBPK model into two compartments: peripheral distribution and central distribution. This is in contrast to PBPK models proposed by Gaohua et al. and Andrews et al., which propose 13 and 20 maternal compartments, respectively.20,21 While our model may not fully describe pregnancy-related changes in drug distribution, this simplification allows us to focus on the roles of the relevant organs during this initial stage of model development.
Model development
The original semi-mechanistic drug metabolism model was developed based on midazolam pharmacokinetics in healthy volunteers taking part in drug–drug interaction studies.7,8,9 The healthy volunteer model incorporated the key organs involved in the absorption, metabolism, and distribution of oral drugs: intestinal lumen, intestinal wall, liver, portal circulation, and systemic circulation. Other physiologic compartments, which are not key determinants of drug clearance, were collapsed into the central and peripheral compartments. Compartments associated with drug metabolism are assumed to be part of the fast-equilibrating compartment, and their volumes are therefore deducted from that of the central compartment. Drug metabolism is described semi-mechanistically, based on the well-stirred model, enabling the interrogation of factors that may affect drug pharmacokinetics (e.g., blood flow, drug binding, and enzymatic activity). The original model was validated by its adequate prediction of plasma concentration–time curves for midazolam in postpartum settings (Figure 2a). It also indicates that, by 6–10 weeks postpartum, the factors controlling midazolam pharmacokinetics have largely returned to those expected in the absence of pregnancy.
In order to describe the pharmacokinetics of midazolam in pregnancy, the original model was altered to incorporate physiologic changes associated with pregnancy: additional placental and fetal compartments with metabolic potential, increased volume of distribution, decreased plasma protein binding, and increased CYP3A clearance. The initial model assumed that both intestinal and hepatic CYP3A activity increased twofold.11 However, it underestimated observed Cmax (Figure 2b). Several factors can affect Cmax, including bioavailability, absorption rate, volume of distribution, and drug clearance. Changes in systemic clearance may result from changes in hepatic blood flow, protein binding, or CYP3A intrinsic clearance. Using sensitivity analyses and knowledge of physiologic changes in pregnancy, we explored the impact of each of these factors on midazolam disposition.
Sensitivity analysis
Intestinal absorption of drugs may be affected in pregnancy by alterations in gastric motility and pH.22 Being a high-permeability drug, midazolam is nearly completely absorbed from the gastrointestinal tract. However, changes in motility can impact the rate of intestinal absorption (i.e., ka), altering both Cmax and time to reach Cmax (tmax). Sensitivity analyses indicated that increasing the ka of midazolam improved prediction by the obstetric model (Figure 3b).
There are some data, though limited, to suggest that pregnancy has little effect on hepatic blood flow.12 In addition, the sensitivity analysis results suggested that changes in hepatic blood flow would have little effect on the AUC of oral midazolam (Figure 3a). Therefore, it does not appear that changes in hepatic blood flow are responsible for the increased clearance of midazolam during pregnancy.
Midazolam is highly bound to albumin, and therefore even small changes in protein binding are capable of producing marked changes in the drug's systemic clearance. This is shown in the sensitivity analysis, where a decrease in fu from 0.048 to 0.024 has an effect on midazolam pharmacokinetics similar to that resulting from a twofold increase in hepatic Vmax (Figure 3 and Table 2). In pregnancy, albumin concentrations decrease by ~20%, with average albumin concentrations in the third trimester being 38.8 ± 2.9 g/l as compared to 47.7 ± 2.5 g/l in nonpregnant women.23 Consequently, the protein binding of drugs that bind primarily to albumin, such as midazolam and nifedipine, is expected to decrease, leading to increased clearance. However, the effect of protein binding cannot fully account for the twofold increase in clearance observed in pregnant women.
Given that changes in liver blood flow and protein binding do not completely account for increased midazolam clearance, it is possible that altered metabolism, either in the liver or fetal/placental unit, may play a role. The presence of CYP3A activity in the placenta is still a matter of debate. CYP3A4, CYP3A5, and CYP3A7 mRNA have been identified in preterm placenta, but the amounts are much lower than those found in the liver.24,25,26,27 However, only one study27 has observed CYP3A activity in placenta. It is unknown whether other placental enzymes (e.g., CYP19) are capable of midazolam metabolism. Because of the incompleteness of current knowledge, we did not consider placenta as a potential driver of midazolam clearance in our model. Fetal liver expresses CYP3A7 to a greater extent than CYP3A4 or CYP3A5.28 In vitro studies of midazolam with CYP3A isoforms,13 and knowledge of CYP3A7 expression in the fetal liver, provided the data for estimating the contribution of the fetal liver to midazolam disposition. As shown in Figure 3f, metabolism of midazolam in the fetal liver had a negligible effect on maternal plasma midazolam concentrations.
Consequently, it appears that the increased midazolam clearance in pregnancy is largely caused by increased hepatic CYP3A activity. Interestingly, even a twofold increase in CYP3A activity in the model, both in the gut wall and in the liver, led to an underestimation of Cmax and AUC. Assuming a 20% increase in fu, hepatic CYP3A activity appears to increase 1.6-fold in pregnancy. Independent regulation of CYP3A activity in the liver and intestines is consistent with results of studies in mice, which showed that hepatic CYP3a activity increased 2.2- and 2.5-fold on gestational days 15 and 19, respectively, but that no change was observed in intestinal CYP3a activity or expression.29 However, studies in the nonhuman primate Macaca nemestrina30 have not found increases in hepatic or intestinal CYP3A expression or midazolam clearance; the mechanism underlying the increase in hepatic CYP3A activity in humans is unknown.
Iterative analyses indicated that maintaining Vmax,GW at nonpregnancy values while increasing Vmax,H, fu, and ka from the initial healthy volunteer parameter estimates resulted in the best fit of the observed pregnancy data. In order to achieve a better fit of the absorption profile, ka was estimated to increase to 2.5 h–1. As this increase in absorption rate was unexpected, we further interrogated the postpartum data. Changing ka in the healthy volunteer model from 1.17 to 2.5 h–1 appeared equally capable of predicting the postpartum data (see Supplementary Figure S3 online). Therefore the increase in ka appears to be attributable to the sample population, and is not an effect of pregnancy.
Validation with nifedipine
The pregnancy-induced changes proposed by the obstetric semi-mechanistic metabolism model were validated using nifedipine. Two-compartment pharmacokinetic parameters for nifedipine administered to healthy volunteers were obtained from the literature15,16 and adapted to fit the semi-mechanistic metabolism model by incorporation of in vitro Km and Vmax (see Supplementary Data online). Predictive checks of the model against published concentration–time profiles (Figure 4) indicated that the healthy volunteer model described the data adequately. Changes in the volume of distribution, protein binding and hepatic CYP3A activity equivalent to those in the obstetrical midazolam model were applied. The model predictions of the mean nifedipine concentrations in subjects taking 20 mg nifedipine for preterm labor were within 15% of observed data. Several concentration values near the early points were overpredicted by the model, possibly because of the high interindividual variation observed in absorption profiles.31
Obstetric PBPK models of pregnancy have typically focused on toxicokinetics and risk assessment.5 However, physiologic and drug-specific parameters can be incorporated into pharmacokinetics models to enhance understanding of drug metabolism in pregnancy. The eventual goal of such work is to develop a “virtual pregnant woman” model to predict the pharmacokinetics and effects of drugs in pregnancy. However, such models require the incorporation of gestational-age-dependent physiologic changes. While some of these physiologic changes can be estimated on the basis of gestational age or weight-based algorithms,32 quantitative knowledge of other changes is limited and more difficult to acquire. Further in vitro and in vivo studies of drug metabolism and transport during pregnancy are required to fully populate these virtual models. Future development of this model will incorporate gestational-age-related growth changes. Additionally, the effects of pregnancy on drug distribution and absorption, as well as other routes of elimination, will be incorporated into future models.
A limitation of this and other pharmacokinetic models is the difficulty in model validation. In practice, drug concentrations can be determined only in the central compartment (plasma). Pharmacokinetic models enable prediction of drug concentrations in other compartments, e.g., at the site of drug action or in the fetus. However, validation of these concentrations is unlikely to be carried out in humans. Fetal concentration values can be obtained noninvasively at birth from umbilical cord blood. However, these values are limited to a single time point after maternal dosing, and may not reflect drug exposures at earlier ages of gestation. To complicate matters, both mother and fetus are exposed to drugs taken by the mother, requiring consideration of both therapeutic effectiveness and the potential for toxicity.
In conclusion, we have shown that our midazolam semi-mechanistic metabolism model predicts midazolam pharmacokinetics in women in a postpartum setting. By incorporating physiologic changes associated with pregnancy, we show that increasing midazolam hepatic clearance 1.6-fold in the model leads to accurate estimates of the clearance of oral midazolam in pregnant women. This was confirmed using data from a second CYP3A substrate, nifedipine. However, further study would be required to fully validate the lack of change in intestinal clearance despite increased hepatic clearance. For example, a pharmacokinetics study involving semi-simultaneous intravenous and oral administration of a probe drug would be informative. The application of this model to substrates metabolized by other enzymes will allow us to better understand the changes in drug pharmacokinetics that are associated with pregnancy. This model is a first step in the development of predictive models for individualized dosing in pregnancy.
Methods
Semi-mechanistic metabolism midazolam pharmacokinetic model. The semi-mechanistic metabolism pharmacokinetic model for midazolam in nonpregnant healthy subjects has been reported previously.7,8,9 For the obstetric model, two additional compartments were added to represent the placenta and fetus (Figure 1).
Elimination of midazolam occurs in the maternal gastrointestinal tract, liver, and kidney. In addition, the placenta and fetal liver have drug metabolizing capabilities. However, placental activity of CYP3A isoforms is minimal,24,25,26,27 and no evidence exists to indicate that midazolam is metabolized by the placenta. Therefore the placenta was not considered an organ of elimination in our model.
Maternal hepatic metabolism occurs through CYP3A-mediated and other enzymatic clearance pathways, e.g., glucuronidation. The CYP3A- and non-CYP3A-mediated intrinsic clearances of midazolam in the liver are estimated as:
where Vmax,3A and Vmax,non3A are the maximum velocity of metabolism by CYP3A and non-CYP3A mechanisms, Km is the Michaelis–Menten constant, fu is the unbound fraction of midazolam, and CH is the hepatic drug concentration.
Intrinsic clearance of midazolam within the gut wall (CLint,GW) is assumed to be entirely by CYP3A; therefore,
where Vmax,GW is the maximum velocity of metabolism by CYP3A in the gut wall. The Km value for the gut wall is assumed to be equivalent to hepatic Km.
The fetal liver primarily expresses CYP3A7. The metabolism of midazolam in the fetal liver, as in the adult liver, can be estimated by scaling up in vitro Vmax and Km values to estimate Vmax, using Equation (4):33
where Vmax,Fet is the maximal reaction velocity in the fetal liver, Vmax,3A7 is the maximal velocity determined in rCYP3A7 in vitro, ISEF is an intersystem extrapolation factor,34 CYP3A7 abundance is the amount of CYP3A7 enzyme per mg microsomal protein, and MPPGL is the amount of microsomal protein in mg per gram of liver. Williams et al. reported the in vitro Vmax and Km values of midazolam hydroxylation by recombinant CYP3A7 as 4 nmol/min/nmol and 114 µmol/l, respectively.13 It is estimated that fetal liver microsomes contain 0.3 nmol CYP450 protein per mg.14 Based on ICRP data, the fetal liver weighs 130 g at 38 weeks gestation.12 MPPGL is estimated to be 26 mg/g.35 In the absence of data, ISEF was assumed to be 1. Therefore, Vmax,Fet is estimated as 88 mg/h.
Volumes of distribution established from compartmental pharmacokinetic analyses are used to estimate the volumes of the central and peripheral compartments. Liver, portal vein, gut, placenta, and fetal volumes were based on reported physiologic volume estimates,12,36 and these volumes are subtracted from the central distribution compartment. During pregnancy, women gain 12.5 kg of body weight on average.12 In the semi-mechanistic metabolism midazolam model, the portion of weight gain not accounted for by the fetus and placenta is divided between the central and peripheral compartments. The assumption is that extracellular fluid and blood volume reside in the central compartment, and that changes in other tissues and fat stores are a component of the peripheral compartment.
While total cardiac output increases in pregnancy, the value of QH is assumed to be similar to that in the nonpregnant state,12 and is calculated as an allometric expression of total body weight:37
Hepatic artery flow (QHA) and portal venous blood flow (QPV) represent 25 and 75% of QH, respectively. Placental (Qplac) and umbilical venous (Quv) blood flows were estimated at 30 weeks of gestational age.38
Calibration data. Hebert et al. determined maternal plasma midazolam concentrations after a single 2 mg oral dose in women at 28–32 weeks of gestation.10 The study was repeated in the women at least 6 weeks postpartum. Individual plasma concentration–time profiles of these subjects were compared with the predicted concentration–time profiles for 500 individuals.
Sensitivity analysis. Sensitivity analysis was conducted to examine the effects of Vmax,H, Vmax,GW, QH, VC, VP, and fetal metabolism (Vmax,fet) on the model. Starting from the initial model, each parameter was individually increased and decreased twofold, except for Vmax,fet which was reduced to 0 and increased 20-fold (Table 2).
Parameter optimization. Based on results of the sensitivity analysis, individual parameters or subsets of them were manually adjusted without taking interindividual variability into account, and holding all other parameters constant. Predicted concentrations were compared with the mean concentration–time curve data reported by Hebert et al.10 The optimal model was chosen based on visual inspection of the concentration vs. time plot and minimization of the residual sum of squares.
Nifedipine semi-mechanistic metabolism model. A healthy volunteer model and an obstetric semi-mechanistic metabolism pharmacokinetic model were constructed for another CYP3A substrate, nifedipine, using the same model structure as for midazolam. Central and peripheral volumes of distribution and ka were determined from published two-compartment analyses of nifedipine pharmacokinetics in healthy volunteers (Table 1).15,16 Nifedipine plasma concentrations in healthy volunteers after oral administration of 10 mg immediate-release capsules were extracted from mean concentration values reported in eight different studies involving healthy volunteers, using Grab-It Graph Digitizer (Datatrend Software, Raleigh, NC).16, In vitro Vmax and Km values of nifedipine in human liver microsomes were obtained from published studies (see Supplementary Data online). All other physiologic parameters, i.e., blood flows and physiologic organ volumes, were identical to the midazolam healthy volunteer semi-mechanistic metabolism model.
Nifedipine obstetric semi-mechanistic metabolism model. The pregnancy-associated changes in CYP3A activity, volume, and plasma protein binding identified in the midazolam semi-mechanistic metabolism obstetric pharmacokinetic model were applied to the corresponding model for nifedipine. Given that protein binding of midazolam and nifedipine are similar, changes in fu and central and peripheral volumes of distribution were applied as per the final midazolam model. Hepatic Vmax was increased 1.6-fold to account for increased CYP3A activity in pregnancy.
Nifedipine validation data. Predicted concentration–time curves of nifedipine after 20 mg oral dosing to pregnant women were compared to data obtained as part of the Under-studied Drugs in Pregnancy protocol conducted by the Obstetric-Fetal Pharmacology Research Unit network.17 The study examined nifedipine pharmacokinetics in 20 women (24–36 weeks gestation) who were receiving 10–20 mg immediate-release nifedipine every 4–8 h for preterm labor, as clinically indicated. Given that the majority of women were taking 20 mg every 6 h, the 10 mg doses were normalized to 20 mg for visualization and comparison. The study was approved by the institutional review board at each study site (Georgetown University, University of Pittsburgh, University of Texas Medical Branch-Galveston, and University of Washington), and written informed consent was obtained from all the participants.
Software and model analysis. All simulations were performed in R v 2.13.1,46 using the odesolve package. Noncompartmental analysis was performed using the PK package47 to obtain AUC0–∞ (midazolam) or AUC0–6 (nifedipine). The example code is provided as online supplementary information (see Supplementary Data online). Predictions were compared with observed data using visual predictive checks and calculation of percentage error ([predicted – observed/observed] and residual sum of squares.
Author Contributions
S.K.Q. wrote the manuscript. S.K.Q., M.F.H., D.M.H., S.C., J.G.U., S.N.C., and L.L. designed the research. S.K.Q., A.N.M., M.F.H., D.M.H., S.C., J.G.U., and S.N.C. performed the research. S.K.Q. and A.N.M. analyzed the data. S.K.Q. contributed new reagents/analytical tools.
Conflict of Interest
The authors declared no conflict of interest.
Study Highlights

Acknowledgments
This project was supported by grants P50HD0444, U10HD063094-01, 5U10HD047892, 5U10HD047890, 2U10HD04789107, and 1U10HD057753 from the Eunice Kennedy Shriver National Institute of Child Health & Human Development, and the National Institute of Health/National Center for Research Resources grants M01RR00037, UL1RR025014, and UL1RR031975. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Eunice Kennedy Shriver National Institute of Child Health & Human Development or the National Institutes of Health.
References
- Daw J.R., Hanley G.E., Greyson D.L., &, Morgan S.G. Prescription drug use during pregnancy in developed countries: a systematic review. Pharmacoepidemiol. Drug Saf. 2011;20:895–902. doi: 10.1002/pds.2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough L.B., Coverdale J.H., &, Chervenak F.A. A comprehensive ethical framework for responsibly designing and conducting pharmacologic research that involves pregnant women. Am. J. Obstet. Gynecol. 2005;193:901–907. doi: 10.1016/j.ajog.2005.06.020. [DOI] [PubMed] [Google Scholar]
- Lyerly A.D., Little M.O., &, Faden R. The second wave: Toward responsible inclusion of pregnant women in research. Int. J. Fem. Approaches Bioeth. 2008;1:5–22. doi: 10.1353/ijf.0.0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland M., Balant L., &, Peck C. Physiologically based pharmacokinetics in drug development and regulatory science: a workshop report (Georgetown University, Washington, DC, May 29-30, 2002) AAPS PharmSci. 2004;6:E6. doi: 10.1208/ps060106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corley R.A., Mast T.J., Carney E.W., Rogers J.M., &, Daston G.P. Evaluation of physiologically based models of pregnancy and lactation for their application in children's health risk assessments. Crit. Rev. Toxicol. 2003;33:137–211. doi: 10.1080/713611035. [DOI] [PubMed] [Google Scholar]
- Zhao P.et al. Applications of physiologically based pharmacokinetic (PBPK) modeling and simulation during regulatory review Clin. Pharmacol. Ther 89259–267.2011 [DOI] [PubMed] [Google Scholar]
- Quinney S.K., Zhang X., Lucksiri A., Gorski J.C., Li L., &, Hall S.D. Physiologically based pharmacokinetic model of mechanism-based inhibition of CYP3A by clarithromycin. Drug Metab. Dispos. 2010;38:241–248. doi: 10.1124/dmd.109.028746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Quinney S.K., Gorski J.C., Jones D.R., &, Hall S.D. Semiphysiologically based pharmacokinetic models for the inhibition of midazolam clearance by diltiazem and its major metabolite. Drug Metab. Dispos. 2009;37:1587–1597. doi: 10.1124/dmd.109.026658. [DOI] [PubMed] [Google Scholar]
- Chien J.Y., Lucksiri A., Ernest C.S. 2nd,, Gorski J.C., Wrighton S.A., &, Hall S.D. Stochastic prediction of CYP3A-mediated inhibition of midazolam clearance by ketoconazole. Drug Metab. Dispos. 2006;34:1208–1219. doi: 10.1124/dmd.105.008730. [DOI] [PubMed] [Google Scholar]
- Hebert M.F.et al. Effects of pregnancy on CYP3A and P-glycoprotein activities as measured by disposition of midazolam and digoxin: a University of Washington specialized center of research study Clin. Pharmacol. Ther 84248–253.2008 [DOI] [PubMed] [Google Scholar]
- Tracy T.S., Venkataramanan R., Glover D.D., &, Caritis S.N., National Institute for Child Health and Human Development Network of Maternal-Fetal-Medicine Units Temporal changes in drug metabolism (CYP1A2, CYP2D6 and CYP3A Activity) during pregnancy. Am. J. Obstet. Gynecol. 2005;192:633–639. doi: 10.1016/j.ajog.2004.08.030. [DOI] [PubMed] [Google Scholar]
- Basic anatomical and physiological data for use in radiological protection: reference values. A report of age- and gender-related differences in the anatomical and physiological characteristics of reference individuals. ICRP Publication 89. Ann ICRP. 2002;32:5–265. [PubMed] [Google Scholar]
- Williams J.A.et al. Comparative metabolic capabilities of CYP3A4, CYP3A5, and CYP3A7 Drug Metab. Dispos 30883–891.2002 [DOI] [PubMed] [Google Scholar]
- Shimada T.et al. Characterization of microsomal cytochrome P450 enzymes involved in the oxidation of xenobiotic chemicals in human fetal liver and adult lungs Drug Metab. Dispos 24515–522.1996 [PubMed] [Google Scholar]
- Kleinbloesem C.H., van Brummelen P., van de Linde J.A., Voogd P.J., &, Breimer D.D. Nifedipine: kinetics and dynamics in healthy subjects. Clin. Pharmacol. Ther. 1984;35:742–749. doi: 10.1038/clpt.1984.105. [DOI] [PubMed] [Google Scholar]
- Raemsch K.D., &, Sommer J. Pharmacokinetics and metabolism of nifedipine. Hypertension. 1983;5:II18–II24. doi: 10.1161/01.hyp.5.4_pt_2.ii18. [DOI] [PubMed] [Google Scholar]
- Haas D.M., for the Obstetric-Fetal Pharmacology Research Units Network et al. Nifedipine pharmacokinetics are influenced by CYP3A5 genotype when used as a preterm labor tocolytic. Am. J. Perinatol. 2012. [DOI] [PMC free article] [PubMed]
- Schmiedlin-Ren P., Thummel K.E., Fisher J.M., Paine M.F., Lown K.S., &, Watkins P.B. Expression of enzymatically active CYP3A4 by Caco-2 cells grown on extracellular matrix-coated permeable supports in the presence of 1alpha,25-dihydroxyvitamin D3. Mol. Pharmacol. 1997;51:741–754. doi: 10.1124/mol.51.5.741. [DOI] [PubMed] [Google Scholar]
- Kim R.B.et al. Interrelationship between substrates and inhibitors of human CYP3A and P-glycoprotein Pharm. Res 16408–414.1999 [DOI] [PubMed] [Google Scholar]
- Gaohua L., Abduljalil K., Jamei M., Johnson T.N., &, Rostami-Hodjegan A. A Pregnancy Physiologically-Based Pharmacokinetic (p-PBPK) Model for Disposition of Drugs Metabolized by CYP1A2, CYP3A4 and CYP2D6. Br. J. Clin. Pharmacol. 2012. [DOI] [PMC free article] [PubMed]
- Andrews J.et al. Comparative study of the metabolism of drug substrates by human cytochrome P450 3A4 expressed in bacterial, yeast and human lymphoblastoid cells Xenobiotica 32937–947.2002 [DOI] [PubMed] [Google Scholar]
- Pavek P., Ceckova M., &, Staud F. Variation of drug kinetics in pregnancy. Curr. Drug Metab. 2009;10:520–529. doi: 10.2174/138920009788897993. [DOI] [PubMed] [Google Scholar]
- Bacq Y.et al. Liver function tests in normal pregnancy: a prospective study of 103 pregnant women and 103 matched controls Hepatology 231030–1034.1996 [DOI] [PubMed] [Google Scholar]
- Schuetz J.D., Kauma S., &, Guzelian P.S. Identification of the fetal liver cytochrome CYP3A7 in human endometrium and placenta. J. Clin. Invest. 1993;92:1018–1024. doi: 10.1172/JCI116607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakkola J.et al. Detection of cytochrome P450 gene expression in human placenta in first trimester of pregnancy Biochem. Pharmacol 52379–383.1996 [DOI] [PubMed] [Google Scholar]
- Hakkola J.et al. Expression of xenobiotic-metabolizing cytochrome P450 forms in human full-term placenta Biochem. Pharmacol 51403–411.1996 [DOI] [PubMed] [Google Scholar]
- Maezawa K., Matsunaga T., Takezawa T., Kanai M., Ohira S., &, Ohmori S. Cytochrome P450 3As gene expression and testosterone 6 beta-hydroxylase activity in human fetal membranes and placenta at full term. Biol. Pharm. Bull. 2010;33:249–254. doi: 10.1248/bpb.33.249. [DOI] [PubMed] [Google Scholar]
- Stevens J.C.et al. Developmental expression of the major human hepatic CYP3A enzymes J. Pharmacol. Exp. Ther 307573–582.2003 [DOI] [PubMed] [Google Scholar]
- Zhang H., Wu X., Wang H., Mikheev A.M., Mao Q., &, Unadkat J.D. Effect of pregnancy on cytochrome P450 3a and P-glycoprotein expression and activity in the mouse: mechanisms, tissue specificity, and time course. Mol. Pharmacol. 2008;74:714–723. doi: 10.1124/mol.107.043851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.et al. Pregnancy does not increase CYP3A or P-glycoprotein activity in the non-human primate, Macaca nemestrina J. Pharmacol. Exp. Ther 330586–595.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nader, A.M., Caritis, S.N., Hebert, M.F., Clark, S.M., Flockhart, D.A., &, Quinney, S.K. Compartmental pharmacokinetics of immediate release nifedipine in pregnancy: variability in absorption. Clin. Pharmacol. Ther. 2012;91:PIII–109. [Google Scholar]
- Young J.F., Branham W.S., Sheehan D.M., Baker M.E., Wosilait W.D., &, Luecke R.H. Physiological “constants” for PBPK models for pregnancy. J. Toxicol. Environ. Health. 1997;52:385–401. doi: 10.1080/00984109708984072. [DOI] [PubMed] [Google Scholar]
- Howgate E.M., Rowland Yeo K., Proctor N.J., Tucker G.T., &, Rostami-Hodjegan A. Prediction of in vivo drug clearance from in vitro data. I: impact of inter-individual variability. Xenobiotica. 2006;36:473–497. doi: 10.1080/00498250600683197. [DOI] [PubMed] [Google Scholar]
- Proctor N.J., Tucker G.T., &, Rostami-Hodjegan A. Predicting drug clearance from recombinantly expressed CYPs: intersystem extrapolation factors. Xenobiotica. 2004;34:151–178. doi: 10.1080/00498250310001646353. [DOI] [PubMed] [Google Scholar]
- Pelkonen O., Kaltiala E.H., Larmi T.K., &, Kärki N.T. Comparison of activities of drug-metabolizing enzymes in human fetal and adult livers. Clin. Pharmacol. Ther. 1973;14:840–846. doi: 10.1002/cpt1973145840. [DOI] [PubMed] [Google Scholar]
- Wosilait W.D., Luecke R.H., &, Young J.F. A mathematical analysis of human embryonic and fetal growth data. Growth. Dev. Aging. 1992;56:249–257. [PubMed] [Google Scholar]
- Brown R.P., Delp M.D., Lindstedt S.L., Rhomberg L.R., &, Beliles R.P. Physiological parameter values for physiologically based pharmacokinetic models. Toxicol. Ind. Health. 1997;13:407–484. doi: 10.1177/074823379701300401. [DOI] [PubMed] [Google Scholar]
- Lees C., Albaiges G., Deane C., Parra M., &, Nicolaides K.H. Assessment of umbilical arterial and venous flow using color Doppler. Ultrasound Obstet. Gynecol. 1999;14:250–255. doi: 10.1046/j.1469-0705.1999.14040250.x. [DOI] [PubMed] [Google Scholar]
- Fukuda T.et al. CYP3A5 genotype did not impact on nifedipine disposition in healthy volunteers Pharmacogenomics J 434–39.2004 [DOI] [PubMed] [Google Scholar]
- Odou P.et al. Grapefruit juice-nifedipine interaction: possible involvement of several mechanisms J. Clin. Pharm. Ther 30153–158.2005 [DOI] [PubMed] [Google Scholar]
- Yu K.S.et al. Ethnic differences and relationships in the oral pharmacokinetics of nifedipine and erythromycin Clin. Pharmacol. Ther 70228–236.2001 [DOI] [PubMed] [Google Scholar]
- Lang C.C., Jamal S.K., Mohamed Z., Mustafa M.R., Mustafa A.M., &, Lee T.C. Evidence of an interaction between nifedipine and nafcillin in humans. Br. J. Clin. Pharmacol. 2003;55:588–590. doi: 10.1046/j.1365-2125.2003.01789.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhr U., Beckmann-Knopp S., Jetter A., Lück H., &, Mengs U. The effect of silymarin on oral nifedipine pharmacokinetics. Planta Med. 2007;73:1429–1435. doi: 10.1055/s-2007-990256. [DOI] [PubMed] [Google Scholar]
- Wang X.D.et al. Rapid and simultaneous determination of nifedipine and dehydronifedipine in human plasma by liquid chromatography-tandem mass spectrometry: Application to a clinical herb-drug interaction study J. Chromatogr. B Analyt. Technol. Biomed. Life Sci 852534–544.2007 [DOI] [PubMed] [Google Scholar]
- Chien S.C., Uang Y.S., Lin H.Y., &, Hsu K.Y. Pharmacokinetics of nifedipine in Taiwanese. Biopharm. Drug Dispos. 2004;25:77–84. doi: 10.1002/bdd.386. [DOI] [PubMed] [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical ComputingVienna, Austria R Foundation for Statistical Computing, 2011
- Jaki, T, Wolfsegger, M.J. Estimation of pharmacokinetic parameters with the R package PK. Pharmaceutical Statistics. 2011;10:284–288. [Google Scholar]