

Abstract
Significance: Stroke, a leading cause of death and disability, poses a substantial burden for patients, relatives, and our healthcare systems. Only one drug is approved for treating stroke, and more than 30 contraindications exclude its use in 90% of all patients. Thus, new treatments are urgently needed. In this review, we discuss oxidative stress as a pathomechanism of poststroke neurodegeneration and the inhibition of its source, type 4 nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX4), as a conceptual breakthrough in stroke therapy. Recent Advances: Among potential sources of reactive oxygen species (ROS), the NOXes stand out as the only enzyme family that is solely dedicated to forming ROS. In rodents, three cerebrovascular NOXes exist: the superoxide-forming NOX1 and 2 and the hydrogen peroxide-forming NOX4. Studies using NOX1 knockout mice gave conflicting results, which overall do not point to a role for this isoform. Several reports find NOX2 to be relevant in stroke, albeit to variable and moderate degrees. In our hands, NOX4 is, by far, the major source of oxidative stress and neurodegeneration on ischemic stroke. Critical Issues: We critically discuss the tools that have been used to validate the roles of NOX in stroke. We also highlight the relevance of different animal models and the need for advanced quality control in preclinical stroke research. Future Directions: The development of isoform-specific NOX inhibitors presents a precious tool for further clarifying the role and drugability of NOX homologues. This could pave the avenue for the first clinically effective neuroprotectant applied poststroke, and even beyond this, stroke could provide a proof of principle for antioxidative stress therapy. Antioxid. Redox Signal. 18, 1418–1427.
Relevance of Stroke and Lack of Treatment Options
Stroke is one of the most common pathologies in the industrialized world and ranks as the second frequent cause of death after heart disease. Due to an aging population, absolute numbers are further increasing, even though prevalence is slightly decreasing. Apart from the individual suffering and burden to partners and families, each stroke patient costs the U.S. healthcare system about U.S. $140,048 for initial care, rehabilitation, and follow-up care of lasting disabilities (51).
There are various risk factors for stroke, including nonmodifiable ones such as age, race, or genetic background, and modifiable ones such as hypertension, poor diet, smoking, lack of exercise, obesity, hypercholesterolemia, and diabetes (22). In the clinic, stroke is presented in two major forms: either ischemic (∼85% of the cases), in which a vessel becomes occluded by a blood clot or by vascular dissection, or hemorrhagic caused by intracerebral (∼10%) or subarachnoidal (∼3%) bleedings. In this review, we will focus on the more common ischemic stroke.
Already 5 min after the onset of ischemia, neurons begin to die. Therefore, blood flow should be restored as early as possible. For this, recombinant tissue plasminogen activator (rt-PA) is applied intravenously, in order to lyse the occluding blood clot. Indeed, it is the only drug that is currently approved for acute stroke treatment and, significantly, neuroprotective therapies are not available at all. Moreover, only a limited number of patients benefit from rt-PA, as rt-PA may be administered to patients unless several requirements are met. First, a computed tomography (CT) scan should be performed to exclude a hemorrhagic stroke, in which case rt-PA would exacerbate bleeding. Second, rt-PA should be administered within the first 3–4.5 h after the cerebrovascular incident. This, however, represents a challenge given the very narrow time window to recognize the stroke, transportation to the hospital, and completing the CT examination. Third, several additional contra-indications exist, for example, an age older than 80 years, increased risk of bleeding, previous surgeries, and so on. Thus, the use of rt-PA is finally restricted to only approximately 10% of all stroke patients. For this reason, other improved thromobolytic drugs are currently under investigation. They are characterized by longer half life and higher fibrin specificity and, hence, a possibly lower risk of bleeding. However, recent clinical trials were disappointing (38). Therefore, in order to find effective treatment options in stroke, we need to identify novel mechanism-based targets. One such promising candidate mechanism is oxidative stress.
The Oxidative Stress Hypothesis
Oxidative stress refers to a relative surplus of reactive oxygen species (ROS) caused by excessive ROS generation and/or impaired ROS degradation. Under conditions of oxidative stress, the overload of ROS becomes detrimental and possibly contributes to various acute and chronic diseases, such as myocardial infarction, hypertension, atherosclerosis, cancer, neurodegeneration, and inflammation. Consequently, oxidative stress has also been suggested as a key underlying mechanism of ischemic stroke (11).
The brain is particularly sensitive to oxidative stress because of its high oxygen consumption and low oxidative defence capacity (20). In stroke, ischemia-derived nutrient and oxygen deficiency lead to cellular energy failure and neuronal damage. Reperfusion even aggravates this damage, because the freshly arriving oxygen will serve as a substrate for new ROS production (10).
Based on the hypothesis that oxidative stress plays a major role in neuronal damage caused by ischemic stroke, clinical trials using antioxidant treatments in an attempt to scavenge ROS were performed. However, despite promising preclinical results and even a first rather promising, although underpowered, Phase III trial (37), these studies eventually failed to show a significant amelioration in stroke patients (56). Possible explanations for the negative outcome of antioxidants in clinical trials are detailed elsewhere (71).
However, one important consideration is that ROS are not always associated with deleterious effects and under physiological conditions, they appear to also have essential functions, for example, within the innate immune response (24), cellular signaling, control of cellular proliferation and differentiation, oxygen sensing, vascular tone, and angiogenesis. Consequently, interfering with these beneficial roles of ROS in a too broad and unspecific manner can result in pathologies as well, a phenomenon termed as reductive stress.
Since these nonspecific antioxidative therapies were unsuccessful in the clinic, the oxidative stress hypothesis still remains unproven. Nevertheless, the failure of antioxidant trials, even in stroke patients (15), does not disprove the involvement of ROS in ischemia-reperfusion (I/R) injury. Rather, it may demonstrate that a much deeper understanding of the underlying mechanisms and more focused, molecular approaches on relevant molecular sources of oxidative stress are needed.
NADPH Oxidases As a Relevant Source of Oxidative Stress in Stroke
A superior approach for using antioxidants and scavenge ROS in a generalized, nonselective manner would be to specifically inhibit the disease-relevant enzymatic source(s) of ROS formation and leave physiological ROS signaling intact. In the central nervous system, ROS can derive from different sources, including mitochondria, xanthine oxidase, uncoupled nitric oxide synthase (NOS), and cyclooxygenase (68). However, NADPH oxidases are the only known enzyme family with the single known function of producing ROS. All other enzymes produce ROS as a by-product or on uncoupling.
NADPH oxidases are multicomponent protein complexes containing a catalytic NOX subunit that transfers electrons from NADPH to oxygen, thereby forming ROS (6). Five NOX isoforms exist, NOX1-5. Two additional proteins, DUOX1 and DUOX2, also contain an oxidase domain, which in this case is linked to a peroxidase-like domain. Their roles in vascular biology are unclear. NOX homologues differ not only in their tissue and (sub)cellular localization, which is an important determinant of their function, but also in their requirements for regulatory subunits and the nature of the ROS produced (46). Since many reviews on NADPH oxidases have been published, we will not discuss in great detail the characteristics of the different NOX homologues but rather refer to recent reviews (49, 59).
Of the five NOX isoforms, NOX3 is most likely not relevant in I/R injury, as it is mainly expressed in the inner ear but not in blood vessels. In contrast, NOX1, NOX2 (previously termed gp91phox), and NOX4 are expressed under physiological conditions in the central nervous system, including intracranial vessels and neuronal tissues (1, 28). Based on mRNA levels, NOX4 is the most widely distributed NOX isoform in the vasculature. Its expression is an order of magnitude higher in cerebral arteries compared with peripheral blood vessels (42). With regard to the NOX5 isoform, it should be noted that NOX5 is not expressed in rats and mice and is, therefore, functionally less characterized than others. Nevertheless, NOX5 is of considerable interest, as it is both expressed in the human vasculature and regulated in a unique manner, that is, direct binding of calcium, which may be of pathomechanistic relevance, as an imbalance in calcium homeostasis appears to be decisive for neuronal cell death after an ischemic stroke. The absence of NOX5 in the rat and mouse genome is, therefore, a noteworthy limitation of all preclinical studies in these species.
Since specific antibodies for the various isoforms are scarce, most papers are limited to measuring NOX mRNA levels. Since NOX activities are also highly regulated at a post-translational level, protein expression and activity data are much more favorable. For example, increased NOX activity does not necessarily require increased subunit expression, although correlation of mRNA levels of NOX2 or NOX4 and NADPH oxidase activity has been observed in transgenic mice (4). After an ischemic stroke, NOX2 and NOX4 are up-regulated within 24 h. It appears that in this process, NOX2 protein levels increase within microglia (23), and NOX4 protein levels increase in neurons and endothelial cells (33, 65) (Fig. 1). This up-regulation might give a first indication of the implication of NOX2 and 4 in stroke pathology, and, hence, these two NOX isoforms present a potential target in stroke therapy. Therefore, several research groups started investigating the role of NADPH oxidases in stroke.
FIG. 1.

Cerebral expression pattern of NOX isoforms implicated in stroke. NOX2 is highly expressed in inflammatory cells such as resident microglia and peripheral neutrophils, whereas NOX4 is rather expressed in neurons. Both endothelial cells and astrocytes seem to express NOX homologues. Cerebral NOX activation and subsequent reactive oxygen species (ROS) generation contribute to blood-brain barrier (BBB) disruption, inflammation, and postischemic neuronal injury [adapted from Ref. (49)].
Lack of Quality Control in Preclinical Stroke Research
In 1996, rt-PA treatment has been approved for clot lysis in stroke. Since then, no innovative stroke treatment has made it to the clinic. O'Collins et al. provided an overview of the numerous attempts of stroke researchers to develop neuroprotective drugs. They come to the alarming conclusion that 1026 experimental treatments have been investigated, from which 114 have even been clinically tested in stroke patients, but that none of them has fulfilled their initial expectations (44). The main reason is a quality issue with most, if not all, preclinical stroke studies.
In preclinical models, stroke is induced artificially in young, healthy animals; whereas in humans, stroke happens in older patients, often as a consequence of several underlying pathologies (e.g., hypertension, atherosclerosis, atrial fibrillation, etc.) (19), and spontaneous recanalization can occur. Thus, neither permanent nor transient animal models of ischemic stroke fully mimic the clinical situation.
The majority of studies are performed in rodents due to economical and ethical reasons, whereas pigs and primates would be better suited because of their brains' higher proportion of white matter and gyrencephalic structure, which is more similar to humans (26). Moreover, commonly used preclinical ischemic stroke models are diverse. The method of vessel occlusion, for example, differs from mechanical or thermal to embolic or chemical (Fig. 2). Most preclinical studies use the mechanical model of transient middle cerebral artery occlusion. In addition, in many studies, the observation period stops after about 24 h poststroke. This per definition will not address long-term morbidity, mortality, and relevant neurological outcomes.
FIG. 2.

Schematic overview of preclinical stroke models. Rodent stroke models have been established with the aim of generating as reproducible infarct sizes as possible. Each stroke model tries to capture elements of human stroke, though with many well-recognized limits. The choice of the experimental stroke model is optional, but it is recommended to use at least one permanent and one transient ischemic stroke model. The use of mice presenting comorbidities is another recommended option.
Significantly, these limitations are not specific to stroke research and hold true for almost all cardiovascular indications, where progress is poor in general. All major drug groups have run out of patent, documenting an obvious 20 year-long innovation block. Thus, the successful translation of preclinical research, in general, requires conceptual changes and quality improvement similar to clinical research in the 70s.
Quality Management in Preclinical Stroke Studies
A group of basic and clinical researchers along with representatives from the pharmaceutical industry formed the Stroke Therapy Academic Industry Roundtable (STAIR) to design preclinical study criteria that should be followed to improve the quality of stroke studies and their later translation into practice (60). Since human populations are more heterogeneous compared with laboratory animals and most stroke patients have diverse comorbidities, experimental animals should ideally also present comorbidities in order to better mimic clinical conditions. Furthermore, old animals should be included, because stroke patients are also mostly elderly people. At present, however, preclinical studies mainly use young animals, presumably to reduce time and costs. Significantly, not only age but also gender plays a role in stroke pathology and/or drug metabolism. Thus, both male and female animals should be assessed in stroke studies. Further aspects that may have hampered translation from animal to clinical studies are the endpoints that are assessed in animals. While the infarct size is often the major investigated parameter in animals, neurological outcome is the most important endpoint in humans. Accordingly, functional studies should be performed up to several weeks postcerebral I/R and survival monitored to meet the criteria of clinical trials, rather than stopping the observation period 24 h after ischemia. In addition, at least one permanent and one transient model should be performed to validate a stroke mechanism. Other quality parameters include the verification of reduced blood flow on vessel occlusion (<0.12 ml/g/min) and/or reperfusion, as well as monitoring rectal temperature, because hypothermia likely has neuroprotective effects in experimental models (26), although there is actually no evidence from clinical trials (14). The most important quality criteria for preclinical stroke studies are summarized in Figure 3. Of course, these strict requirements only apply for true translational and not for early proof of principle or purely mechanistic studies.
FIG. 3.

Recommended quality criteria for preclinical stroke research. To date, animal stroke studies vary in quality and reliability. Thus, when performing preclinical stroke experiments, the quality criteria listed above should be followed. Adherence to these criteria can be used to assess the quality of experimental studies.
Similar to all preclinical experiments, randomization (only performed in 36% of all stroke studies), allocation concealment (11%), and blinded analysis of the results (29%) should be performed (54). Mortalities per experimental group need to be explicitly stated, as this is both a quality criterion for the animal surgery and may distort data representation when only the phenotype of the surviving animals is reported.
Moreover, it has recently been recommended to conduct preclinical studies both internationally and multicenter (16, 17). Finally, another aspect that should not be neglected is publication bias (55). Negative findings are often not publishable or more difficult to publish, and the literature is, thus, in favor of positive results, which may overestimate efficacy of any experimental therapy.
Knockout Models to Validate the Role of NOX in Stroke
Knockout (KO) models are powerful tools for determining the role of different NOX isoforms, allowing assessment of a causality-effect relationship. A general artifact is that in KO mice the gene is deleted from the onset of stroke whereas any therapy is likely to initiate only hours after the ischemic event. Apart from this, even truncated proteins or splice variants with residual activity may be generated, depending on the KO strategy. When taking the example of our own NOX4 KO model, we deleted the exons that code for the protein region which is essential for NADPH binding (Fig. 4). Thus, NADPH oxidase activity will be zero, irrespective of any truncated protein being present (33). In addition, abolishing the expression of one NOX isoform may result in counter-regulation of other NOX isoforms and ROS formation. Moreover, most NOX proteins have several binding partners such as the highly abundant p22phox. It is unclear whether p22phox's sole function relates to NOX. If not, the lack of one binding partner of p22phox may have bystander effects on other p22phox-binding proteins. Moreover genes flanking the target gene or genetic background (due to random recombination) may also lead to misinterpretations (18). Most genetically modified mice have been generated on a 129 substrain, which was the first to be easily genetically manipulated but has poor breeding efficacy. Therefore, embryonic stem cells from a 129 strain are implanted into blastocysts of the well-breeding C57BL6 strain. The resultant chimeric mouse will always have some remaining genetic information from the 129 strain, even after extensive backcrossing onto C57BL6 mice and extensive SNP analysis. Thus, it cannot be excluded that some NOX KO mice display a phenotype that is mistakenly attributed to the deleted NOX gene. In this context, also the sub-strain on which the mice have been backcrossed, that is, C57BL6J versus C57BL6N showing different mitochondrial abnormalities, may represent an important factor. Finally, not all publications clearly state whether littermate mice have been used as wild-type controls.
FIG. 4.
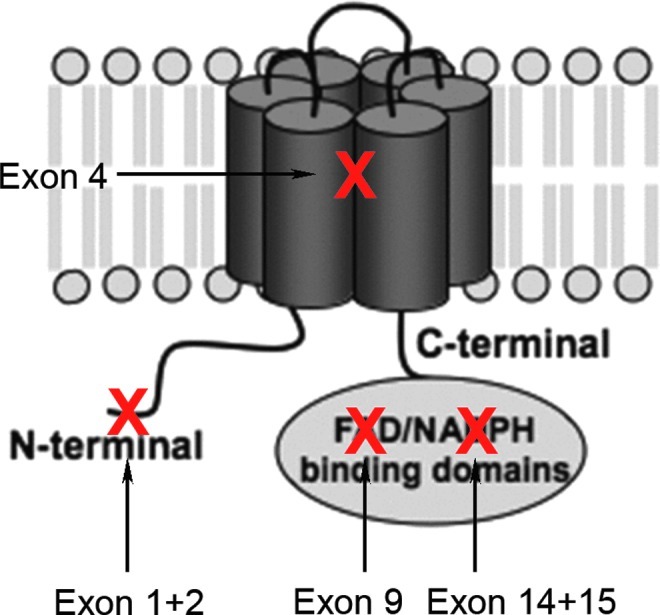
Existing NOX4 knockout (KO) models. The NOX4 protein is composed of six transmembrane domains and cytosolic binding domains for flavin adenine dinucleotide (FAD) and nicotinamide adenine dinucleotide phosphate (NADPH) at the C-terminus. Several groups have generated NOX4 KO models by deleting different exons of the NOX4 gene. Our group's NOX4 KO mouse was generated by deleting exons 14 and 15, which correspond to the NADPH binding domain. This likely results in the expression of a nonfunctional enzyme (33). Another NOX4 KO mouse was generated by conditionally deleting exon 9 of NOX4 in cardiomyocytes, thereby deleting the FAD binding domain (35). The splice variants of NOX4 in different cell types and tissues are not known. However, independent of which residual protein would be expressed, these two KOs would be biochemically inactive. In a third NOX4 KO model, exons 1 and 2 were deleted in an attempt to delete the NOX4 protein completely (72). Another NOX4 KO model deleted exon 4 (9). Depending on alternative splicing, these may bear the risk of truncated splice variants being expressed, which contain the NADPH/FAD reductase domain. This domain is able to generate ROS independently of the double heme domain and may, thus, in some cells not eliminate NOX4-dependent ROS formation, target it to different subcellular compartments with different or no regulation. However, this is a hypothetical risk, as the splicing in wild-type and KO mice is not fully mapped. Alternatively, truncated NOX4 proteins, that is, without NADPH and/or FAD binding sites, may affect other NOX isoforms in a dominant negative fashion, for example, by scavenging p22phox. However, there is no evidence as yet for this possibility (7).
No implication of NOX1
With regard to a possible role of NOX1 in stroke, three studies have been published that all used the same NOX1 KO mouse strain (30, 31, 33), although not all studies reported details according to the STAIR criteria. Jackman et al. concluded that NOX1 is relevant for angiotensin-stimulated ROS production but has no impact on overall ROS production, stroke size, and neurological outcome (30). However, after a subgroup analysis, the authors postulated that cortical strokes were four times increased in NOX1 KO versus control mice, possibly indicating that NOX1 may play a protective role by limiting cortical damage. Another group reported opposite findings, that is, reduced infarct size in NOX1 KO mice (31). This effect was only visible when the ischemic period lasted 1 h but not when lasting longer. Interestingly, antioxidant treatment showed additional neuroprotective effects in NOX1 KO mice and reduced infarct size to the same extent as in apocynin-treated wild-type mice. Thus, the effect of scavenging ROS by antioxidants was unlikely mediated by NOX1 but rather by other NOX isoforms or non-NOX sources of ROS. We observed neither a difference in infarct volume nor in neurological behavior when investigating NOX1 KO mice in stroke (33). Collectively, NOX1 appears to have no or only a very minor effect on neurological damage after stroke. This is consistent with the fact that NOX1, at least at the mRNA level, is not up-regulated after cerebral I/R.
NOX4, the key player
Using our NOX4 KO mice, we unambiguously showed that NOX4 is a pathologically relevant NOX isoform in ischemic stroke in mice (33). In this study, we tried to adhere to the STAIR criteria as much as possible, for example, by using mice of different ages and both gender, transient and permanent stroke models, and a long follow-up period, including evaluation by magnetic resonance imaging. The deletion of NOX4 dramatically protected mice from brain damage, resulting in a 75% reduction in infarct size, as did intrathecal application of a pharmacological NOX inhibitor in wild-type mice (33). Significantly, the drug was applied after the induction of ischemia, which from a clinical point is therapeutically much more relevant than the prevention of brain damage before the onset of ischemia as is the case in KO mice. Certainly, the intrathecal route of administration is not optimal, and an intravenous administration would be much more desirable but was not possible due to pharmacokinetic limitations of VAS2870, the inhibitor used in our study. Moreover, this compound may have potential off-target effects (61), which may make it less attractive for further development into a drug.
However, that NOX4 is a promising target in stroke was recently confirmed by another group using a transgenic mouse model overexpressing NOX4 in endothelial cells (5). Their findings are supportive of those seen in our global NOX4 KO in that they found increased infarcts when compared with wild-type mice. Mechanistically, this was hypothesized to be due to suppression of endothelial NOS by NOX4 (5). In accordance with these findings and the role of endothelial NOX4, infarcts are smaller in endothelial cell-specific NOX4 KO mice compared with wild-type littermates (Radermacher, Kleinschnitz and Schmidt, unpublished observations).
It was recently suggested that NOX4-derived hydrogen peroxide might also have a beneficial role in the cerebral circulation by mediating cerebral vasodilatation and increasing blood flow. However, this effect should be selective for the ischemic zone, as a general cerebral vasodilation would rather be harmful because of the “steal effect,” a phenomenon that refers to the natural principle of the flow collateralization of arterial occlusions. To weigh the degree of potential harm versus the benefit of inhibiting NOX4 and to approve or reject this hypothesis, the effects of different NOX isoform-specific inhibitors at different time points after stroke and also in different species will be of interest. At present, all available data clearly point toward an essential pathophysiological role of NOX4 in ischemia-induced brain damage.
An accessory role for NOX2?
NOX2, a key enzyme of the innate immune and inflammatory response (48), is thought to be involved in almost every cardiovascular disease setting (58), including I/R injury (39). NOX2 was the first NOX homologue that was discovered. Several studies have been performed to study the role of NOX2 in stroke (8, 12, 13, 29, 32–34, 63, 66). These studies used the same commercially available NOX2 KO mouse strain (strain No. 002365 from Jackson), which had already been generated in 1995 (47). Two of the studies (34, 66) reported the use of wild-type littermates, while the remaining groups used the recommended control animals from Jackson (strain No. 000664).
The purpose of the first published NOX2 stroke study was to elucidate the role of NADPH oxidases in neutrophil-derived ROS in stroke injury. The authors investigated whether deleterious NOX2 originates from influx of peripheral leucocytes and/or from activation of on-site microglia. Although brain lesions were reduced by 46% in NOX2 KO mice when compared with wild-type littermates, the amount of neutrophils migrating into the brain was the same in NOX2 KO and wild-type mice. This raised the question whether NOX2 implicated in brain damage originates from brain tissue (neuronal or vascular origin) or from inflammatory cells. Therefore, the authors transplanted bone marrow from wild-type into NOX2 KO mice and vice versa, thus eliminating the influence from circulating neutrophil-derived NOX2. Either way, they observed no difference in infarct size, concluding that the detrimental role of NOX2 involves the presence of peripheral neutrophils in the infarcted brain (66). Fourteen years later, Tang et al. repeated these experiments (63). Unlike the procedure used in the first study of Walder et al. (66), Tang et al. assessed different time points after bone marrow transplantation and shielded the animal's head to avoid a confounding induction of neuronal damage and microglia activation by irradiation. They concluded that NOX2 from peripheral immune cells contributes more to neurodegeneration than does microglia-derived NOX2 (63).
In NOX2 KO mice, the peak levels of ROS formation (72 h poststroke) were attenuated and so were infarct volumes (34). However, ROS were still detectable at that time, and other sources of ROS (such as other NOX homologues) may still play a role. With regard to infarct size observed in NOX2 KO animals, the published data are conflicting. Most studies suggest a protective effect of NOX2 deletion on stroke outcomes. Initially, we could not reproduce these findings, although we used a high number of animals and followed the STAIR guidelines (33). More recently, we performed further stroke experiments in an older cohort of NOX2 KO mice (20 weeks) in two different labs, amounting to a total of n=21 wild-type and n=14 KO mice. Pooling data from both centers, we observed a small reduction in infarct size (Radermacher et al., submitted for publication). Thus, the NOX2 isoform seems to play a role in stroke pathology in elderly mice, probably via its role in inflammatory processes. However, the role of NOX2 appears to be more modest when compared with that of NOX4. This and a possible publication bias against studies with no or smaller effect sizes (45) may explain some of the conflicting literature. Based on these divergent results, a meta-analysis including all stroke studies performed in NOX2 KO mice would be of interest. It may be that there are other studies that did not find any protection by deleting NOX2, which have not been published. Significantly, such a potential publication bias should be revealed by a funnel plot. This graphical method plots the result of each study (positive or negative) against the sample size of the same study, which—in the ideal case—results in an inverted funnel shape peaking at the real effect size. An asymmetry of this funnel indicates that studies are missing, either because they were not found by the search strategy of the systematic review or as they have not been published. This is important, because a meta-analysis will only pool published data, thereby making a null effect seem real. This analysis should not only be applied for the case of NOX2 KO mice, but would also be interesting for NOX1 and NOX4 KO mice, once more stroke studies have been published.
One important confounder that may explain the qualitatively different results could be inflammation. In human stroke, the inflammatory response is thought to be at least partly initiated to remove dead tissue. Factors that could potentially influence the extent of inflammation and contribution of NOX2 are the degree of aseptic surgery or a housing effect (NOX2 KO mice are immune compromised and develop granulomas). The more prominent the inflammation is, the more prominent the role of NOX2 may become. Possibly, the inflammation may also have the opposite effect by distracting neutrophils that are removed from dead tissue to fight bacteria. Interestingly, a paradoxical anti-inflammatory effect of NOX2 was observed in autoimmune diseases (27), which further complicates the considerations just mentioned.
After the publication of updated STAIR criteria, some research groups started re-investigating the role of NOX2 under influence of gender or comorbidities. Brait et al. (8) showed that NOX2-dependent ROS production from T-lymphocytes after stroke is increased during reperfusion, and that NOX2 only affects lesion size in male but not in female mice, possibly because of estrogen having anti-inflammatory effects. Since hypercholesterolemia is a frequent comorbidity in stroke patients, another group tested the neuroprotective drug betulinic acid in hypercholesterolemia-developing ApoE KO mice, which had larger infarcts than wild-type mice. Betulinic acid seemed to protect against stroke, probably via NOX2, neuronal NOS, and inflammatory NOS down-regulation (40).
In conclusion, it is likely that both isoforms, NOX2 and NOX4, are, to some extent, implicated in cerebral I/R injury, depending on as yet unclear parameters. However, due to higher neuroprotection that we have observed in NOX4 KO mice and due to potential immunosuppressive effects of NOX2 inhibition, we believe that NOX4 is the more promising target for stroke therapy.
Pharmacological Approaches to Inhibit NOX Activity
In addition to using genetic animal models, another and clinically more relevant option for validating NOX enzymes as therapeutic targets is the use of pharmacological compounds that selectively suppress NADPH oxidase activity. The most widely used substances that inhibit NOX have been apocynin and diphenylene iodonium (DPI) (Fig. 5). DPI already showed neuroprotection in vivo when administered poststroke (43). However, its usefulness, initially demonstrated as an inhibitor of phagocyte-related NOX, is limited, as it also interacts with NOS and other flavoproteins (69). Apocynin, a polyphenolic molecule with anti-inflammatory properties, is thought to block the assembly of p47phox and NOX2 and has to be activated by myeloperoxidases, which are not present in all cells. The interpretation of studies using apocynin is further complicated by its antioxidant capacity (25) and inhibition of Rho kinase (52). Thus, classical NOX inhibitors are not specific for NADPH oxidases, and, therefore, any results on the role of NADPH oxidase obtained by using apocynin or DPI should be questioned (43, 57, 69, 71). A more specific NOX inhibitor is the triazolopyrimidine VAS2870 (Fig. 5), which already proved its protective effects in preclinical stroke experiments (33). VAS2870 and its derivate VAS3947 likely inhibit all NOX isoforms and are neither general flavoprotein inhibitors nor ROS scavengers (64, 69). Mechanistically, VAS2870 seems to be a pan-NOX inhibitor (70), as it inhibits assembly or conformational changes to active NOX complexes (3). When a complex assembly of NOX2 with its cytosolic binding proteins was induced before the addition of VAS2870, no inhibition of NOX activity was observed (3, 21). When VAS2870 was added before complex assembly induction, inhibition of NOX2 activity was observed (3). Therefore, the order by which a NOX complex assembly activator and a NOX inhibitor are added in cell-free assays is crucial. Very recently, potential off-target effects of VAS2870 have been published, suggesting that the compound alkylates cysteine thiol residues (61). The authors suggest that thio-alkylation of cysteine residues may play a role in NOX inhibition. Nevertheless, the importance of this off-target alkylation in the presence of physiological glutathione concentrations and the occurrence of other off-target effects of VAS2870 need to be further analyzed in a more extended off-target screen.
FIG. 5.

NOX inhibitors assessed for their neuroprotective potential. The figure displays chemical structures of NOX-inhibiting compounds that have been tested as therapeutics in cerebral infarction. All of them have been shown to successfully reduce infarct size after ischemic stroke. Since apocynin and diphenylene iodonium (DPI) are not sufficiently specific for NADPH oxidases, their application in vivo is limited due to many off-target effects. VAS2870 seems to be more specific for NOX, although off-target effects cannot be excluded and due to poor solubility, the intrathecal mode of administration is used, which is not optimal.
Beside VAS compounds, which are general NOX inhibitors, NOX isoform-specific inhibitors may represent a better tool for assessing the role of individual NOX isoforms and also for developing experimental therapeutics. One approach was to use peptides, such as the chimeric peptide, gp91ds-tat (50), which combines the p47phox docking sequence of NOX2 with the cell penetrating tat peptide (Fig. 6). This peptide not only prevents NOX assembly and is relatively specific for NOX2, but also potentially inhibits NOX1 (50). However, off-target effects of gp91ds-tat have been described in vivo (2). More recently, a selective NOX1/4 inhibitor, the pyrazolopyridine derivate GKT136901, was introduced (Fig. 6) (36).
FIG. 6.

Promising NOX inhibitors to be tested in preclinical stroke research. The VAS2870 derivate VAS3947 does not differ from VAS2870 in its NOX inhibition profile, but it may be better suited for in vivo application because of its better solubility. The peptidic inhibitor gp91ds-tat interferes with the assembly between NOX2 and p47phox, which renders it NOX2 selective. An inhibition of NOX1 could also be imaginable, as the NOX1 activity also depends on subunit regulation by p47phox. Another promising NOX inhibitor, GKT136901, has been reported to mainly inhibit NOX1 and NOX4.
To validate NADPH oxidases as a target for stroke therapy, apocynin has mostly been used as NOX inhibitor. Jackman et al. (29) showed that apocynin given prestroke caused a reduction in infarct size, but not when administered poststroke. Indeed, in most studies, apocynin was given before stroke onset (32, 62, 67), a procedure that does not allow statements on therapeutic relevance in a clinical setting, where any therapy has to work poststroke. Tang et al. did not find any additional neuroprotection in apocynin-treated mice on top of NOX2 gene deletion (63). Does this exclude a role for another NOX isoform in ischemia-induced brain damage? This is probably not the case, as apocynin interferes with p47phox binding to NOX and NOX activation. However, NOX4 does not seem to depend on p47phox (41). Neuroprotective effects of the NOX inhibitor VAS2870 have been demonstrated when injected intrathecally into the spinal liquor space of wild-type mice, but did not mediate any further protection in NOX4 KO mice, suggesting that NOX4 is the major detrimental player (33). Nevertheless, stroke experiments with other, more isoform-specific NOX inhibitors would be of great interest. Alternatively, NOX activity can also be repressed by using small interfering RNAs (3), but the interference of siRNA with other than the intended NOX isoform has to be excluded (53).
Conclusion
In conclusion, NOX4 and, possibly to a lesser extent, NOX2 appear to play a role in ischemic stroke. Both are up-regulated in human stroke, and experiments with experimental compounds suggest that NOX inhibition may be relevant for a highly innovative neuroprotective poststroke therapy. Clearly, further studies using different KO models and inhibitors in different species are needed to fully validate the NOX isoform that is the most promising target and a possible time window for further development.
Abbreviations Used
- BBB
blood-brain barrier
- CT
computed tomography
- DPI
diphenylene iodonium
- FAD
flavin adenine dinucleotide
- I/R
ischemia-reperfusion
- KO
knockout
- MCAO
middle cerebral artery occlusion
- NADPH
nicotinamide adenine dinucleotide phosphate
- NOS
nitric oxide synthase
- NOX
catalytic subunit of NADPH oxidases
- ROS
reactive oxygen species
- rt-PA
recombinant tissue plasminogen activator
- STAIR
Stroke Therapy Academic Industry Roundtable
Acknowledgments
The authors wish to acknowledge national and international competitive grant support by the NHMRC, EU and ERC (HHHW), the Brain Foundation of the Netherlands (KR), and DFG (CK). Work performed at the German Mouse Clinic was funded by grants from the German Federal Ministry of Education and Research (BMBF, NGFN-Plus grants No. 01GS0850, 01GS0851, and 01GS0854).
Author Disclosure Statement
H.H.H.W.S. is a cofounder of Vasopharm GmbH, which is developing NOX inhibitors. C.K. has received funding from Novartis Pharma GmbH, Nürnberg, Germany, to conduct research on NADPH oxidase.
References
- 1.Ago T. Kitazono T. Kuroda J. Kumai Y. Kamouchi M. Ooboshi H. Wakisaka M. Kawahara T. Rokutan K. Ibayashi S. Iida M. NAD(P)H oxidases in rat basilar arterial endothelial cells. Stroke. 2005;36:1040–1046. doi: 10.1161/01.STR.0000163111.05825.0b. [DOI] [PubMed] [Google Scholar]
- 2.Aldieri E. Riganti C. Polimeni M. Gazzano E. Lussiana C. Campia I. Ghigo D. Classical inhibitors of NOX NAD(P)H oxidases are not specific. Curr Drug Metab. 2008;9:686–696. doi: 10.2174/138920008786049285. [DOI] [PubMed] [Google Scholar]
- 3.Altenhofer S. Kleikers PW. Radermacher KA. Scheurer P. Rob Hermans JJ. Schiffers P. Ho H. Wingler K. Schmidt HH. The NOX toolbox: validating the role of NADPH oxidases in physiology and disease. Cell Mol Life Sci. 2012;69:2327–2343. doi: 10.1007/s00018-012-1010-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anilkumar N. Weber R. Zhang M. Brewer A. Shah AM. Nox4 and nox2 NADPH oxidases mediate distinct cellular redox signaling responses to agonist stimulation. Arterioscler Thromb Vasc Biol. 2008;28:1347–1354. doi: 10.1161/ATVBAHA.108.164277. [DOI] [PubMed] [Google Scholar]
- 5.Arimura K. Ago T. Kuroda J. Ishitsuka K. Nishimura A. Sugimori H. Kamouchi M. Sasaki T. Kitazono T. Role of NADPH oxidase 4 in Brain Endothelial cells after Ischemic Stroke. Stroke. 2012;43:A2514. [Google Scholar]
- 6.Bedard K. Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 7.Ben Mkaddem S. Pedruzzi E. Werts C. Coant N. Bens M. Cluzeaud F. Goujon JM. Ogier-Denis E. Vandewalle A. Heat shock protein gp96 and NAD(P)H oxidase 4 play key roles in Toll-like receptor 4-activated apoptosis during renal ischemia/reperfusion injury. Cell Death Differ. 2010;17:1474–1485. doi: 10.1038/cdd.2010.26. [DOI] [PubMed] [Google Scholar]
- 8.Brait VH. Jackman KA. Walduck AK. Selemidis S. Diep H. Mast AE. Guida E. Broughton BR. Drummond GR. Sobey CG. Mechanisms contributing to cerebral infarct size after stroke: gender, reperfusion, T lymphocytes, and Nox2-derived superoxide. J Cereb Blood Flow Metab. 2010;30:1306–1317. doi: 10.1038/jcbfm.2010.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carnesecchi S. Deffert C. Donati Y. Basset O. Hinz B. Preynat-Seauve O. Guichard C. Arbiser JL. Banfi B. Pache JC. Barazzone-Argiroffo C. Krause KH. A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxid Redox Signal. 2011;15:607–619. doi: 10.1089/ars.2010.3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan PH. Role of oxidants in ischemic brain damage. Stroke. 1996;27:1124–1129. doi: 10.1161/01.str.27.6.1124. [DOI] [PubMed] [Google Scholar]
- 11.Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21:2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- 12.Chen H. Kim GS. Okami N. Narasimhan P. Chan PH. NADPH oxidase is involved in post-ischemic brain inflammation. Neurobiol Dis. 2011;42:341–348. doi: 10.1016/j.nbd.2011.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen H. Song YS. Chan PH. Inhibition of NADPH oxidase is neuroprotective after ischemia-reperfusion. J Cereb Blood Flow Metab. 2009;29:1262–1272. doi: 10.1038/jcbfm.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Den Hertog HM. van der Worp HB. Tseng MC. Dippel DW. Cooling therapy for acute stroke. Cochrane Database Syst Rev. 2009:CD001247. doi: 10.1002/14651858.CD001247.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diener HC. Lees KR. Lyden P. Grotta J. Davalos A. Davis SM. Shuaib A. Ashwood T. Wasiewski W. Alderfer V. Hardemark HG. Rodichok L. NXY-059 for the treatment of acute stroke: pooled analysis of the SAINT I and II Trials. Stroke. 2008;39:1751–1758. doi: 10.1161/STROKEAHA.107.503334. [DOI] [PubMed] [Google Scholar]
- 16.Dirnagl U. Fisher M. International, multicenter randomized preclinical trials in translational stroke research: It's time to act. J Cereb Blood Flow Metab. 2012;32:933–935. doi: 10.1038/jcbfm.2012.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dirnagl U. Fisher M. REPRINT: international, multicenter randomized preclinical trials in translational stroke research: it is time to act. Stroke. 2012;43:1453–1454. doi: 10.1161/STROKEAHA.112.653709. [DOI] [PubMed] [Google Scholar]
- 18.Eisener-Dorman AF. Lawrence DA. Bolivar VJ. Cautionary insights on knockout mouse studies: the gene or not the gene? Brain Behav Immun. 2009;23:318–324. doi: 10.1016/j.bbi.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fisher M. Feuerstein G. Howells DW. Hurn PD. Kent TA. Savitz SI. Lo EH. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke. 2009;40:2244–2250. doi: 10.1161/STROKEAHA.108.541128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flamm ES. Demopoulos HB. Seligman ML. Poser RG. Ransohoff J. Free radicals in cerebral ischemia. Stroke. 1978;9:445–447. doi: 10.1161/01.str.9.5.445. [DOI] [PubMed] [Google Scholar]
- 21.Gatto GJ. Ao Z. Kearse MG. Zhou M. Morales CR. Daniels E. Bradley BT. Goserud MT. Goodman KB. Douglas SA. Harpel MR. Johns DG. NADPH oxidase-dependent and -independent mechanisms of reported inhibitors of reactive oxygen generation. J Enzyme Inhibit Med Chem. 2011 doi: 10.3109/14756366.2011.636360. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 22.Goldstein LB. Bushnell CD. Adams RJ. Appel LJ. Braun LT. Chaturvedi S. Creager MA. Culebras A. Eckel RH. Hart RG. Hinchey JA. Howard VJ. Jauch EC. Levine SR. Meschia JF. Moore WS. Nixon JV. Pearson TA. Guidelines for the primary prevention of stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42:517–584. doi: 10.1161/STR.0b013e3181fcb238. [DOI] [PubMed] [Google Scholar]
- 23.Green SP. Cairns B. Rae J. Errett-Baroncini C. Hongo JA. Erickson RW. Curnutte JT. Induction of gp91-phox, a component of the phagocyte NADPH oxidase, in microglial cells during central nervous system inflammation. J Cereb Blood Flow Metab. 2001;21:374–384. doi: 10.1097/00004647-200104000-00006. [DOI] [PubMed] [Google Scholar]
- 24.Halliwell B. Phagocyte-derived reactive species: salvation or suicide? Trends Biochem Sci. 2006;31:509–515. doi: 10.1016/j.tibs.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 25.Heumuller S. Wind S. Barbosa-Sicard E. Schmidt HH. Busse R. Schroder K. Brandes RP. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008;51:211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]
- 26.Howells DW. Porritt MJ. Rewell SS. O'Collins V. Sena ES. van der Worp HB. Traystman RJ. Macleod MR. Different strokes for different folks: the rich diversity of animal models of focal cerebral ischemia. J Cereb Blood Flow Metab. 2010;30:1412–1431. doi: 10.1038/jcbfm.2010.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hultqvist M. Olsson LM. Gelderman KA. Holmdahl R. The protective role of ROS in autoimmune disease. Trends Immunol. 2009;30:201–208. doi: 10.1016/j.it.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 28.Infanger DW. Sharma RV. Davisson RL. NADPH oxidases of the brain: distribution, regulation, and function. Antioxid Redox Signal. 2006;8:1583–1596. doi: 10.1089/ars.2006.8.1583. [DOI] [PubMed] [Google Scholar]
- 29.Jackman KA. Miller AA. De Silva TM. Crack PJ. Drummond GR. Sobey CG. Reduction of cerebral infarct volume by apocynin requires pretreatment and is absent in Nox2-deficient mice. Br J Pharmacol. 2009;156:680–688. doi: 10.1111/j.1476-5381.2008.00073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jackman KA. Miller AA. Drummond GR. Sobey CG. Importance of NOX1 for angiotensin II-induced cerebrovascular superoxide production and cortical infarct volume following ischemic stroke. Brain Res. 2009;1286:215–220. doi: 10.1016/j.brainres.2009.06.056. [DOI] [PubMed] [Google Scholar]
- 31.Kahles T. Kohnen A. Heumueller S. Rappert A. Bechmann I. Liebner S. Wittko IM. Neumann-Haefelin T. Steinmetz H. Schroeder K. Brandes RP. NADPH oxidase Nox1 contributes to ischemic injury in experimental stroke in mice. Neurobiol Dis. 2010;40:185–192. doi: 10.1016/j.nbd.2010.05.023. [DOI] [PubMed] [Google Scholar]
- 32.Kahles T. Luedike P. Endres M. Galla HJ. Steinmetz H. Busse R. Neumann-Haefelin T. Brandes RP. NADPH oxidase plays a central role in blood-brain barrier damage in experimental stroke. Stroke. 2007;38:3000–3006. doi: 10.1161/STROKEAHA.107.489765. [DOI] [PubMed] [Google Scholar]
- 33.Kleinschnitz C. Grund H. Wingler K. Armitage ME. Jones E. Mittal M. Barit D. Schwarz T. Geis C. Kraft P. Barthel K. Schuhmann MK. Herrmann AM. Meuth SG. Stoll G. Meurer S. Schrewe A. Becker L. Gailus-Durner V. Fuchs H. Klopstock T. de Angelis MH. Jandeleit-Dahm K. Shah AM. Weissmann N. Schmidt HH. Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol. 2010;8:e1000479. doi: 10.1371/journal.pbio.1000479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kunz A. Anrather J. Zhou P. Orio M. Iadecola C. Cyclooxygenase-2 does not contribute to postischemic production of reactive oxygen species. J Cereb Blood Flow Metab. 2007;27:545–551. doi: 10.1038/sj.jcbfm.9600369. [DOI] [PubMed] [Google Scholar]
- 35.Kuroda J. Ago T. Matsushima S. Zhai P. Schneider MD. Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci U S A. 2010;107:15565–15570. doi: 10.1073/pnas.1002178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laleu B. Gaggini F. Orchard M. Fioraso-Cartier L. Cagnon L. Houngninou-Molango S. Gradia A. Duboux G. Merlot C. Heitz F. Szyndralewiez C. Page P. First in class, potent, and orally bioavailable NADPH oxidase isoform 4 (Nox4) inhibitors for the treatment of idiopathic pulmonary fibrosis. J Med Chem. 2010;53:7715–7730. doi: 10.1021/jm100773e. [DOI] [PubMed] [Google Scholar]
- 37.Lees KR. Zivin JA. Ashwood T. Davalos A. Davis SM. Diener HC. Grotta J. Lyden P. Shuaib A. Hardemark HG. Wasiewski WW. NXY-059 for acute ischemic stroke. N Engl J Med. 2006;354:588–600. doi: 10.1056/NEJMoa052980. [DOI] [PubMed] [Google Scholar]
- 38.Levy DE. del Zoppo GJ. Demaerschalk BM. Demchuk AM. Diener HC. Howard G. Kaste M. Pancioli AM. Ringelstein EB. Spatareanu C. Wasiewski WW. Ancrod in acute ischemic stroke: results of 500 subjects beginning treatment within 6 hours of stroke onset in the ancrod stroke program. Stroke. 2009;40:3796–3803. doi: 10.1161/STROKEAHA.109.565119. [DOI] [PubMed] [Google Scholar]
- 39.Loukogeorgakis SP. van den Berg MJ. Sofat R. Nitsch D. Charakida M. Haiyee B. de Groot E. MacAllister RJ. Kuijpers TW. Deanfield JE. Role of NADPH oxidase in endothelial ischemia/reperfusion injury in humans. Circulation. 2010;121:2310–2316. doi: 10.1161/CIRCULATIONAHA.108.814731. [DOI] [PubMed] [Google Scholar]
- 40.Lu Q. Xia N. Xu H. Guo L. Wenzel P. Daiber A. Munzel T. Forstermann U. Li H. Betulinic acid protects against cerebral ischemia-reperfusion injury in mice by reducing oxidative and nitrosative stress. Nitric Oxide Biol Chem. 2011;24:132–138. doi: 10.1016/j.niox.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 41.Martyn KD. Frederick LM. von Loehneysen K. Dinauer MC. Knaus UG. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal. 2006;18:69–82. doi: 10.1016/j.cellsig.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 42.Miller AA. Drummond GR. Schmidt HH. Sobey CG. NADPH oxidase activity and function are profoundly greater in cerebral versus systemic arteries. Circ Res. 2005;97:1055–1062. doi: 10.1161/01.RES.0000189301.10217.87. [DOI] [PubMed] [Google Scholar]
- 43.Nagel S. Genius J. Heiland S. Horstmann S. Gardner H. Wagner S. Diphenyleneiodonium and dimethylsulfoxide for treatment of reperfusion injury in cerebral ischemia of the rat. Brain Res. 2007;1132:210–217. doi: 10.1016/j.brainres.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 44.O'Collins VE. Donnan GA. Howells DW. History of animal models of stroke. Stroke. 2011;6:77–78. doi: 10.1111/j.1747-4949.2010.00543.x. [DOI] [PubMed] [Google Scholar]
- 45.O'Collins VE. Macleod MR. Donnan GA. Horky LL. van der Worp BH. Howells DW. 1,026 experimental treatments in acute stroke. Ann Neurol. 2006;59:467–477. doi: 10.1002/ana.20741. [DOI] [PubMed] [Google Scholar]
- 46.Opitz N. Drummond GR. Selemidis S. Meurer S. Schmidt HH. The ‘A's and ‘O's of NADPH oxidase regulation: a commentary on “Subcellular localization and function of alternatively spliced Noxo1 isoforms”. Free Radic Biol Med. 2007;42:175–179. doi: 10.1016/j.freeradbiomed.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 47.Pollock JD. Williams DA. Gifford MA. Li LL. Du X. Fisherman J. Orkin SH. Doerschuk CM. Dinauer MC. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet. 1995;9:202–209. doi: 10.1038/ng0295-202. [DOI] [PubMed] [Google Scholar]
- 48.Rada B. Leto TL. Oxidative innate immune defenses by Nox/Duox family NADPH oxidases. Contrib Microbiol. 2008;15:164–187. doi: 10.1159/000136357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Radermacher KA. Wingler K. Kleikers PW. Altenhofer SA. Hermans JR. Kleinschnitz C. Schmidt HH. The 1027th target candidate in stroke: Will NADPH oxidase hold up? Exp Transl Stroke Med. 2012;4:11. doi: 10.1186/2040-7378-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rey FE. Cifuentes ME. Kiarash A. Quinn MT. Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(-) and systolic blood pressure in mice. Circ Res. 2001;89:408–414. doi: 10.1161/hh1701.096037. [DOI] [PubMed] [Google Scholar]
- 51.Roger VL. Go AS. Lloyd-Jones DM. Benjamin EJ. Berry JD. Borden WB. Bravata DM. Dai S. Ford ES. Fox CS. Fullerton HJ. Gillespie C. Hailpern SM. Heit JA. Howard VJ. Kissela BM. Kittner SJ. Lackland DT. Lichtman JH. Lisabeth LD. Makuc DM. Marcus GM. Marelli A. Matchar DB. Moy CS. Mozaffarian D. Mussolino ME. Nichol G. Paynter NP. Soliman EZ. Sorlie PD. Sotoodehnia N. Turan TN. Virani SS. Wong ND. Woo D. Turner MB. Heart disease and stroke statistics—2012 update: a report from the american heart association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schluter T. Steinbach AC. Steffen A. Rettig R. Grisk O. Apocynin-induced vasodilation involves Rho kinase inhibition but not NADPH oxidase inhibition. Cardiovasc Res. 2008;80:271–279. doi: 10.1093/cvr/cvn185. [DOI] [PubMed] [Google Scholar]
- 53.Schroder K. Wandzioch K. Helmcke I. Brandes RP. Nox4 acts as a switch between differentiation and proliferation in preadipocytes. Arterioscler Thromb Vasc Biol. 2009;29:239–245. doi: 10.1161/ATVBAHA.108.174219. [DOI] [PubMed] [Google Scholar]
- 54.Sena E. van der Worp HB. Howells D. Macleod M. How can we improve the pre-clinical development of drugs for stroke? Trends Neurosci. 2007;30:433–439. doi: 10.1016/j.tins.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 55.Sena ES. van der Worp HB. Bath PM. Howells DW. Macleod MR. Publication bias in reports of animal stroke studies leads to major overstatement of efficacy. PLoS Biol. 2010;8:e1000344. doi: 10.1371/journal.pbio.1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shuaib A. Lees KR. Lyden P. Grotta J. Davalos A. Davis SM. Diener HC. Ashwood T. Wasiewski WW. Emeribe U. NXY-059 for the treatment of acute ischemic stroke. N Engl J Med. 2007;357:562–571. doi: 10.1056/NEJMoa070240. [DOI] [PubMed] [Google Scholar]
- 57.Simonyi A. Serfozo P. Lehmidi TM. Cui J. Gu Z. Lubahn DB. Sun AY. Sun GY. The neuroprotective effects of apocynin. Front Biosci. 2012;4:2183–2193. doi: 10.2741/535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sirker A. Zhang M. Shah AM. NADPH oxidases in cardiovascular disease: insights from in vivo models and clinical studies. Basic Res Cardiol. 2011;106:735–747. doi: 10.1007/s00395-011-0190-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sorce S. Krause KH. NOX enzymes in the central nervous system: from signaling to disease. Antioxid Redox Signal. 2009;11:2481–2504. doi: 10.1089/ars.2009.2578. [DOI] [PubMed] [Google Scholar]
- 60.STAIR. Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke. 1999;30:2752–2758. doi: 10.1161/01.str.30.12.2752. [DOI] [PubMed] [Google Scholar]
- 61.Sun QA. Hess DT. Wang B. Miyagi M. Stamler JS. Off-target thiol alkylation by the NADPH oxidase inhibitor 3-benzyl-7-(2-benzoxazolyl)thio-1,2,3-triazolo[4,5-d]pyrimidine (VAS2870) Free Radic Biol Med. 2012;52:1897–1902. doi: 10.1016/j.freeradbiomed.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tang XN. Cairns B. Cairns N. Yenari MA. Apocynin improves outcome in experimental stroke with a narrow dose range. Neuroscience. 2008;154:556–562. doi: 10.1016/j.neuroscience.2008.03.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tang XN. Zheng Z. Giffard RG. Yenari MA. Significance of marrow-derived nicotinamide adenine dinucleotide phosphate oxidase in experimental ischemic stroke. Ann Neurol. 2011;70:606–615. doi: 10.1002/ana.22476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.ten Freyhaus H. Huntgeburth M. Wingler K. Schnitker J. Baumer AT. Vantler M. Bekhite MM. Wartenberg M. Sauer H. Rosenkranz S. Novel Nox inhibitor VAS2870 attenuates PDGF-dependent smooth muscle cell chemotaxis, but not proliferation. Cardiovasc Res. 2006;71:331–341. doi: 10.1016/j.cardiores.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 65.Vallet P. Charnay Y. Steger K. Ogier-Denis E. Kovari E. Herrmann F. Michel JP. Szanto I. Neuronal expression of the NADPH oxidase NOX4, and its regulation in mouse experimental brain ischemia. Neuroscience. 2005;132:233–238. doi: 10.1016/j.neuroscience.2004.12.038. [DOI] [PubMed] [Google Scholar]
- 66.Walder CE. Green SP. Darbonne WC. Mathias J. Rae J. Dinauer MC. Curnutte JT. Thomas GR. Ischemic stroke injury is reduced in mice lacking a functional NADPH oxidase. Stroke. 1997;28:2252–2258. doi: 10.1161/01.str.28.11.2252. [DOI] [PubMed] [Google Scholar]
- 67.Wang Q. Tompkins KD. Simonyi A. Korthuis RJ. Sun AY. Sun GY. Apocynin protects against global cerebral ischemia-reperfusion-induced oxidative stress and injury in the gerbil hippocampus. Brain Res. 2006;1090:182–189. doi: 10.1016/j.brainres.2006.03.060. [DOI] [PubMed] [Google Scholar]
- 68.Warner DS. Sheng H. Batinic-Haberle I. Oxidants, antioxidants and the ischemic brain. J Exp Biol. 2004;207:3221–3231. doi: 10.1242/jeb.01022. [DOI] [PubMed] [Google Scholar]
- 69.Wind S. Beuerlein K. Eucker T. Muller H. Scheurer P. Armitage ME. Ho H. Schmidt HH. Wingler K. Comparative pharmacology of chemically distinct NADPH oxidase inhibitors. Br J Pharmacol. 2010;161:885–898. doi: 10.1111/j.1476-5381.2010.00920.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wingler K. Altenhoefer SA. Kleikers PW. Radermacher KA. Kleinschnitz C. Schmidt HH. VAS2870 is a pan-NADPH oxidase inhibitor. Cell Mol Life Sci. 2012;69:3159–3160. doi: 10.1007/s00018-012-1107-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wingler K. Hermans J. Schiffers P. Moens A. Paul M. Schmidt H. NOX 1, 2, 4, 5: Counting out oxidative stress. Br J Pharmacol. 2011;164:866–883. doi: 10.1111/j.1476-5381.2011.01249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang M. Brewer AC. Schroder K. Santos CX. Grieve DJ. Wang M. Anilkumar N. Yu B. Dong X. Walker SJ. Brandes RP. Shah AM. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc Natl Acad Sci U S A. 2010;107:18121–18126. doi: 10.1073/pnas.1009700107. [DOI] [PMC free article] [PubMed] [Google Scholar]