

Summary
The oncogenic epithelial–mesenchymal transition (EMT) contributes to tumor progression in various context-dependent ways, including increased metastatic potential, expansion of cancer stem cell subpopulations, chemo-resistance and disease recurrence. One of the hallmarks of EMT is resistance of tumor cells to anoikis. This resistance contributes to metastasis and is a defining property not only of EMT but also of cancer stem cells. Here, we review the mechanistic coupling between EMT and resistance to anoikis. The discussion focuses on several key aspects. First, we provide an update on new pathways that lead from the loss of E-cadherin to anoikis resistance. We then discuss the relevance of transcription factors that are crucial in wound healing in the context of oncogenic EMT. Next, we explore the consequences of the breakdown of cell-polarity complexes upon anoikis sensitivity, through the Hippo, Wnt and transforming growth factor β (TGF-β) pathways, emphasizing points of crossregulation. Finally, we summarize the direct regulation of cell survival genes through EMT-inducing transcription factors, and the roles of the tyrosine kinases focal adhesion kinase (FAK) and TrkB neurotrophin receptor in EMT-related regulation of anoikis. Emerging from these studies are unifying principles that will lead to improvements in cancer therapy by reprogramming sensitivity of anoikis.
Key words: EMT, Anoikis, Cancer
Introduction
Epithelial tissues with a high turnover, for example, in the gastrointestinal tract and skin, shed billions of epithelial cells per minute. Intuitively, there is a need for cells that are shed in this manner to be eliminated as, otherwise, we would be in constant danger that they attach at inappropriate sites, leading to widespread neoplastic growth. This problem would be exacerbated by precancerous mutations or epigenetic changes that occurred in the epithelial cells prior to shedding.
About eighteen years ago, two publications showed that epithelial cells and endothelial cells depend on appropriate cell–matrix interactions for their survival, confirming this intuition (Frisch and Francis, 1994; Meredith et al., 1993). The transformation of epithelial cells by oncogenes was further shown to confer resistance to the apoptosis phenomenon termed anoikis, which occurs when cells lost contact with their extracellular matrix. This definition of anoikis was later broadened to include any apoptosis that is inhibited by appropriate cell–matrix interactions. Intriguingly, a ubiquitously acting tumor suppressor, adenovirus E1a, was shown to restore sensitivity to anoikis (Frisch and Francis, 1994).
Further extension and refinement of research since then has demonstrated that, in the context of cancer, resistance to anoikis, a hallmark of the oncogenic epithelial–mesenchymal transition (EMT), is required for tumor metastasis (Guadamillas et al., 2011; Tiwari et al., 2012). The mechanistic connections between EMT and anoikis resistance are the focus of this Commentary. Here, we address the specific question that is posed in Fig. 1: how does the oncogenic EMT confer resistance to anoikis, and can this be exploited to develop novel modalities for cancer therapy? We focus on the interactions between various signaling pathways that regulate the cellular commitment to EMT, in turn modulating cell-survival-related gene expression programs that adjust the setpoint for anoikis.
Fig. 1.
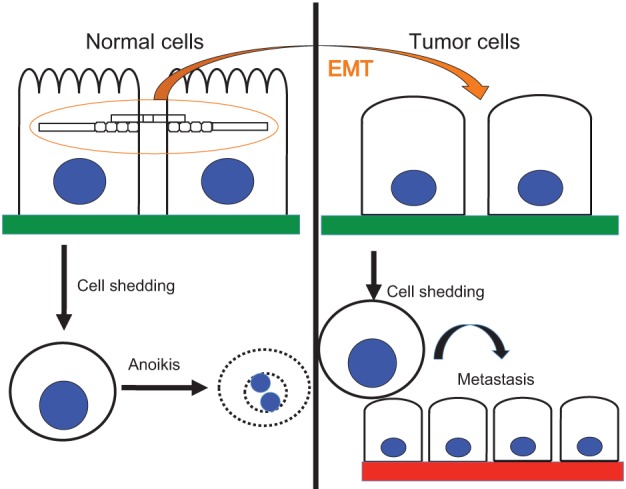
EMT programs anoikis resistance. Mechanisms that connect these phenomena are discussed in this Commentary. The intercellular bridge protein depicted in this diagram is a generalized intercellular component of junctional complexes (e.g. E-cadherin, desmoplakin, desmoglein, claudin) and a generalized cytoplasmic or cytoskeletal element is shown transducing its signals.
EMT
The defining characteristic of epithelial cells is their propensity to exist as a layer of extensively communicating cells, which manifests itself as the familiar cobblestone pattern that can be seen in two-dimensional cell culture. Specific epithelial proteins such as E-cadherin, occludins, claudins, desmogleins and desmocollins are located at cellular junctions, which promote and maintain the integrity of epithelial layers (Baum and Georgiou, 2011; Dusek and Attardi, 2011; Turksen and Troy, 2011). In vivo epithelial cells are typically polarized with one side attached to extracellular matrix and adjacent cells, and the other facing the lumen of a vessel. Maintenance of the polarity is achieved by the tight junctions through claudins and occludins, which prevent integral membrane proteins from diffusing between the apical and basolateral membranes (Yeaman et al., 1999). The appropriate localization of these junctional proteins in epithelia is regulated by epithelial-specific cytosolic proteins, such as ankyrin G and Rab25 (Kizhatil et al., 2007a; Kizhatil et al., 2007b; Senga et al., 2012). The master regulators of the epithelial gene expression program have recently been identified; among these are the transcription factors grainyhead-like protein 2 homolog (GRHL2) and GATA3, the mRNA splicing factors ESRP1 and ESRP2, and the mir200 family of micro-RNAs (Cieply et al., 2012; Gregory et al., 2008; Kouros-Mehr et al., 2008; Warzecha et al., 2010; Werth et al., 2010).
Separation of a normal epithelial cell from its matrix results in anoikis. Epithelial cells can be re-programmed, however, so that they no longer require these attachments for their survival, through a process known as EMT. The defining characteristics of EMT in culture are (i) deviation from the cobblestone morphology, owing to loss of cell adhesion molecules such as E-cadherin; (ii) loss of epithelium-specific genes; (iii) gain of mesenchymum-specific genes and (iv) loss of sensitivity to anoikis (Kalluri and Weinberg, 2009; Scheel and Weinberg, 2011; Tiwari et al., 2012). Enhanced migratory capacity of cells is commonly used as a marker of EMT; however this feature is context-dependent as epithelial sheets can migrate as well or better than individual mesenchymal cells (Matise et al., 2012). EMT is observed in vivo during the delamination of the neural crest or in the invasive front of carcinoma, which are both examples of cells that detach from their epithelial layer and migrate (Brabletz et al., 2001; Theveneau and Mayor, 2012). The factors and microenvironmental cues that regulate EMT, and its significance to development and disease have been reviewed extensively elsewhere (Guarino et al., 2007; Kalluri and Weinberg, 2009; Klymkowsky and Savagner, 2009; Thiery et al., 2009; Weinberg, 2008). Here, we focus on the gene expression and cell signaling events that are associated with EMT and that contribute to anoikis resistance.
Anoikis
The term anoikis is used to distinguish a scenario of apoptosis – for example, the response to cell–matrix detachment – not a mechanism. In fact, many of the apoptotic mechanisms that occur in response to other cellular stresses apply to anoikis, as noted below. Subsequently, we discuss upstream regulators of the apoptotic machinery that are likely to be unique to anoikis.
In anoikis, the ubiquitous regulatory proteins of the Bcl-2 family control mitochondrial outer-membrane permeabilization, cytochrome c release and apoptosome assembly just as they do in other apoptotic scenarios. Specific roles for the upregulation of the pro-apoptotic BH3-only protein Bim in detached cells of the mammary epithelial cell line MCF10a, or degradation of the anti-apoptotic protein Mcl1 in detached 3T3 cells has been noted (Reginato et al., 2003; Woods et al., 2007).
Activation of the phosphoinositide 3-kinase (PI3K)/Akt pathway has a widespread role in cell survival signaling in general, and anoikis is no exception. Accordingly, activation of insulin receptor or insulin-like growth factor receptors upstream of PI3K/Akt is important in the resistance of tumor cells to anoikis (Martin et al., 2006). A clinically important connection between anoikis and systemic metabolic changes in response to diet, exercise and growth hormones is implied by this observation, and warrants further investigation.
Cellular metabolic regulation of the levels of crucial energy carriers – such as nicotinamide adenine dinucleotide phosphate (NADP+) or its reduced form NADPH and ATP, and that of reactive oxygen species (ROS) is also an important feature in the regulation of cell survival. Correspondingly, integrin signaling partly regulates anoikis by controlling the pentose phosphate pathway and, specifically, NADPH and ROS (Schafer et al., 2009). Interestingly, this work sounds a cautionary note because antioxidants that are considered as chemopreventive agents might inhibit anoikis, thus acting as tumor promoters.
In addition to these factors, other components of the core apoptotic machinery (e.g. death receptors) have also been implicated in anoikis. For example, The FAS-associated death domain protein (FADD) has been shown to contribute to anoikis through unknown mechanisms, suggesting a potential role for death receptors in the process (Frisch, 1999). Nevertheless, a role for death receptors in anoikis has been shown only in certain cell lines, or following experimental manipulations that are only remotely related to anoikis, raising questions about the generality of this role.
As a general mediator of stress- and genotoxin-induced apoptosis, p53 might intuitively be expected to promote anoikis. However, this has so far been shown only in a limited context, i.e. in specific cell lines with PI3K-activating mutations, in which salt-inducible kinase 1 (SIK1)</emph> regulates anoikis through p53 (Cheng et al., 2009). A more general role of p53 in anoikis has not been shown. However, a protective role of the p53-related protein p63 (Tp63) through induction of extracellular matrix and integrin gene expression has been demonstrated (Carroll et al., 2006).
The role of E-cadherin in anoikis
The publication that first described anoikis reported that Madin-Darby canine kidney epithelial cells (MDCK) show an increased sensitivity to anoikis when they are grown to confluence (Frisch and Francis, 1994). The exact basis for this phenomenon in MDCK is still not fully elucidated and may be due to multiple factors. Two studies strongly support the contention that E-cadherin-mediated cell interactions have a significant role in sensitizing epithelial cells to anoikis. The conditional knockout of E-cadherin in a p53-dominant-negative tumor cell model enhances tumor metastasis substantially and the derived cultured cells are highly resistant to anoikis compared with E-cadherin-expressing control cells (Derksen et al., 2006). A subsequent, more mechanistic, study reported similar findings by using the immortalized human mammary epithelial (HMLE) cell line (Onder et al., 2008). Here, stable knockdown of E-cadherin confers anoikis resistance to the cells that is accompanied by EMT. This effect has been attributed to the decreased phosphorylation of β-catenin and, therefore, its increased activation, resulting in Wnt activation (Onder et al., 2008). Consistent with this, anoikis sensitivity can be partially restored by simultaneous knockdown of β-catenin. Total β-catenin levels were not, however, affected, and although there is clear evidence for activation of the Wnt pathway, this work could not exclude the existence of additional pathways that link E-cadherin to anoikis resistance.
Subsequently, an additional pathway was found, that involved an unexpected factor ankyrin-G (Kumar et al., 2011). Ankyrin-G is the widely expressed epithelial isoform of the ankyrin family of cytoskeletal linker proteins. Ankyrin-G bridges the cytoskeleton, through spectrin and actin complexes, with the cell membrane, through diverse transmembrane proteins including CD44, Na+/K+-ATPase, L1CAM and, importantly, E-cadherin (Bennett and Healy, 2008; Kizhatil et al., 2007a). In addition, ankyrin-G is downregulated in diverse types of tumor, and is included in a signature – consisting of eleven genes – that correlates with poor prognosis following chemotherapy of cancer patients (Glinsky et al., 2005). Moreover, ankyrin-G has two domains that are implicated in apoptosis: a death domain and a ZU5 domain. Together, they comprise a ‘supermodule’ of a defined structure that is also present in the apoptosis-regulating netrin receptor Unc5 (Wang et al., 2009). The ZU5 domain of ankyrin-G, similar to that of Unc5, has been found to interact with the neurotrophin-receptor-associated MAGE homolog (MAGED1, also known as and hereafter referred to as NRAGE), a member of the melanoma antigen family of proteins that has been previously shown to regulate apoptosis in conjunction with Unc5 (Williams et al., 2003). Ankyrin-G has been shown to sequester NRAGE in the cytoplasm (Kumar et al. 2011). In the absence of ankyrin-G (e.g. after the loss of E-cadherin accompanying EMT), a fraction of NRAGE translocates to the nucleus, where it interacts with the oncogenic transcriptional repressor protein TBX2, forming a repressor/co-repressor complex that attenuates the gene expression of the tumor suppressor p14ARF (CDKN2A) (Kumar et al., 2011). In HMLE cells, p14ARF promotes anoikis (Kumar et al., 2011). Consequently, the repression of p14ARF confers anoikis resistance, suggesting a novel pathway that regulates anoikis through E-cadherin–ankyrin-G complexes (Kumar et al., 2011), as depicted in Fig. 2.
Fig. 2.
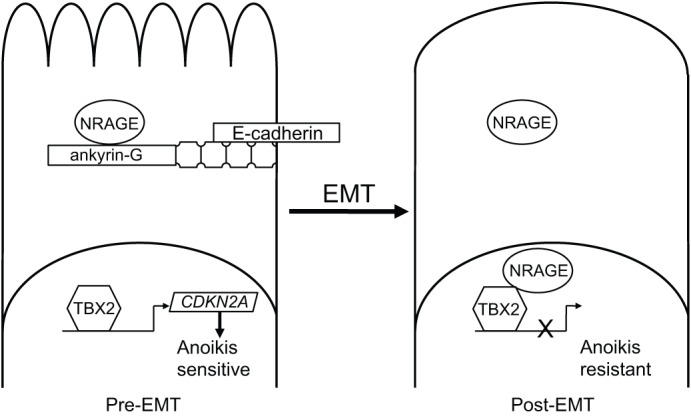
E-cadherin and ankyrin-G are lost coordinately during EMT, conferring anoikis resistance. This occurs because the ankyrin-G-interacting protein NRAGE is no longer sequestered by ankyrin-G. This, in turn, permits translocation of NRAGE to the nucleus, where it forms a repressor/co-repressor complex with TBX2, thereby blocking the expression of the p14ARF gene (CDKN2A) and attenuating anoikis.
It is worth noting that MCF10a cells, which are widely used to study anoikis, do not express p14ARF. Anoikis occurs far more slowly in this cell line, compared with other mammary epithelial cell lines (∼48 hours for commonly used MCF10a variants, versus 4–6 hours for HMLE cells, to reach a threshold where cell death can be detected by a DNA-fragmentation assay), and the ectopic expression of low levels of p14ARF dramatically sensitizes these cells to anoikis. This observation, and the correlation between the expression of ankyrin-G and p14ARF in breast cancer cell lines, suggests that p14ARF is an important determinant of anoikis sensitivity (Kumar et al., 2011). This may also constitute an underlying reason for the widespread loss of p14ARF in human cancers and the observation that even heterozygosity for wild-type p14ARF correlates strongly with tumor incidence (Hewitt et al., 2002; Moreno-Miralles et al., 2005). Although p14ARF was originally shown to induce apoptosis through p53, p53-independent mechanisms that involve its interaction with the Myc proto-oncogene, the generalized EMT-mediating co-repressor protein, C-terminal binding protein (CTBP) or the mitochondrial p32 protein have now been shown (Itahana and Zhang, 2008; Paliwal et al., 2006; Qi et al., 2004).
Another suggestion arising from this report is that the downregulation of ankyrin-G is so prevalent that it could be considered as a marker for EMT (Kumar et al., 2011). However, it cannot be excluded that ankyrin-G regulates anoikis through additional mechanism that are mediated by its death domain and/or the solenoid conformation of the ankyrin-repeat domain, which has been shown to act as a mechanotransducer (Lee et al., 2006).
In addition, another pathway linking E-cadherin to the control of anoikis is emerging. Homophilic interactions between E-cadherins of neighbouring cells is an important initiator of contact inhibition, and this effect was shown to involve the regulation of phosphorylation of Yes-associated protein (YAP), a key component of the Hippo signaling pathway. The role of the Hippo pathway in anoikis has been further demonstrated more recently (discussed below), indicating that E-cadherin is likely to control anoikis through this additional pathway.
Taken together, the regulation of anoikis by E-cadherin is surprisingly complex, involving at least three pathways: Wnt – through β-catenin and T-cell-specific transcription factors (TCFs), the ankyrin–NRAGE–TBX2–p14ARF signaling axis, and the Hippo pathway, with others probably still awaiting discovery. In addition, these pathways are also likely to interact, as discussed below.
The role of cell polarity in anoikis
Epithelial cell polarity is compromised during oncogenic EMT through diverse transcriptional and signaling effects (Godde et al., 2010; Moreno-Bueno et al., 2008; Xu et al., 2009). Three polarity complexes, crumbs</emph>, Par and scribble, contribute to polarity in normal epithelial cells and aberrations in each of these affect tumor progression (Ellenbroek et al., 2012). Below, we discuss the demonstrated or inferred roles of these complexes in the regulation of anoikis.
As a preface to this, it is important to understand that those signaling pathways that are crucial for EMT, i.e. the Hippo pathway, the transforming growth factor β (TGF-β) pathway and the Wnt pathway, are highly interconnected and that there is a substantial degree of cross-talk between them (see recent review by Varelas and Wrana, 2012). TGF-β and Wnt signaling induce EMT in a cooperative manner and drive anoikis resistance (Gauger et al., 2011; Scheel et al., 2011), whereas Hippo signaling has been viewed as the conductor that orchestrates the TGF-β–Wnt ‘symphony’ (Varelas and Wrana, 2012).
Further insights into the role of Hippo signaling in anoikis have emerged recently from two studies. First, the p53 effector protein ASPP1 (also known as PPP1R13B) was found to activate the two principal transcriptional co-activator proteins of the Hippo pathway, YAP and TAZ, by inhibiting their interaction with the inactivating kinases LATS1 or LATS2, which normally leads to nuclear export and degradation of YAP and TAZ (Fig. 3) (Vigneron et al., 2010). Directly or indirectly, activation of YAP and TAZ results in the downregulation of the pro-apoptotic protein Bim (also known as BCL2L11), thus protecting cells against anoikis. This result indicates a novel role for ASPP1 as a potentially oncogenic promoter of cell survival, although it also raises the question of how the transcriptional co-activators YAP and TAZ repress Bim gene expression.
Fig. 3.

The epithelial specific cell polarity proteins and maintain anoikis sensitivity by regulating the Hippo, Wnt and TGF-β pathways. In normal, interacting epithelial cells (left panel), the cell polarity complexes, crumbs (Crb) and scribble (Scrib) stimulate the phosphorylation of YAP and TAZ through the LATS kinase. This maintains YAP and TAZ in the cytoplasm, sensitizing cells to anoikis. In addition, cytoplasmic YAP and TAZ interact with Smad3 and prevent its nuclear translocation, even in the presence of active TGF-β receptors. Furthermore, cytoplasmic TAZ interacts with disheveled (Dsh), inhibiting canonical Wnt signaling. Expression or activation of CTBP; ZEB1/2,Snail, Twist, FAK, ILK, NF-κB or TrkB induce EMT (right panel), which – in turn – compromises cell polarity complexes, promotes the nuclear translocation of YAP, TAZ and Smad3, and induces expression of cell survival genes. The absence of cytoplasmic TAZ allows Dsh to be activated by CK1, which inhibits glycogen synthase kinase 3 β (GSK3β), thus allowing β-catenin to transactivate pro-survival genes in the nucleus. Alternatively, the Akt–GSK3β–β-catenin axis can be stimulated through activation of TrkB.
A recent report demonstrates that YAP phosphorylation is regulated by integrin–matrix interactions in a manner that is dependent on the actin and tubulin cytoskeleton (Zhao et al., 2012a). The authors show that cell–matrix detachment activates LATS1 and LATS2, thereby inactivating YAP. The functional roles of these factors were demonstrated by manipulating their expression in normal or tumor cell lines, and the observation that LATS1 and LATS2 are downregulated in metastatic prostate tumors.
The above findings suggest a function for Hippo in anoikis and are complemented by other work that links Hippo signaling to cell polarity proteins. For instance, pronephros development in zebrafish is controlled in part by the interaction between the cadherin family member Fat1, which is an upstream component of the Hippo pathway, and scribble. The resulting phenotype (cyst formation) is also affected by mutations in YAP, underscoring the importance of interactions between the Hippo and the cell polarity pathways (Skouloudaki et al., 2009).
In mammalian epithelial cells, YAP and TAZ also interact with scribble or crumbs (Varelas and Wrana, 2012). In their unphosphorylated forms, YAP and TAZ are localized in the nucleus, where they interact with Smad complexes (transducers of TGF-β signaling) to promote the nuclear localization of Smad and the activation of its target genes. Upon activation of the Hippo pathway, kinases LATS1 and LATS2, and YAP and TAZ are phosphorylated and excluded from the nucleus, so that they can interact with cell polarity proteins – such as membrane-localized scribble and crumbs, which stably sequester YAP and TAZ to the membrane (Varelas and Wrana, 2012).
During EMT, polarity proteins fail to localize correctly to the membrane, relieving the sequestration of Smads and promoting TGF-β signaling, which, in turn, stabilizes the EMT phenotype and the consequent resistance to anoikis (Humbert et al., 2008; Varelas et al., 2010b) (Fig. 3). An example of this is the inhibitory interaction between the crumbs polarity complex and Smads that is mediated by YAP and TAZ. The phosphorylated forms of YAP and TAZ interact with crumbs, thereby sequestering Smads and inhibiting both pathways (Varelas et al., 2010b). Although not yet tested directly, this is predicted to maintain anoikis sensitivity in normal epithelial cells, as the role of YAP and TAZ and that of TGF-β signaling in anoikis is established (Cieply et al., 2012; Zhao et al., 2012a).
Another example of the interplay between the cell polarity pathways is the interaction between the cell polarity protein scribble and the Hippo pathway co-activator TAZ (also known as WWTR1), which, in its phosphorylated form, is found in the cytoplasm (Cordenonsi et al., 2011). TAZ activity correlates with a cancer stem cell phenotype in mammary tumors, as reflected by the expression profile of TAZ target genes, and is functionally important for maintaining this phenotype in cell culture models (Cordenonsi et al., 2011). Manipulating TAZ gene expression does not affect EMT, but is required for mammosphere generation, which is a marker of cancer stem cells that clearly requires a transition to anoikis resistance. Scribble is frequently defective in tumor cells and, following EMT, is aberrantly localized to the cytoplasm instead of at the basolateral membrane (Humbert et al., 2008). Interestingly, scribble normally inhibits TAZ activation in epithelial cells, which has been attributed to its role in promoting an inhibitory LATS1–TAZ complex, but when it is mislocalized in cells with a mesenchymal phenotype, it is unable to inhibit TAZ by this means, resulting in hyperactive TAZ and promoting the anoikis resistance that facilitates mammosphere formation (Cordenonsi et al., 2011).
Interestingly, YAP and TAZ interact also with the disheveled homolog Dvl, inhibit its activation through casein kinase I (CK1), and the subsequent activation of the canonical Wnt pathway, which is crucial in the control of anoikis (Onder et al., 2008; Varelas et al., 2010a). This observation provides a mechanism by which cell polarity complexes might regulate anoikis through a YAP(TAZ)–Dvl-Wnt axis (Fig. 3).
It should be emphasized that different polarity complexes may have distinct, cell-type specific roles. For example, in a keratinocyte cell line (HaCat), depletion of scribble compromises cell–cell adhesion and increases matrigel invasiveness, without having a significant effect on anoikis. By contrast, the depletion of the discs large homologue 1 (Dlg1) in these cells maintains cell-cell adhesion and suppresses invasiveness, but profoundly decreases the occurrence of anoikis (Massimi et al., 2012). Thus, specific molecular components of polarity complexes may regulate anoikis in certain instances instead of cell polarity per se.
Finally, several lines of evidence point to a role of Jun N-terminal kinase (JNK) signaling as another possible link between cell polarity and anoikis. First, anoikis and cell polarity in three-dimensional MCF10a acinar cultures both depend on JNK signaling (McNally et al., 2011). Second, the mammary-gland-specific knockout of JNK1 and JNK2 (MAPK8 and MAPK9, respectively) demonstrates that they have tumor-suppressive activities that are accompanied by defective luminal clearing, a sign of defective anoikis (Cellurale et al., 2010). From the cell polarity perspective, scribble enforces anoikis-driven luminal clearing in MCF10a acini through Rac–JNK signaling (Zhan et al., 2008). Although JNK is a clear candidate for linking polarity with anoikis, it is almost certain that there are also other signals that yet await discovery.
In summary, cell polarity-determining adhesion molecules are frequently downregulated by transcription factors that meditate EMT. In turn, the loss of correctly localized cell-polarity complexes alters signaling through Wnt, TGF-β and Hippo signaling pathways and, thus, reinforces the EMT phenotype and enables tumor cells to evade anoikis.
Control of cell survival genes by EMT transcription factors
As mentioned earlier, anoikis utilizes the canonical apoptotic machinery, including Bcl-2 family members, caspases and, perhaps, death receptor signaling components. In this section, we describe the gene regulation of these proteins by transcription factors that drive EMT.
The E-box-binding factors Snail, Slug, Twist and zinc finger E-box-binding homeoboxes 1 and 2 (ZEB1 and ZEB2, respectively) were originally characterized as transcriptional repressors of cell adhesion genes that encode components of adherens junctions, desmosomes and tight junctions (Thiery et al., 2009; Tiwari et al., 2012). By downregulating junctional assembly, they interfere with cell polarity and may suppress anoikis through the pathways described above. Additionally, these factors can control cell survival genes. For example, ZEB1 represses the pro-apoptotic tumor protein p73 (Tp73) gene through direct interaction with the E-box sites that are present in intron 1 (Fontemaggi et al., 2001). Other apoptosis-regulating target genes for ZEB1 and Snail have been proposed (Sayan et al., 2009; Wu and Zhou, 2010). Apoptosis induced by Myc is dependent upon induction of p14ARF gene expression, which results in the highly pro-apoptotic Myc–ARF complex (Qi et al., 2004). Twist directly represses the expression of the p14ARF gene, thereby suppressing apoptosis by blocking the formation of this complex and collaborating with Myc to transform cells (Valsesia-Wittmann et al., 2004). It is noteworthy that Myc promotes apoptosis during luminal clearing in MCF10a acinar development and, thus, might also contribute to anoikis (our own unpublished data), providing a molecular pathway by which Twist might suppress anoikis (Zhan et al., 2008).
NF-κB is crucial for the induction of EMT in some contexts, which provides interesting conceptual links of EMT and anoikis to inflammatory cytokines (Li et al., 2012; Min et al., 2008; Pantuck et al., 2010). Several studies have demonstrated a significant role for NF-κB in anoikis, particularly in intestinal epithelial cells (Toruner et al., 2006; Yan et al., 2005). NF-κB is also important in cell survival signaling by activating its target genes, which include two members of the inhibitor of apoptosis (IAP) family, survivin and XIAP, as well as CFLAR, bcl-xl (BCL2L1), bcl-2 (BCL2) and osteoprotegerin (TNFRSF11B). Other targets are cytokine genes such as tumor necrosis factor (TNF) and interleukin-6 (IL6) that participate in a positive feedback loop to further activate NF-κB. In this context, survivin has recently shown to have a crucial role in regulating anoikis: a complex containing survivin and XIAP activates NF-κB, which leads to an increased expression of fibronectin, that, in turn, establishes integrin clustering under detached conditions and results in rescue from anoikis (Mehrotra et al., 2010). This study clearly illustrates the concept that increased expression of matrix proteins during EMT is a mechanism underlying anoikis-rescue through constitutive integrin clustering, which has also been proposed for certain mammary epithelial cells (Zahir et al., 2003) and might also apply more generally.
Finally, the transcriptional co-repressor CTBP is also required for most, if not all, cases of EMT and it attenuates anoikis (Grooteclaes et al., 2003); it might act with ZEB1 to repress gene expression of E-cadherin and other epithelial genes (Grooteclaes and Frisch, 2000). Interestingly, CTBP also represses the promoter of Bcl-2-interacting killer (Bik), a member of the BH3 family of pro-apoptotic genes, a function that is antagonized by the alternative reading frame (ARF) tumor suppressor, which normally promotes anoikis (Kovi et al., 2010; Kumar et al., 2011). Conversely, the wound-healing regulatory transcription factor GRHL2 has recently been found to suppress EMT, in part, by repressing ZEB1 expression and TGF-β transcriptional signaling. Accordingly, GRHL2 restores sensitivity of tumor cells to anoikis and attenuates two cancer stem cell phenotypes, mammosphere generation and CD44 expression (Cieply et al., 2012). However, for both CTBP and GRHL2, the mechanisms that link EMT to anoikis resistance have yet to be determined. The expression of phosphatase and tensin homolog (PTEN), an antagonist of Akt signaling, is repressed by CTBP and activated by GRHL3 (a protein highly related to GRHL2 both structurally and functionally) and PTEN could, thus, be an important interface between EMT and anoikis (Darido et al., 2011; Paliwal et al., 2007).
The role of FAK and ILK in coupling anoikis resistance with EMT
The roles of focal adhesion kinase (FAK) and integrin-linked protein kinase (ILK) in suppressing anoikis are well established (Atwell et al., 2000; Frisch et al., 1996). FAK and ILK colocalize with integrins at sites of cell attachment to the extracellular matrix, and integrin-ligand interactions stimulate the function of these proteins (Hannigan et al., 1996; Lipfert et al., 1992; Schaller et al., 1992). Attachment of epithelial cells to collagen I can induce EMT, and both FAK and ILK have been implicated in the control of EMT in response to adhesion to collagen I (Medici and Nawshad, 2010; Shintani et al., 2008). FAK and ILK are also activated in response to growth factors, including ligands of growth factor receptors that induce EMT, and they have been implicated in the control of growth-factor-induced EMT, for example in response to TGF-β (Cicchini et al., 2008; Serrano et al., 2013). Conventionally, FAK activates PI3K and mitogen-activated protein kinase (MAPK). These pathways stimulate the Ets and SP1 transcription factors (Wang et al., 2008). FAK-mediated increase in SP1 activity directly results in an elevated expression of another transcription factor, Krüppel-like factor 8 (KLF8) (Wang et al., 2008; Zhao et al., 2003), which is a driver of EMT under some conditions (Wang et al., 2007). Ectopic expression of KLF8 represses the expression of E-cadherin by directly binding to a GT-box in the E-cadherin promoter (Wang et al., 2007). Thus, one potential signaling pathway through which FAK might drive EMT is the FAK–PI3K–MAPK–SP1–KLF8 axis; however, this hypothesis has not yet been firmly established. FAK has also been implicated in regulating the gene expression of Snail, Zeb1, Zeb2 and Twist in fibroblasts (Li et al., 2011), which requires activity of MAPK and PI3K. Moreover, expression of these transcription factors is also crucial to maintain the mesenchymal phenotype of murine fibroblasts, including suppression of E-cadherin gene expression (Li et al., 2011). Although FAK is involved in regulating the expression of the E-cadherin gene under some conditions, in other models of EMT FAK does not have a role in regulating the expression of epithelial markers, including that of the E-cadherin gene, but has been implicated in inducing the expression of mesenchymal genes (Cicchini et al., 2008). FAK also has a role in regulating the surface expression of E-cadherin, as impairment of FAK attenuates internalization of E-cadherin upon EMT induction (Cicchini et al., 2008). Furthermore, impaired E-cadherin internalization can be seen when FAK is inhibited in a mouse model (Canel et al., 2010), but the underlying mechanism remains to be determined.
Ectopic expression of ILK in lung cancer cells promotes morphological changes that are associated with EMT, such as downregulation of the E-cadherin gene and elevated expression of mesenchymal genes (Chen et al., 2013). Conversely, inhibition of ILK expression in bladder carcinoma cells, breast cancer cell lines and TGF-β stimulated mammary epithelial cells impairs the gene expression of mesenchymal markers and results in elevated expression of the E-cadherin gene (Serrano et al., 2013; Zhu et al., 2012). Mesenchymal markers that are regulated by ILK include the transcription factors Snail and Slug (Chen et al., 2013; Medici and Nawshad, 2010; Serrano et al., 2013; Zhu et al., 2012). These results may be clinically relevant, as elevated protein expression of ILK correlates with increased gene expression of Snail and decreased expression of the E-cadherin gene in oral squamous cell carcinoma tumor samples (Zhao et al., 2012b), examples of transcriptional regulation that are thought to be mediated by the regulation of NF-κB activity through ILK (Chen et al., 2013; Medici and Nawshad, 2010).
The role of TrkB in anoikis and EMT
Originally isolated as an oncogenic fusion protein (Martin-Zanca et al., 1986), tropomyosin-related kinase A (Trk, also known as NTRK1) is the founding member of a family of proteins that have a crucial role in the nervous system. Acquired expression of some Trk family members (TrkA and TrkC) in various types of cancer can be a good prognostic indicator (Thiele et al., 2009). However, acquired expression of TrkB (NTRK2) has been associated with poor patient survival (Thiele et al., 2009). TrkB contributes to disease progression by inhibiting anoikis and promoting EMT. A role of TrkB in preventing anoikis emerged from an unbiased screen of rat intestinal epithelial (RIE) cells for genes that are capable of suppressing anoikis (Douma et al., 2004). Stimulation with brain-derived neurotrophic factor (BDNF), a natural ligand for TrkB, enhances TrkB-mediated cell survival. The kinase activity of TrkB is essential for protection against anoikis, and PI3K appears to be an important downstream target of this cell-survival pathway (Douma et al., 2004; Geiger and Peeper, 2007). Expression of TrkB in several epithelial cell lines induces a profound morphological change. Although the details vary depending on which cell line is used, expression of epithelial markers is always reduced, whereas that of mesenchymal markers is always enhanced (Smit et al., 2009). Pharmacological studies suggest that MAPK, but not PI3K, is crucial for the observed changes in morphology and alterations in the expression of EMT markers. Three EMT-associated transcription factors, Twist, Snail and ZEB1 are essential for TrkB-induced EMT and downregulation of E-cadherin gene expression is also crucial (Smit et al., 2009; Smit and Peeper, 2011). These studies suggest that TrkB has an important role in promoting survival under conditions that trigger anoikis, and in inducing EMT. Additional studies that confirmi the underlying mechanisms are required, but the current evidence that PI3K activity is required for protection against anoikis and that MAPK activity is required for the induction of EMT suggests that different signaling pathways are important for the induction of these two biological processes through TrkB. Interestingly, TrkB has been identified as a target of the microRNA miR-200c, and a TrkB construct that is resistant to miR200c prevents it from inducing anoikis (Howe et al., 2011). This is an intriguing initial observation, but further investigation is required to completely elucidate the underlying mechanism, given that downstream components of TrkB signalling, such as ZEB1, are also targets of the miR200 family.
Concluding remarks
Although the molecular mechanisms that underlie anoikis and EMT may have appeared hopelessly diverse at the inception, unifying principles are now emerging. In particular, transcription factors that are widespread or, quite possibly, ubiquitous mediators (such as CTBP) or suppressors (e.g. GRHL2) of EMT have been identified. This gives hope to the notion that EMT can be reversed and anoikis sensitivity be restored in a generalized fashion through drugs that target these factors. In addition, the cell polarity complexes, which can now be viewed as both upstream and downstream of EMT, have substantial effects on at least three signaling pathways (Hippo, Wnt, TGF-β) for which pharmacologically relevant compounds already exist or are being developed. Suppression of EMT through this new category of agents might also restore anoikis sensitivity. If this is the case, suppression of metastasis and of disease recurrence that is due to an EMT-related conversion of tumor cells to cancer stem cells, could reasonably be anticipated and would constitute a major therapeutic advance.
Acknowledgments
The author thank J. Farris and P. Pifer for the critical reading of the manuscript.
Footnotes
Funding
S.M.F. was supported by a National Institutes of Health grant [grant number RO1CA123359]. Deposited in PMC for release after 12 month.
References
- Attwell S., Roskelley C., Dedhar S. (2000). The integrin-linked kinase (ILK) suppresses anoikis. Oncogene 19, 3811–3815 10.1038/sj.onc.1203711 [DOI] [PubMed] [Google Scholar]
- Baum B., Georgiou M. (2011). Dynamics of adherens junctions in epithelial establishment, maintenance, and remodeling. J. Cell Biol. 192, 907–917 10.1083/jcb.201009141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett V., Healy J. (2008). Organizing the fluid membrane bilayer: diseases linked to spectrin and ankyrin. Trends Mol. Med. 14, 28–36 10.1016/j.molmed.2007.11.005 [DOI] [PubMed] [Google Scholar]
- Brabletz T., Jung A., Reu S., Porzner M., Hlubek F., Kunz–Schughart L. A., Knuechel R., Kirchner T. (2001). Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc. Natl. Acad. Sci. USA 98, 10356–10361 10.1073/pnas.171610498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canel M., Serrels A., Miller D., Timpson P., Serrels B., Frame M. C., Brunton V. G. (2010). Quantitative in vivo imaging of the effects of inhibiting integrin signaling via Src and FAK on cancer cell movement: effects on E-cadherin dynamics. Cancer Res. 70, 9413–9422 10.1158/0008-5472.CAN-10-1454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll D. K., Carroll J. S., Leong C. O., Cheng F., Brown M., Mills A. A., Brugge J. S., Ellisen L. W. (2006). p63 regulates an adhesion programme and cell survival in epithelial cells. Nat. Cell Biol. 8, 551–561 10.1038/ncb1420 [DOI] [PubMed] [Google Scholar]
- Cellurale C., Weston C. R., Reilly J., Garlick D. S., Jerry D. J., Sluss H. K., Davis R. J. (2010). Role of JNK in a Trp53-dependent mouse model of breast cancer. PLoS ONE 5, e12469 10.1371/journal.pone.0012469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D., Zhang Y., Zhang X., Li J., Han B., Liu S., Wang L., Ling Y., Mao S., Wang X. (2013). Overexpression of integrin-linked kinase correlates with malignant phenotype in non-small cell lung cancer and promotes lung cancer cell invasion and migration via regulating epithelial-mesenchymal transition (EMT)-related genes. Acta Histochem 115, 128–136 10.1016/j.acthis.2012.05.004 [DOI] [PubMed] [Google Scholar]
- Cheng H., Liu P., Wang Z. C., Zou L., Santiago S., Garbitt V., Gjoerup O. V., Iglehart J. D., Miron A., Richardson A. L.et al. (2009). SIK1 couples LKB1 to p53-dependent anoikis and suppresses metastasis. Sci. Signal. 2, ra35 10.1126/scisignal.2000369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicchini C., Laudadio I., Citarella F., Corazzari M., Steindler C., Conigliaro A., Fantoni A., Amicone L., Tripodi M. (2008). TGFbeta-induced EMT requires focal adhesion kinase (FAK) signaling. Exp. Cell Res. 314, 143–152 10.1016/j.yexcr.2007.09.005 [DOI] [PubMed] [Google Scholar]
- Cieply B., Riley P., 4th, Pifer P. M., Widmeyer J., Addison J. B., Ivanov A. V., Denvir J., Frisch S. M. (2012). Suppression of the epithelial-mesenchymal transition by Grainyhead-like-2. Cancer Res. 72, 2440–2453 10.1158/0008-5472.CAN-11-4038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordenonsi M., Zanconato F., Azzolin L., Forcato M., Rosato A., Frasson C., Inui M., Montagner M., Parenti A. R., Poletti A.et al. (2011). The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 147, 759–772 10.1016/j.cell.2011.09.048 [DOI] [PubMed] [Google Scholar]
- Darido C., Georgy S. R., Wilanowski T., Dworkin S., Auden A., Zhao Q., Rank G., Srivastava S., Finlay M. J., Papenfuss A. T.et al. (2011). Targeting of the tumor suppressor GRHL3 by a miR-21-dependent proto-oncogenic network results in PTEN loss and tumorigenesis. Cancer Cell 20, 635–648 10.1016/j.ccr.2011.10.014 [DOI] [PubMed] [Google Scholar]
- Derksen P. W., Liu X., Saridin F., van der Gulden H., Zevenhoven J., Evers B., van Beijnum J. R., Griffioen A. W., Vink J., Krimpenfort P.et al. (2006). Somatic inactivation of E-cadherin and p53 in mice leads to metastatic lobular mammary carcinoma through induction of anoikis resistance and angiogenesis. Cancer Cell 10, 437–449 10.1016/j.ccr.2006.09.013 [DOI] [PubMed] [Google Scholar]
- Douma S., Van Laar T., Zevenhoven J., Meuwissen R., Van Garderen E., Peeper D. S. (2004). Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB. Nature 430, 1034–1039 10.1038/nature02765 [DOI] [PubMed] [Google Scholar]
- Dusek R. L., Attardi L. D. (2011). Desmosomes: new perpetrators in tumour suppression. Nat. Rev. Cancer 11, 317–323 10.1038/nrc3051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellenbroek S. I., Iden S., Collard J. G. (2012). Cell polarity proteins and cancer. Semin. Cancer Biol. 22, 208–215 10.1016/j.semcancer.2012.02.012 [DOI] [PubMed] [Google Scholar]
- Fontemaggi G., Gurtner A., Strano S., Higashi Y., Sacchi A., Piaggio G., Blandino G. (2001). The transcriptional repressor ZEB regulates p73 expression at the crossroad between proliferation and differentiation. Mol. Cell. Biol. 21, 8461–8470 10.1128/MCB.21.24.8461-8470.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch S. M. (1999). Evidence for a function of death-receptor-related, death-domain-containing proteins in anoikis. Curr. Biol. 9, 1047–1049 10.1016/S0960-9822(99)80455-2 [DOI] [PubMed] [Google Scholar]
- Frisch S. M., Francis H. (1994). Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol. 124, 619–626 10.1083/jcb.124.4.619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch S. M., Vuori K., Ruoslahti E., Chan–Hui P. Y. (1996). Control of adhesion-dependent cell survival by focal adhesion kinase. J. Cell Biol. 134, 793–799 10.1083/jcb.134.3.793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauger K. J., Chenausky K. L., Murray M. E., Schneider S. S. (2011). SFRP1 reduction results in an increased sensitivity to TGF-β signaling. BMC Cancer 11, 59 10.1186/1471-2407-11-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger T. R., Peeper D. S. (2007). Critical role for TrkB kinase function in anoikis suppression, tumorigenesis, and metastasis. Cancer Res. 67, 6221–6229 10.1158/0008-5472.CAN-07-0121 [DOI] [PubMed] [Google Scholar]
- Glinsky G. V., Berezovska O., Glinskii A. B. (2005). Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J. Clin. Invest. 115, 1503–1521 10.1172/JCI23412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godde N. J., Galea R. C., Elsum I. A., Humbert P. O. (2010). Cell polarity in motion: redefining mammary tissue organization through EMT and cell polarity transitions. J. Mammary Gland Biol. Neoplasia 15, 149–168 10.1007/s10911-010-9180-2 [DOI] [PubMed] [Google Scholar]
- Gregory P. A., Bert A. G., Paterson E. L., Barry S. C., Tsykin A., Farshid G., Vadas M. A., Khew–Goodall Y., Goodall G. J. (2008). The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 10, 593–601 10.1038/ncb1722 [DOI] [PubMed] [Google Scholar]
- Grooteclaes M. L., Frisch S. M. (2000). Evidence for a function of CtBP in epithelial gene regulation and anoikis. Oncogene 19, 3823–3828 10.1038/sj.onc.1203721 [DOI] [PubMed] [Google Scholar]
- Grooteclaes M., Deveraux Q., Hildebrand J., Zhang Q., Goodman R. H., Frisch S. M. (2003). C-terminal-binding protein corepresses epithelial and proapoptotic gene expression programs. Proc. Natl. Acad. Sci. USA 100, 4568–4573 10.1073/pnas.0830998100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guadamillas M. C., Cerezo A., Del Pozo M. A. (2011). Overcoming anoikis—pathways to anchorage-independent growth in cancer. J. Cell Sci. 124, 3189–3197 10.1242/jcs.072165 [DOI] [PubMed] [Google Scholar]
- Guarino M., Rubino B., Ballabio G. (2007). The role of epithelial-mesenchymal transition in cancer pathology. Pathology 39, 305–318 10.1080/00313020701329914 [DOI] [PubMed] [Google Scholar]
- Hannigan G. E., Leung–Hagesteijn C., Fitz–Gibbon L., Coppolino M. G., Radeva G., Filmus J., Bell J. C., Dedhar S. (1996). Regulation of cell adhesion and anchorage-dependent growth by a new beta 1-integrin-linked protein kinase. Nature 379, 91–96 10.1038/379091a0 [DOI] [PubMed] [Google Scholar]
- Hewitt C., Lee Wu C., Evans G., Howell A., Elles R. G., Jordan R., Sloan P., Read A. P., Thakker N. (2002). Germline mutation of ARF in a melanoma kindred. Hum. Mol. Genet. 11, 1273–1279 10.1093/hmg/11.11.1273 [DOI] [PubMed] [Google Scholar]
- Howe E. N., Cochrane D. R., Richer J. K. (2011). Targets of miR-200c mediate suppression of cell motility and anoikis resistance. Breast Cancer Res. 13, R45 10.1186/bcr2867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert P. O., Grzeschik N. A., Brumby A. M., Galea R., Elsum I., Richardson H. E. (2008). Control of tumourigenesis by the Scribble/Dlg/Lgl polarity module. Oncogene 27, 6888–6907 10.1038/onc.2008.341 [DOI] [PubMed] [Google Scholar]
- Itahana K., Zhang Y. (2008). Mitochondrial p32 is a critical mediator of ARF-induced apoptosis. Cancer Cell 13, 542–553 10.1016/j.ccr.2008.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R., Weinberg R. A. (2009). The basics of epithelial-mesenchymal transition. J. Clin. Invest. 119, 1420–1428 10.1172/JCI39104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizhatil K., Davis J. Q., Davis L., Hoffman J., Hogan B. L., Bennett V. (2007a). Ankyrin-G is a molecular partner of E-cadherin in epithelial cells and early embryos. J. Biol. Chem. 282, 26552–26561 10.1074/jbc.M703158200 [DOI] [PubMed] [Google Scholar]
- Kizhatil K., Yoon W., Mohler P. J., Davis L. H., Hoffman J. A., Bennett V. (2007b). Ankyrin-G and beta2-spectrin collaborate in biogenesis of lateral membrane of human bronchial epithelial cells. J. Biol. Chem. 282, 2029–2037 10.1074/jbc.M608921200 [DOI] [PubMed] [Google Scholar]
- Klymkowsky M. W., Savagner P. (2009). Epithelial-mesenchymal transition: a cancer researcher's conceptual friend and foe. Am. J. Pathol. 174, 1588–1593 10.2353/ajpath.2009.080545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouros–Mehr H., Kim J. W., Bechis S. K., Werb Z. (2008). GATA-3 and the regulation of the mammary luminal cell fate. Curr. Opin. Cell Biol. 20, 164–170 10.1016/j.ceb.2008.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovi R. C., Paliwal S., Pande S., Grossman S. R. (2010). An ARF/CtBP2 complex regulates BH3-only gene expression and p53-independent apoptosis. Cell Death Differ. 17, 513–521 10.1038/cdd.2009.140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Park S. H., Cieply B., Schupp J., Killiam E., Zhang F., Rimm D. L., Frisch S. M. (2011). A pathway for the control of anoikis sensitivity by E-cadherin and epithelial-to-mesenchymal transition. Mol. Cell. Biol. 31, 4036–4051 10.1128/MCB.01342-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G., Abdi K., Jiang Y., Michaely P., Bennett V., Marszalek P. E. (2006). Nanospring behaviour of ankyrin repeats. Nature 440, 246–249 10.1038/nature04437 [DOI] [PubMed] [Google Scholar]
- Li X. Y., Zhou X., Rowe R. G., Hu Y., Schlaepfer D. D., Ilić D., Dressler G., Park A., Guan J. L., Weiss S. J. (2011). Snail1 controls epithelial-mesenchymal lineage commitment in focal adhesion kinase-null embryonic cells. J. Cell Biol. 195, 729–738 10.1083/jcb.201105103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C. W., Xia W., Huo L., Lim S. O., Wu Y., Hsu J. L., Chao C. H., Yamaguchi H., Yang N. K., Ding Q.et al. (2012). Epithelial-mesenchymal transition induced by TNF-α requires NF-κB-mediated transcriptional upregulation of Twist1. Cancer Res. 72, 1290–1300 10.1158/0008-5472.CAN-11-3123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipfert L., Haimovich B., Schaller M. D., Cobb B. S., Parsons J. T., Brugge J. S. (1992). Integrin-dependent phosphorylation and activation of the protein tyrosine kinase pp125FAK in platelets. J. Cell Biol. 119, 905–912 10.1083/jcb.119.4.905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. J., Melnyk N., Pollard M., Bowden M., Leong H., Podor T. J., Gleave M., Sorensen P. H. (2006). The insulin-like growth factor I receptor is required for Akt activation and suppression of anoikis in cells transformed by the ETV6-NTRK3 chimeric tyrosine kinase. Mol. Cell. Biol. 26, 1754–1769 10.1128/MCB.26.5.1754-1769.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin–Zanca D., Hughes S. H., Barbacid M. (1986). A human oncogene formed by the fusion of truncated tropomyosin and protein tyrosine kinase sequences. Nature 319, 743–748 10.1038/319743a0 [DOI] [PubMed] [Google Scholar]
- Massimi P., Zori P., Roberts S., Banks L. (2012). Differential regulation of cell-cell contact, invasion and anoikis by hScrib and hDlg in keratinocytes. PLoS ONE 7, e40279 10.1371/journal.pone.0040279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matise L. A., Palmer T. D., Ashby W. J., Nashabi A., Chytil A., Aakre M., Pickup M. W., Gorska A. E., Zijlstra A., Moses H. L. (2012). Lack of transforming growth factor-β signaling promotes collective cancer cell invasion through tumor-stromal crosstalk. Breast Cancer Res. 14, R98 10.1186/bcr3217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally S., McArdle E., Gilligan E., Napoletano S., Gajewska M., Bergin O., McCarthy S., Whyte J., Bianchi A., Stack J.et al. (2011). c-Jun N-terminal kinase activity supports multiple phases of 3D-mammary epithelial acinus formation. Int. J. Dev. Biol. 55, 731–744 10.1387/ijdb.113374sm [DOI] [PubMed] [Google Scholar]
- Medici D., Nawshad A. (2010). Type I collagen promotes epithelial-mesenchymal transition through ILK-dependent activation of NF-kappaB and LEF-1. Matrix Biol. 29, 161–165 10.1016/j.matbio.2009.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrotra S., Languino L. R., Raskett C. M., Mercurio A. M., Dohi T., Altieri D. C. (2010). IAP regulation of metastasis. Cancer Cell 17, 53–64 10.1016/j.ccr.2009.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith J. E., Jr, Fazeli B., Schwartz M. A. (1993). The extracellular matrix as a cell survival factor. Mol. Biol. Cell 4, 953–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min C., Eddy S. F., Sherr D. H., Sonenshein G. E. (2008). NF-kappaB and epithelial to mesenchymal transition of cancer. J. Cell. Biochem. 104, 733–744 10.1002/jcb.21695 [DOI] [PubMed] [Google Scholar]
- Moreno–Bueno G., Portillo F., Cano A. (2008). Transcriptional regulation of cell polarity in EMT and cancer. Oncogene 27, 6958–6969 10.1038/onc.2008.346 [DOI] [PubMed] [Google Scholar]
- Moreno–Miralles I., Pan L., Keates–Baleeiro J., Durst–Goodwin K., Yang C., Kim H. G., Thompson M. A., Klug C. A., Cleveland J. L., Hiebert S. W. (2005). The inv(16) cooperates with ARF haploinsufficiency to induce acute myeloid leukemia. J. Biol. Chem. 280, 40097–40103 10.1074/jbc.M506855200 [DOI] [PubMed] [Google Scholar]
- Onder T. T., Gupta P. B., Mani S. A., Yang J., Lander E. S., Weinberg R. A. (2008). Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 68, 3645–3654 10.1158/0008-5472.CAN-07-2938 [DOI] [PubMed] [Google Scholar]
- Paliwal S., Pande S., Kovi R. C., Sharpless N. E., Bardeesy N., Grossman S. R. (2006). Targeting of C-terminal binding protein (CtBP) by ARF results in p53-independent apoptosis. Mol. Cell. Biol. 26, 2360–2372 10.1128/MCB.26.6.2360-2372.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paliwal S., Kovi R. C., Nath B., Chen Y. W., Lewis B. C., Grossman S. R. (2007). The alternative reading frame tumor suppressor antagonizes hypoxia-induced cancer cell migration via interaction with the COOH-terminal binding protein corepressor. Cancer Res. 67, 9322–9329 10.1158/0008-5472.CAN-07-1743 [DOI] [PubMed] [Google Scholar]
- Pantuck A. J., An J., Liu H., Rettig M. B. (2010). NF-kappaB-dependent plasticity of the epithelial to mesenchymal transition induced by Von Hippel-Lindau inactivation in renal cell carcinomas. Cancer Res. 70, 752–761 10.1158/0008-5472.CAN-09-2211 [DOI] [PubMed] [Google Scholar]
- Qi Y., Gregory M. A., Li Z., Brousal J. P., West K., Hann S. R. (2004). p19ARF directly and differentially controls the functions of c-Myc independently of p53. Nature 431, 712–717 10.1038/nature02958 [DOI] [PubMed] [Google Scholar]
- Reginato M. J., Mills K. R., Paulus J. K., Lynch D. K., Sgroi D. C., Debnath J., Muthuswamy S. K., Brugge J. S. (2003). Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nat. Cell Biol. 5, 733–740 10.1038/ncb1026 [DOI] [PubMed] [Google Scholar]
- Sayan A. E., Griffiths T. R., Pal R., Browne G. J., Ruddick A., Yagci T., Edwards R., Mayer N. J., Qazi H., Goyal S.et al. (2009). SIP1 protein protects cells from DNA damage-induced apoptosis and has independent prognostic value in bladder cancer. Proc. Natl. Acad. Sci. USA [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer Z. T., Grassian A. R., Song L., Jiang Z., Gerhart–Hines Z., Irie H. Y., Gao S., Puigserver P., Brugge J. S. (2009). Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 461, 109–113 10.1038/nature08268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller M. D., Borgman C. A., Cobb B. S., Vines R. R., Reynolds A. B., Parsons J. T. (1992). pp125FAK a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc. Natl. Acad. Sci. USA 89, 5192–5196 10.1073/pnas.89.11.5192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel C., Weinberg R. A. (2011). Phenotypic plasticity and epithelial-mesenchymal transitions in cancer and normal stem cells? Int. J. Cancer 129, 2310–2314 10.1002/ijc.26311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel C., Eaton E. N., Li S. H., Chaffer C. L., Reinhardt F., Kah K. J., Bell G., Guo W., Rubin J., Richardson A. L.et al. (2011). Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 145, 926–940 10.1016/j.cell.2011.04.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senga K., Mostov K. E., Mitaka T., Miyajima A., Tanimizu N. (2012). Grainyhead-like 2 regulates epithelial morphogenesis by establishing functional tight junctions through the organization of a molecular network among claudin3, claudin4, and Rab25. Mol. Biol. Cell 23, 2845–2855 10.1091/mbc.E12-02-0097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano I., McDonald P. C., Lock F. E., Dedhar S. (2013). Role of the integrin-linked kinase (ILK)/Rictor complex in TGFbeta-1-induced epithelial-mesenchymal transition (EMT). Oncogene 32, 50–60 10.1038/onc.2012.30 [DOI] [PubMed] [Google Scholar]
- Shintani Y., Fukumoto Y., Chaika N., Svoboda R., Wheelock M. J., Johnson K. R. (2008). Collagen I-mediated up-regulation of N-cadherin requires cooperative signals from integrins and discoidin domain receptor 1. J. Cell Biol. 180, 1277–1289 10.1083/jcb.200708137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skouloudaki K., Puetz M., Simons M., Courbard J. R., Boehlke C., Hartleben B., Engel C., Moeller M. J., Englert C., Bollig F.et al. (2009). Scribble participates in Hippo signaling and is required for normal zebrafish pronephros development. Proc. Natl. Acad. Sci. USA 106, 8579–8584 10.1073/pnas.0811691106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit M. A., Peeper D. S. (2011). Zeb1 is required for TrkB-induced epithelial-mesenchymal transition, anoikis resistance and metastasis. Oncogene 30, 3735–3744 10.1038/onc.2011.96 [DOI] [PubMed] [Google Scholar]
- Smit M. A., Geiger T. R., Song J. Y., Gitelman I., Peeper D. S. (2009). A Twist-Snail axis critical for TrkB-induced epithelial-mesenchymal transition-like transformation, anoikis resistance, and metastasis. Mol. Cell. Biol. 29, 3722–3737 10.1128/MCB.01164-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theveneau E., Mayor R. (2012). Neural crest delamination and migration: from epithelium-to-mesenchyme transition to collective cell migration. Dev. Biol. 366, 34–54 10.1016/j.ydbio.2011.12.041 [DOI] [PubMed] [Google Scholar]
- Thiele C. J., Li Z., McKee A. E. (2009). On Trk—the TrkB signal transduction pathway is an increasingly important target in cancer biology. Clin. Cancer Res. 15, 5962–5967 10.1158/1078-0432.CCR-08-0651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery J. P., Acloque H., Huang R. Y., Nieto M. A. (2009). Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890 10.1016/j.cell.2009.11.007 [DOI] [PubMed] [Google Scholar]
- Tiwari N., Gheldof A., Tatari M., Christofori G. (2012). EMT as the ultimate survival mechanism of cancer cells. Semin. Cancer Biol. 22, 194–207 10.1016/j.semcancer.2012.02.013 [DOI] [PubMed] [Google Scholar]
- Toruner M., Fernandez–Zapico M., Sha J. J., Pham L., Urrutia R., Egan L. J. (2006). Antianoikis effect of nuclear factor-kappaB through up-regulated expression of osteoprotegerin, BCL-2, and IAP-1. J. Biol. Chem. 281, 8686–8696 10.1074/jbc.M512178200 [DOI] [PubMed] [Google Scholar]
- Turksen K., Troy T. C. (2011). Junctions gone bad: claudins and loss of the barrier in cancer. Biochim. Biophys. Acta 1816, 73–79 [DOI] [PubMed] [Google Scholar]
- Valsesia–Wittmann S., Magdeleine M., Dupasquier S., Garin E., Jallas A. C., Combaret V., Krause A., Leissner P., Puisieux A. (2004). Oncogenic cooperation between H-Twist and N-Myc overrides failsafe programs in cancer cells. Cancer Cell 6, 625–630 10.1016/j.ccr.2004.09.033 [DOI] [PubMed] [Google Scholar]
- Varelas X., Wrana J. L. (2012). Coordinating developmental signaling: novel roles for the Hippo pathway. Trends Cell Biol. 22, 88–96 10.1016/j.tcb.2011.10.002 [DOI] [PubMed] [Google Scholar]
- Varelas X., Miller B. W., Sopko R., Song S., Gregorieff A., Fellouse F. A., Sakuma R., Pawson T., Hunziker W., McNeill H.et al. (2010a). The Hippo pathway regulates Wnt/beta-catenin signaling. Dev. Cell 18, 579–591 10.1016/j.devcel.2010.03.007 [DOI] [PubMed] [Google Scholar]
- Varelas X., Samavarchi–Tehrani P., Narimatsu M., Weiss A., Cockburn K., Larsen B. G., Rossant J., Wrana J. L. (2010b). The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-β-SMAD pathway. Dev. Cell 19, 831–844 10.1016/j.devcel.2010.11.012 [DOI] [PubMed] [Google Scholar]
- Vigneron A. M., Ludwig R. L., Vousden K. H. (2010). Cytoplasmic ASPP1 inhibits apoptosis through the control of YAP. Genes Dev. 24, 2430–2439 10.1101/gad.1954310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Zheng M., Liu G., Xia W., McKeown–Longo P. J., Hung M. C., Zhao J. (2007). Krüppel-like factor 8 induces epithelial to mesenchymal transition and epithelial cell invasion. Cancer Res. 67, 7184–7193 10.1158/0008-5472.CAN-06-4729 [DOI] [PubMed] [Google Scholar]
- Wang X., Urvalek A. M., Liu J., Zhao J. (2008). Activation of KLF8 transcription by focal adhesion kinase in human ovarian epithelial and cancer cells. J. Biol. Chem. 283, 13934–13942 10.1074/jbc.M709300200 [DOI] [PubMed] [Google Scholar]
- Wang R., Wei Z., Jin H., Wu H., Yu C., Wen W., Chan L. N., Wen Z., Zhang M. (2009). Autoinhibition of UNC5b revealed by the cytoplasmic domain structure of the receptor. Mol. Cell 33, 692–703 10.1016/j.molcel.2009.02.016 [DOI] [PubMed] [Google Scholar]
- Warzecha C. C., Jiang P., Amirikian K., Dittmar K. A., Lu H., Shen S., Guo W., Xing Y., Carstens R. P.2010). An ESRP-regulated splicing programme is abrogated during the epithelial-mesenchymal transition. EMBO J. 293286–3300 10.1038/emboj.2010.195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg R. A. (2008). Twisted epithelial-mesenchymal transition blocks senescence. Nat. Cell Biol. 10, 1021–1023 10.1038/ncb0908-1021 [DOI] [PubMed] [Google Scholar]
- Werth M., Walentin K., Aue A., Schönheit J., Wuebken A., Pode–Shakked N., Vilianovitch L., Erdmann B., Dekel B., Bader M.et al. (2010). The transcription factor grainyhead-like 2 regulates the molecular composition of the epithelial apical junctional complex. Development 137, 3835–3845 10.1242/dev.055483 [DOI] [PubMed] [Google Scholar]
- Williams M. E., Strickland P., Watanabe K., Hinck L. (2003). UNC5H1 induces apoptosis via its juxtamembrane region through an interaction with NRAGE. J. Biol. Chem. 278, 17483–17490 10.1074/jbc.M300415200 [DOI] [PubMed] [Google Scholar]
- Woods N. T., Yamaguchi H., Lee F. Y., Bhalla K. N., Wang H. G. (2007). Anoikis, initiated by Mcl-1 degradation and Bim induction, is deregulated during oncogenesis. Cancer Res. 67, 10744–10752 10.1158/0008-5472.CAN-07-3148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y., Zhou B. P. (2010). Snail: More than EMT. Cell Adh. Migr. 4, 199–203 10.4161/cam.4.2.10943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Lamouille S., Derynck R. (2009). TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 19, 156–172 10.1038/cr.2009.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan S. R., Joseph R. R., Rosen K., Reginato M. J., Jackson A., Allaire N., Brugge J. S., Jobin C., Stadnyk A. W. (2005). Activation of NF-kappaB following detachment delays apoptosis in intestinal epithelial cells. Oncogene 24, 6482–6491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeaman C., Grindstaff K. K., Hansen M. D., Nelson W. J. (1999). Cell polarity: Versatile scaffolds keep things in place. Curr. Biol. 9, R515–R517 10.1016/S0960-9822(99)80324-8 [DOI] [PubMed] [Google Scholar]
- Zahir N., Lakins J. N., Russell A., Ming W., Chatterjee C., Rozenberg G. I., Marinkovich M. P., Weaver V. M. (2003). Autocrine laminin-5 ligates alpha6beta4 integrin and activates RAC and NFkappaB to mediate anchorage-independent survival of mammary tumors. J. Cell Biol. 163, 1397–1407 10.1083/jcb.200302023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan L., Rosenberg A., Bergami K. C., Yu M., Xuan Z., Jaffe A. B., Allred C., Muthuswamy S. K. (2008). Deregulation of scribble promotes mammary tumorigenesis and reveals a role for cell polarity in carcinoma. Cell 135, 865–878 10.1016/j.cell.2008.09.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J., Bian Z. C., Yee K., Chen B. P., Chien S., Guan J. L. (2003). Identification of transcription factor KLF8 as a downstream target of focal adhesion kinase in its regulation of cyclin D1 and cell cycle progression. Mol. Cell 11, 1503–1515 10.1016/S1097-2765(03)00179-5 [DOI] [PubMed] [Google Scholar]
- Zhao B., Li L., Wang L., Wang C. Y., Yu J., Guan K. L. (2012a). Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 26, 54–68 10.1101/gad.173435.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D., Tang X. F., Yang K., Liu J. Y., Ma X. R. (2012b). Over-expression of integrin-linked kinase correlates with aberrant expression of Snail, E-cadherin and N-cadherin in oral squamous cell carcinoma: implications in tumor progression and metastasis. Clin. Exp. Metastasis 29, 957–969 10.1007/s10585-012-9485-1 [DOI] [PubMed] [Google Scholar]
- Zhu J., Pan X., Zhang Z., Gao J., Zhang L., Chen J. (2012). Downregulation of integrin-linked kinase inhibits epithelial-to-mesenchymal transition and metastasis in bladder cancer cells. Cell. Signal. 24, 1323–1332 10.1016/j.cellsig.2012.02.013 [DOI] [PubMed] [Google Scholar]