

Introduction
Dilated cardiomyopathy (DCM) is an important cause of sudden cardiac death (SCD) and heart failure (HF) and is the leading indication for cardiac transplantation in children and adults worldwide.1 It is characterized by ventricular chamber enlargement and systolic dysfunction with normal left ventricular wall thickness. Different etiologies can lead to DCM, including inherited, infectious and inflammatory diseases. However the majority of cases remain unexplained after a thorough review for secondary cause.2 Discoveries made over the past 20 years have revealed a genetic basis in both inherited and hitherto idiopathic forms, with many different genes implicated.3–8 These developments have enlightened both clinical diagnosis and investigation, and fostered informed therapeutic decisions.
Genetic Background
Genetic etiologies are often suspected based on the presence of familial clustering of disease. Although initial estimates of familial involvement in DCM were as low as 2%,9 such ascertainment was derived from early studies that were limited to retrospective analyses of medical records. Recent studies, which assessed for hereditary disease with prospective pedigree analysis and echocardiography, have suggested that up to 40% of DCM may be inherited.10, 11 Accordingly, the importance of considering familial disease in DCM has been emphasized in recent consensus statements.12, 13
The study of affected kindreds has revealed familial DCM to be mostly inherited in an autosomal dominant fashion with characteristic age-dependent penetrance and variable clinical expression (Figure 1). Autosomal recessive, X-linked and mitochondrial forms have been described in a limited number of cases, often with associated skeletal myopathy.10 Although DCM may develop at any age, it usually does not present until adolescence or young adulthood (i.e. age dependent penetrance). Clinical manifestations can vary tremendously, even within individual families, where a proportion develop end-stage HF as infants while others may survive to the 7th decade with mild subclinical DCM (i.e. variable clinical expression).14 In a subset of familial cardiomyopathy, there are associated clinical features which are inherited with DCM.15 However, the majority of cases present with isolated DCM.
Figure 1.

Inheritance and Expression Patterns in Familial DCM. Disease transmission from father to son supports autosomal dominant inheritance (arrow). Incomplete penetrance is demonstrated by individual A, who has no evidence of DCM but is an obligate carrier of a disease variant by virtue of having an affected father and son. Variable clinical expression is evident with clinical manifestations differing amongst affected individuals within the kindred, which may obscure recognition of shared genetic disease. In this family, the presence of arrhythmias, conduction disease and DCM are consistent with disease caused by a mutation in LMNA, which was subsequently identified in individual
B. Black symbols = clinically affected individuals; white symbols = clinically unaffected individuals; circle=female; square=male; slash=deceased. + = LMNA mutation positive; − = LMNA mutation negative; AF = atrial fibrillation; AVB = atrioventricular block; LVEF = left ventricular ejection fraction; SCD = sudden cardiac death.
Genetic etiologies of DCM were initially identified through the study of affected families. However, sporadic, non-familial DCM may have a genetic etiology as frequently as hereditary presentation.16 Accordingly, genetic etiologies should be considered equally in familial and non-familial DCM. Mutations in over 40 different genes have been implicated in the pathogenesis of DCM. This concept of locus heterogeneity is further compounded by allelic heterogeneity, whereby different mutations within the same gene have been shown to cause DCM, with most being private to individual families.
DCM Disease Genes
Mutations in different genes, encoding a diverse array of proteins, may cause DCM,17 and largely have been identified through genome wide linkage analysis,4 and candidate gene sequencing.14 More recently, new technologies have enabled whole-exome sequencing,18 which is being increasingly used for gene discovery. Genes implicated to cause DCM encode components of the sarcomere, cytoskeleton (e.g. Z-disk proteins), nuclear envelope, and sarcolemma. In addition to these structural elements, mutations have also been identified in genes important for calcium cycling (PLN5), RNA splicing (RBM2019) and protein trafficking (BAG318). A comprehensive review of confirmed and putative disease genes has been described in several recent publications;17, 20 an abbreviated list of disease genes is provided on table 1.
Table 1.
Dilated cardiomyopathy – Selected Disease Genes
| Gene protein | Class | Inheritance | Prevalence* | Conduction Disease | Skeletal Myopathy |
|---|---|---|---|---|---|
|
TTN Titin |
Sarcomere | AD | 15–25% | − | rare |
|
MYH7 β-Myosin Heavy chain |
Sarcomere | AD | 4–8% | − | rare |
|
TNNT2 cTroponin-T |
Sarcomere | AD | 3–6% | − | − |
|
TPM1 A-tropomyosin |
Sarcomere | AD | 2–4% | − | − |
|
LMNA Lamin A/C |
Nuclear Lamina | AD, AR | 4–8%† | ++ | +/− |
|
EMD Emerin |
Nuclear Lamina | XL | <1%† | ++ | +++ |
|
SCN5A Nav1.5 |
Ion Channel | AD | 1–2%† | ++ | − |
|
DES Desmin |
Intermediate filament | AD, AR | <1% | ++ | ++/− |
|
ZNF9 DM2 |
Nucleic acid-binding | AD | <1% | ++ | +++ |
|
DMD Dystrophin |
Dystrophin | XL | − | + | +++ |
|
DSP Desmoplakin |
Desmosome | AD, AR | 1–3% | − | − |
|
RBM20 RNA Binding Motif 20 |
Spliceosome | AD | 3–6% | +/− | − |
|
BAG3 BCL2-associated athanogene 3 |
Cochaperones | AD | 2–4% | − | +/− |
|
PLN Phospholamban |
Calcium homeostasis | AD | <1% | ||
|
VCL Vinculin |
Z-Disk | AD | − | − | − |
Non-comprehensive list of DCM associated disease genes. Selected for inclusion were genes with strong data supporting pathogenesis or important associated phenotypes.
estimated based on limited studies;
prevalence higher (~30%) in setting of concomitant conduction disease or arrhythmias.
AD = autosomal dominant; AR = autosomal recessive; XL = X-linked.
The principle genetic etiologies of DCM have been reported in sequencing studies of moderate sized cohorts (n~300) comprised of both familial and sporadic cases. These studies suggest that mutations in sarcomeric genes, including TTN (titin), MYH7 (myosin heavy chain), TNNT2 (cardiac troponin T) and TPM1 (α-tropomyosin) are the most common etiologies, collectively accounting for ~30% of cases.8, 16, 21–23
Mutations in LMNA, which encodes Lamin A/C, a nuclear envelope protein, have been shown to cause different phenotypes associated with DCM, including DCM with limb-girdle or Emery-Dreifuss muscular dystrophy. Patients with LMNA mutations and DCM also have electrical instability (DCM+E), with supraventricular arrhythmias and atrioventricular block (AVB) often present before systolic dysfunction.3, 24 In large DCM cohorts, LMNA mutations are present in ~5% of patients25, however they are more prevalent (~33%) in patients with a DCM+E phenotype.26
Desmosome gene mutations are a known cause of arrhythmogenic right ventricular cardiomyopathy (ARVC), but may also play a role in DCM. Some patients with ARVC may present with left ventricular predominant disease, often caused by mutations in the desmosomal gene DSP (desmoplakin).27 Affected patients may present with an increased burden of ventricular tachyarrhythmia and/or coarse hair with palmoplantar hyperkeratosis (cardiocutaneous syndrome).27 Autosomal recessive cardiocutaneous syndrome with primary LV involvement is termed Carvajal syndrome,28 although autosomal dominant forms have been described.29 Alternatively, DCM caused by desmosomal mutations may present without any apparent RV involvement or excess arrhythmia.30
DCM is a relatively common feature in several forms of inherited skeletal myopathies, including those caused by mutations in DMD (dystrophin),31 DES (desmin),32 EMD (emerin)33, and ZNF9.34 Mutations in these genes may cause cardiomyopathy without apparent skeletal myopathy,35 especially in female carriers of mutations in the DMD gene which resides on the X-chromosome.36
Clinical Presentation
Heart failure, sudden death or thromboembolism may be the presenting manifestation of DCM. Alternatively, patients may be diagnosed with subclinical DCM identified in the process of family evaluations. As described in the preceding section, clinical manifestations vary substantially even within an individual family. In families where the pathogenic mutation has been identified, most, but not all carriers of the disease-mutation ultimately develop overt cardiomyopathy (i.e. incomplete penetrance). DCM caused by mutations in the sarcomere genes MYH7, TNNT2 and TPM1 may present at any age, however adverse outcomes appear to be more common with pediatric presentations.4, 14 TTN associated DCM typically does not present until adulthood, although adverse outcomes occur earlier in males.8 Alternatively, LMNA associated heart disease typically does not manifest until the 3rd decade, usually with conduction disease or arrhythmia that precedes DCM.37
Associated phenotypes
Most patients with familial DCM have isolated heart muscle disease, however extra-cardiomyopathic phenotypes are present in a minority of cases (Table 2).
Table 2.
Associated Clinical Features which are Present in a Minority of Patients with Familial Dilated Cardiomyopathy
| Associated Phenotype | Clinical Features | Comment | Associated Gene* |
|---|---|---|---|
|
| |||
| Conduction disease | Sinus arrest AV block Interventricular block |
May precede DCM |
DES DMD EMD LMNA SCN5A |
|
| |||
| Supraventricular arrhythmia prior to DCM | Premature atrial contraction Atrial fibrillation |
Often with slow ventricular response |
EMD LMNA SCN5A |
|
| |||
| Skeletal Myopathy | Limb Girdle | Proximal muscle weakness | LMNA |
| Emery-Dreifuss | Contractures, skeletal myopathy and wasting | EMD, LMNA | |
| Myotonic Dystrophy | Myotonia, weakness, baldness and cataracts. | ZNF9, DMPK1 | |
| Duchenne/Becker | Progressive X-linked proximal myopathy | DMD | |
| Myofibrillar Myopathy | Slowly progressive proximal and distal weakness | DES | |
|
| |||
| Hearing loss | Sensorineural hearing loss | Hearing loss typically occurs in 1st and 2nd decade of life | EYA |
|
| |||
| Palmoplantar keratoderma | Increased thickness of the palms and soles with woolly or excessively curly hair | May precede cardiac involvement | DSP |
Selected, incomplete list of associated disease genes.
AVB = atrioventricular block.
Associated or preceding conduction disease and supraventricular arrhythmia (DCM+E) are characteristic of DCM associated with LMNA,37 EMD or SCN5A38 mutations. AVB typically manifests as PR prolongation which may progress to compete heart block. Electrophysiologic studies (EPS) have revealed heart block to be intra-nodal in LMNA associated cardiomyopathy.39 Alternatively, interventricular conduction disease or sinus node dysfunction may be the presenting manifestations of DCM+E. Supraventricular tachyarrhythmias may begin as isolated premature atrial contractions (PAC) which progress to permanent atrial fibrillation.3 These patients also appear to have a greater burden of ventricular tachyarrhytmias than would be anticipated based on the severity of systolic dysfunction.40
The presence of left ventricle hypertrabeculation without overt systolic dysfunction or congenital anomaly defines isolated left ventricular noncompaction (LVNC). However, kindreds with both DCM and LVNC have been described, and both can be caused by sarcomeric gene mutations.41
Although rare, sensorineural hearing loss may be an associated DCM phenotype in the context of dominant mutations in the transcriptional coactivator, EYA4.42 Hearing loss is reported to precede cardiac involvement by decades in affected families.43
DCM may present with several types of inherited skeletal myopathy, including limb-girdle, Emery-Dreifuss, myotonic, myofibrillar and dystrophin associated muscular dystrophies.44 Patients with overt skeletal myopathy may have subclinical DCM, with “masking” of cardiac symptoms due to the activity imitations imposed by skeletal myopathy.45 Alternatively, patients with cardiomyopathy may have unnoticed skeletal myopathy which may ultimately complicate advanced therapies such as cardiac transplantation.46 Elevated serum creatine kinase (CK) concentrations may indicate subclinical skeletal myopathy, regardless of type. Atrial arrhythmias and conduction disease may be the only cardiac manifestations in patients with skeletal myopathy, or may precede DCM.
Clinical Evaluations of Familial DCM
The identification of left ventricular chamber dilation and systolic dysfunction with echocardiography is diagnostic of DCM, and can allow consideration of secondary causes (e.g. regional wall motion abnormalities in coronary heart disease). Isolated borderline left ventricular dilation and/or systolic dysfunction may represent an early stage of disease,11, 47 and when seen within the context of a family history, can be diagnostic. Echocardiography is recommended for screening asymptomatic patients for DCM in affected families and for providing clinical imaging follow-up of affected individuals.13, 48
Electrocardiographic findings are non-specific in most familial DCM, however associated conduction disease should be considered. Prolongation of the PR interval is often the earliest manifestation of genetic DCM+E, and less common manifestations such as atrial standstill may develop with progressive disease.49 Likewise, frequent PACs may be present early and may progress to permanent AF prior to the development of overt DCM.
Stress testing serves a dual role in the evaluation of a patient with presumed familial dilated cardiomyopathy; detection of coronary heart disease and quantification of exercise capacity. Individuals with genetic cardiomyopathy remain susceptible to coronary disease, which may result in two different causes of systolic dysfunction in the same patient. Alternatively, ischemic cardiomyopathy may be the cause of heart disease in patient who did not inherit the genetic cause of primary cardiomyopathy in an affected family (phenocopy). Exercise stress testing also allows for the objective quantification of functional capacity, which is a powerful determinant of survival amongst patients with heart failure,50 and can prioritize consideration for advanced therapies (e.g. cardiac transplantation).
Ambulatory electrocardiography can be useful for symptom evaluation and risk stratification, especially in patients with a family history of SCD or frequent ventricular ectopy. In cases of suspected ARVC, PVCs in excess of 500/24 hours represent a minor criterion for diagnosis based upon the Modified 2010 Task Force Criteria, although no diagnostic standard for ambient ectopy burden has yet been established for non-ARVC cases of suspected DCM+E. In some cases of frequent ventricular ectopy associated with LV dysfunction, catheter ablation or pharmacologic therapy has been associated with reversal of cardiomyopathy, and the origin of the pathogenic PVCs in these cases would be considered idiopathic rather than secondary to a myocardial process.51
CMR provides accurate assessment of ventricular chamber size, wall thickness and systolic function. CMR can be especially valuable in patients with poor echocardiographic image quality, but can also provide non-invasive tissue characterization. Delayed hyperenhancement (DE) of the CMR perfusion agent gadolinium indicates expansion of the extracellular space, which may be secondary to intramyocardial scar formation. The pattern of DE can differentiate coronary versus non-coronary heart disease with good specificity.52 In particular a subepicardial or midmyocardial pattern of DE in a non-coronary distribution suggests non-ischemic cardiomyopathy (figure 2). Specific patterns of DE have been purported to indicate a particular genetic association, however these findings are non-specific and are also present in viral myocarditis or cardiac sarcoidosis.24
Figure 2.
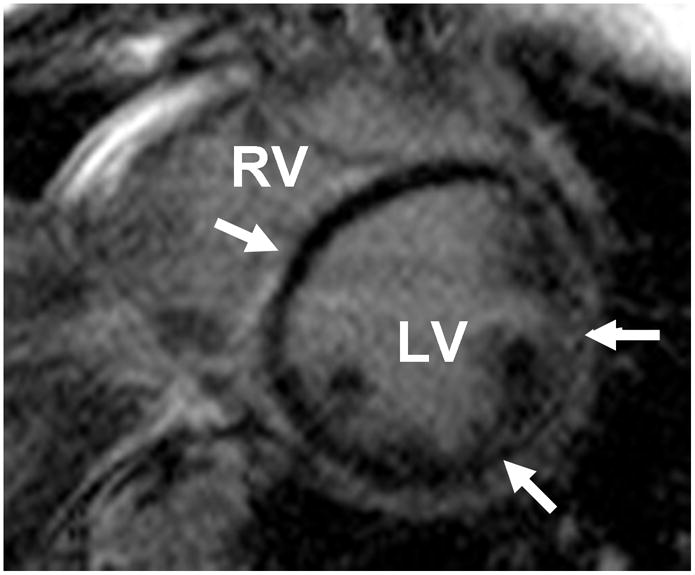
Non-Coronary Pattern of Delayed Gadolinium Enhancement imaged with Cardiac MRI in Familial DCM. Short axis Cardiac MRI (CMR) images reveal circumferential subepicardial delayed enhancement (DE) of gadolinium (arrows) in a young patient with familial DCM and palmoplantar keratoderma caused by a DSP mutation. The pattern of delayed enhancement of gadolinium is not specific for genetic heart disease, but is distinct from ischemic cardiomyopathy.
LV = left ventricle; RV = right ventricle.
Images courtesy of Ravi Shah, MD and Raymond Kwong, MD, Brigham and Women’s Hospital, Boston, MA.
Management
Lifestyle recommendations
As with other forms of heart failure, patients with familial DCM are advised to limit excess dietary sodium and to avoid alcohol ingestion or exposure to other cardiotoxins – in particular cocaine, amphetamines and certain anti-neoplastic agents.12 Observational studies have found that competitive athletes have an increased rate of adverse events in LMNA associated DCM.37 Accordingly, it would be reasonable to advise against sustained endurance training in affected patients, even with minimal disease manifestations. Competitive sport is generally proscribed in DCM, consistent with consensus recommendations.53
Medical therapy
In patients with DCM, significant clinical benefit, including increased survival, has been associated with the use of angiotensin converting enzyme inhibitors (ACEi), angiotensin receptor blockers (ARB), beta adrenoreceptor blockers (BB), aldosterone antagonists, and vasodilators. As described in consensus guidelines, these medications should be titrated to the dose used in clinical trials unless limited by side effect.12
Patients with DCM were included in the pivotal trials which established the survival benefit associated with ACEi, ARB and BB use in a broad spectrum of patients with DCM; from severe54, 55 to moderate56, 57 heart failure to asymptomatic LV systolic dysfunction.58, 59 The benefit conferred by aldosterone antagonists in patients with mild60 and severe heart failure61 was similar in patients with both non-ischemic and ischemic cardiomyopathy. Therapy with the vasodilator combination of hydralazine and isosorbide dinitrate improved survival in patients with DCM. 62, 63 However, not all vasodilators are beneficial, including doxazosin62 and dihydropyridine calcium channel blockers64 which did not improve outcomes in DCM.
The proportion of study subjects with familial or genetic DCM was not specified in the large HF clinical trials. In a small randomized, placebo controlled trial of non-genotyped patients with suspected early familial DCM, carvedilol did not improve cardiac function, although longer term follow-up suggested benefit.47 Nonrandomized trials have supported the use of BB and ACEi in patients with Becker and Duchenne Muscular Dystrophy.31
Sudden death risk stratification and implantable cardiac device therapy
The prevention of SCD is a primary concern in patients with inherited cardiomyopathy. Medical therapies, especially BB and aldosterone antagonists,57, 61 reduce the risk of cardiac arrest in patients with DCM and should be used in accordance with guideline recommendations as described in the previous section. ICD therapy can offer incremental prevention of SCD and is advised in selected patients (table 3).65 The use of ICD for secondary prevention is non-controversial, as patients with prior cardiac arrest or sustained ventricular tachycardia benefit from ICD placement.65
Table 3.
Accepted Indications for ICD therapy in Familial Dilated Cardiomyopathy
| Indication | ICD Recommendation* |
|---|---|
| Resuscitated sudden cardiac death from VF or VT | Class I |
| Sustained VT and significant LV dysfunction† | Class I |
| LVEF≤35% and class II–III heart failure on optimal medical therapy | Class I |
| Unexplained syncope and significant LV systolic dysfunction† with inducible VT/VF at EPS | Class I |
| Unexplained syncope and significant LV systolic dysfunction† | Class IIa |
| Sustained VT and normal or near normal LVEF | Class IIa |
| LVEF≤30–35% and class I heart failure on optimal medical therapy | Class IIb |
| Family history of sudden death | Class IIb |
| LV hypertrabeculation/Noncompaction | Class IIb |
2008 AHA/ACC/HRS consensus guideline recommendations.
Class I: Should be performed; Class IIa: Reasonable to perform; Class IIb: May be considered. ICD therapy is not advised for patients without reasonable expectation of survival with good functional status for more than 1 year.
EPS = electrophysiological study.
The indication for primary prevention of SCD with ICD therapy in DCM is largely based on the severity of systolic dysfunction. The risk of SCD increases with decline in systolic function in coronary heart disease,66 non-genotyped DCM,67 and genetic DCM.68 There is strong evidence to support the use of ICD therapy for primary prevention in patients with severe left ventricular systolic dysfunction. Evidence is most robust in patients with ischemic heart disease. However, trials have also been conducted in exclusively non-ischemic DCM. In the DEFINITE trial, ICD therapy was studied in 458 patients with DCM, LVEF≤35%, NYHA class I–III heart failure, and ambient ventricular arrhythmias (e.g. NSVT on ambulatory electrocardiographic monitoring).69 The risk of SCD was significantly reduced (HR 0.20; 0.06–0.71, p=0.006) and there was an associated trend towards reduced all caused mortality with ICD therapy. The SCD-HeFT study of primary prevention ICD therapy in patients with LVEF≤35% and NYHA class II–III heart failure included a prespecified subgroup analysis in the 1,213 patients with DCM.70 There was a non-significant trend towards reduced mortality (HR 0.73; CI 0.50–1.07, p=0.06) in patients with DCM. Importantly, both of these studies were performed in patients receiving recommended BB and ACEi therapy. Consensus guidelines recommend primary prevention ICD placement in patients who have severe systolic dysfunction (LVEF≤30–35%), are receiving optimal medical therapy and have reasonable 1 year survival. Recommendations are strongest for patients with heart failure symptoms (NYHA class II and III).71 The role of ICD therapy for primary prevention of SCD is less well established in patients with lesser degrees of systolic dysfunction (e.g. LVEF≥40%), although recent studies indicate increased risk of arrhythmic events in LMNA mutation carriers with lesser degrees of systolic dysfunction (LVEF<45%).72
Beyond systolic dysfunction, there are accepted (Table 3) and emerging (Table 4) risk factors which can identify patients at increased risk of SCD who may benefit from ICD therapy. Consensus guidelines recommend ICD placement for patients with DCM and unexplained syncope or a family history of SCD.71 However, ascertainment of a family history of SCD may be challenging. Anecdotally, patients often refer to these family events as “heart attacks” which may falsely imply ischemic heart disease. Accordingly, consideration of other factors (age at death, concomitant atherosclerotic risk factors) and review of autopsy data may be needed to clarify the family history.
Table 4.
Potential/Emerging Indications for ICD therapy in Familial Dilated Cardiomyopathy
| Indication |
|---|
| Need for pacemaker |
| Nonsustained VT on ambulatory electrocardiographic monitoring |
| Delayed enhancement of Gadolinium present on CMR |
| Gene based diagnosis* |
LMNA, DES, SCN5A, Desmosomal.
CMR = cardiac magnetic resonance imaging.
In a non-genotyped cohort of individuals with NSVT and DCM, the number and length of NSVT runs is predictive of major ventricular arrhythmias only in cases where LVEF exceeds 35%.73
Compelling data do not support, and guidelines do not advise, the use of genetic testing for SCD risk stratification in patients with DCM.71 However, genetic testing for DCM is still in its infancy and thus, robust genotype-phenotype relationships are still rare. Due to the private nature of mutations in DCM it is difficult to assess risk on a mutation basis. However, the type of mutation (e.g. missense versus frame shift) may be predictive of events. For example, compared to missense mutations, splice-site mutations have been associated with increased risk of life threatening ventricular arrhythmias and SCD in patients with LMNA associated heart disease.37, 72 In addition to mutation type, male gender, mildly reduced LVEF (<45%) and the presence of non-sustained ventricular tachycardia signified increased risk of SCD in a cohort 269 carriers of LMNA mutations, independent of the degree of systolic dysfunction.72 Malignant ventricular arrhythmias did not occur in this cohort, unless a patient had at least 2 of these risk factors. Invasive EPS has not effectively risk stratified patients with non-ischemic or genetic DCM40 and is not routinely advised.
Preliminary reports suggest that DE identified with CMR can identify patients at increased risk of SCD.74 However, electroanatomic mapping has emerged as potentially superior to CMR DE for identification of myocardial scar substrates in patients with ventricular arrhythmias of RV origin75, 76 A comparison of CMR DE with electroanatomic mapping in LV cardiomyopathies has not yet been performed. The results of small studies notwithstanding, the role of genetic testing, ambulatory electrocardiographic monitoring, invasive EPS and advanced imaging for SCD risk stratification in DCM requires further study.
As described above, certain genetic causes of DCM also cause AV block and sinus arrest (DCM+E), often before cardiomyopathy develops. Symptomatic bradyarrhythmias in these patients are appropriately treated with pacing. In patients with concomitant neuromuscular disease, such as myotonic dystrophy, placement of a permanent pacemaker is indicated for third degree or advanced second degree AV block (Class I recommendation), although implant can also be considered for any degree of AV block due to the unpredictable progression of heart block (Class IIb recommendation).71 These patients with established DCM+E, regardless of concomitant neuromuscular disease, remain at risk of SCD68 and consideration should be given to placement of an ICD in lieu of pacing therapy alone.40 In a multicenter registry of 406 patients with genetically confirmed myotonic dystrophy type 1, 46 patients (11.3%) underwent permanent pacemaker implant and 21 (5.2%) received an ICD. Seven patients treated with pacemakers (15.2%) died of SCD. In comparison, 3/21 (14.3%) of ICD patients received appropriate therapies for ventricular arrhythmias, including patients without severe systolic dysfunction.77 In a small prospective study, Meune et al. evaluated the efficacy of ICD therapy in patients (n=19) with LMNA associated heart disease who had symptomatic bradyarrhythmias but preserved systolic function (LVEF 58 ±12%).40 After 33.9±21.0 months follow-up, appropriate ICD therapies for VT and VF occurred in 42% of patients. However, larger studies have not confirmed these findings,37, 72 and the role of ICD therapy in patients with genetic DCM who otherwise require pacing remains undefined. A reasonable approach would be to incorporate a patients individual risk (LVEF, ambient ventricular arrhythmia, family history) when selecting device therapy for heart block or bradyarrhythmia. Assessment of myocardial scar using either CMR or electroanatomic mapping to guide device selection in these patients has not yet been explored but merits further study.
Cardiac resynchronization therapy (CRT) improves survival and reduces heart failure symptoms in patients with systolic dysfunction, QRS prolongation and mild78 to severe79 heart failure symptoms. Although clinical trials of CRT therapy did not exclusively enroll patients with non-ischemic DCM, subgroup analysis suggested similar benefit of CRT regardless of the etiology of systolic dysfunction. Indeed, recent multivariate analysis has found a greater benefit in patient with non-ischemic DCM and greater degree of QRS prolongation (>150 ms).80 In genetically confirmed DCM with advanced AVB but with mild to no impairment of LV function (EF≥40%), CRT may be reasonable to consider given the deleterious effects of chronic RV pacing on LV performance and heart failure symptoms.81
Advanced Therapies
In spite of appropriate medical and device therapy, a proportion of patients with genetic/familial DCM may develop progressive pump failure. As with other causes of heart failure, cardiac transplantation and mechanical circulatory support are appropriate therapies for selected patients with end-stage DCM.
Genetic testing and family evaluations
Genetic testing for DCM has been clinically available for several years. A “panel” based approach, whereby multiple disease genes are re-sequenced, is advised owing to the considerable genetic diversity of DCM. Multiple CLIA approved labs offer genetic testing for DCM utilizing this approach, a complete list of which can be found at www.genetests.org. Although precise data are lacking, approximately 30–40% of patients currently referred for clinical genetic testing will be found to have a pathogenic genetic variant (aka mutation). In general, clinical and demographic factors, including family history, are not predictive of the results of genetic testing.16 However, the likelihood of finding a mutation appears less likely in older patients (> 40 years) with non-familial disease.23
Ordering providers should be aware of the limitations of genetic testing for DCM, especially as they apply to indeterminate and “negative” test results in index patients. Importantly, not all genetic variation identified by genetic testing is pathogenic, and clinical decisions should not be predicated on genetic variants of uncertain or indeterminate clinical significance. In order to consider a particular DNA variant pathogenic, it is important that several lines of evidence support an association with disease. Evidence supportive of pathogenicity can include absence of the variant from large race-matched control populations, evolutionary conservation of the mutated amino acid, co-segregation of genotype and phenotype within a kindred, functional analysis and computational (i.e. in silico) analysis. Secondly, a “negative” genetic test result in the index patient does not imply that the patient does not have genetic disease. Rather, they do not have a mutation in one of the tested genes. As our knowledge of the genetics of DCM is incomplete, there remains the possibility that a mutation could be present in a non-tested gene.
Notwithstanding cost and the limitations thus enumerated, when should genetic testing be performed in DCM? First, genetic testing is warranted if the identification of a genetic cause can inform clinical management of the index patient. This may be of most use in DCM+E where non-genetic heart disease (e.g. cardiac sarcoid) may have a similar clinical presentation but is managed differently (e.g. high dose corticosteroid therapy).24 In these cases, the diagnosis of genetic heart disease can allow deferral of evaluations for alternate diagnoses (e.g. endomyocardial biopsy). Second, genetic testing is a requisite for preimplantation genetic diagnosis. When performed with in vitro fertilization, preimplantation genetic diagnosis and selective transfer of genetically unaffected embryos can prevent the transmission of disease to offspring.82 Nevertheless, it is in the prioritization of family evaluations where the clinical value of genetic testing is likely greatest.
Recognition that a patient has a familial or genetic cardiomyopathy is accompanied with the responsibility to consider the risk of disease in the patient’s family. Privacy concerns and medical ethics preclude direct communication with at-risk family members, although the relevant recommendations can be relayed through the index patient. Because familial DCM can present early in life, consensus guidelines recommend screening first degree relatives with clinical examination, echocardiography and ECG +/− measurement of serum CK beginning in childhood. Screening for DCM should be repeated every 3 to 5 years owing to age-dependent penetrance, which is typical of genetic DCM.13 When genetic testing identifies a pathogenic DNA variant (mutation) in the index patient, clinical screening can be restricted to family members who have inherited the mutation.13 The use of genetic testing to prioritize family evaluations is anticipated to halve the cost of screening and limits evaluations only to those at risk of developing disease.13 When genetic testing of the index patient fails to identify a pathogenic variant, all first degree family members should be clinically screened, especially when familial disease is present. In cases where a variant of indeterminate significance was identified in the index patient, combined clinical and genetic testing of their relatives can clarify variant pathogenicity.
Prior to offering predictive genetic testing to clinically unaffected family members, the risks of genetic testing should be explained, including implications for the procurement of life insurance. Family members should also be counseled to the limitations of genetic testing, and the possibility that current interpretations of variant pathogenicity are subject to change which could lead to reassessment of their risk. Accordingly, genetic counseling is a recommended component of the management of families with DCM and should be performed by an experienced professional (e.g. genetics counselor and/or clinical geneticist). 20, 83
Future directions
Dramatic improvements in sequencing technology and greater insight into cardiomyopathy pathogenesis will influence the diagnosis, evaluation and management of familial DCM. The use of genetic testing in clinical practice has begun, however the cost of sequencing and relative insensitivity of clinical assays has limited widespread acceptance. This is expected to change in the near future as the cost of sequencing the entire genome is anticipated to fall below $1,000 dollars in the next 3 years.84 Sophisticated information systems and decision support will be necessary for providers to incorporate genome sequencing in clinical practice.85 Inexpensive and accurate genetic testing will increase the number of DCM “patients” identified to have pre-clinical disease. These are hitherto unaffected relatives of patients with overt DCM who carry a pathogenic mutation. Evaluations performed on these preclinical patients suggest that disease mechanisms are activated early and can be detected with contemporary diagnostic modalities.86 The characterization of the preclinical DCM phenotype will inform our pathophysiologic models. Ideally, this will translate into preventative therapies which limit the future burden of heart failure and arrhythmia. Beyond prevention, patients with overt disease will stand to benefit from gene and pathway specific therapies. Small molecule inhibitors of the ERK1/2 and JNK pathways slow the progression of LMNA associated DCM in animal models.87 Antisense oligonucleotides developed to treat Duchenne muscular dystrophy, have already shown promise in small clinical studies, however the effects of this therapy on cardiac function are not currently known.88
Summary
Familial DCM has diverse genetic etiologies and is an important cause of HF and SCD. An important minority of patients with familial DCM will present with associated clinical features, including conduction disease, arrhythmia and skeletal myopathy. Evidenced-based medical and device therapies improve clinical outcomes. The application of genetic testing has begun to inform the clinical management of these patients, however it has not yet dramatically influenced therapy.
Acknowledgments
The authors appreciate Sunu S. Thomas, MD for his thoughtful review of this manuscript.
Footnotes
Conflict of Interest Disclosures: None
References
- 1.Taylor DO, Edwards LB, Boucek MM, Trulock EP, Aurora P, Christie J, Dobbels F, Rahmel AO, Keck BM, Hertz MI. Registry of the International Society for Heart and Lung Transplantation: twenty-fourth official adult heart transplant report--2007. J Heart Lung Transplant. 2007;26:769–781. doi: 10.1016/j.healun.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 2.Felker GM, Thompson RE, Hare JM, Hruban RH, Clemetson DE, Howard DL, Baughman KL, Kasper EK. Underlying Causes and Long-Term Survival in Patients with Initially Unexplained Cardiomyopathy. N Engl J Med. 2000;342:1077–1084. doi: 10.1056/NEJM200004133421502. [DOI] [PubMed] [Google Scholar]
- 3.Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, Atherton J, Vidaillet HJ, Jr, Spudich S, De Girolami U, Seidman JG, Seidman C, Muntoni F, Muehle G, Johnson W, McDonough B. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341:1715–1724. doi: 10.1056/NEJM199912023412302. [DOI] [PubMed] [Google Scholar]
- 4.Kamisago M, Sharma SD, DePalma SR, Solomon S, Sharma P, McDonough B, Smoot L, Mullen MP, Woolf PK, Wigle ED, Seidman JG, Seidman CE. Mutations in Sarcomere Protein Genes as a Cause of Dilated Cardiomyopathy. N Engl J Med. 2000;343:1688–1696. doi: 10.1056/NEJM200012073432304. [DOI] [PubMed] [Google Scholar]
- 5.Schmitt JP, Kamisago M, Asahi M, Li GH, Ahmad F, Mende U, Kranias EG, MacLennan DH, Seidman JG, Seidman CE. Dilated Cardiomyopathy and Heart Failure Caused by a Mutation in Phospholamban. Science. 2003;299:1410–1413. doi: 10.1126/science.1081578. [DOI] [PubMed] [Google Scholar]
- 6.Olson TM, Michels VV, Ballew JD, Reyna SP, Karst ML, Herron KJ, Horton SC, Rodeheffer RJ, Anderson JL. Sodium Channel Mutations and Susceptibility to Heart Failure and Atrial Fibrillation. JAMA. 2005;293:447–454. doi: 10.1001/jama.293.4.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li D, Morales A, Gonzalez-Quintana J, Norton N, Siegfried JD, Hofmeyer M, Hershberger RE. Identification of novel mutations in RBM20 in patients with dilated cardiomyopathy. Clin Transl Sci. 2010;3:90–97. doi: 10.1111/j.1752-8062.2010.00198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, Teodorescu DL, Cirino AL, Banner NR, Pennell DJ, Graw S, Merlo M, Di Lenarda A, Sinagra G, Bos JM, Ackerman MJ, Mitchell RN, Murry CE, Lakdawala NK, Ho CY, Barton PJ, Cook SA, Mestroni L, Seidman JG, Seidman CE. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–628. doi: 10.1056/NEJMoa1110186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fuster V, Gersh BJ, Giuliani ER, Tajik AJ, Brandenburg RO, Frye RL. The natural history of idiopathic dilated cardiomyopathy. Am J Cardiol. 1981;47:525–531. doi: 10.1016/0002-9149(81)90534-8. [DOI] [PubMed] [Google Scholar]
- 10.Burkett EL, Hershberger RE. Clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol. 2005;45:969–981. doi: 10.1016/j.jacc.2004.11.066. [DOI] [PubMed] [Google Scholar]
- 11.Mahon NG, Murphy RT, MacRae CA, Caforio AL, Elliott PM, McKenna WJ. Echocardiographic evaluation in asymptomatic relatives of patients with dilated cardiomyopathy reveals preclinical disease. Ann Intern Med. 2005;143:108–115. doi: 10.7326/0003-4819-143-2-200507190-00009. [DOI] [PubMed] [Google Scholar]
- 12.Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG, Jessup M, Konstam MA, Mancini DM, Michl K, Oates JA, Rahko PS, Silver MA, Stevenson LW, Yancy CW Writing Committee M. 2009 Focused Update Incorporated Into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults. Circulation. 2009;119:e391–e479. doi: 10.1161/CIRCULATIONAHA.109.192065. [DOI] [PubMed] [Google Scholar]
- 13.Hershberger RE, Lindenfeld J, Mestroni L, Seidman CE, Taylor MR, Towbin JA. Genetic evaluation of cardiomyopathy--a Heart Failure Society of America practice guideline. J Card Fail. 2009;15:83–97. doi: 10.1016/j.cardfail.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 14.Lakdawala NK, Dellefave L, Redwood CS, Sparks E, Cirino AL, Depalma S, Colan SD, Funke B, Zimmerman RS, Robinson P, Watkins H, Seidman CE, Seidman JG, McNally EM, Ho CY. Familial dilated cardiomyopathy caused by an alpha-tropomyosin mutation: the distinctive natural history of sarcomeric dilated cardiomyopathy. J Am Coll Cardiol. 2010;55:320–329. doi: 10.1016/j.jacc.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sparks EA, Boudoulas KD, Raman SV, Sasaki T, Graber HL, Nelson SD, Seidman CE, Boudoulas H. Heritable cardiac conduction and myocardial disease: from the clinic to the basic science laboratory and back to the clinic. Cardiology. 2011;118:179–186. doi: 10.1159/000328638. [DOI] [PubMed] [Google Scholar]
- 16.Hershberger RE, Parks SB, Kushner JD, Li D, Ludwigsen S, Jakobs P, Nauman D, Burgess D, Partain J, Litt M. Coding sequence mutations identified in MYH7, TNNT2, SCN5A, CSRP3, LBD3, and TCAP from 313 patients with familial or idiopathic dilated cardiomyopathy. Clin Transl Sci. 2008;1:21–26. doi: 10.1111/j.1752-8062.2008.00017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dellefave L, McNally EM. The genetics of dilated cardiomyopathy. Curr Opin Cardiol. 2010;25:198–204. doi: 10.1097/HCO.0b013e328337ba52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Norton N, Li D, Rieder MJ, Siegfried JD, Rampersaud E, Zuchner S, Mangos S, Gonzalez-Quintana J, Wang L, McGee S, Reiser J, Martin E, Nickerson DA, Hershberger RE. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am J Hum Genet. 2011;88:273–282. doi: 10.1016/j.ajhg.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brauch KM, Karst ML, Herron KJ, de Andrade M, Pellikka PA, Rodeheffer RJ, Michels VV, Olson TM. Mutations in Ribonucleic Acid Binding Protein Gene Cause Familial Dilated Cardiomyopathy. J Am Coll Cardiol. 2009;54:930–941. doi: 10.1016/j.jacc.2009.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hershberger RE, Siegfried JD. Update 2011: clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol. 2011;57:1641–1649. doi: 10.1016/j.jacc.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hershberger RE, Cowan J, Morales A, Siegfried J. Progress With Genetic Cardiomyopathies: Screening, Counseling, and Testing in Dilated, Hypertrophic, and Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Circ Heart Fail. 2009;2:253–261. doi: 10.1161/CIRCHEARTFAILURE.108.817346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hershberger RE, Norton N, Morales A, Li D, Siegfried JD, Gonzalez-Quintana J. Coding Sequence Rare Variants Identified in MYBPC3, MYH6, TPM1, TNNC1 and TNNI3 from 312 Patients with Familial or Idiopathic Dilated Cardiomyopathy. Circ Cardiovasc Genet. 2010;3:155–161. doi: 10.1161/CIRCGENETICS.109.912345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lakdawala NK, Funke BH, Baxter S, Cirino AL, Roberts AE, Judge DP, Johnson N, Mendelsohn NJ, Morel C, Care M, Chung WK, Jones C, Psychogios A, Duffy E, Rehm HL, White E, Seidman JG, Seidman CE, Ho CY. Genetic testing for dilated cardiomyopathy in clinical practice. J Card Fail. 2012;18:296–303. doi: 10.1016/j.cardfail.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lakdawala NK, Givertz MM. Dilated cardiomyopathy with conduction disease and arrhythmia. Circulation. 2010;122:527–534. doi: 10.1161/CIRCULATIONAHA.109.892240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parks SB, Kushner JD, Nauman D, Burgess D, Ludwigsen S, Peterson A, Li D, Jakobs P, Litt M, Porter CB, Rahko PS, Hershberger RE. Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Am Heart J. 2008;156:161–169. doi: 10.1016/j.ahj.2008.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arbustini E, Pilotto A, Repetto A, Grasso M, Negri A, Diegoli M, Campana C, Scelsi L, Baldini E, Gavazzi A, Tavazzi L. Autosomal dominant dilated cardiomyopathy with atrioventricular block: a lamin A/C defect-related disease. J Am Coll Cardiol. 2002;39:981–990. doi: 10.1016/s0735-1097(02)01724-2. [DOI] [PubMed] [Google Scholar]
- 27.Sen-Chowdhry S, Syrris P, Prasad SK, Hughes SE, Merrifield R, Ward D, Pennell DJ, McKenna WJ. Left-Dominant Arrhythmogenic Cardiomyopathy: An Under-Recognized Clinical Entity. J Am Coll Cardiol. 2008;52:2175–2187. doi: 10.1016/j.jacc.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 28.Norgett EE, Hatsell SJ, Carvajal-Huerta L, Ruiz Cabezas J-C, Common J, Purkis PE, Whittock N, Leigh IM, Stevens HP, Kelsell DP. Recessive mutation in desmoplakin disrupts desmoplakin “intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet. 2000;9:2761–2766. doi: 10.1093/hmg/9.18.2761. [DOI] [PubMed] [Google Scholar]
- 29.Norgett EE, Lucke TW, Bowers B, Munro CS, Leigh IM, Kelsell DP. Early Death from Cardiomyopathy in a Family with Autosomal Dominant Striate Palmoplantar Keratoderma and Woolly Hair Associated with a Novel Insertion Mutation in Desmoplakin. J Invest Dermatol. 2006;126:1651–1654. doi: 10.1038/sj.jid.5700291. [DOI] [PubMed] [Google Scholar]
- 30.Elliott P, O’Mahony C, Syrris P, Evans A, Rivera Sorensen C, Sheppard MN, Carr-White G, Pantazis A, McKenna WJ. Prevalence of Desmosomal Protein Gene Mutations in Patients With Dilated Cardiomyopathy/Clinical Perspective. Circ Cardiovasc Genet. 2010;3:314–322. doi: 10.1161/CIRCGENETICS.110.937805. [DOI] [PubMed] [Google Scholar]
- 31.Jefferies JL, Eidem BW, Belmont JW, Craigen WJ, Ware SM, Fernbach SD, Neish SR, Smith EOB, Towbin JA. Genetic Predictors and Remodeling of Dilated Cardiomyopathy in Muscular Dystrophy. Circulation. 2005;112:2799–2804. doi: 10.1161/CIRCULATIONAHA.104.528281. [DOI] [PubMed] [Google Scholar]
- 32.Dalakas MC, Park K-Y, Semino-Mora C, Lee HS, Sivakumar K, Goldfarb LG. Desmin Myopathy, a Skeletal Myopathy with Cardiomyopathy Caused by Mutations in the Desmin Gene. N Engl J Med. 2000;342:770–780. doi: 10.1056/NEJM200003163421104. [DOI] [PubMed] [Google Scholar]
- 33.Bonne G, Leturcq F, Ben Yaou R. Emery-Dreifuss Muscular Dystrophy. Gene Reviews. 2010 [PubMed] [Google Scholar]
- 34.Bushby K, Muntoni F, Bourke JP. 107th ENMC International Workshop: the management of cardiac involvement in muscular dystrophy and myotonic dystrophy. 7th–9th June 2002, Naarden, the Netherlands. Neuromuscular Disorders. 2003;13:166–172. doi: 10.1016/s0960-8966(02)00213-4. [DOI] [PubMed] [Google Scholar]
- 35.Feng J, Yan JY, Buzin CH, Sommer SS, Towbin JA. Comprehensive mutation scanning of the dystrophin gene in patients with nonsyndromic X-linked dilated cardiomyopathy. J Am Coll Cardiol. 2002;40:1120–1124. doi: 10.1016/s0735-1097(02)02126-5. [DOI] [PubMed] [Google Scholar]
- 36.Politano L, Nigro V, Nigro G, Petretta VR, Passamano L, Papparella S, Di Somma S, Comi LI. Development of cardiomyopathy in female carriers of Duchenne and Becker muscular dystrophies. JAMA. 1996;275:1335–1338. [PubMed] [Google Scholar]
- 37.Pasotti M, Klersy C, Pilotto A, Marziliano N, Rapezzi C, Serio A, Mannarino S, Gambarin F, Favalli V, Grasso M, Agozzino M, Campana C, Gavazzi A, Febo O, Marini M, Landolina M, Mortara A, Piccolo G, Viganò M, Tavazzi L, Arbustini E. Long-Term Outcome and Risk Stratification in Dilated Cardiolaminopathies. J Am Coll Cardiol. 2008;52:1250–1260. doi: 10.1016/j.jacc.2008.06.044. [DOI] [PubMed] [Google Scholar]
- 38.McNair WP, Sinagra G, Taylor MRG, Di Lenarda A, Ferguson DA, Salcedo EE, Slavov D, Zhu X, Caldwell JH, Mestroni L Familial Cardiomyopathy Registry Research G. SCN5A Mutations Associate With Arrhythmic Dilated Cardiomyopathy and Commonly Localize to the Voltage-Sensing Mechanism. J Am Coll Cardiol. 2011;57:2160–2168. doi: 10.1016/j.jacc.2010.09.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Otomo JUN, Kure S, Shiba T, Karibe A, Shinozaki T, Yagi T, Naganuma H, Tezuka F, Miura M, Ito M, Watanabe JUN, Matsubara Y, Shirato K. Electrophysiological and Histopathological Characteristics of Progressive Atrioventricular Block Accompanied by Familial Dilated Cardiomyopathy Caused by a Novel Mutation of Lamin A/C Gene. J Cardiovasc Electrophysiol. 2005;16:137–145. doi: 10.1046/j.1540-8167.2004.40096.x. [DOI] [PubMed] [Google Scholar]
- 40.Meune C, Van Berlo JH, Anselme F, Bonne G, Pinto YM, Duboc D. Primary Prevention of Sudden Death in Patients with Lamin A/C Gene Mutations. N Engl J Med. 2006;354:209-a–210. doi: 10.1056/NEJMc052632. [DOI] [PubMed] [Google Scholar]
- 41.Probst S, Oechslin E, Schuler P, Greutmann M, Boye P, Knirsch W, Berger F, Thierfelder L, Jenni R, Klaassen S. Sarcomere Gene Mutations in Isolated Left Ventricular Noncompaction Cardiomyopathy Do Not Predict Clinical Phenotype. Circ Cardiovasc Genet. 2011;4:367–374. doi: 10.1161/CIRCGENETICS.110.959270. [DOI] [PubMed] [Google Scholar]
- 42.Schonberger J, Wang L, Shin JT, Kim SD, Depreux FFS, Zhu H, Zon L, Pizard A, Kim JB, MacRae CA, Mungall AJ, Seidman JG, Seidman CE. Mutation in the transcriptional coactivator EYA4 causes dilated cardiomyopathy and sensorineural hearing loss. Nat Genet. 2005;37:418–422. doi: 10.1038/ng1527. [DOI] [PubMed] [Google Scholar]
- 43.Schonberger J, Levy H, Grunig E, Sangwatanaroj S, Fatkin D, MacRae C, Stacker H, Halpin C, Eavey R, Philbin EF, Katus H, Seidman JG, Seidman CE. Dilated Cardiomyopathy and Sensorineural Hearing Loss : A Heritable Syndrome That Maps to 6q23–24. Circulation. 2000;101:1812–1818. doi: 10.1161/01.cir.101.15.1812. [DOI] [PubMed] [Google Scholar]
- 44.Verhaert D, Richards K, Rafael-Fortney JA, Raman SV. Cardiac Involvement in Patients With Muscular Dystrophies. Circ Cardiovasc Imaging. 2011;4:67–76. doi: 10.1161/CIRCIMAGING.110.960740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nigro G, Comi LI, Politano L, Bain RJI. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int J Cardiol. 1990;26:271–277. doi: 10.1016/0167-5273(90)90082-g. [DOI] [PubMed] [Google Scholar]
- 46.Piccolo G, Azan G, Tonin P, Arbustini E, Gavazzi A, Banfi P, Mora M, Morandi L, Tedeschi S. Dilated cardiomyopathy requiring cardiac transplantation as initial manifestation of Xp21 Becker type muscular dystrophy. Neuromuscular Disorders. 1994;4:143–146. doi: 10.1016/0960-8966(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 47.Yeoh T, Hayward C, Benson V, Sheu A, Richmond Z, Feneley MP, Keogh AM, Macdonald P, Fatkin D. A Randomised, Placebo-controlled Trial of Carvedilol in Early Familial Dilated Cardiomyopathy. Heart, Lung and Circulation. 2011;20:566–573. doi: 10.1016/j.hlc.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 48.Douglas PS, Garcia MJ, Haines DE, Lai WW, Manning WJ, Patel AR, Picard MH, Polk DM, Ragosta M, Parker Ward R, Weiner RB. ACCF/ASE/AHA/ASNC/HFSA/HRS/SCAI/SCCM/SCCT/SCMR 2011 Appropriate Use Criteria for Echocardiography. A Report of the American College of Cardiology Foundation Appropriate Use Criteria Task Force, American Society of Echocardiography, American Heart Association, American Society of Nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, Society of Critical Care Medicine, Society of Cardiovascular Computed Tomography, Society for Cardiovascular Magnetic Resonance American College of Chest Physicians. J Am Soc Echocardiogr. 2011;24:229–267. doi: 10.1016/j.echo.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 49.Buckley AE, Dean J, Mahy IR. Cardiac involvement in Emery Dreifuss muscular dystrophy: a case series. Heart. 1999;82:105–108. doi: 10.1136/hrt.82.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mancini DM, Eisen H, Kussmaul W, Mull R, Edmunds LH, Wilson JR. Value of peak exercise oxygen consumption for optimal timing of cardiac transplantation in ambulatory patients with heart failure. Circulation. 1991;83:778–786. doi: 10.1161/01.cir.83.3.778. [DOI] [PubMed] [Google Scholar]
- 51.Bogun F, Crawford T, Reich S, Koelling TM, Armstrong W, Good E, Jongnarangsin K, Marine JE, Chugh A, Pelosi F, Oral H, Morady F. Radiofrequency ablation of frequent, idiopathic premature ventricular complexes: comparison with a control group without intervention. Heart Rhythm. 2007;4:863–867. doi: 10.1016/j.hrthm.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 52.McCrohon JA, Moon JCC, Prasad SK, McKenna WJ, Lorenz CH, Coats AJS, Pennell DJ. Differentiation of Heart Failure Related to Dilated Cardiomyopathy and Coronary Artery Disease Using Gadolinium-Enhanced Cardiovascular Magnetic Resonance. Circulation. 2003;108:54–59. doi: 10.1161/01.CIR.0000078641.19365.4C. [DOI] [PubMed] [Google Scholar]
- 53.Maron BJ, Ackerman MJ, Nishimura RA, Pyeritz RE, Towbin JA, Udelson JE. Task Force 4: HCM and other cardiomyopathies, mitral valve prolapse, myocarditis, and Marfan syndrome. J Am Coll Cardiol. 2005;45:1340–1345. doi: 10.1016/j.jacc.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 54.Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). The CONSENSUS Trial Study Group. N Engl J Med. 1987;316:1429–1435. doi: 10.1056/NEJM198706043162301. [DOI] [PubMed] [Google Scholar]
- 55.Packer M, Coats AJ, Fowler MB, Katus HA, Krum H, Mohacsi P, Rouleau JL, Tendera M, Castaigne A, Roecker EB, Schultz MK, DeMets DL. Effect of carvedilol on survival in severe chronic heart failure. N Engl J Med. 2001;344:1651–1658. doi: 10.1056/NEJM200105313442201. [DOI] [PubMed] [Google Scholar]
- 56.Granger CB, McMurray JJ, Yusuf S, Held P, Michelson EL, Olofsson B, Ostergren J, Pfeffer MA, Swedberg K. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function intolerant to angiotensin-converting-enzyme inhibitors: the CHARM-Alternative trial. Lancet. 2003;362:772–776. doi: 10.1016/S0140-6736(03)14284-5. [DOI] [PubMed] [Google Scholar]
- 57.Committees C-IIa. The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): a randomised trial. Lancet. 1999;353:9–13. [PubMed] [Google Scholar]
- 58.Jong P, Yusuf S, Rousseau MF, Ahn SA, Bangdiwala SI. Effect of enalapril on 12-year survival and life expectancy in patients with left ventricular systolic dysfunction: a follow-up study. Lancet. 2003;361:1843–1848. doi: 10.1016/S0140-6736(03)13501-5. [DOI] [PubMed] [Google Scholar]
- 59.Colucci WS, Kolias TJ, Adams KF, Armstrong WF, Ghali JK, Gottlieb SS, Greenberg B, Klibaner MI, Kukin ML, Sugg JE. Metoprolol reverses left ventricular remodeling in patients with asymptomatic systolic dysfunction: the REversal of VEntricular Remodeling with Toprol-XL (REVERT) trial. Circulation. 2007;116:49–56. doi: 10.1161/CIRCULATIONAHA.106.666016. [DOI] [PubMed] [Google Scholar]
- 60.Zannad F, McMurray JJ, Krum H, van Veldhuisen DJ, Swedberg K, Shi H, Vincent J, Pocock SJ, Pitt B. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med. 2011;364:11–21. [Google Scholar]
- 61.Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, Wittes J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999;341:709–717. doi: 10.1056/NEJM199909023411001. [DOI] [PubMed] [Google Scholar]
- 62.Cohn JN, Archibald DG, Ziesche S, Franciosa JA, Harston WE, Tristani FE, Dunkman WB, Jacobs W, Francis GS, Flohr KH, Goldman S, Cobb FR, Shah PM, Saunders R, Fletcher RD, Loeb HS, Hughes VC, Baker B. Effect of vasodilator therapy on mortality in chronic congestive heart failure. Results of a Veterans Administration Cooperative Study. N Engl J Med. 1986;314:1547–1552. doi: 10.1056/NEJM198606123142404. [DOI] [PubMed] [Google Scholar]
- 63.Taylor AL, Ziesche S, Yancy C, Carson P, D’Agostino R, Jr, Ferdinand K, Taylor M, Adams K, Sabolinski M, Worcel M, Cohn JN. Combination of isosorbide dinitrate and hydralazine in blacks with heart failure. N Engl J Med. 2004;351:2049–2057. doi: 10.1056/NEJMoa042934. [DOI] [PubMed] [Google Scholar]
- 64.Thackray S, Witte K, Clark AL, Cleland JG. Clinical trials update: OPTIME-CHF, PRAISE-2, ALL-HAT. Eur J Heart Fail. 2000;2:209–212. doi: 10.1016/s1388-9842(00)00080-5. [DOI] [PubMed] [Google Scholar]
- 65.Epstein AE, DiMarco JP, Ellenbogen KA, Estes NA, 3rd, Freedman RA, Gettes LS, Gillinov AM, Gregoratos G, Hammill SC, Hayes DL, Hlatky MA, Newby LK, Page RL, Schoenfeld MH, Silka MJ, Stevenson LW, Sweeney MO, Smith SC, Jr, Jacobs AK, Adams CD, Anderson JL, Buller CE, Creager MA, Ettinger SM, Faxon DP, Halperin JL, Hiratzka LF, Hunt SA, Krumholz HM, Kushner FG, Lytle BW, Nishimura RA, Ornato JP, Page RL, Riegel B, Tarkington LG, Yancy CW. ACC/AHA/HRS 2008 Guidelines for Device-Based Therapy of Cardiac Rhythm Abnormalities: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the ACC/AHA/NASPE 2002 Guideline Update for Implantation of Cardiac Pacemakers and Antiarrhythmia Devices) developed in collaboration with the American Association for Thoracic Surgery and Society of Thoracic Surgeons. J Am Coll Cardiol. 2008;51:e1–62. doi: 10.1016/j.jacc.2008.02.032. [DOI] [PubMed] [Google Scholar]
- 66.Solomon SD, Zelenkofske S, McMurray JJV, Finn PV, Velazquez E, Ertl G, Harsanyi A, Rouleau JL, Maggioni A, Kober L, White H, Van de Werf F, Pieper K, Califf RM, Pfeffer MA. Sudden Death in Patients with Myocardial Infarction and Left Ventricular Dysfunction, Heart Failure, or Both. N Engl J Med. 2005;352:2581–2588. doi: 10.1056/NEJMoa043938. [DOI] [PubMed] [Google Scholar]
- 67.Borleffs CJW, van Welsenes GH, van Bommel RJ, van der Velde ET, Bax JJ, van Erven L, Putter H, van der Bom JG, Rosendaal FR, Schalij MJ. Mortality risk score in primary prevention implantable cardioverter defibrillator recipients with non-ischaemic or ischaemic heart disease. Eur Heart J. 2010;31:712–718. doi: 10.1093/eurheartj/ehp497. [DOI] [PubMed] [Google Scholar]
- 68.Nelson SD, Sparks EA, Graber HL, Boudoulas H, Mehdirad AA, Baker P, Wooley C. Clinical characteristics of sudden death victims in heritable (chromosome 1p1-1q1) conduction and myocardial disease. J Am Coll Cardiol. 1998;32:1717–1723. doi: 10.1016/s0735-1097(98)00424-0. [DOI] [PubMed] [Google Scholar]
- 69.Kadish A, Dyer A, Daubert JP, Quigg R, Estes NAM, Anderson KP, Calkins H, Hoch D, Goldberger J, Shalaby A, Sanders WE, Schaechter A, Levine JH. Prophylactic Defibrillator Implantation in Patients with Nonischemic Dilated Cardiomyopathy. N Engl J Med. 2004;350:2151–2158. doi: 10.1056/NEJMoa033088. [DOI] [PubMed] [Google Scholar]
- 70.Bardy GH, Lee KL, Mark DB, Poole JE, Packer DL, Boineau R, Domanski M, Troutman C, Anderson J, Johnson G, McNulty SE, Clapp-Channing N, Davidson-Ray LD, Fraulo ES, Fishbein DP, Luceri RM, Ip JH. Amiodarone or an Implantable Cardioverter-Defibrillator for Congestive Heart Failure. N Engl J Med. 2005;352:225–237. doi: 10.1056/NEJMoa043399. [DOI] [PubMed] [Google Scholar]
- 71.Zipes DP, Camm AJ, Borggrefe M, Buxton AE, Chaitman B, Fromer M, Gregoratos G, Klein G, Moss AJ, Myerburg RJ, Priori SG, Quinones MA, Roden DM, Silka MJ, Tracy C. ACC/AHA/ESC 2006 Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: A Report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death): Developed in Collaboration With the European Heart Rhythm Association and the Heart Rhythm Society. Circulation. 2006;114:e385–484. doi: 10.1161/CIRCULATIONAHA.106.178233. [DOI] [PubMed] [Google Scholar]
- 72.van Rijsingen IAW, Arbustini E, Elliott PM, Mogensen J, Hermans-van Ast JF, van der Kooi AJ, van Tintelen JP, van den Berg MP, Pilotto A, Pasotti M, Jenkins S, Rowland C, Aslam U, Wilde AAM, Perrot A, Pankuweit S, Zwinderman AH, Charron P, Pinto YM. Risk Factors for Malignant Ventricular Arrhythmias in Lamin A/C Mutation Carriers: A European Cohort Study. J Am Coll Cardiol. 2012;59:493–500. doi: 10.1016/j.jacc.2011.08.078. [DOI] [PubMed] [Google Scholar]
- 73.Zecchin M, Di Lenarda A, Gregori D, Merlo M, Pivetta A, Vitrella G, Sabbadini G, Mestroni L, Sinagra G. Are nonsustained ventricular tachycardias predictive of major arrhythmias in patients with dilated cardiomyopathy on optimal medical treatment? Pacing Clin Electrophysiol. 2008;31:290–299. doi: 10.1111/j.1540-8159.2008.00988.x. [DOI] [PubMed] [Google Scholar]
- 74.Iles L, Pfluger H, Lefkovits L, Butler MJ, Kistler PM, Kaye DM, Taylor AJ. Myocardial Fibrosis Predicts Appropriate Device Therapy in Patients With Implantable Cardioverter-Defibrillators for Primary Prevention of Sudden Cardiac Death. J Am Coll Cardiol. 2011;57:821–828. doi: 10.1016/j.jacc.2010.06.062. [DOI] [PubMed] [Google Scholar]
- 75.Perazzolo Marra M, Leoni L, Bauce B, Corbetti F, Zorzi A, Migliore F, Silvano M, Rigato I, Tona F, Tarantini G, Cacciavillani L, Basso C, Buja G, Thiene G, Iliceto S, Corrado D. Imaging Study of Ventricular Scar in Arrhythmogenic Right Ventricular Cardiomyopathy: Comparison of Three-Dimensional Electroanatomic Voltage Mapping and Contrast-Enhanced CARDIAC Magnetic Resonance. Circ Arrhythm Electrophysiol. 2011 doi: 10.1161/CIRCEP.111.964635. [DOI] [PubMed] [Google Scholar]
- 76.Santangeli P, Hamilton-Craig C, Russo AD, Pieroni M, Casella M, Pelargonio G, Biase LD, Smaldone C, Bartoletti S, Narducci ML, Tondo C, Bellocci F, Natale A. Imaging of Scar in Patients with Ventricular Arrhythmias of Right Ventricular Origin: Cardiac Magnetic Resonance Versus Electroanatomic Mapping. J Cardiovasc Electrophysiol. 2011;22:1359–1366. doi: 10.1111/j.1540-8167.2011.02127.x. [DOI] [PubMed] [Google Scholar]
- 77.Bhakta D, Shen C, Kron J, Epstein AE, Pascuzzi RM, Groh WJ. Pacemaker and Implantable Cardioverter-Defibrillator Use in a US Myotonic Dystrophy Type 1 Population. J Cardiovasc Electrophysiol. 2011;22:1369–1375. doi: 10.1111/j.1540-8167.2011.02200.x. [DOI] [PubMed] [Google Scholar]
- 78.Tang ASL, Wells GA, Talajic M, Arnold MO, Sheldon R, Connolly S, Hohnloser SH, Nichol G, Birnie DH, Sapp JL, Yee R, Healey JS, Rouleau JL. Cardiac-Resynchronization Therapy for Mild-to-Moderate Heart Failure. N Engl J Med. 2010;363:2385–2395. doi: 10.1056/NEJMoa1009540. [DOI] [PubMed] [Google Scholar]
- 79.Cleland JGF, Daubert J-C, Erdmann E, Freemantle N, Gras D, Kappenberger L, Tavazzi L. The Effect of Cardiac Resynchronization on Morbidity and Mortality in Heart Failure. N Engl J Med. 2005;352:1539–1549. doi: 10.1056/NEJMoa050496. [DOI] [PubMed] [Google Scholar]
- 80.Goldenberg I, Moss AJ, Hall WJ, Foster E, Goldberger JJ, Santucci P, Shinn T, Solomon S, Steinberg JS, Wilber D, Barsheshet A, McNitt S, Zareba W, Klein H. Predictors of Response to Cardiac Resynchronization Therapy in the Multicenter Automatic Defibrillator Implantation Trial With Cardiac Resynchronization Therapy (MADIT-CRT)/Clinical Perspective. Circulation. 2011;124:1527–1536. doi: 10.1161/CIRCULATIONAHA.110.014324. [DOI] [PubMed] [Google Scholar]
- 81.Steinberg JS, Fischer A, Wang P, Schuger C, Daubert J, McNitt S, Andrews M, Brown M, Hall WJ, Zareba W, Moss AJ. The clinical implications of cumulative right ventricular pacing in the multicenter automatic defibrillator trial II. J Cardiovasc Electrophysiol. 2005;16:359–365. doi: 10.1046/j.1540-8167.2005.50038.x. [DOI] [PubMed] [Google Scholar]
- 82.Mastenbroek S, Twisk M, van der Veen F, Repping S. Preimplantation genetic screening: a systematic review and meta-analysis of RCTs. Hum Reprod Update. 2011;17:454–466. doi: 10.1093/humupd/dmr003. [DOI] [PubMed] [Google Scholar]
- 83.Richards CS, Bale S, Bellissimo DB, Das S, Grody WW, Hegde MR, Lyon E, Ward BE. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet Med. 2008;10:294–300. doi: 10.1097/GIM.0b013e31816b5cae. [DOI] [PubMed] [Google Scholar]
- 84.Collins F. Has the revolution arrived? Nature. 2010;464:674–675. doi: 10.1038/464674a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Venter JC. Multiple personal genomes await. Nature. 2010;464:676–677. doi: 10.1038/464676a. [DOI] [PubMed] [Google Scholar]
- 86.Koikkalainen JR, Antila M, Lotjonen JM, Helio T, Lauerma K, Kivisto SM, Sipola P, Kaartinen MA, Karkkainen ST, Reissell E, Kuusisto J, Laakso M, Oresic M, Nieminen MS, Peuhkurinen KJ. Early familial dilated cardiomyopathy: identification with determination of disease state parameter from cine MR image data. Radiology. 2008;249:88–96. doi: 10.1148/radiol.2491071584. [DOI] [PubMed] [Google Scholar]
- 87.Lu JT, Muchir A, Nagy PL, Worman HJ. LMNA cardiomyopathy: cell biology and genetics meet clinical medicine. Dis Model Mech. 2011;4:562–568. doi: 10.1242/dmm.006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Goemans NM, Tulinius M, van den Akker JT, Burm BE, Ekhart PF, Heuvelmans N, Holling T, Janson AA, Platenburg GJ, Sipkens JA, Sitsen JM, Aartsma-Rus A, van Ommen GJ, Buyse G, Darin N, Verschuuren JJ, Campion GV, de Kimpe SJ, van Deutekom JC. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N Engl J Med. 2011;364:1513–1522. doi: 10.1056/NEJMoa1011367. [DOI] [PubMed] [Google Scholar]