

Abstract
The present report describes clinical variability in an affected dizygotic twin pair. Twin 1 showed classical features of the congenital myasthenic syndromes (CMS), that is, ptosis, dysphonia, asthenia and hypotonia. In twin 2, these clinical signs were less pronounced, but subtle resulting in severe lumbar hyperlordosis. Molecular analysis, performed for both twins, revealed the presence of three polymorphisms in the heterozygous form in RAPSN gene. The present report highlights the clinical variability of the CMS.
Background
Congenital myasthenic syndromes (CMS) are uncommon disorders of neuromuscular transmission, classified into three subgroups according to the affected component of the neuromuscular junction: presynaptic, postsynaptic or synaptic basal lamina-associated.1
We herein report on dizygotic twins, heterozygous for RAPSN gene mutation, affected by postsynaptic CMS who showed clinical variability and different course.
Case presentation
The male twins were born at 34 weeks of gestation with two chorions. Parents were healthy and non-consanguineous.
Twin 1
The child was born with talipes equino-varus, treated with Ponseti casts. Since the first months of life, the child showed bilateral ptosis more evident in the right, dysphonia and dysphagia, smooth voice and respiratory failure. Afterwards he, also, complained of hypotonia, frequent fall down and muscle pain. During the subsequent years some clinical signs such as ptosis, dysphagia and smooth voice spontaneously became less frequent while hypotonia, muscular fatigue persisted.
The child was first admitted at the Pediatric Unity Hospital Vittorio-Emanuele of Catania, Italy at 6 years of age. Myasthenic syndrome was soon suspected. Laboratory test showed no alteration in the blood cell count, erythrocyte sedimentation rate, reactive protein, ammonia and lactate. Cranial MRI, electroencephalography and abdominal ultrasonography were normal as were sensory nerve conduction and motor conduction velocity and electromyography. A decrease on the 3 and 5 Hz repetitive stimulations in abductor digiti minimi and abductor pollicis brevis muscles was observed.
Acetylcholine receptor (AChR) antibodies were negative. The tensilon test was positive and treatment with pyridostigmine therapy was started with dosage of 60 mg three times daily. Treatment was initially unsatisfactory and a few months later, the child was submitted to muscle biopsy which gave normal results. The increase of the treatment of pyridostigmine four times a day resulted in an improvement of the clinical signs.
The patient was again admitted at our unit at the age of 9.5 years. Physical examination was normal. He showed good school performances, with an IQ of 98. Recently, at the treatment with pyridostigmine was joined bromide pyridostigmine 3/4 tablet. This resulted in an improvement in his clinical condition during the night. Delay of treatment causes asthenia, muscular fatigue and sometimes ptosis after 20 min.
Twin 2
The parents reported that he had begun to complain of muscle fatigue and weakness at the age of 2.5 years, but less intense than his twin. He had frequent fall down episodes and difficulty in running. He also showed waddling gait, internal rotation of the right foot, difficulty in jumping and dyspnoea after exertion. By the age of 8 years, he had developed mild kyphosis with pelvic and scapular asymmetry with the right side higher than the left.
He came at our observation at the age of 7.5 years. Sensory nerve conduction and motor conduction velocity and electromyography were normal. No decrease in the repetitive muscular stimulation was observed. AChR antibodies were negative. Tensilon test was positive and the treatment was started with pyridostigmine 60 mg every 6 h. The scholastic performance was good.
At the present age of 11 he showed a severe lumbar hyperlordosis (figure 1). As his brother, he complains of asthenia and signs of fatigue, but about 1 h after delay in the treatment.
Figure 1.
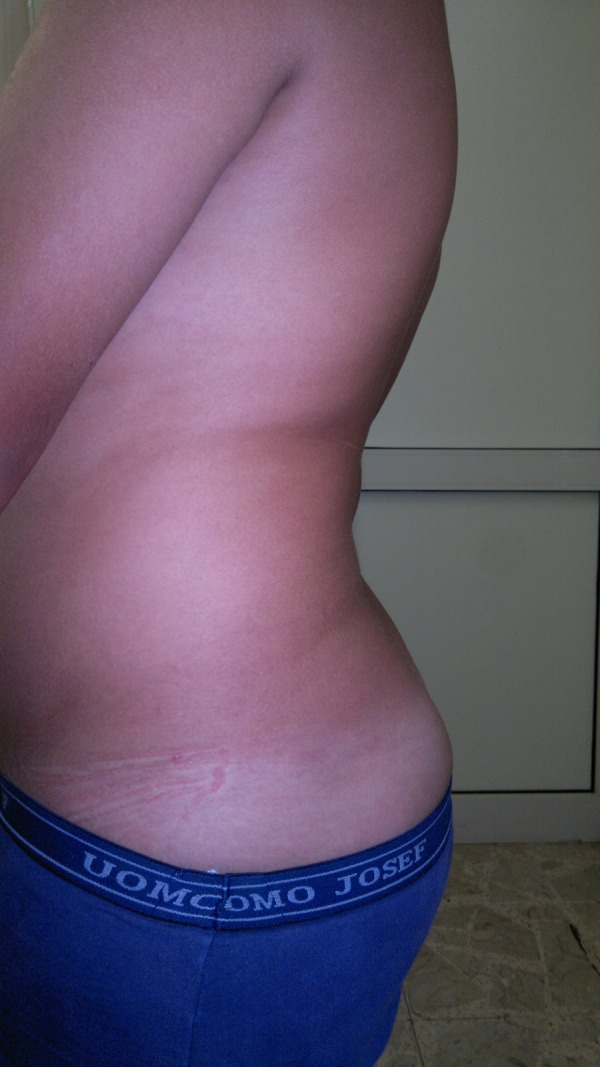
Severe hyperlordosis in twin 2 at the age of 11.
Molecular analysis
Parents were negative for known mutations.
In the twins analysis for the detection of the known RAPSN gene (11p11.2) mutations A25V, R91L, L361R and K373del in TE671 cells, and mutations in CHRNA1, CHRNE and the CHRNE promoter was negative. Three polymorphisms were detected: c.456t>C (pY152Y) in exon 2; c.1143T>C (p.P81P) in exon 7 and IVS3-11delC in intron 3.
Discussion
The dizygotic twins were affected by CMS of postsynaptic type. The diagnosis was made on personal history, on dynamic signs and symptoms and positive response to pyridostigmine, while other diagnostic results (electrophysiological and muscle biopsy) were inconclusive.
Clinical features of CMS in twin 1 were more evident than in twin 2, and disease onset was earlier (see table 1). Furthermore, twin 1 showed features absent in twin 2, such as, bilateral ptosis, dysphonia, dysphagia and episodes of respiratory insufficiency. Clinical course was progressive in twin 1 and mildly progressive in twin 2. However, even if the clinical muscular signs were more relevant in twin 1 its progresses more subtly in the twin 2 causing a severe lumbar hyperlordosis.
Table 1.
Clinical features of the twins
| Follow-up at 11 years | |||||||
|---|---|---|---|---|---|---|---|
| Twin | Clinical signs at birth | Clinical signs at 1 year | Age at diagnosis Clinical signs |
Complementary laboratory findings | Treatment | Response to treatment | Onset of clinical signs after delay of treatment (min) |
| 1 | Talipes equino-varus | 6 years | Decrease on the 3 and 5 Hz repetitive stimulations in abductor digiti minimi and abductor pollicis brevis muscles | Pyridostigmine and pyridostigmine bromure | Good | 20 | |
| Bilateral ptosis | Muscle fatigue | ||||||
| Smooth voice | Unilateral ptosis | ||||||
| Dysphonia and dysphagia | Muscle pain | ||||||
| Respiratory failure | Asthenia | ||||||
| 7.5 years | |||||||
| 2 | – | – | Muscle fatigue | – | Pyridostigmine | Good | 60 |
| Hypotonia | |||||||
As happened in the twins, the results of ancillary tests (muscle biopsy or electrophysiological studies) may be misleading. In fact, muscle biopsy often reveals non-specific myopathic features,2 3 while neurophysiological studies may fail to demonstrate a positive response to repetitive nerve stimulation (RNS), in about 25% of the cases.4 5 Although single fibre electromyography-jitter may be more sensitive for the detection of a disturbance of the neuromuscular junction than RNS, the results of the examination in affected cases may be within the normal range.4
The majority of CMS patients present with mutations within the RAPSN coding region and are usually homozygous for N88K or heteroallelic for a second mutation;6–8 rapsyn binds to the long cytoplasmatic loop of the AChR subunits and is essential for the clustering and anchoring of the AChR within the postsynaptic membrane. Mutations in rapsyn cause endplate AChR deficiency and thus compromise the safety margin of neuromuscular transmission.
The molecular analysis performed in the twins disclosed the presence of three polymorphisms (c.456t>C (pY152Y) in exon 2; c.1143T>C (p.P81P) in exon 7 and IVS3-11delC in intron 3) in heterozygous form.
The molecular polymorphism reported in our patients seems not to correlate with the disease presented by the twins. In fact, two of the polymorphisms in exons 7 and 2 do not cause amino acid changes and thus cannot be related with the effect on the disease. The third mutation, being intronic, may have acted in the twins as transcriptional or conformational changes of the protein, but this would need further investigations, not performed in our patients.
Reported cases of CMS in twins are rare. Nishida et al9 described two siblings with congenital myopathy, myasthenic features and cataracts. In these patients the muscle weakness fluctuated, and was alleviated by edrophonium chloride.
In our patients, remarkable was the variability of the clinical signs and the different clinical course even if the response to the treatment was effective in both the children.
Learning points.
Congenital myasthenia is a rare disease. Prevalence is estimated in 1–2/500 000 newborns.
The patients were twins born from two chorions. They showed clinical variability, one of them being more affected.
Initial clinical signs in twin 1 were smooth voice, easy fatigability and bilateral ptosis; in twin 2 the clinical signs appeared later and were less evident but had subtle course resulting in progressive lumbar hyperlordosis.
In the twin molecular analysis revealed the presence of three polymorphisms in heterozygosis in RAPSN gene.
Treatment with pyridostigmine was diagnostic and efficacy in both twins.
Acknowledgments
We wish to thank Professor Raffaella Brugnoni, Laboratory NBS Biotech, Fondazione Istituto Neruologico Carlo Besta, Milan, for molecular analysis and Bioedit Ltd for editing the final version of the manuscript.
Footnotes
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Engel AG, Sine SM. Current understanding of congenital myasthenic syndromes. Curr Opin Pharmacol 2005;5:308–21 [DOI] [PubMed] [Google Scholar]
- 2.Gurnett CA, Bodnar JA, Neil J, et al. Congenital myasthenic syndrome: presentation, electrodiagnosis, and muscle biopsy. J Child Neurol 2004;19:175–82 [PubMed] [Google Scholar]
- 3.Bonifati DM, Willcox N, Vincent A, et al. Lack of association between acetylcholine receptor epsilon polymorphisms and early-onset myasthenia gravis. Muscle Nerve 2004;29:436–9 [DOI] [PubMed] [Google Scholar]
- 4.Pitt M. Neurophysiological strategies for the diagnosis of disorders of the neuromuscular junction in children. Dev Med Child Neurol 2008;50:328–33 [DOI] [PubMed] [Google Scholar]
- 5.Rabie M, Jossiphov J, Nevo Y. Electromyography (EMG) accuracy compared to muscle biopsy in childhood. J Child Neurol 2007;22:803–8 [DOI] [PubMed] [Google Scholar]
- 6.Müller JS, Mildner G, Müller-Felber W, et al. Rapsyn N88K is a frequent cause of congenital myasthenic syndromes in European patients. Neurology 2003;60:1805–10 [DOI] [PubMed] [Google Scholar]
- 7.Milone M, Shen XM, Selcen D, et al Myasthenic syndrome due to defects in rapsyn: clinical and molecular findings in 39 patients. Neurology 2009;73:228–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaudon K, Pénisson-Besnier I, Chabrol B, et al Multiexon deletions account for 15% of congenital myasthenic syndromes with RAPSN mutations after negative DNA sequencing. J Med Genet 2010;47:795–6 [DOI] [PubMed] [Google Scholar]
- 9.Nishida Y, Kobayashi T, Machi M, et al Congenital myopathy with myasthenic features and congenital cataract in two siblings. J Neurol 1989;236:161. [DOI] [PubMed] [Google Scholar]