

Abstract
A 56-year-old man, presented with bilateral chronic visual loss associated with generalised skin lesions. He had undergone multiple penetrating keratoplasties in his right eye for recurrent corneal infections and perforations. On ocular examination, his left eye was phthisical and his right eye had light perception vision owing to a failed and vascularised corneal graft. Dermatological evaluation revealed multiple hyperpigmented and hypopigmented lesions along with thickening of skin on nose, scalp and dorsum of hands. Skin biopsy showed focal areas of deposition of faint periodic acid Schiff-positive diastase-resistant perivascular material. The high-performance liquid chromatography assessment revealed increased presence of porphyrins in blood and urine, thus confirming a diagnosis of porphyria cutanea tarda. The patient's vision in the right eye improved after undergoing Boston type 1 keratoprosthesis along with general photoprotective measures for the exposed parts of the body.
Background
Porphyria is a rare metabolic disorder that is characterised by the accumulation of photosensitive, toxic intermediates of the heme metabolic pathway in various organs of the body including the skin, eye and neural tissue.1 Porphyria as a potential cause for corneal ulceration, opacification and vascularisation is very infrequently emphasised in the literature,2 probably due to the relatively rare occurrence and lack of well-documented cases. Herein, we report a case of porphyria cutanea tarda (PCT), in which Boston keratoprosthesis was carried out to restore useful vision in the only salvageable eye with multiple failed grafts following recurrent corneal ulceration and infection.
Case presentation
A 56-year-old gentleman, known diabetic and hypertensive, born of non-consanguineous parentage reported with 5 years loss of vision in the right eye and 7 months loss of vision in the left eye. His medical history revealed that he had undergone penetrating keratoplasty (PK) twice in the right eye for presumed infective keratitis. Eight months following the first PK, his corneal graft developed melt and fungal infection with graft failure. He underwent a repeat PK in the right eye in January 2011. On subsequent follow-up, he presented with corneal infiltrate which revealed fungal filaments on microbiological evaluation. The infection resolved, but the corneal graft failed due to scarring and endothelial rejection. It is at this stage that the patient reported to our institute for further management.
On physical examination, the patient had multiple hyperpigmented and hypopigmented lesions along with thickening of skin on nose, scalp and dorsum of hands for 12 years. The patient's visual acuity was light perception with accurate projection of rays in the right eye. His left eye was phthisical secondary to spontaneous corneal perforation. Ocular examination of the right eye revealed a failed and vacularised corneal graft (figure 1) with localised exudative retinal and choroidal detachment (RD and CD) on B-scan ultrasonography (Opro: OTI-300). Based on the above clinical features, a trial of intravenous corticosteroid (methylprednisolone sodium succinate; Solu-Medrol) with intravenous alkylating agent (cyclophosphamide; Neosar) was advised. Serial B-scan showed resolution of both RD and CD, thereby confirming the exudative nature of retinal detachment.
Figure 1.
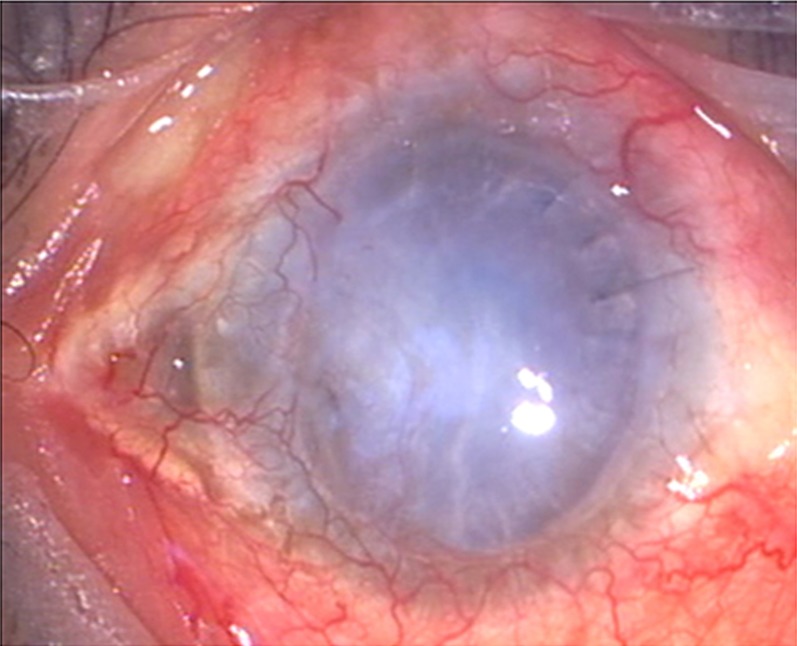
Preoperative photograph showing failed vascularised graft.
Investigations
Suspecting porphyria, we evaluated his urine, blood and skin. The high-performance liquid chromatography assessment revealed increased coproporphyrin, uroporphyrin, porphobilinogen and aminolevulinic acid in urine, and increased protoporphyrin in blood. Skin biopsy showed focal areas of faint periodic acid Schiff-positive material in papillary dermis which appeared to be diastase resistant.
Based on the clinical, biochemical and dermatological evaluation, a diagnosis of PCT was made.
Treatment
Considering multiple failed grafts with four quadrants of corneal deep vascularisation, it was planned to perform Boston type 1 keratoprosthesis (Massachusetts Eye and Ear Infirmary, Boston, MA, USA) in this patient. Aphakic Boston keratoprosthesis (axial length: 20.40 mm, power: 60 dioptre, optic cylinder: 3 mm, backplate: 8.5 mm in diameter) along with intraocular ocular lens explanation, anterior vitrectomy and lateral paramedian tarsorrhapy was performed in March 2012.
Outcome and follow-up
Postoperatively, on day 1, it was found that his unaided vision was 20/125p with a stable keratoprosthesis assembly. He was started on a tapering dosage of topical steroid and topical antibiotics with regular follow-ups. Nine months after the procedure, the patient was highly contended with his best corrected vision of 20/125p (due to changes at retinal pigment epithelium secondary to cystoid macular oedema) with stable Boston type 1 keratoprosthesis (figure 2). Moreover, the patient was advised to take general protective measures of the exposed parts of the body.
Figure 2.
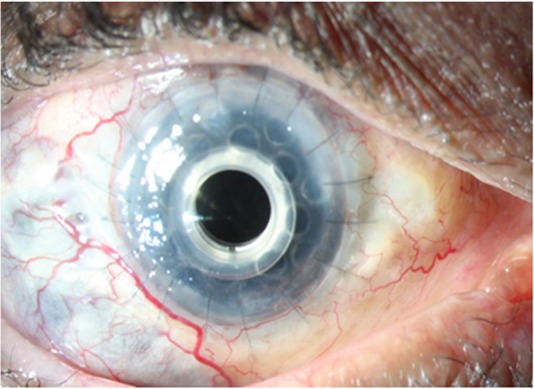
Postoperative photograph showing stable Boston keratoprosthesis.
Discussion
PCT, the most common form of porphyria,3 is a metabolic disorder that is caused by reduced activity of the enzyme uroporphyrinogen decarboxylase in the liver. Clinical manifestations are predominantly dermatological with photoactive porphyrins depositing in the skin causing bullae, hyperpigmentation and hypopigmentation, pseudoscleroderma and hypertrichosis in sun-exposed areas.4 Ocular involvement, though rare, may manifest as lid scarring, ectropion,5 pingueculae, pterygium,6 acute scleritis with scleral necrosis,7 8 corneal thinning and corneal perforation.9
The exact mechanism by which ocular damage occurs in porphyria is still unclear; however, a constellation of photic damage, ischaemia and inflammation secondary to accumulation of porphyrins has been stipulated as the underlying cause.10
What we believe is that the presence of porphyrin precursors in the tears or in the corneal tissue incite inflammatory response secondary to phototoxicity. It is this inflammatory response that manifests as corneal thinning, corneal ulceration with subsequent superadded infection, opacification and vascularisation. A similar phenomenon was seen in our patient in whom superadded infection secondary to corneal involvement occurred once in native cornea and thrice in graft thereby leading to repeated graft failure.
Aldave et al11 have reported a favourable outcome of Boston keratoprosthesis in patients with multiple failed grafts. Although, after 6 months, the keratoprosthesis is doing well in our patient, our main concern is its long-term survivability in view of PCT.
This case report highlights one of the rarest ocular complications of PCT which is sparingly described in the literature. Also, it has shown the fate of grafts secondary to repeated ulceration and infection. Considering the poor fate of grafts in PCT, Boston keratoprosthesis has emerged as a viable option, though a long-term follow-up is required. To the best of our knowledge, this is the first reported attempt to salvage an eye having multiple failed grafts secondary to PCT using a keratoprosthesis.
Learning points.
Ocular complications of porphyria cutanea tarda (PCT) are rare, and occurrence of corneal ulceration and infection is the rarest.
Corneal grafts fail as a long-term solution due to recurrent ocular surface problems and secondary graft infections.
Boston type 1 keratoprosthesis is a viable alternative to corneal transplantation for visual restoration in cases of bilateral corneal blindness due to PCT.
Footnotes
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Venkatesh P, Garg SP, Kumaran E, et al. Congenital porphyria with necrotizing scleritis in a 9-year-old child. Clin Exp Ophthalmol 2000;28:314–18 [DOI] [PubMed] [Google Scholar]
- 2.Steiner G, Arffa RC. Psoriasis, ichthyosis, and porphyria. Int Ophthalmol Clin 1997;37:41–61 [DOI] [PubMed] [Google Scholar]
- 3.Yachimski P, Shah N, Chung RT. Porphyria cutanea tarda. Clin Gastroenterol Hepatol 2007;5:e6. [DOI] [PubMed] [Google Scholar]
- 4.Grossman ME, Bickers DR, Poh-Fitzpatrick MB, et al. Porphyria cutaneatarda. Clinical features and laboratory findings in forty patients. Am J Med 1979;67:277–81 [DOI] [PubMed] [Google Scholar]
- 5.Sober AJ, Grove AS, Muhlbauer JE. Cicatricial ectropion and lacrimal obstruction associated with the sclerodermoid variant of porphyria cutaneatarda. Am J Ophthalmol 1981;91:396–400 [DOI] [PubMed] [Google Scholar]
- 6.Hammer H, Korom I. Photodamage of the conjunctiva in patients with porphyria cutaneatarda. Br J Ophthalmol 1992;76:592–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Veenashree MP, Sangwan VS, Vemuganti GK, et al. Acute scleritis as a manifestation of congenital erythropoietic porphyria. Cornea 2012;21:530–1 [DOI] [PubMed] [Google Scholar]
- 8.Altiparmak UE, Oflu Y, Kocaoglu FA, et al. Ocular complications in 2 cases with porphyria. Cornea 2008;27:1093–6 [DOI] [PubMed] [Google Scholar]
- 9.Zaborowski AG, Paulson GH, Peters AL. Sight threatening complications in porphyria cutaneatarda. Eye 2004;18:949–50 [DOI] [PubMed] [Google Scholar]
- 10.Siddique SS, Gonzalez LA, Thakuria P, et al. Scleral necrosis in a patient with congenital erythropoieticpurpura. Cornea 2011;30:97–9 [DOI] [PubMed] [Google Scholar]
- 11.Aldave AJ, Sangwan VS, Basu S, et al. International results with the Boston type I keratoprosthesis. Ophthalmology 2012;119:1530–8 [DOI] [PubMed] [Google Scholar]