

Abstract
Neuroinflammation is an integral component of neurodegenerative disorders, CNS infection and trauma. Astroglial chemokines, such as CXCL10, are instrumental in neuroinflammatory signaling as well as neurotoxicity. We have utilized proinflammatory-induced CXCL10 expression in normal human astrocytes (NHA) as a model in which to assess the anti-inflammatory actions of the selective, mu-opioid receptor (MOR) antagonist, β-funaltrexamine (β-FNA). Interferon (IFN)γ + HIV-1 Tat-induced CXCL10 expression (secreted protein and mRNA) was inhibited by co-treatment with β-FNA. Neither the MOR-selective antagonist, D-Phe-Cys-Tyr-D-Trp-Arg-Pen-Thr-NH2 (CTAP) nor the nonselective opioid receptor antagonist, naltrexone inhibited IFNγ + HIV-1 Tat-induced CXCL10 expression. Furthermore, co-treatment with excess CTAP or naltrexone did not prevent β-FNA mediated inhibition of IFNγ + HIV-1 Tat-induced CXCL10 expression. Additionally, we utilized an inhibitor of NF-κB activation (SN50) to demonstrate that IFNγ + HIV-1 Tat-induced CXCL10 expression is NF-κB-dependent in NHA. Subsequent experiments revealed that β-FNA did not significantly affect NF-κB activation. Interestingly, we discovered that β-FNA inhibited p38 activation as indicated by decreased expression of phospho-p38. Together, these findings suggest that the inhibitory actions of β-FNA are MOR-independent and mediated, in part, via a transcriptional mechanism. These findings add to our understanding of the mechanism by which chemokine expression is inhibited by β-FNA. In conjunction with future investigations, these novel findings are expected to provide insights into the development of safe and effective treatments for neuroinflammation.
Keywords: astroglial, HIV-1, mu opioid receptor, neuroinflammation, interferon γ
1. Introduction
HIV-1 enters the central nervous system (CNS) within in a matter of weeks following systemic infection (Gray et al., 1993) and can presumably persist for years (Kramer-Hammerle et al., 2005; Thompson et al., 2011). Approximately 50–60% of HIV-1 infected individuals suffer from HIV-associated neurocognitive disorders (HAND) (Giunta et al., 2006; Ozdener, 2005). The incidence of the most severe form of neurocognitive involvement, HIV-1 associated dementia (HAD), has been reduced with the advent of antiretroviral therapy; however, reports of more subtle forms of CNS impairment are increasing (Ances and Ellis, 2007; Cysique and Brew, 2009; Fischer-Smith and Rappaport, 2005). The exact mechanism by which HIV-1 causes these neuropathologies is not completely understood; however increasing evidence suggests neuronal damage results in part from microglial and astroglial derived cytokines and chemokines (Deshpande et al., 2005; Gonzalez-Scarano and Martin-Garcia, 2005; Minagar et al., 2002; Navia et al., 1986; Zhou et al., 2004).
The chemokine interferon γ inducible protein-10 (CXCL10) is among the proinflammatory molecules implicated in HIV-1 associated neuropathogenesis (Chang et al., 2004; Cinque et al., 2005; Eugenin et al., 2006; Lane et al., 2003; van Marle et al., 2004). CXCL10 is a member of the CXC or α-chemokine family; all members of this family contain four highly conserved cysteine residues with the first two separated by a single amino acid (Bajetto et al., 2002; Luster et al., 1985). CXCL10 is also subclassified as ELR-negative due to the lack of a conserved glutamate-leucine-arginine (ELR) motif at the N-terminus. CXCL10 is a small secreted protein, instrumental in physiological and pathological processes, but most fully characterized as a chemoattractant for activated T cells (Taub et al., 1993), monocytes/macrophages (Taub et al., 1993) and microglia (Flynn et al., 2003). In addition to recruiting inflammatory cells, CXCL10 induces astroglial proliferation (Flynn et al., 2003) and is directly neurotoxic (Sui et al., 2004; Sui et al., 2006). Substantial evidence has been documented in terms of a role for CXCL10 in HIV-1 neuropathogenesis. For instance, CXCL10 is elevated in the CNS of patients with HIV-1 associated dementia (HAD) and HIV-1 encephalitis (Conant et al., 1998; Kelder et al., 1998; Kolb et al., 1999). Also, CXCL10 levels in the CSF of HIV-1 infected individuals are positively correlated with neurological deficits (Kolb et al., 1999). In autopsy brain tissue, CXCL10 protein expression is greater in HIV+ individuals compared to HIV- subjects and is detected primarily in astrocytes located in proximity to microglial nodules and activated microglia (Sanders et al., 1998). Furthermore, CXCL10 stimulates HIV-1 replication in macrophage and peripheral blood lymphocytes (Lane et al., 2003).
Viral proteins released from HIV-1 infected cells are also involved in the pathogenesis of HAD (Ensoli et al., 1993; Ghafouri et al., 2006; Westendorp et al., 1995). For example, the virally encoded transactivator protein, Tat, is directly neurotoxic (King et al., 2006), but also enters uninfected astrocytes and microglia and induces the expression of numerous proinflammatory molecules (Nath, 2002). HIV-1 Tat induces the expression of chemokines, including CXCL10, in astrocytes (Khiati et al., 2010; Kutsch et al., 2000). Furthermore, HIV-1 Tat-induced CXCL10 in human astrocytes is potentiated by cytokines, including TNFα and IFNγ (Williams et al., 2009).
While antiretroviral therapy (ART) inhibits HIV-1 replication in the periphery, few of these therapeutic agents readily enter the CNS or reduce neuroinflammatory responses associated with HIV-1 (Chang et al., 2003; Chang et al., 2004; Nath and Sacktor, 2006). In fact, there is ongoing neuroinflammation in ART treated patients (Anthony et al., 2005). Therefore, there has been considerable interest in identifying agents which attenuate neuroinflammatory responses activated by HIV-1 infection (Agrawal et al., 2007; Chen et al., 2003; Dou et al., 2005; Wilson et al., 2006). For example, treatment of SIV-infected macaques with the antibiotic minocycline reduced severity of encephalitis and decreased expression of several neuroinflammatory molecules including chemokines (Zink et al., 2005). Chemokines, such as CXCL10, are potentially key molecules which could be targeted as a means of reducing HIV-1 associated neuropathogenesis (Ansari et al., 2006; Biber et al., 2006; Eugenin et al., 2006; Lane et al., 2003). While not specifically targeted to chemokines, another intriguing strategy that has been employed to attenuate inflammation-mediated neuropathogenesis is treatment with naloxone (Liao et al., 2003; Liu et al., 2000; Liu et al., 2002; Liu and Hong, 2003). Naloxone is well characterized as a non-selective opioid receptor antagonist; however, naloxone reduces neuroinflammation via mechanisms that do not require binding to opioid receptors. Such opioid receptor independent actions of naloxone include prevention of bacterial lipopolysaccharide (LPS)-binding to microglia (Liu et al., 2000) and reduced microglial superoxide production (Liu et al., 2002). Furthermore, we more recently determined that TNFα induced CXCL10 protein expression in human astroglial cells is dose-dependently inhibited by the selective, mu-opioid receptor (MOR) antagonist, β-funaltrexamine (β-FNA). These findings were particularly interesting given that β-FNA inhibited TNFα-induced CXCL10 expression in part through a MOR-independent mechanism (Davis et al., 2007). Given that β-FNA is centrally penetrating (Labuz et al., 2007), it holds promise as a therapeutic agent for select neuropathologies.
The mechanism of action of β-FNA in both behavioral (Ward et al., 1982) and in vitro assays (Liu-Chen et al., 1990) is initially reversible kappa-opioid receptor (KOR) agonism followed by irreversible MOR antagonism. However, our studies suggest that it is not the interaction of β-FNA with classical opioid receptors that causes the inhibition of proinflammatory-induced CXCL10 expression (Davis et al., 2007). Prior to the present report, our studies have focused on β-FNA effects on inflammatory signaling in the human A172 astroglial cell line (Davis et al., 2007; 2008). However, we have now expanded our studies and have determined that β-FNA also has anti-inflammatory actions in normal human astrocytes (NHA). Furthermore, the data support our previous findings that the anti-inflammatory actions of β-FNA not mediated through the MOR.
2. Materials and methods
2.1. Cell culture
Normal human astrocytes (NHA, cat# HA1800; ScienCell Research Laboratories, Carlsbad, CA) were cultured using reagents and procedures provided by the supplier. Cultures were maintained in Astrocyte Medium (cat.# 1801) containing 2% fetal bovine serum, growth supplement (cat.# 1852), and penicillin/streptomycin solution (cat.# 0503). All cultures were maintained in a humidified incubator at 37°C, 5% CO2 and 95% air. Experimental cultures (passages 1–3) were seeded at a cell density (1 × 104/cm2) that provided 80–90% confluence at the time of treatment. Culture medium was replenished every 48 h unless otherwise noted.
2.2. Proinflammatory stimulation
In order to induce CXCL10 expression and nuclear translocation of NF-κB, growth medium in astroglial cultures was replaced with serum free medium containing human recombinant IFNγ (10 ng/ml; Peprotech, Rocky Hill, NJ) and HIV-1 Tat1–72 (100 nM; Prepared by Phil Ray, University of Kentucky Neuroscience, Department of Neurology). The duration of stimulation was 24 h for induction of CXCL10 protein expression, 8 h for CXCL10 mRNA expression and 10–270 min for NF-κB activation.
2.3. Opioid compounds
The MOR-selective antagonists included β-FNA (10 μM; obtained from the NIDA reagent supply program) and D-Phe-Cys-Tyr-D-Trp-Arg-Pen-Thr-NH2 (CTAP; 30, 100 μM; Sigma, St. Louis, MO). Naltrexone (30, 100 μM; Sigma) was used as a nonselective opioid receptor antagonist. In all experiments, opioid reagents were added to cell cultures at the time of proinflammatory stimulation.
2.4. CXCL10 protein expression
A standard dual-antibody solid phase immunoassay (ELISA Development Kit; Peprotech, Rocky Hill, NJ) was used for quantitation of secreted CXCL10 in cell culture supernatants, according to the manufacturer’s instructions and as previously described (Davis et al., 2007).
2.5. CXCL10 mRNA expression
Total RNA was isolated with TRIzol reagent (Invitrogen, Carlsbad, CA) and CXCL10 mRNA assessed by real-time PCR using the SYBR Green detection method using the basic protocol previously described (Davis et al., 2008). The RT-PCR primer pair sets were obtained from Invitrogen and amplification from first strand cDNA was performed as instructed by the supplier. The primer sequences for CXCL10 were AACCTCCAGTCTCAGCACCATGAA (forward) and AGGTACAGCGTAAGGTTCTAGAGAG (reverse); the primer pairs for GAPDH were GAGTCAACGGATTTGGTCGT (forward) and TTGATTTTGGAGGGATCTCG (reverse). Relative quantification of gene expression was evaluated using the comparative cycle threshold (CT) method (Hettinger et al., 2001) as previously described (Davis et al., 2008). Expression of CXCL10 mRNA was normalized to the expression of the endogenous control, GAPDH mRNA. Relative expression was calculated using the comparative Δ ΔCT method and fold change calculated using 2− ΔΔCT.
2.6. Inhibiton of NF-κB activation
To assess the role of NF-κB in IFN-γ + Tat-induced CXCL10 expression, NHA were stimulated in the presence of SN50 (Biomol; Farmingdale, NY), a cell permeable inhibitory peptide containing the nuclear localization sequence of NF-κB p50, which prevents nuclear translocation of the active NF-κB complex (Lin et al., 1995). As done previously, (Pahan et al., 2001; Yang et al., 2005), cells were pre-incubated with 50 μM SN50 or 50 μM of the inactive mutant peptide SN50M, for 1 h prior to stimulation.
2.7. Nuclear translocation of NF-κB p65
NF-κB activation, as determined by increased levels of the active (DNA-binding) form of NF-κB p65 in the nucleus, was assessed using the p65 Transcription Factor kits (Thermo Scientific, Rockford, IL) as we previously described (Tousi et al., 2010). Nuclear protein was obtained according to the protocol described in our previous report (Davis and Syapin, 2004) and luminescence of the labeled NF-κB-DNA product was then measured.
2.8. p38 activation
Western blotting was done as previously described (Das et al., 2011) to determine phospho-p38 levels, as a measure of p38 activation. Following experimental treatments, cells were washed twice with PBS and cytosolic extracts prepared using Cytoplasmic Extraction Reagent (Thermo Scientific, Rockford, IL). Briefly, fifty micrograms of total protein were loaded on 10% SDS-PAGE gel, electrophoresed and then transferred to PVDF membrane. Antibodies used were anti-Phospho-p38 and anti-p38 (both from Cell Signaling, Danvers, MA) and were used in the dilution range of 1:1000. The blots were scanned in a phosphoimager Typhoon 9410 (GE Healthcare, Uppsala, Sweden) using enhanced chemifluorescence reagent (GE Healthcare, UK). Densitometric analysis of western blot protein bands was done using Image J (National Institute of Health).
2.9. Determination of total protein content
Total cellular/nuclear protein levels were determined using the bicinchoninic acid (BCA) protein assay as previously described (Davis et al., 2002) in order to normalize data when appropriate.
2.10. Statistical analysis
Prism™ version 4.0 software (GraphPad Inc., San Diego, CA) was used for figure presentation and statistical analysis. Analyses included one-way analysis of variance (ANOVA) with Neuman-Kuels multiple comparisons or a Dunnett’s comparison. Additionally, a two-ANOVA was used, followed by Bonferroni’s multiple comparisons. Data are presented as mean + S.E.M and a probability (p) of < 0.05 was accepted as demonstrating statistically significant differences between groups. The n values and number of independent experiments from which the data were obtained are provided in the individual figure legends.
3. Results
3.1. Effects of β-FNA on IFNγ and Tat-induced CXCL10 expression
Initially we assessed the induction of CXCL10 expression following stimulation with IFNγ and Tat alone, and in combination. Next we determined the effect of β-FNA on induced CXCL10 expression. Constitutive expression of CXCL10 protein in NHA was negligible; whereas, 24 h exposure to either IFNγ or Tat alone induced low levels of CXCL10 protein (192 ± 73 and 136 ± 49 pg/ml, respectively; Fig. 1A). Concurrent exposure to IFNγ and Tat resulted in a synergistic increase in CXCL10 protein expression (2711 ± 411 pg/ml; Fig. 1A). Given the robust induction of CXCL10 protein expression by IFNγ + Tat, subsequent experiments used only this proinflammatory combination for stimulation of NHA. Co-exposure to β-FNA significantly inhibited IFNγ + Tat-induced CXCL10 protein expression.
Fig. 1.

β-FNA inhibits proinflammatory-induced CXCL10 expression in normal human astrocytes. Panel A) β-FNA effects on CXCL10 protein expression: In the presence or absence or β-FNA (10 μM), normal human astrocytes were exposed to human recombinant IFNγ (10 ng/ml), HIV-1 Tat1–72 (100 nM), IFNγ + HIV-1 Tat1–72, or serum free medium alone for 24 h. CXCL10 protein levels in the media were then measured by ELISA. The data represent mean + S.E.M of 9 independent experiments. Two-way ANOVA (β-FNA treatment × stimulus) indicated a significant effect of β-FNA (p < 0.009), stimulus (p < 0.0001) and an interaction (p < 0.0005). Bonferroni’s multiple comparisons indicated that β-FNA significantly (* p < 0.001) inhibited IFNγ + HIV-1 Tat1–72-induced CXCL10 expression. Panel B) β-FNA effects on CXCL10 mRNA expression: In the presence or absence or β-FNA (10 μM), normal human astrocytes were exposed to human recombinant IFNγ (10 ng/ml) + HIV-1 Tat1–72 (100 nM) or in serum free medium alone for 8 h. Total RNA was isolated and CXCL10 and GAPDH mRNA assessed by real-time PCR. The data represent mean + S.E.M of 6 independent experiments. Analysis of ΔCT values by one-way ANOVA followed by Newman-Keuls multiple comparisons indicated that β-FNA significantly inhibited IFNγ + HIV-1 Tat1–72-induced CXCL10 mRNA expression. * p < 0.05 vs. IFNγ + HIV-1 Tat1–72
Consistent with CXCL10 protein expression data, constitutive CXCL10 mRNA levels were negligible in NHA, whereas, exposure to IFNγ + Tat resulted in approximately a 1000-fold increase in CXCL10 mRNA expression 8 h post-stimulation (Fig. 1B). However, co-exposure to β-FNA significantly inhibited IFNγ + Tat-induced CXCL10 mRNA expression.
3.2. Comparison of the effects of select MOR antagonists on IFNγ + Tat-induced CXCL10 protein expression
Firstly, this series of experiments was performed to determine whether the anti-inflammatory actions are similar among different MOR antagonists. Secondly, we aimed to determine whether MOR signaling was involved in the anti-inflammatory actions of β-FNA. Thus, we were interested to know whether the effects of β-FNA could be antagonized by co-exposure to a second opioid receptor antagonist (in relative excess). Similar to our previous experiments, 10 μM β-FNA significantly inhibited IFNγ + Tat-induced CXCL10 protein expression (Fig. 2). Conversely, exposure to considerably higher concentrations (30 and 100 μM) of either naltrexone (nonselective opioid receptor antagonist) or CTAP (MOR-selective antagonist) did not significantly affect IFNγ + Tat-induced CXCL10 protein expression. Co-treatment with naltrexone (30 or 100 μM) did not block the inhibition of CXCL10 protein expression caused by β-FNA (Fig. 2A). Likewise, co-treatment with CTAP (30 or 100 μM) did not block the inhibition of CXCL10 protein expression caused by β-FNA (Fig. 2B).
Fig. 2.

β-FNA inhibition of IFNγ + HIV-1 Tat1–72-induced CXCL10 protein expression in normal human astrocytes is not affected by co-exposure to either naltrexone (Panel A) or CTAP (Panel B). Normal human astrocytes were exposed to human recombinant IFNγ (10 ng/ml) + HIV-1 Tat1–72 (100 nM) alone or in the presence of β-FNA (10 μM), naltrexone or CTAP (30 and 100 μM), or β-FNA + naltrexone or CTAP for 24 h. CXCL10 protein levels in the media were then measured by ELISA. The data are expressed as % control (relative to IFNγ + HIV-1 Tat1–72) and represent mean + S.E.M of 2–4 independent experiments; triplicate measures (wells) in each experiment. Data were analyzed using one-way ANOVA and Dunnett’s post test comparison. * p < 0.05 vs. IFNγ + HIV-1 Tat1–72
3.3. Role of NF-κB in IFNγ + Tat -induced CXCL10 protein expression
To confirm that IFNγ + Tat induces CXCL10 expression in NHA via an NF-κB-dependent mechanism, we utilized SN50, a well characterized inhibitor of NF-κB nuclear translocation. Pretreatment of NHA with SN50 significantly inhibited IFNγ + Tat-induced CXCL10 protein expression (Fig. 3). Conversely, pretreatment of NHA with the mutated form of this inhibitory molecule, SN50 Mut, had no effect on IFNγ + Tat-induced CXCL10 protein expression (Fig. 3).
Fig. 3.
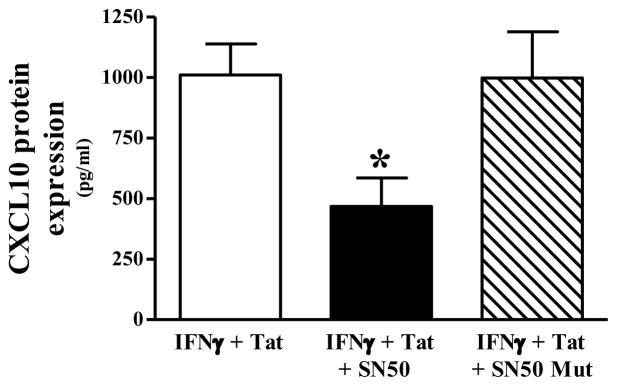
IFNγ + HIV-1 Tat1–72-induced CXCL10 protein expression in normal human astrocytes is NF-κB- dependent. CXCL10 expression was induced in normal human astrocytes by stimulating with human recombinant IFNγ (10 ng/ml) + HIV-1 Tat1–72 (100 nM) for 24 h. To assess the role of NF-κB, the inhibitor of NF-κB nuclear translocation, 50 μM SN50 (or 50 μM of the inactive control peptide, SN50 Mut), was added to cell cultures 1 h prior to stimulation. The data represent mean + S.E.M of 2 independent experiments; triplicate measures (wells) in each experiment. Data were analyzed using one-way ANOVA and Newman-Keuls post test comparison. * p < 0.05 vs. IFNγ + HIV-1 Tat1–72
3.4. Effect of β-FNA on IFNγ + Tat -induced of NF-κB activation
This experiment was performed to determine whether β-FNA inhibits NF-κB activation, a process that is necessary for IFNγ + Tat-induced CXCL10 protein expression. IFNγ + Tat induced a time-dependent increase in NF-κB activation (Fig. 4). IFNγ + Tat-induced NF-κB activation steadily increased over the first 90 min of stimulation, and then returned toward baseline by 270 min (Fig. 4). Two-ANOVA (β-FNA treatment × time) followed by Bonferroni’s multiple comparisons indicated that NF-κB activity was significantly elevated at 90 min (p < 0.05 vs. 10 min). Two-ANOVA also indicated a significant effect of time (p < 0.004) on IFNγ + Tat-induced NF-κB activation; however, β-FNA did not significantly affect NF-κB activation (p = 0.59) nor was an interaction detected (p = 0.62) (Fig. 4).
Fig. 4.

β-FNA does not inhibit IFNγ + HIV-1 Tat1–72, -induced NF-κB p65-DNA binding activity in normal human astrocytes. In the presence or absence or β-FNA (10 μM), normal human astrocytes were exposed to human recombinant IFNγ (10 ng/ml) + HIV-1 Tat1–72 for 10–270 min. NF-κB activation was indicated by increased levels of the active (DNA-binding) form of NF-κB p65 in the nucleus. The data represent mean + S.E.M of 3 independent experiments. Two-way ANOVA (β-FNA treatment × time) indicated a significant effect of time (p < 0.004); whereas, there was neither a significant effect of β-FNA (p = 0.59), nor an interaction (p = 0.62). * p < 0.05 vs. 10 min as indicated by Bonferroni’s multiple comparisons. Binding activity in unstimulated cells was 43 ± 7 arbitrary units/μg protein.
3.5. Effect of β-FNA on IFNγ + Tat -induced of p38 activation
Activation of p38 is instrumental in the induction of CXCL10 expression, therefore, we were interested in determining whether this pathways is disrupted by β-FNA. Exposure to IFNγ + Tat induced p38 activation as indicated by significantly increased phosphorylation of p38 following 10 min of stimulation (Fig. 5). Co-exposure to β-FNA significantly inhibited proinflammatory induced p38 activation as indicated by lower levels of phospho-p38 at 10 and 30 min post-stimulation.
Fig. 5.

β-FNA inhibits IFNγ + HIV-1 Tat1–72, -induced p38 activation in normal human astrocytes. In the presence or absence or β-FNA (10 μM), normal human astrocytes were exposed to human recombinant IFNγ (10 ng/ml) + HIV-1 Tat1–72 for 10–270 min. Panel A): Western blot was used to determine levels of total p38 and phosphorylated-p38 (p-p38) in the cytosolic fraction. Panel B): Data for the densitometric analysis represent mean + S.E.M of 3–5 independent experiments. Integrated density values are presented as a ratio of p-p38/total p38 and expressed as fold change relative to unstimulated control.. Data were analyzed using one-way ANOVA and Newman-Keuls post test comparison. ** p < 0.01 vs. IFN γ + HIV- 1 Tat1–72 at 10 min; *** p < 0.001 vs. IFNγ + HIV-1 Tat1–72 at 30 min.
4. Discussion
Neuroinflammation is associated with neurodegenerative disorders as well as CNS infection and trauma (Hirsch and Hunot, 2009; Kadiu et al., 2005; Persidsky and Gendelman, 2003; Poluektova et al., 2005; Vlodavsky et al., 2006). Chemokines are among the inflammatory mediators which are instrumental in response to CNS insults and the expression levels are frequently elevated with neuropathology (Cinque et al., 2005; Gonzalez et al., 2003; Kolb et al., 1999; Xia et al., 2000). The relative contribution of these chemokines to neuroprotection or neurotoxicity depends largely on the temporal and cellular context; in fact there is still much to learn about the specific involvement of individual chemokines in the various types of neuropathology. The chemokine CXCL10 can be directly neurotoxic and the involvement of this chemokine in HIV-1 associated neuropathogenesis has been well documented (Kolb et al., 1999; Sui et al., 2004; Sui et al., 2006).
Neutralization of CXCL10 as a means of reducing neuroinflammatory damage is of interest to our group as well as others (Davis et al., 2007; Glaser et al., 2004; Sorensen, 2004). We are particularly interested in the anti-inflammatory actions of β-FNA (Davis et al., 2007; 2008). We previously reported that β-FNA inhibits TNFα-induced CXCL10 expression in the human A172 astroglial cell line (Davis et al., 2007). We have expanded upon these previous studies and used primary astrocytes and proinflammatory stimuli germane to HIV-1 infection, IFNγ and Tat, in order to study the anti-inflammatory effects of β-FNA. Exposure to IFNγ + Tat synergistically enhanced CXCL10 expression in NHA. However, co-exposure to β-FNA inhibited IFNγ + Tat-induced induced CXCL10 expression (protein and mRNA), which suggests that the inhibitory actions of β-FNA are, in part, via a transcriptional mechanism. Together with our previous report (Davis et al., 2007), these findings suggest that inhibition of proinflammatory-induced CXCL10 expression by β-FNA is not unique to human A172 astroglial cells, as inhibition also occurs in NHA.
Astrocytes can express multiple opioid receptor types and these receptors are differentially expressed among brain regions (Ruzicka et al., 1995; Stiene-Martin et al., 1998). With regards to its mechanism of action, β-FNA exhibits irreversible antagonism at MOR (Ward et al., 1982). We were the first to report the anti-inflammatory actions of β-FNA in human astroglial cells (Davis et al., 2007; 2008), and the data indicated a MOR-independent mechanism of action. For instance, we found that the relatively potent MOR agonist, fentanyl, dose-dependently inhibited TNFα-induced CXCL10 expression in A172 cells. However, neither the non-selective MOR antagonist, naltrexone, nor the highly selective MOR antagonist, β-FNA, blocked the fentanyl-mediated inhibition of CXCL10 expression (Davis et al., 2007). In fact, β-FNA was a more potent inhibitor of CXCL10 expression in A172 cells than was fentanyl (Davis et al., 2007). Subsequently, we performed a series of experiments to further address the potential involvement of opioid receptors, including MOR, KOR and nociceptin/orphanin FQ receptor (ORL). We found that β-FNA actions are not modulated by another MOR selective antagonist, CTAP (at a concentration 3-fold higher than β-FNA), further suggesting that inhibition of CXCL10 expression by β-FNA is not through actions at the MOR (unpublished data). Similarly, KOR and ORL do not appear to be involved in β-FNA effects on CXCL10 expression as nor-binaltorphimine and [Nphe1]-nociceptin1–13 amide, respectively (at concentrations 3-fold higher than β-FNA), failed to block β-FNA effects (unpublished data).
Rather unexpected effects of β-FNA on chemokine (IL-8 and MIP-1β) expression in human astrocytes have been reported by others (Mahajan et al., 2002). In addition to blocking morphine-mediated suppression of constitutive chemokine expression, β-FNA alone up-regulated chemokine expression in human astrocytes (Mahajan et al., 2002). These investigators suggested that β-FNA may be activating at another type of opioid family receptor such as ORL or another as yet unidentified site. Others have reported that naloxone has anti-inflammatory and neuroprotective actions that are unrelated to MOR antagonism (Dutta et al., 2008; Liu et al., 2000; Liu et al., 2002; Zhang et al., 2010). For instance, LPS-stimulated microglial-induced neuronal toxicity is blocked by naloxone, in part, by blocking LPS binding to microglia. These neuroprotective actions were mediated by both the inactive and active stereoisomers (with respect to MOR antagonism), (+) naloxone and (−)-naloxone, respectively (Liu et al., 2000). This same group later demonstrated that Aβ-induced neurotoxicity is reduced by naloxone, reportedly by inhibiting microglial superoxide production in a MOR-independent manner (Liu et al., 2002). More recently, it was reported that in neuron-glia cultures, MnCl2 + LPS-induced dopaminergic toxicity was prevented by pre-treatment with naloxone (Zhang et al., 2010). Furthermore, cytokine release and generation of reactive oxygen species by microglia and astrocytes was reduced by naloxone pre-treatment (Zhang et al., 2010).
In the present study, again we used co-exposure to excess (3- and 10-fold relative to β-FNA) opioid receptor antagonists as a means of assessing the potential involvement of opioid receptors in β-FNA mediated inhibition of CXCL10 in NHA. The fact that neither a non-selective opioid receptor antagonist (naltrexone) nor a MOR-selective antagonist (CTAP) blocked β-FNA inhibition of IFNγ + Tat-induced CXCL10 expression, further supports a MOR-independent mechanism of action in human astrocytes. It is also important to note that neither naltrexone nor CTAP alone inhibit IFNγ + Tat-induced CXCL10 expression, further highlighting the novel anti-inflammatory actions of β-FNA. Interestingly, there was a trend for CTAP alone to increase IFNγ + Tat-induced CXCL10 expression. Thus, the slight attenuation of β-FNA-inhibition of IFNγ + Tat-induced CXCL10 expression by 100 μM CTAP likely reflects the stimulatory action of CTAP rather than antagonism at MOR.
Given the apparent MOR-independent nature of the anti-inflammatory actions of β-FNA, we investigated another potential intracellular target important in CXCL10 induction, NF-κB. While proinflammatory induction of CXCL10 has been shown to be NF-κB-dependent we needed to confirm this in our experimental model. Indeed, utilizing the inhibitor of NF-κB nuclear translocation, SN50, we demonstrated that IFNγ + Tat-induced CXCL10 expression is NF-κB-dependent in NHA. NF-κB activation, as determined by levels of NF-κB p65-DNA binding activity, was not inhibited by β-FNA. Likewise, NF-κB p50-DNA binding activity was not significantly affected by β-FNA (data not shown). These data suggest that β-FNA inhibition of CXCL10 expression is not through an NF-κB p65 mediated mechanism. These findings are consistent with our observations in A172 cells where β-FNA did not significantly affect nuclear levels NF-κB p65 (as determined by immunoblot) (Davis et al., 2007). Additionally, it may be that β-FNA enhances the degradation or nuclear export of NF-κB proteins. However, before a role for NF-κB signaling is completely abandoned as a target of β-FNA, future investigations should assess other proteins in the NF-κB pathway, as well as more functional measures including dimerization of the various NF-κB subunits and transcriptional activation. For instance, we have found that TNFα-induced NF-κB driven promoter activity in THP-1 monocytes is inhibited by β-FNA (unpublished data). These findings also hint at the possibility that β-FNA acts at multiple sites, resulting in stimulus-dependent effects.
Given the importance of p38 activation in inflammatory signaling in glia, including CXCL10 expression (Williams et al., 2009), were also investigated the effects of β-FNA on this signaling molecule. Importantly, we discovered that β-FNA inhibits IFNγ + Tat-induced p38 activation in human astroglial cells. This finding provides insight into the mechanism underlying the anti-inflammatory actions of β-FNA. It remains to be determined whether β-FNA alters phosphorylation per se or targets upstream signaling events. Ongoing and future investigations are expected to provide new details about the mechanism of action for the anti-inflammatory actions of β-FNA. However, we have demonstrated that the inhibitory actions of β-FNA on CXCL10 expression are MOR-independent and, in part, are transcriptional.
CXCL10 is not the only inflammatory molecule affected by β-FNA. For instance, we previously reported that expression of inducible nitric oxide synthase is transcriptionally inhibited by β-FNA (Davis et al., 2008). Interestingly, the expression of other inflammatory molecules such as CXCL10/IL-8 and CCL2/MCP-1, are much less sensitive to the inhibitory actions of β-FNA (unpublished data). Further investigation of β-FNA effects on CXCL8, CCL2, and other inflammatory mediators are therefore warranted.
The anti-inflammatory actions of β-FNA identified support and add to the findings of others (Liu et al., 2000; Liu and Hong, 2003) which have indicated MOR-independent, anti-inflammatory actions of naloxone (a non-specific, opioid receptor antagonist). These investigators found that lipopolysaccharide (LPS)-stimulated microglia mediated pronounced neuronal toxicity when added to rat mesencephalic mixed neuron-glia cultures (Liu et al., 2000). Naloxone reportedly provided neuroprotection by blocking LPS binding to microglia (Liu et al., 2000). Furthermore, it was determined that the inactive stereoisomer, (+)-naloxone, was equally effective as (−)-naloxone (Liu et al., 2000). These investigators also determined that Aβ (1–42)-induced neurotoxicity was inhibited by naloxone in an opioid receptor-independent mechanism, reduced microglial superoxide production (Liu et al., 2002). Furthermore, additional information about opioid actions that are not mediated through classical opioid receptors has been recently reviewed (Hutchinson et al., 2011). For instance, LPS-induced toll-like receptor-4 (TLR4) activation is inhibited by (+)-naloxone as well as (+)-naltrexone (Hutchinson et al., 2008).
Thus, there is ongoing interest in the MOR-independent actions of opioid compounds. The findings presented herein add to this knowledge base and highlight the potential for β-FNA as adjunct therapy for neuropathologies involving neuroinflammation. However, further investigation is warranted in order to more fully define the mechanism of action for the anti-inflammatory actions of β-FNA.
Highlights.
IFNγ + HIV-1 Tat induces CXCL10 expression in human astrocytes
β-FNA inhibits IFNγ + HIV-1 Tat induced CXCL10 expression
inhibitory actions of β-FNA are not mediated through the mu-opioid receptor
β-FNA does not inhibit IFNγ + HIV-1 Tat induced NF-κB activation
β-FNA inhibits IFNγ + HIV-1 Tat induced p38 activation
Acknowledgments
This work was supported in part by NIH grants NS 062664 (RLD) and DA 012448 (CWS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agrawal L, Louboutin JP, Strayer DS. Preventing HIV-1 tat-induced neuronal apoptosis using antioxidant enzymes: Mechanistic and therapeutic implications. Virology. 2007;363:462–472. doi: 10.1016/j.virol.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Ances BM, Ellis RJ. Dementia and neurocognitive disorders due to HIV-1 infection. Semin Neurol. 2007;27:86–92. doi: 10.1055/s-2006-956759. [DOI] [PubMed] [Google Scholar]
- Ansari AW, Bhatnagar N, Dittrich-Breiholz O, Kracht M, Schmidt RE, Heiken H. Host chemokine (C-C motif) ligand-2 (CCL2) is differentially regulated in HIV type 1 (HIV-1)-infected individuals. Int Immunol. 2006;18:1443–1451. doi: 10.1093/intimm/dxl078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony IC, Ramage SN, Carnie FW, Simmonds P, Bell JE. Influence of HAART on HIV-related CNS disease and neuroinflammation. J Neuropathol Exp Neurol. 2005;64:529–536. doi: 10.1093/jnen/64.6.529. [DOI] [PubMed] [Google Scholar]
- Bajetto A, Bonavia R, Barbero S, Schettini G. Characterization of chemokines and their receptors in the central nervous system: physiopathological implications. J Neurochem. 2002;82:1311–1329. doi: 10.1046/j.1471-4159.2002.01091.x. [DOI] [PubMed] [Google Scholar]
- Biber K, de Jong EK, van Weering HR, Boddeke HW. Chemokines and their receptors in central nervous system disease. Curr Drug Targets. 2006;7:29–46. doi: 10.2174/138945006775270196. [DOI] [PubMed] [Google Scholar]
- Chang L, Ernst T, St Hillaire C, Conant K. Antiretroviral treatment alters relationship between MCP-1 and neurometabolites in HIV patients. Antivir Ther. 2004;9:431–440. doi: 10.1177/135965350400900302. [DOI] [PubMed] [Google Scholar]
- Chang L, Ernst T, Witt MD, Ames N, Walot I, Jovicich J, DeSilva M, Trivedi N, Speck O, Miller EN. Persistent brain abnormalities in antiretroviral-naive HIV patients 3 months after HAART. Antivir Ther. 2003;8:17–26. [PubMed] [Google Scholar]
- Chen X, Yang L, Zhang N, Turpin JA, Buckheit RW, Osterling C, Oppenheim JJ, Howard OM. Shikonin, a component of chinese herbal medicine, inhibits chemokine receptor function and suppresses human immunodeficiency virus type 1. Antimicrob Agents Chemother. 2003;47:2810–2816. doi: 10.1128/AAC.47.9.2810-2816.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinque P, Bestetti A, Marenzi R, Sala S, Gisslen M, Hagberg L, Price RW. Cerebrospinal fluid interferon-gamma-inducible protein 10 (IP-10, CXCL10) in HIV-1 infection. J Neuroimmunol. 2005;168:154–163. doi: 10.1016/j.jneuroim.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Conant K, Garzino-Demo A, Nath A, McArthur JC, Halliday W, Power C, Gallo RC, Major EO. Induction of monocyte chemoattractant protein-1 in HIV-1 Tat-stimulated astrocytes and elevation in AIDS dementia. Proc Natl Acad Sci U S A. 1998;95:3117–3121. doi: 10.1073/pnas.95.6.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cysique LA, Brew BJ. Neuropsychological functioning and antiretroviral treatment in HIV/AIDS: a review. Neuropsychol Rev. 2009;19:169–185. doi: 10.1007/s11065-009-9092-3. [DOI] [PubMed] [Google Scholar]
- Das S, Kelschenbach J, Charboneau R, Barke RA, Roy S. Morphine withdrawal stress modulates lipopolysaccharide-induced interleukin 12 p40 (IL-12p40) expression by activating extracellular signal-regulated kinase 1/2, which is further potentiated by glucocorticoids. J Biol Chem. 2011;286:29806–29817. doi: 10.1074/jbc.M111.271460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RL, Buck DJ, Saffarian N, Mohan S, DeSilva U, Fernando SC, Stevens CW. Beta-funaltrexamine inhibits inducible nitric-oxide synthase expression in human astroglial cells. J Neuroimmune Pharmacol. 2008;3:150–153. doi: 10.1007/s11481-008-9102-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RL, Buck DJ, Saffarian N, Stevens CW. The opioid antagonist, beta-funaltrexamine, inhibits chemokine expression in human astroglial cells. J Neuroimmunol. 2007;186:141–149. doi: 10.1016/j.jneuroim.2007.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RL, Dertien J, Syapin PJ. Ethanol-induced modulation of inducible nitric-oxide synthase activity in human A172 astrocytoma cells. Alcohol Clin Exp Res. 2002;26:1404–1411. doi: 10.1097/01.ALC.0000030841.92766.80. [DOI] [PubMed] [Google Scholar]
- Davis RL, Syapin PJ. Ethanol increases nuclear factor-kappa B activity in human astroglial cells. Neurosci Lett. 2004;371:128–132. doi: 10.1016/j.neulet.2004.08.051. [DOI] [PubMed] [Google Scholar]
- Deshpande M, Zheng J, Borgmann K, Persidsky R, Wu L, Schellpeper C, Ghorpade A. Role of activated astrocytes in neuronal damage: potential links to HIV-1-associated dementia. Neurotox Res. 2005;7:183–192. doi: 10.1007/BF03036448. [DOI] [PubMed] [Google Scholar]
- Dou H, Ellison B, Bradley J, Kasiyanov A, Poluektova LY, Xiong H, Maggirwar S, Dewhurst S, Gelbard HA, Gendelman HE. Neuroprotective mechanisms of lithium in murine human immunodeficiency virus-1 encephalitis. J Neurosci. 2005;25:8375–8385. doi: 10.1523/JNEUROSCI.2164-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta G, Zhang P, Liu B. The lipopolysaccharide Parkinson’s disease animal model: mechanistic studies and drug discovery. Fundam Clin Pharmacol. 2008;22:453–464. doi: 10.1111/j.1472-8206.2008.00616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ensoli B, Buonaguro L, Barillari G, Fiorelli V, Gendelman R, Morgan RA, Wingfield P, Gallo RC. Release, uptake, and effects of extracellular human immunodeficiency virus type 1 Tat protein on cell growth and viral transactivation. J Virol. 1993;67:277–287. doi: 10.1128/jvi.67.1.277-287.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugenin EA, Osiecki K, Lopez L, Goldstein H, Calderon TM, Berman JW. CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood-brain barrier: a potential mechanism of HIV-CNS invasion and NeuroAIDS. J Neurosci. 2006;26:1098–1106. doi: 10.1523/JNEUROSCI.3863-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer-Smith T, Rappaport J. Evolving paradigms in the pathogenesis of HIV-1-associated dementia. Expert Rev Mol Med. 2005;7:1–26. doi: 10.1017/S1462399405010239. [DOI] [PubMed] [Google Scholar]
- Flynn G, Maru S, Loughlin J, Romero IA, Male D. Regulation of chemokine receptor expression in human microglia and astrocytes. J Neuroimmunol. 2003;136:84–93. doi: 10.1016/s0165-5728(03)00009-2. [DOI] [PubMed] [Google Scholar]
- Ghafouri M, Amini S, Khalili K, Sawaya BE. HIV-1 associated dementia: symptoms and causes. Retrovirology. 2006;3:28. doi: 10.1186/1742-4690-3-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giunta B, Obregon D, Hou H, Zeng J, Sun N, Nikolic V, Ehrhart J, Shytle D, Fernandez F, Tan J. EGCG mitigates neurotoxicity mediated by HIV-1 proteins gp120 and Tat in the presence of IFN-gamma: role of JAK/STAT1 signaling and implications for HIV-associated dementia. Brain Res. 2006;1123:216–225. doi: 10.1016/j.brainres.2006.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser J, Gonzalez R, Perreau VM, Cotman CW, Keirstead HS. Neutralization of the chemokine CXCL10 enhances tissue sparing and angiogenesis following spinal cord injury. J Neurosci Res. 2004;77:701–708. doi: 10.1002/jnr.20204. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5:69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- Gonzalez R, Glaser J, Liu MT, Lane TE, Keirstead HS. Reducing inflammation decreases secondary degeneration and functional deficit after spinal cord injury. Exp Neurol. 2003;184:456–463. doi: 10.1016/s0014-4886(03)00257-7. [DOI] [PubMed] [Google Scholar]
- Gray F, Hurtrel M, Hurtrel B. Early central nervous system changes in human immunodeficiency virus (HIV)-infection. Neuropathol Appl Neurobiol. 1993;19:3–9. doi: 10.1111/j.1365-2990.1993.tb00399.x. [DOI] [PubMed] [Google Scholar]
- Hettinger AM, Allen MR, Zhang BR, Goad DW, Malayer JR, Geisert RD. Presence of the acute phase protein, bikunin, in the endometrium of gilts during estrous cycle and early pregnancy. Biol Reprod. 2001;65:507–513. doi: 10.1095/biolreprod65.2.507. [DOI] [PubMed] [Google Scholar]
- Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 2009;8:382–397. doi: 10.1016/S1474-4422(09)70062-6. [DOI] [PubMed] [Google Scholar]
- Hutchinson MR, Shavit Y, Grace PM, Rice KC, Maier SF, Watkins LR. Exploring the neuroimmunopharmacology of opioids: an integrative review of mechanisms of central immune signaling and their implications for opioid analgesia. Pharmacol Rev. 2011;63:772–810. doi: 10.1124/pr.110.004135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Zhang Y, Brown K, Coats BD, Shridhar M, Sholar PW, Patel SJ, Crysdale NY, Harrison JA, Maier SF, Rice KC, Watkins LR. Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: involvement of toll-like receptor 4 (TLR4) Eur J Neurosci. 2008;28:20–29. doi: 10.1111/j.1460-9568.2008.06321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadiu I, Glanzer JG, Kipnis J, Gendelman HE, Thomas MP. Mononuclear phagocytes in the pathogenesis of neurodegenerative diseases. Neurotox Res. 2005;8:25–50. doi: 10.1007/BF03033818. [DOI] [PubMed] [Google Scholar]
- Kelder W, McArthur JC, Nance-Sproson T, McClernon D, Griffin DE. Beta-chemokines MCP-1 and RANTES are selectively increased in cerebrospinal fluid of patients with human immunodeficiency virus-associated dementia. Ann Neurol. 1998;44:831–835. doi: 10.1002/ana.410440521. [DOI] [PubMed] [Google Scholar]
- Khiati A, Chaloin O, Muller S, Tardieu M, Horellou P. Induction of monocyte chemoattractant protein-1 (MCP-1/CCL2) gene expression by human immunodeficiency virus-1 Tat in human astrocytes is CDK9 dependent. J Neurovirol. 2010;16:150–167. doi: 10.3109/13550281003735691. [DOI] [PubMed] [Google Scholar]
- King JE, Eugenin EA, Buckner CM, Berman JW. HIV tat and neurotoxicity. Microbes Infect. 2006;8:1347–1357. doi: 10.1016/j.micinf.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Kolb SA, Sporer B, Lahrtz F, Koedel U, Pfister HW, Fontana A. Identification of a T cell chemotactic factor in the cerebrospinal fluid of HIV-1-infected individuals as interferon-gamma inducible protein 10. J Neuroimmunol. 1999;93:172–181. doi: 10.1016/s0165-5728(98)00223-9. [DOI] [PubMed] [Google Scholar]
- Kramer-Hammerle S, Rothenaigner I, Wolff H, Bell JE, Brack-Werner R. Cells of the central nervous system as targets and reservoirs of the human immunodeficiency virus. Virus Res. 2005;111:194–213. doi: 10.1016/j.virusres.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Kutsch O, Oh JW, Nath A, Benveniste EN. Induction of the chemokines interleukin-8 and IP-10 by human immunodeficiency virus type 1 Tat in astrocytes. J Virol. 2000;74:9214–9221. doi: 10.1128/jvi.74.19.9214-9221.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labuz D, Mousa SA, Schafer M, Stein C, Machelska H. Relative contribution of peripheral versus central opioid receptors to antinociception. Brain Res. 2007;1160:30–38. doi: 10.1016/j.brainres.2007.05.049. [DOI] [PubMed] [Google Scholar]
- Lane BR, King SR, Bock PJ, Strieter RM, Coffey MJ, Markovitz DM. The C-X-C chemokine IP-10 stimulates HIV-1 replication. Virology. 2003;307:122–134. doi: 10.1016/s0042-6822(02)00045-4. [DOI] [PubMed] [Google Scholar]
- Liao SL, Chen WY, Raung SL, Chen CJ. Neuroprotection of naloxone against ischemic injury in rats: role of mu receptor antagonism. Neurosci Lett. 2003;345:169–172. doi: 10.1016/s0304-3940(03)00540-8. [DOI] [PubMed] [Google Scholar]
- Lin YZ, Yao SY, Veach RA, Torgerson TR, Hawiger J. Inhibition of nuclear translocation of transcription factor NF-kappa B by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J Biol Chem. 1995;270:14255–14258. doi: 10.1074/jbc.270.24.14255. [DOI] [PubMed] [Google Scholar]
- Liu-Chen LY, Li SX, Tallarida RJ. Studies on kinetics of [3H]beta-funaltrexamine binding to mu opioid receptor. Mol Pharmacol. 1990;37:243–250. [PubMed] [Google Scholar]
- Liu B, Du L, Hong JS. Naloxone protects rat dopaminergic neurons against inflammatory damage through inhibition of microglia activation and superoxide generation. J Pharmacol Exp Ther. 2000;293:607–617. [PubMed] [Google Scholar]
- Liu B, Hong JS. Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J Pharmacol Exp Ther. 2003;304:1–7. doi: 10.1124/jpet.102.035048. [DOI] [PubMed] [Google Scholar]
- Liu Y, Qin L, Wilson BC, An L, Hong JS, Liu B. Inhibition by naloxone stereoisomers of beta-amyloid peptide (1–42)-induced superoxide production in microglia and degeneration of cortical and mesencephalic neurons. J Pharmacol Exp Ther. 2002;302:1212–1219. doi: 10.1124/jpet.102.035956. [DOI] [PubMed] [Google Scholar]
- Luster AD, Unkeless JC, Ravetch JV. Gamma-interferon transcriptionally regulates an early-response gene containing homology to platelet proteins. Nature. 1985;315:672–676. doi: 10.1038/315672a0. [DOI] [PubMed] [Google Scholar]
- Mahajan SD, Schwartz SA, Shanahan TC, Chawda RP, Nair MP. Morphine regulates gene expression of alpha- and beta-chemokines and their receptors on astroglial cells via the opioid mu receptor. J Immunol. 2002;169:3589–3599. doi: 10.4049/jimmunol.169.7.3589. [DOI] [PubMed] [Google Scholar]
- Minagar A, Shapshak P, Fujimura R, Ownby R, Heyes M, Eisdorfer C. The role of macrophage/microglia and astrocytes in the pathogenesis of three neurologic disorders: HIV-associated dementia, Alzheimer disease, and multiple sclerosis. J Neurol Sci. 2002;202:13–23. doi: 10.1016/s0022-510x(02)00207-1. [DOI] [PubMed] [Google Scholar]
- Nath A. Human immunodeficiency virus (HIV) proteins in neuropathogenesis of HIV dementia. J Infect Dis. 2002;186(Suppl 2):S193–198. doi: 10.1086/344528. [DOI] [PubMed] [Google Scholar]
- Nath A, Sacktor N. Influence of highly active antiretroviral therapy on persistence of HIV in the central nervous system. Curr Opin Neurol. 2006;19:358–361. doi: 10.1097/01.wco.0000236614.51592.ca. [DOI] [PubMed] [Google Scholar]
- Navia BA, Cho ES, Petito CK, Price RW. The AIDS dementia complex: II. Neuropathology Ann Neurol. 1986;19:525–535. doi: 10.1002/ana.410190603. [DOI] [PubMed] [Google Scholar]
- Ozdener H. Molecular mechanisms of HIV-1 associated neurodegeneration. J Biosci. 2005;30:391–405. doi: 10.1007/BF02703676. [DOI] [PubMed] [Google Scholar]
- Pahan K, Sheikh FG, Liu X, Hilger S, McKinney M, Petro TM. Induction of nitric-oxide synthase and activation of NF-kappaB by interleukin-12 p40 in microglial cells. J Biol Chem. 2001;276:7899–7905. doi: 10.1074/jbc.M008262200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persidsky Y, Gendelman HE. Mononuclear phagocyte immunity and the neuropathogenesis of HIV-1 infection. J Leukoc Biol. 2003;74:691–701. doi: 10.1189/jlb.0503205. [DOI] [PubMed] [Google Scholar]
- Poluektova L, Meyer V, Walters L, Paez X, Gendelman HE. Macrophage-induced inflammation affects hippocampal plasticity and neuronal development in a murine model of HIV-1 encephalitis. Glia. 2005;52:344–353. doi: 10.1002/glia.20253. [DOI] [PubMed] [Google Scholar]
- Ruzicka BB, Fox CA, Thompson RC, Meng F, Watson SJ, Akil H. Primary astroglial cultures derived from several rat brain regions differentially express mu, delta and kappa opioid receptor mRNA. Brain Res Mol Brain Res. 1995;34:209–220. doi: 10.1016/0169-328x(95)00165-o. [DOI] [PubMed] [Google Scholar]
- Sanders VJ, Pittman CA, White MG, Wang G, Wiley CA, Achim CL. Chemokines and receptors in HIV encephalitis. Aids. 1998;12:1021–1026. [PubMed] [Google Scholar]
- Sorensen TL. Targeting the chemokine receptor CXCR3 and its ligand CXCL10 in the central nervous system: potential therapy for inflammatory demyelinating disease? Curr Neurovasc Res. 2004;1:183–190. doi: 10.2174/1567202043480143. [DOI] [PubMed] [Google Scholar]
- Stiene-Martin A, Zhou R, Hauser KF. Regional, developmental, and cell cycle-dependent differences in mu, delta, and kappa-opioid receptor expression among cultured mouse astrocytes. Glia. 1998;22:249–259. [PMC free article] [PubMed] [Google Scholar]
- Sui Y, Potula R, Dhillon N, Pinson D, Li S, Nath A, Anderson C, Turchan J, Kolson D, Narayan O, Buch S. Neuronal apoptosis is mediated by CXCL10 overexpression in simian human immunodeficiency virus encephalitis. Am J Pathol. 2004;164:1557–1566. doi: 10.1016/S0002-9440(10)63714-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui Y, Stehno-Bittel L, Li S, Loganathan R, Dhillon NK, Pinson D, Nath A, Kolson D, Narayan O, Buch S. CXCL10-induced cell death in neurons: role of calcium dysregulation. Eur J Neurosci. 2006;23:957–964. doi: 10.1111/j.1460-9568.2006.04631.x. [DOI] [PubMed] [Google Scholar]
- Taub DD, Lloyd AR, Conlon K, Wang JM, Ortaldo JR, Harada A, Matsushima K, Kelvin DJ, Oppenheim JJ. Recombinant human interferon-inducible protein 10 is a chemoattractant for human monocytes and T lymphocytes and promotes T cell adhesion to endothelial cells. J Exp Med. 1993;177:1809–1814. doi: 10.1084/jem.177.6.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson KA, Cherry CL, Bell JE, McLean CA. Brain cell reservoirs of latent virus in presymptomatic HIV-infected individuals. Am J Pathol. 2011;179:1623–1629. doi: 10.1016/j.ajpath.2011.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tousi NS, Buck DJ, Zecca L, Davis RL. Neuromelanin inhibits CXCL10 expression in human astroglial cells. Neurosci Lett. 2010;486:47–50. doi: 10.1016/j.neulet.2010.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Marle G, Henry S, Todoruk T, Sullivan A, Silva C, Rourke SB, Holden J, McArthur JC, Gill MJ, Power C. Human immunodeficiency virus type 1 Nef protein mediates neural cell death: a neurotoxic role for IP-10. Virology. 2004;329:302–318. doi: 10.1016/j.virol.2004.08.024. [DOI] [PubMed] [Google Scholar]
- Vlodavsky E, Palzur E, Soustiel JF. Hyperbaric oxygen therapy reduces neuroinflammation and expression of matrix metalloproteinase-9 in the rat model of traumatic brain injury. Neuropathol Appl Neurobiol. 2006;32:40–50. doi: 10.1111/j.1365-2990.2005.00698.x. [DOI] [PubMed] [Google Scholar]
- Ward SJ, Portoghese PS, Takemori AE. Pharmacological characterization in vivo of the novel opiate, beta-funaltrexamine. J Pharmacol Exp Ther. 1982;220:494–498. [PubMed] [Google Scholar]
- Westendorp MO, Frank R, Ochsenbauer C, Stricker K, Dhein J, Walczak H, Debatin KM, Krammer PH. Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature. 1995;375:497–500. doi: 10.1038/375497a0. [DOI] [PubMed] [Google Scholar]
- Williams R, Dhillon NK, Hegde ST, Yao H, Peng F, Callen S, Chebloune Y, Davis RL, Buch SJ. Proinflammatory cytokines and HIV-1 synergistically enhance CXCL10 expression in human astrocytes. Glia. 2009;57:734–743. doi: 10.1002/glia.20801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson ME, Dimayuga FO, Reed JL, Curry TE, Anderson CF, Nath A, Bruce-Keller AJ. Immune modulation by estrogens: role in CNS HIV-1 infection. Endocrine. 2006;29:289–297. doi: 10.1385/ENDO:29:2:289. [DOI] [PubMed] [Google Scholar]
- Xia MQ, Bacskai BJ, Knowles RB, Qin SX, Hyman BT. Expression of the chemokine receptor CXCR3 on neurons and the elevated expression of its ligand IP-10 in reactive astrocytes: in vitro ERK1/2 activation and role in Alzheimer’s disease. J Neuroimmunol. 2000;108:227–235. doi: 10.1016/s0165-5728(00)00285-x. [DOI] [PubMed] [Google Scholar]
- Yang YT, Ju TC, Yang DI. Induction of hypoxia inducible factor-1 attenuates metabolic insults induced by 3-nitropropionic acid in rat C6 glioma cells. J Neurochem. 2005;93:513–525. doi: 10.1111/j.1471-4159.2005.03032.x. [DOI] [PubMed] [Google Scholar]
- Zhang P, Lokuta KM, Turner DE, Liu B. Synergistic dopaminergic neurotoxicity of manganese and lipopolysaccharide: differential involvement of microglia and astroglia. J Neurochem. 2010;112:434–443. doi: 10.1111/j.1471-4159.2009.06477.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BY, Liu Y, Kim B, Xiao Y, He JJ. Astrocyte activation and dysfunction and neuron death by HIV-1 Tat expression in astrocytes. Mol Cell Neurosci. 2004;27:296–305. doi: 10.1016/j.mcn.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Zink MC, Uhrlaub J, DeWitt J, Voelker T, Bullock B, Mankowski J, Tarwater P, Clements J, Barber S. Neuroprotective and anti-human immunodeficiency virus activity of minocycline. JAMA. 2005;293:2003–2011. doi: 10.1001/jama.293.16.2003. [DOI] [PubMed] [Google Scholar]