

Abstract
Inhibition of Sonic hedgehog (Shh) signaling is of great clinical interest. Here we exploit Hedgehog acyltransferase (Hhat)-mediated Shh palmitoylation, a modification critical for Shh signaling, as a novel target for Shh pathway inhibition. A target-oriented high-throughput screen was used to identify small-molecule inhibitors of Hhat. In cells, these Hhat inhibitors specifically block Shh palmitoylation and inhibit autocrine and paracrine Shh signaling.
Shh is a secreted signaling protein that is essential for proper embryonic development1,2. In adults, aberrant Shh signaling drives initiation and maintenance of medulloblastoma and basal cell carcinoma, and has been implicated in the progression of prostate cancer, gastrointestinal tumors, and pancreatic cancer3. The mature Shh signaling protein is formed via a series of post-translational processing reactions. Following removal of the signal peptide, Shh undergoes autocleavage to produce a 19 kDa N-terminal product, ShhN. During this reaction, cholesterol is attached to the C terminus of ShhN4. In a separate reaction, Hhat catalyzes attachment of palmitate to the N-terminal cysteine of ShhN via an amide bond4,5. Palmitoylation of Shh plays a critical role in regulating the signaling potency of Shh in cells6,7. Hhat knockout mice and palmitoylation-deficient Shh transgenic mice exhibit developmental defects similar to those observed in Shh knockout mice7. Thus, Hhat presents an attractive, novel target to block Shh signaling.
Hhat is a member of the membrane bound O-acyl transferase (MBOAT) family of proteins8. Due to the presence of multiple transmembrane domains, molecular and structural characterization of this family in general, and Hhat in particular, has been limited5,9. In an effort to discover a small-molecule inhibitor of Hhat, we conducted a high-throughput screen using a peptide-based assay to monitor Hhat-mediated Shh palmitoylation. We screened a library of 63,885 unique structures (Supplementary Results, Supplementary Table 1). A secondary screen was performed on 648 molecules, using the peptide-based assay and an orthogonal cell viability assay, to yield 95 confirmed hits. Four compounds, RU-SKI 39 (1), 41 (2), 43 (3) and 50 (4), were selected based on their low IC50 values and drug-like scaffold (Table 1, Supplementary Figs. 1 and 2).
Table 1.
Structures and IC50 values of the Hhat inhibitor hit compounds.
| Number | Compound ID | Structure | IC50 μM |
|---|---|---|---|
| 1 | RU-SKI 39 |
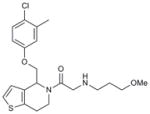
|
3.07 |
| 2 | RU-SKI 41 |
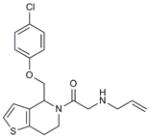
|
0.18 |
| 3 | RU-SKI 43 |
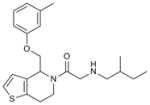
|
0.85 |
| 4 | RU-SKI 50 |
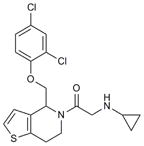
|
5.44 |
The four Hhat inhibitors were re-tested in an in vitro palmitoylation assay using ShhN protein. Each compound at 12.5 μM inhibited Hhat-mediated palmitoylation of ShhN by 40–80% (Fig. 1a). ShhN C24A, a mutant Shh protein that cannot incorporate palmitate, and Hhat D339A, an inactive Hhat mutant9, served as negative controls. Inhibition of ShhN palmitoylation was specific to the RU-SKI compounds, since two structurally related molecules, C-1 (5) and C-2 (6; Supplementary Fig. 3), did not affect ShhN palmitoylation (Fig. 1a). We next analyzed the kinetics of RU-SKI 43 inhibition of ShhN palmitoylation in vitro using purified Hhat and ShhN. RU-SKI 43 behaved as an uncompetitive inhibitor (Ki=7.4 μM) with respect to Shh, and as a noncompetitive inhibitor (Ki=6.9 μM) with respect to 125I-iodo-palmitoylCoA (Fig. 1b).
Figure 1. RU-SKI 43 inhibits Hhat.

a) RU-SKIs inhibit Shh palmitoylation in vitro. Membranes from cells transfected with wild-type Hhat or the inactive Hhat D339A mutant were preincubated with DMSO or 12.5μM RU-SKIs, C-1 or C-2, then incubated with ShhN (wild-type or C24A) and 125I-iodo-palmitoylCoA. 125I-iodo-palmitoyl incorporation was normalized to that of ShhN + DMSO; each bar is Mean±SD (n=2–4). b) RU-SKI 43 inhibition kinetics with purified Hhat; each point is Mean ±SD (n=2). c,d) RU-SKIs inhibit Shh protein palmitoylation in cells. COS-1 cells expressing HA-Hhat and Shh were treated with DMSO or RU-SKIs (c, 25 μM, 16 h; d, 0, 10 or 20 μM RU-SKI 43, 5 h) and labeled with 125I-iodo-palmitate. Anti-Shh immunoprecipitates were analyzed by SDS-PAGE and phosphorimaging (upper panel) or Western blotting (c, lower panel; d, middle panel). Total cell lysates (TCL) were analyzed by Western blotting. e) RU-SKI 43 exhibits specificity for Hhat. COS-1 cells expressing H-Ras N17, Fyn or c-Src, were treated with RU-SKI 43 and labeled with the indicated fatty acids. f) RU-SKI 43 does not inhibit Porcupine. Mouse L Wnt-3a cells were transfected with FLAG-Porcupine or empty vector (EV), treated with DMSO, 10 μM RU-SKI-43, or 100 nM Wnt C59, and labeled with 125I-iodo-pentadecanoate. Full, uncut gel images are presented in Supplementary Figures 9–12.
Hhat inhibition was then analyzed using COS-1 cells expressing HA-tagged Hhat and Shh. Cells were pre-treated for 16 h with 25 μM compound or DMSO, then labeled with 125I- iodo-palmitate, a radiolabeled palmitate analog5,10,11. RU-SKI 39, 41 and 43 strongly inhibited radiolabel incorporation into ShhN; RU-SKI 50 was less effective in cells (Fig. 1c, Supplementary Fig. 4a). Based on its ability to function as an Hhat inhibitor in vitro and in cells, we focused on RU-SKI 43. Dose-dependent inhibition of Shh palmitoylation was observed following only 5 h of treatment (Fig. 1d, Supplementary Fig. 4c). Importantly, no effect on Shh palmitoylation was observed when cells were incubated with 10 μM C-2 (Supplementary Fig. 4 b,c).
Several lines of evidence suggest that inhibition by RU-SKI 43 is specific to Shh palmitoylation. Neither palmitoylation of H-Ras and Fyn nor myristoylation of c-Src was affected by treatment of cells with the compound (Fig. 1e). Treatment of cells with RU-SKI 43 had no effect on fatty acylation of Wnt3a12 by Porcupine, another member of the MBOAT family, whereas Wnt C59 (a Porcupine inhibitor) blocked radiolabel incorporation (Fig. 1f). Overexpression of Hhat reduced the ability of RU-SKI 43 to inhibit Shh palmitoylation in transfected COS-1 cells, whereas overexpression of Porcupine had no effect (Supplementary Fig. 5). Moreover, RU-SKI 43 inhibited palmitoylation of Shh by endogenous Hhat in COS-1 cells (Supplementary Fig. 6). Finally, RU-SKI 43 did not alter Shh autoprocessing, steady-state levels of Shh and Hhat, or subcellular localization of Shh and Hhat (Fig. 1d, Supplementary Fig. 7). Taken together, these data support the contention that RU-SKI 43 specifically inhibits Hhat but not other fatty acyl transferases.
Inhibition of Hhat is predicted to block Shh signaling in cells. We used three cell-based systems to test the specificity of RU-SKI 43 for the Shh pathway. First, NIH 3T3 cells were cotransfected with plasmids encoding Shh, a Gli-responsive Firefly luciferase reporter, and Renilla luciferase as a control. Increased luciferase production was observed, compared to cells transfected with a mutant Gli-luciferase plasmid, indicative of Gli1 activation (Fig. 2a). Importantly, addition of 10 μM RU-SKI 43 or LDE225, a Smoothened (Smo) inhibitor13, blocked luciferase activation, consistent with Shh pathway inhibition, whereas C-2 had no effect (Fig. 2a). These data suggest that RU-SKI 43 blocks autocrine Shh signaling in cells.
Figure 2. RU-SKI 43 blocks Shh signaling.

a) RU-SKI 43 blocks Gli activation. NIH 3T3 cells were cotransfected with vectors encoding 8XGliBS-Firefly luciferase (unless indicated otherwise), Renilla luciferase reporter (pRL-TK) and Shh. Confluent cells were treated with DMSO, 10 μM LDE225, 10 μM RU-SKI 43 or 10 μM C-2. The firefly luciferase (FL)/renilla luciferase (RL) ratio in cell lysates was calculated and normalized to DMSO-treated samples; each bar is Mean±SD (n=2–3). b,c) RU-SKI 43 does not affect exogenous Shh pathway activation. Shh Light II cells were treated with 100 nM SAG (b) or 1μg/mL Shh (C24II) (c) −/+ 10 μM RU-SKI 43. The FL/RL ratio in cell lysates was calculated for each sample, and was normalized to the DMSO or RU-SKI 43-treated samples; each bar represents Mean±SD (n=2). d) RU-SKI 43 does not affect Shh pathway activation in SuFu−/− cells. SuFu−/− cells were transfected with 8XGliBS-FL (unless otherwise indicated) and pRL-TK. Confluent cells were treated with DMSO or 10 μM RU-SKI 43. FL/RL ratios were calculated as in c); each bar is Mean ± SD (n=2–4). e) RU-SKI 43 blocks paracrine Shh signaling in cells. COS-1 cells expressing Hhat and Shh were cocultured with C3H10T1/2 fibroblasts, incubated with DMSO or 10 μM RU-SKI 43, and AP activity was measured; each point is Mean ± SD (n=3).
We then utilized Shh Light II cells that stably express a Gli1 reporter plasmid14. RU-SKI 43 had no effect on the ability of the Smo agonist SAG or Shh (C42II), a recombinant, hydrophobic variant of Shh, to activate Gli1 in Shh Light II cells (Fig. 2b,c). Moreover, RU-SKI 43 had no effect on Shh signaling in SuFu−/−cells, where Shh signaling is activated downstream of Smo (Fig. 2d). These findings imply that RU-SKI 43 inhibits Shh signaling at the level of the Shh ligand.
The effects of Hhat inhibition on paracrine Shh signaling were examined using C3H10T1/2 cells, a Shh-reporter cell line that produces alkaline phosphatase (AP) in response to Shh15. Upon coculture with COS-1 cells expressing Shh and Hhat, C3H10T1/2 cells produced AP (Fig. 2e). AP activity was reduced to baseline when cocultured cells were treated with 10 μM RU-SKI 43 (Fig. 2e). The effect of RU-SKI 43 was not due to inhibition of BMPs, which can cooperate with Shh16, as the BMP inhibitor Noggin had no effect on AP activity. These findings provide proof of concept that Hhat inhibitors block Shh signaling.
In this study, we describe the first example of a small-molecule that inhibits the activity of Hhat in vitro and blocks Hhat-mediated Shh palmitoylation in cells. RU-SKI 43 exhibits specificity for Hhat in cells: it did not affect fatty acylation mediated by other acyltransferases and its inhibitory effect was reversed by overexpression of Hhat (Supplementary Fig. 5). RU-SKI 43 decreased Gli1 activation in a Shh-driven cell-based reporter system, suggesting that Hhat inhibition accounts, at least in part, for the reduction of Shh signaling in cells, although we cannot exclude the possibility that off-target effects also occur. We were not able to rescue the inhibitory effect of RU-SKI 43 in Shh-transfected cells with SAG or Shh (C24II; Supplementary Fig. 8). Even in untreated Shh-expressing cells, neither SAG (Supplementary Fig. 8) nor overexpression of Smo17 activated Gli1 to levels higher than those achieved with Shh alone. Since these cells continuously produce Shh, it is likely that the Shh receptor Patched is saturated with ligand (Patched can bind both palmitoylated and nonpalmitoylated Shh18), and further stimulation with exogenous Shh (C24II) or SAG cannot occur. However, RU-SKI 43 had no effect on Shh signaling in SuFu−/− cells, nor did it affect the ability of either SAG or Shh (C24II) to activate Gli1 in Shh Light II cells (Fig. 2). These findings position Hhat inhibition as a novel method to turn off hedgehog signaling.
There is great interest in Shh signaling as a clinically relevant target in human malignancies3. Several compounds that block Shh signaling have been described, including Robotnikin, which binds to Shh, and inhibitors of Smo and Gli, which are downstream signaling components of the Shh pathway19,20. The Hhat inhibitor is unique, as it hits upstream, directly at palmitoylation of the Shh ligand. As a result, Hhat inhibition could offer a novel treatment modality for pancreatic and gastric cancers and a subset of sarcomas, cancers characterized by Shh overexpression and extremely poor prognoses3. The short half-life of RU-SKI 43 in vivo (t1/2= 17 min in mouse plasma after IV administration) limits our ability to perform toxicity studies in animals. However, in most “normal” adult cells, Shh expression and signaling is turned off. Thus, Hhat inhibition could be a valid option for treatment of cancers characterized by Shh overexpression, and offers several potential advantages. Hhat inhibition would block all downstream Shh signaling, including noncanonical, Smo-independent pathways21,22. Moreover, Hhat is intolerant to a wide variety of mutations9. It is thus possible that drug resistance might be less of a concern, although long-term exposure of cancer cells has not yet been evaluated. Furthermore, Hhat inhibition could be used in combination with Smo inhibition to attack both Shh-producing and Shh-responding cells. This could increase the efficiency of therapies, and potentially delay the development of drug resistance by patients treated with Smo inhibitors23. The work described in this study suggests that there is great potential for producing additional, optimized derivatives of Hhat inhibitors for future therapeutic development.
Online Methods
Reagents and antibodies
Fatty acyl CoA’s, Coenzyme A, CoA synthetase, octylglucoside, mouse monoclonal anti-Flag M2 and rabbit polyclonal anti-HA antibodies, FlagM2 agarose and 3xFlag peptide were purchased from Sigma-Aldrich (St. Louis, MO). [125I]NaI and SPA scintillation beads were obtained from Perkin Elmer (Waltham, MA). Goat polyclonal anti-Shh (N-19) antibody, rabbit polyclonal anti-Shh (H-160) antibody, rabbit polyclonal anti-H-Ras antibody, rat monoclonal anti-H-Ras antibody and protein A/G plus agarose were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Mouse monoclonal anti-Src antibody (clone GD11) was obtained from Millipore (Billerica, MA). Rabbit monoclonal anti-Shh antibody and rabbit monoclonal anti-Wnt3a antibody were purchased from Cell Signaling (Danvers, MA). Mouse monoclonal anti-HA antibody was purchased from Roche (Basel, Switzerland). Goat anti-mouse Alexa568 and goat anti-rabbit Alexa488 antibodies were obtained from Invitrogen (Carlsbad, CA). Anti-Fyn serum was generated as previously reported10. A C-terminally biotinylated peptide containing the first 10 amino acids of mature Shh (CGPGRGFGKR) was synthesized by the MSKCC Microchemistry Core Facility.
Chemical libraries
Compound screening libraries that include marketed drugs, natural products, and combinatorially elaborated active pharmacophores were purchased from AMRI (Albany, NY), Microsource Discovery Systems Incorporated (Gaylordsville, CT), ChemDiv Inc. (San Diego, CA), Cerep SA (Paris, France), Prestwick Inc. (Strasbourg, France), and Sigma-Aldrich (St. Louis, MO). The AMRI library contains 50,000 compounds designed for rapid re-synthesis. The MicroSource Spectrum Collection contains 2,000 biologically active compounds including a diverse set of pure natural products. The ChemDiv Library contains 21,000 structurally diverse compounds based on 125 combinatorial templates. Cerep Odyssey II collection contains 4,000 compounds derived from more than 350 scaffolds onto which biologically relevant pharmacophores were reacted with diverse sets of partners (more than 1,000 diversity points) to obtain final compounds with drug-like properties. The Prestwick library contains 880 compounds, of which over 85% are marketed drugs. Finally, LOPAC1280 is a collection of 1,280 compounds of high purity and well-documented pharmacological activity. A library containing 200 compounds representing spiroketals, polyketides and alkaloid/terpenoid-like polycyclics that was generated by Dr. Derek Tan was also included. All libraries were received as powders, dissolved in DMSO to 5mM, distributed in aliquots and stored at −28°C until use.
Mammalian expression plasmids, cell culture and transfection
Plasmids encoding Shh, HA-Hhat, HhatHAFlagHis, H-Ras, c-Src, and Fyn have been previously described5,24,25. N-terminal FLAG-tagged Porcupine cDNA was generated from a mouse Porcupine clone (a gift from Dr. Joseph Goldstein (UT Southwestern)). The pRL-TK and 8XGliBS-Firefly luciferase cDNAs, and SuFu −/− murine embryonic fibroblasts, were gifts from Dr. Kathryn Anderson (MSKCC). COS-1, 293FT, L-Wnt3a, NIH 3T3, Shh Light II and C3H10T1/2 cells were obtained from the ATCC. Cells in 100mm tissue culture dishes were transfected with 3μg (unless otherwise stated) of the indicated plasmid DNAs using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions.
Synthesis of 125I-iodo-palmitate, 125I-iodo-myristate, and 125I-iodo-pentadecanoate analogues
Radioiodination of iodo-palmitate, iodo-myristate (iodo-tridecanoate), and iodo-pentadecanoate with [125I]NaI and the synthesis of 125I-iodo-palmitoylCoA were carried out as previously described11,26. The final concentration of 125I-iodo-palmitoylCoA was determined from the absorbance at 260nm, based on the extinction coefficient for palmitoylCoA.
Labeling of cells with palmitate and myristate
COS-1 cells transfected with Shh and HA-Hhat, H-Ras, Fyn, or Src were starved for 1h in DMEM containing 2% dialyzed fetal calf serum, and then 15–30 μCi/ml 125I-iodo-palmitate or 30 μCi/ml 125I-iodo-tridecanoate 10,11 was added to the media; the cells were incubated for 4h at 37°C. When indicated, cells were also treated with RU-SKI compounds or DMSO starting the night before or at the beginning of the 5h serum starvation. Cells were washed twice with 2ml of ice cold STE (100mM NaCl, 10mM Tris, 1mM EDTA [pH 7.4]) and lysed in 500μl of RIPA buffer (150mM NaCl, 10mM Tris, pH 7.4, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 1mM EDTA). Cell lysates were clarified by ultracentrifugation at 100,000xg for 15min in a T100.2 rotor (Beckman, Fullerton, CA). SDS-PAGE and Western blot analysis were used to determine protein levels. Immunoprecipitations were performed by incubating clarified cell lysates with 5μl of the indicated antibody and 50μl of A/G plus agarose at 4°C for 16h. The beads were washed twice with 500μl RIPA buffer, and the final pellet was resuspended in 40μl 2X SDS-PAGE sample buffer. Samples were electrophoresed on a 12.5% SDS-PAGE gel. Radioactivity in the dried gel was analyzed on a FLA-700 phosphorimager (Fuji, Tokyo, Japan) and quantified using ImageGauge software. Experiments were performed in duplicate and repeated at least three times.
Labeling of cells expressing Wnt3a
Mouse L-Wnt3a cells were transfected with 6μg of N-terminally FLAG-tagged Porcupine or empty vector. 48h after transfection L Wnt-3a cells were serum starved for 3h in DMEM containing 2% dialyzed fetal calf serum, and either DMSO, 10μM RU-SKI-43, or 100nM Wnt C59. Cells were labeled for 6h with 40μCi of 125I-iodo-pentadecanoate – a palmitate analog that we have determined is suitable for monitoring Wnt3a palmitoleoylation. Cells were lysed and total cell lysates were collected as described for cell labeling with palmitate or myristate. Both Wnt3a and Flag-Porcupine were immunoprecipitated by incubating 60 μL A/G plus agarose with respectively 350μL and 100μL of cell lysate, along with 7 μL rabbit anti-Wnt3a antibody or 5 μL mouse anti-Flag M2 antibody for 16h at 4°C. The beads were washed twice with 500μl RIPA buffer, and the final pellet was resuspended in 60μl 2X SDS-PAGE sample buffer, containing 60mM DTT. Samples were run on a SDS-PAGE gel, and analyzed by Western blot and phosphorimaging analysis, as previously described for palmitate and myristate labeling of proteins. The experiment was performed in duplicate and repeated three times.
Purification of recombinant Shh and HhatHAFlagHis proteins
His-tagged Shh24–197 constructs were expressed in E.coli BL21 (DE3) codon plus (Novagen), and purified as described5. Protein concentrations were determined using the DC protein assay (BioRad). Purification of HhatHAFlagHis from 20×100mm plates of transfected 293FT cells was carried out following the protocol previously shown to result in a single band on a Silver stained gel5. The final concentration of the protein eluted from the Flag column was determined from the absorbance at 280nm using an extinction coefficient of 19,3045cm−1M−1.
In vitro palmitoylation assay
The assay was performed as previously described5. 10μl of HhatHAFlagHis in 20mM HEPES, pH 7.3, 175mM NaCl, 1% octylglucoside, 1% glycerol were incubated with 10μl recombinant Shh (0.2–0.4mg/ml in 20mM MES, pH 6.5, 1mM EDTA, 1mM DTT), and 30μl of reaction buffer (167mM MES, pH 6.5, 1.7mM DTT, 0.083% Triton X-100, 167μM 125I-iodo-palmitoylCoA). The reaction was incubated for 30min at room temperature, then stopped by the addition of 50μl 2X SDS-PAGE sample buffer. Analysis of RU-SKI 43 inhibition kinetics was performed as follows. Since this is a two-substrate system which does not strictly obey Michaelis-Menten kinetics, the levels of either ShhN or 125I-iodo-palmitoylCoA were titrated, with one substrate held at a super-saturating concentration (10 to 30-times Km). Purified Hhat was pre-treated for 20min with RU-SKI 43, then incubated with increasing concentrations of recombinant ShhN in the presence of 30μM 125I-iodo-palmitoylCoA, or with increasing concentrations of 125I-iodo-palmitoylCoA in the presence of 38μM ShhN. Samples were electrophoresed on a 12.5% SDS-PAGE gel. The gel was dried, exposed to a phosphorimager screen for 16h, and analyzed on a FLA-700 phosphorimager. Data were quantified using ImageGauge software. Experiments were performed in duplicate and repeated at least two times.
High-throughput Hhat activity assay
5μl of 10mM MES, pH 6.5 buffer were dispensed in each well of a 384-well white/clear-bottom plate (Greiner Bio-One, Kremsmuenster, Austria) using a Thermo Multi-Drop Combi (Thermo Scientific, Hudson, NH) dispenser and plates were spun for 30sec at 1000×g. Compounds (12.5μM final concentration) were dispensed using a Janus “Varispan” automated syringe pipette (PerkinElmer, Waltham, MA). Next, 3μl of P100 membranes from HA-Hhat transfected 293FT cells were dispensed with the Thermo Multi-Drop Combi dispenser, the plates were centrifuged for 30sec at 1000×g, and then incubated for 20min at room temperature. The reaction was started by the addition of 12μl reaction buffer (167mM MES, pH 6.5, 2mM DTT, 0.083% Triton X-100, 8.3μM 125I-iodo-palmitoylCoA, 5.21μM Shh biotinylated peptide) by a Matrix WellMate dispenser (Thermo Scientific, Hudson, NH). Following a 1h incubation at room temperature, the reaction was stopped by the addition of 70μl SPA beads solution (7.14mg/ml in RIPA buffer), using the Matrix WellMate dispenser. The signal was detected on a Microbeta Trilux reader (PerkinElmer, Waltham, MA). Each plate included high control (DMSO only) and low control (0.125% TFA final concentration) rows.
Cell viability assay
5000 AsPC-1 human pancreatic cancer cells were plated in each well of a 384-well black/clear-bottom tissue culture plate, using Thermo Multi-Drop Combi dispenser. The plates were incubated at 37°C for 24h before compounds were dispensed using a Janus “Varispan” automated syringe pipette at 50μM final concentration. High control (DMSO only) and low control (cell media only) rows were included in each plate. After 48h incubation, Alamar Blue (Invitrogen, Carlsbad, CA) was added to each well in 1:100 ratio. 4h later, cell viability was assessed by measuring fluorescence on a Perkin-Elmer EnVision plate reader (PerkinElmer, Waltham, MA).
High-throughput screening
The Hhat activity assay described above was used to test compounds from the libraries at the Rockefeller University High Throughput Screening Resource Center (New York, NY). For the primary screen, compounds were tested as single points at 12.5μM concentration. Percent inhibition for each experimental point was determined by the formula: . As an assessment of quality27, Z′ values for each plate were calculated using the formula: . Compounds were organized based on their percent inhibition, and those with >60% inhibition, molecular weight <425kDa, no Lipinski violations, and that were on plates with Z′>0.5 were selected for secondary screening. Structures selected from this primary screen were re-tested in duplicate using Hhat activity and cell viability assays. 95 compounds yielded >60% inhibition in both assays and were selected as hits. 11-point 2-fold serial dilutions in DMSO were prepared to yield final concentrations ranging from 12.5μM to 0μM. The Hhat activity assay was performed in duplicate, and the data were used to calculate IC50 values for each compound. The compounds were organized by structural similarity and those with the lowest IC50 values and most promising structure for further drug development were chosen for further analysis.
Compound analytics
RU-SKI 39,41,43,50; C-1, C-2: These compounds were purchased from AMRI as powders with purities of greater than 90%, as detected by LCMS (220 nm) as follows: (1) 95%, (2) 90%, (3) 97%, (4)100%, (5) 97%, (6) 94%. Compound RU-SKI 43 was re-purchased from AMRI in 100 mg quantity and analyzed prior to use in cell-based experiments. Reverse-phase HPLC-ESI-MS analysis yielded a single peak (A220), at retention time of 8.5 min. containing 97.6% of the total area under the spectra. Mass spectrometry yielded one total ion current peak at 8.6 minutes, containing 83% of the area under all non-solvent ion counts with one positive ion of 387.2 amu (predicted 387.6 amu, M+H). Analysis of the same sample in negative ion detection mode yielded a negative ion of 385.2 amu (M−H) (385.5 amu predicted) at retention time 8.5 minutes (Supplementary Fig. 13).
Shh peptide CGPGRGFGKR (PEG-BIOTIN): Reverse-Phase HPLC yielded a single peak (Absorbance 215nm) at retention time of 23.8 min containing 93.97% of the total area under the spectrum. Matrix-Assisted-Laser-Desorption/Ionization (MALDI) MS of the HPLC purified peptide yielded a positive ion at experimental mass, m/z: 1462.571 Da (theoretical average, protonated mass, MH+: 1463.09 Da) (Supplementary Figs. 14 and 15).
Immunofluorescence
COS-1 cells were transfected with plasmids encoding Shh and HA-Hhat. 24h after transfection, the cells were seeded onto glass coverslips at low density. After 24h, the cells were washed with phosphate-buffered saline (PBS) and fixed with 4% (v/v) paraformaldehyde in PBS for 20min at room temperature. The cells were washed twice with PBS and permeabilized with 0.2% Triton X-100 in PBS for 7 min at room temperature, followed by two washes with PBS. Cells were then incubated for 30 min in blocking buffer (3% bovine serum albumin in PBS) and incubated for 1h with primary antibody (rabbit anti-Shh polyclonal antibody 1:100; mouse anti-HA monoclonal antibody 1:200) diluted in blocking buffer. Cells were washed five times with PBS, followed by a 45-min incubation with the secondary antibody (anti-mouse Alexa568 antibody 1:1000; anti-rabbit Alexa488 antibody) and Hoechst dye (1:2000), diluted in blocking buffer. The cells were washed five times in PBS, and coverslips were mounted on slides using ProLong Gold mounting solution (Invitrogen, Carlsbad, CA). Images were acquired on a Zeiss LSM 510 microscope (Zeiss, Germany) using a 63X oil objective. 5–10 images were taken per slide, and at least 2 slides were prepared per condition. The experiment was performed three times.
Luciferase assays
NIH 3T3 cells were transfected with pRL-TK, 8XGliBS-FL (or mutant 8XGliBS-FL), and Shh plasmids as previously described14. 24h after transfection, the cells were seeded in 6-well plates at a density of 1×106 cells/well, and allowed to reach confluence. Cells were treated with DMSO, 10μM LDE225 (Selleckchem, Houston, TX), 10μM RU-SKI 43, or 10μM C-2 for 24 h. Luciferase activity was measured in cell lysates using the Dual-Luciferase®Reporter Assay System (Promega, Madison, WI). FL and RL levels were detected on a Veritas™ Microplate Luminometer (Promega, Madison, WI). The FL/RL ratio was calculated for each sample, and was normalized to the ratio of the DMSO-treated cells.
Shh Light II cells were grown until confluent, transferred to media containing 0.5% serum and treated for 24 h with DMSO or 10μM RU-SKI 43, in the presence or absence of 100nM SAG or 1μg/mL Shh (C24II). Luciferase activity was measured as above. The FL/RL ratio was determined for each sample, and was normalized to the ratio for cells treated with either DMSO or RU-SKI 43 alone. The experiment was performed at least twice.
Sufu −/− cells were transfected with wild-type or mutant 8XGliBS and pRL-TK plasmids. 24h after transfection, the cells were seeded, grown to confluence, and then treated with DMSO, 10μM LDE225 or 10μM RU-SKI 43 for 24h. Luciferase activity was measured as above, and the experiment was performed at least twice.
C3H10T1/2 reporter assay
C3H10T1/2 and COS-1 cells, transfected with HA-Hhat and Shh encoding plasmids, were plated either alone or together (4:1 ratio) in 60mm tissue culture plates. The cells were grown for 24h at 37°C, after which the media was changed to media containing DMSO or 10μM RU-SKI 43. The cells were then incubated for 72h (DMSO/drug was replenished 48h after the initial addition). The SensoLyte®FDP Alkaline Phosphatase Assay Kit (AnaSpec, Fremont, CA) was used to measure alkaline phosphatase levels in the cell lysates by monitoring fluorescence for 30min in 5-minute intervals on a Tecan Infinite F500 plate reader (Männdorf, Switzerland). The experiment was performed two times.
Supplementary Material
Acknowledgments
We thank Raisa Louft-Nisenbaum, Dr. Kathryn Anderson, Dr. Derek Tan, Dr. Emilie Legué and Dr. Alexandra Joyner. This work was supported by NIH grants GM57966 and CA158474, Cycle for Survival, the Geoffrey Beene Cancer Research Foundation, the Sarcoma Foundation of America, Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research, and the Experimental Therapeutics Center of Memorial Sloan-Kettering Cancer Center. The Organic Synthesis Core is partially supported by NCI P30 CA008748-43.
Footnotes
Contributions
E.P. performed all experiments, except for Wnt acylation performed by J.R-E. J.F.G. supervised the high-throughput screening performed by E.P., and hits were analyzed by E.P., O.O., J.F.G and M.D.R. The research plan was designed by E.P., J.F.G and M.D.R. E.P. and M.D.R. wrote the manuscript with input from all authors.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Ingham PW, McMahon AP. Genes Dev. 2001;15:3059–87. doi: 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- 2.Fuccillo M, Joyner AL, Fishell G. Nat Rev Neurosci. 2006;7:772–83. doi: 10.1038/nrn1990. [DOI] [PubMed] [Google Scholar]
- 3.Barakat MT, Humke EW, Scott MP. Trends Mol Med. 2010;16:337–48. doi: 10.1016/j.molmed.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mann RK, Beachy PA. Annu Rev Biochem. 2004;73:891–923. doi: 10.1146/annurev.biochem.73.011303.073933. [DOI] [PubMed] [Google Scholar]
- 5.Buglino JA, Resh MD. J Biol Chem. 2008;283:22076–88. doi: 10.1074/jbc.M803901200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goetz JA, Singh S, Suber LM, Kull FJ, Robbins DJ. J Biol Chem. 2006;281:4087–93. doi: 10.1074/jbc.M511427200. [DOI] [PubMed] [Google Scholar]
- 7.Chen MH, Li YJ, Kawakami T, Xu SM, Chuang PT. Genes Dev. 2004;18:641–59. doi: 10.1101/gad.1185804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hofmann K. Trends Biochem Sci. 2000;25:111–2. doi: 10.1016/s0968-0004(99)01539-x. [DOI] [PubMed] [Google Scholar]
- 9.Buglino JA, Resh MD. PLoS One. 2010;5:e11195. doi: 10.1371/journal.pone.0011195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liang X, et al. J Biol Chem. 2001;276:30987–94. doi: 10.1074/jbc.M104018200. [DOI] [PubMed] [Google Scholar]
- 11.Peseckis SM, Deichaite I, Resh MD. J Biol Chem. 1993;268:5107–14. [PubMed] [Google Scholar]
- 12.Janda CY, Waghray D, Levin AM, Thomas C, Garcia KC. Science. 2012;337:59–64. doi: 10.1126/science.1222879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pan S, et al. ACS Medicinal Chemistry Letters. 2010;1:130–134. doi: 10.1021/ml1000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taipale J, et al. Nature. 2000;406:1005–9. doi: 10.1038/35023008. [DOI] [PubMed] [Google Scholar]
- 15.Wu X, Walker J, Zhang J, Ding S, Schultz PG. Chem Biol. 2004;11:1229–38. doi: 10.1016/j.chembiol.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 16.Yamaguchi A, Komori T, Suda T. Endocr Rev. 2000;21:393–411. doi: 10.1210/edrv.21.4.0403. [DOI] [PubMed] [Google Scholar]
- 17.Rohatgi R, Milenkovic L, Corcoran RB, Scott MP. Proc Natl Acad Sci U S A. 2009;106:3196–201. doi: 10.1073/pnas.0813373106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taylor FR, et al. Biochemistry. 2001;40:4359–71. doi: 10.1021/bi002487u. [DOI] [PubMed] [Google Scholar]
- 19.Stanton BZ, et al. Nat Chem Biol. 2009;5:154–6. doi: 10.1038/nchembio.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peukert S, Miller-Moslin K. ChemMedChem. 2010;5:500–12. doi: 10.1002/cmdc.201000011. [DOI] [PubMed] [Google Scholar]
- 21.Testaz S, et al. Proc Natl Acad Sci U S A. 2001;98:12521–6. doi: 10.1073/pnas.221108698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chinchilla P, Xiao L, Kazanietz MG, Riobo NA. Cell Cycle. 2010;9:570–79. doi: 10.4161/cc.9.3.10591. [DOI] [PubMed] [Google Scholar]
- 23.Metcalfe C, de Sauvage FJ. Cancer Res. 2011;71:5057–61. doi: 10.1158/0008-5472.CAN-11-0923. [DOI] [PubMed] [Google Scholar]
- 24.Donepudi M, Resh MD. Cell Signal. 2008;20:1359–67. doi: 10.1016/j.cellsig.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wolven A, Okamura H, Rosenblatt Y, Resh MD. Mol Biol Cell. 1997;8:1159–73. doi: 10.1091/mbc.8.6.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berthiaume L, Resh MD. J Biol Chem. 1995;270:22399–405. doi: 10.1074/jbc.270.38.22399. [DOI] [PubMed] [Google Scholar]
- 27.Zhang JH, Chung TD, Oldenburg KR. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.