

Abstract
The transthyretin amyloidoses are diseases of protein misfolding characterized by the extracellular deposition of fibrils and other aggregates of the homotetrameric protein transthyretin (TTR) in peripheral nerves, heart and other tissues. Age is the major risk factor for the development of these diseases. We hypothesized that an age-associated increase in protein oxidation could be involved in the onset of the senile forms of the TTR amyloidoses. To test this hypothesis we have produced and characterized relevant age-related oxidative modifications of wild type (WT) and the Val122Ile (V122I) TTR variant, both involved in cardiac TTR deposition in the elderly. Our studies show that methionine/cysteine oxidized TTR and carbonylated TTR either from WT or the V122I variant, are thermodynamically less stable than their non-oxidized counterparts. Moreover, carbonylated WT and carbonylated V122I TTR have a greater propensity to form aggregates and fibrils than WT and V122I TTR, respectively, at physiologically attainable pH. It is well known that TTR tetramer dissociation, the limiting step for aggregation and amyloid fibril formation, can be prevented by small molecules that bind the TTR tetramer interface. Here, we report that carbonylated WT TTR is less amenable to resveratrol-mediated tetramer stabilization than WT TTR. All the oxidized forms of TTR tested are cytotoxic to a human cardiomyocyte cell line known to be a target for cardiac-specific TTR variants. Overall these studies demonstrate that age-related oxidative modifications of TTR can contribute to the onset of the senile forms of the TTR amyloidoses.
Keywords: Transthyretin, amyloid, oxidation, carbonylation, protein stability
The amyloidoses are a subset of protein misfolding diseases characterized by the systemic extracellular deposition of β-sheet rich fibrils and amorphous aggregates in tissues which results in compromised organ function and death (1). Transthyretin (TTR) is one of the ~30 human amyloidogenic proteins thus far identified (2). TTR is a 55kDa homotetramer synthesized mainly in the liver and the choroid plexus of the brain, which circulates in the plasma and the cerebrospinal fluid (CSF). The known functions of TTR are to transport retinol (through retinol-binding protein) and thyroxine (T4) in plasma, and T4 in the CSF. TTR can interact with a large variety of small molecules including peptides such as amyloid β peptide (3), suggesting that its function might also be that of a general detoxifier of unwanted molecules and metabolic by-products (4).
Wild-type (WT) TTR undergoes age-dependent deposition in the heart, gut, carpal tunnel, and lungs, producing the syndrome known as senile systemic amyloidosis (SSA). The average age of onset of SSA is 65–70 years, depending on the population studied (5;6). Autopsy data indicate that 10–25% of people older than 80 are affected by WT TTR-derived amyloidosis and it appears to be the cause of death of 50% of those older than 90 years (5). There are more than 100 amyloidogenic TTR variants that have been associated with various protein deposition syndromes. Val122IIe (V122I) TTR is the most common amyloidogenic mutation worldwide. It produces the syndrome known as familial amyloid cardiomyopathy (FAC), characterized by age-dependent cardiac amyloid deposition resulting in arrhythmias, congestive heart failure and death. The age of onset of FAC due to the V122I TTR mutant is about a decade earlier than that of WT TTR amyloidosis. Three to four per cent of African-Americans are heterozygous for the V122I TTR allele, which produces clinical disease in almost 100% of the carriers (7–9).
Biophysical studies have shown that the loss of TTR quaternary structure, i.e. the disassembly of the native tetramer in to its constituent monomers, is required and is the rate-limiting step for aggregation and subsequent fibril formation in vitro, and presumably in vivo (10). The released monomer misfolds and forms oligomers, soluble aggregates, insoluble aggregates and amyloid fibrils in a downhill polymerization process (11). Many of the amyloidogenic TTR variants studied thus far have lower thermodynamic and/or kinetic stabilities relative to WT TTR, which make the proteins more prone to tetramer disassembly, misfolding and aggregation (12;13).
Although it is well documented that age is the major risk factor for the development of TTR-associated diseases, it is not known which aspects of the aging process contribute to disease onset. Given the well established general increase in protein oxidation with aging (14), we sought to investigate whether oxidative modifications in TTR might change its stability increasing its tendency towards aggregation and fibril formation.
There are several types of oxidative modifications that occur in proteins. Sulfur-containing amino acids such as methionine (Met) and cysteine (Cys) are some of the most susceptible to oxidation (15). The addition of one oxygen atom into the sulfur of Met generates methionine sulfoxide (MetO). Cys oxidation results in the formation of disulfide bonds and Cys sulfenic, sulfinic and sulfonic acids by addition of one, two or three oxygen atoms, respectively (16;17). Both, Met and Cys oxidation processes, are reversible and have biological as well as pathological significance (18;19). Another type of age-related oxidative modification is protein carbonylation, which results in the formation of reactive aldehydes and ketones by a variety of mechanisms (14). Lys, Arg, Pro and Thr residues for example, are susceptible to metal-catalyzed carbonylation of their side chains (20). Protein carbonylation is common, irreversible, and it is considered a universal indicator of oxidative stress or damage (21). It has been established that the content of carbonylated proteins increases dramatically in the last third of life (22) and it is highly correlated with age-related diseases such as Alzheimer’s (23;24) or Parkinson’s Disease (25). Carbonylated TTR has been identified in human plasma of healthy individuals (26;27) as well as in CSF (28). Furthermore, an increase in protein carbonyls has been reported in biopsies of patients with TTR deposits compared to age-matched controls (29), although it is not clear from this report whether TTR itself is carbonylated.
Previously, we showed that H2O2-induced oxidation of WT TTR and the amyloidogenic variant Val30Met (V30M) TTR results in proteins with lower propensities to form aggregates and fibrils at pH 4.4 as measured by turbidity (30). In the present study we have produced and characterized relevant age-related oxidized TTR isoforms. We demonstrate that treatment of WT and V122I TTR with H2O2 results in the transformation of all Met and Cys residues into MetO and Cys sulfonic acid (Cys-SO3H). In addition, we have prepared carbonylated WT and V122I TTR. Aggregation and fibril formation studies at several pH confirm our previous work with respect to H2O2 oxidized WT TTR at pH 4.4. More interestingly, the data show that the oxidized TTR isoforms, particularly carbonylated TTRs, form more insoluble aggregates at pH closer to physiological conditions than their non-oxidized counterparts. Moreover, urea denaturation/renaturation studies show that the oxidative modifications render the TTR tetramers thermodynamically less stable than those of the non-oxidized isoforms. Transgenic mouse models and human biopsies of asymptomatic TTR mutant carriers have shown evidence of tissue damage and cell death well before there is detectable TTR deposition (31). Here, we demonstrate that the oxidized TTR isoforms are cytotoxic to human cardiomyocytes in a tissue culture model that reflects the specificity of cardiac TTR variants (32). On the whole, the data reveal that age-related TTR oxidative modifications can play a role in the onset of the senile forms of the TTR amyloidoses.
EXPERIMENTAL PROCEDURES
Recombinant protein preparation
TTR variants were prepared and purified in an Escherichia coli expression system as described elsewhere (33). The last step of purification was gel filtration chromatography on a Superdex 75 column (GE healthcare) in sodium phosphate buffer (GF buffer: 10 mM sodium phosphate, pH 7.6, 100 mM KCl, 1 mM EDTA) or Hank’s Balanced Salt Solution (HBSS, Mediatech, Manassas) without phenol red. Additional recombinant TTR variants, named rTTRs were produced using published vector and protocols from Dr Lawreen Connors’ laboratory to produce His-tagged TTR (34). During purification the His-tag is cleaved by dipeptidyl aminopeptidase I (Qiagen, USA) yielding TTR lacking Met in position-1. Site directed mutagenesis was used to create C10A TTR, M13I TTR, C10A rTTR, and M13I rTTR. The plasmids obtained were sequenced to confirm that we had introduced the desired point mutations. The identity of the proteins was determined by liquid chromatography-mass spectrometry (LC-MS). The molecular weight of each variant was: WT TTR, 13892 Da; V122I TTR, 13907 Da; C10A TTR, 13860 Da; M13I TTR, 13874 Da; T119M TTR, 13923 Da; F87M/L110M TTR, 13894 Da; rTTR, 13760 Da; C10A rTTR, 13728 Da; and M13I rTTR, 13742 Da. All TTR proteins were stored as aliquots at −80°C at concentrations lower than 2.5 mg/ml. Under these conditions we observed no changes in oxidation or aggregation of the proteins over time.
H2O2 oxidation
TTR variants (1.5–2.5 mg/ml in GF buffer) were treated with 3% hydrogen peroxide (H2O2, H1009, Sigma) at room temperature for 30 min. The oncentration of the H2O2 stock solution was regularly measured by spectrophotometry (ε240 = 43.6 M−1 cm−1 (35)). The oxidized TTRs were then purified by gel filtration on a Superdex 75 column to remove the excess oxidant and any TTR aggregates generated during the oxidative reaction. LC-MS analysis consistently revealed a single species with molecular weight corresponding to the complete transformation of all Met residues into Met sulfoxide (MetO) and the oxidation of the single Cys residue in position 10 to Cys sulfonic acid (Cys-SO3H) (see results).
Iron-Catalyzed Carbonylation
TTR carbonylation was performed as in (36). Briefly, WT or V122I TTR (10 mg/ml) were dialyzed against 25 mM Hepes pH 7.4, 100 mM KCl, 10 mM MgCl2; then incubated in freshly prepared oxidative reaction buffer (25 mM sodium ascorbate, 100 μM FeCl3, solubilized in 25 mM Hepes pH 7.4, 100 mM KCl, 10 mM MgCl2) at 37 °C for 5 hours or overnight. The proteins were re-purified by gel filtration to remove excess reagents and TTR aggregates. The presence of the newly generated carbonyl groups was confirmed using the Oxyblot protein oxidation detection kit (Millipore, S7150). Briefly, the proteins were derivatized with dinitrophenylhydrazine, separated by SDS-PAGE and transferred onto a PVDF membrane. The derivatized proteins were detected using a rabbit anti-diphenylhydrazone antibody (1:150) or by rabbit anti-human TTR antibody (A0002 Dako) followed by alkaline phosphatase-labelled goat anti-rabbit IgG (A-8025 Sigma-Aldrich).
Far and Near UV-Circular Dichroism (CD)
The far and near UV-CD spectra of native WT and V122I TTR and their oxidized isoforms (8 μM in GF buffer) were acquired at 25°C using an AVIV 202SF CD spectrometer (AVIV, Lakewood, NJ) equipped with a Peltier thermostated cell holder. Far UV-CD spectra were recorded from 190 to 250 nm using a 2 mm path length Suprasil quartz cell with 1 nm bandwidth; Supplemental Figure S2 shows the spectra from 205 to 250 nm to omit the noise due to the buffer absorbance at lower wavelengths. Near UV-CD spectra were recorded from 250 to 320 nm in a 1 cm path length quartz cell with 1 nm bandwidth. The wavelength step was set at 0.5 nm with time constants of 100 ms for both far and near UV-CD. Blank spectra corresponding to GF buffer was subtracted from the samples and the data was transformed to mean residue ellipticity, θMRW (deg cm2 dmol−1). Each spectrum represents the average of four scans. No smoothing algorithm was applied to any of the spectra to conserve detail.
Acid-mediated TTR aggregation and fibril formation
pH dependent fibril formation studies were performed as described previously (37). Briefly, TTR solutions in GF buffer (8 μM, ~ 0.4 mg/ml) were diluted 1:1 with acidic buffers (200 mM sodium citrate, acetate or phosphate with 100 mM KCl and 1mM EDTA) to reach various pH (4.04–5.88). The mixtures were incubated at 37 °C for the desired amount of time (up to 21 days) in cluster tubes (Genessee Scientific, San Diego, CA). At the end of the incubation time, the TTR solutions were vortexed for 10 seconds and transferred into ½ area 96-well UV-transparent plates (Corning) in triplicate (50 μL/well). The turbidity of the solutions at 400nm was recorded using a UV-spectrophotometer (SpectramaxPlus, Molecular Devices, USA). Blanks consisting of the corresponding buffer solutions were subtracted from each sample. All experiments were repeated at least twice in triplicate.
Measurement of soluble and insoluble TTR amounts
Four hundred microliters of aggregated TTR solutions (see above) were transferred into fresh Eppendorf tubes and the samples were centrifuged at 20,000 × g for 30 min at 4 °C. The supernatants were carefully transferred into fresh eppendorf tubes and the protein concentration in these solutions was measured using ½ area 96-well UV-transparent plates in triplicate (50 μL/well). To determine the amount of aggregated TTR (insoluble) 200 μL of 8 M guanidinium chloride (GndCl) solution was added to the protein precipitates. The samples were then vortexed briefly and left at r.t. for 5 min to allow the disassembly of TTR aggregates to take place. The TTR concentration was then measured by UV-spectrophotometry in ½ area 96-well UV-transparent plates in triplicate, using 8 M GndCl as blank. The percentages of protein in the supernatant and in the pellet were calculated from the total initial protein content at time 0.
Inhibition of TTR aggregation and fibril formation by resveratrol
TTR solutions (8 μM, 250 μL) in GF buffer were added to 1 μL of resveratrol (4 mM stock in DMSO, 2 equivalents resveratrol/TTR) or to 1 μL DMSO (vehicle control). The samples were vortexed and incubated at r.t. for 30 min to allow resveratrol binding. Two hundred and fifty microliters of acetate buffer (200 mM Na-Acetate pH 4.23, 100 mM KCl, 1 mM EDTA) was added to reach a final pH of 4.4. The mixtures were incubated at 37 °C for 3 days. The samples were centrifuged at 20,000 × g, 30 min at 4 °C and the aggregated TTR was measured as above using 8 M GndCl. The percentage of insoluble TTR in the samples containing resveratrol was calculated relative to the samples containing TTR alone (with DMSO).
Thioflavin T (ThT) binding
TTR aggregation and fibril formation was performed as above for 1 or 3 days at a final pH of 4.4. The solutions were then diluted to 1 μM (tetramer equivalent) in 200 mM Tris, 150 mM NaCl, pH 8.0. Two microliters of ThT stock solution (2 mM in 200 mM Tris, 150 mM NaCl, pH 8.0) were added to 400 μL of the diluted TTR aggregates. The samples were vortexed briefly, dispensed into black 96-well plates (Corning) in triplicate (100 μL/well), and ThT fluorescence was recorded with Exc/Em at 440/482 nm and 5 nm bandwidth using a multiwell spectrofluorimeter (Tecan Safire 2, Austria).
Transmission Electron Microscopy (TEM)
Carbon coated copper grids (400 mesh, Electron Microscopy Sciences, Hatfield PA) were glow-discharged and inverted on 5μL aliquots of TTR subjected to acid-mediated aggregation conditions (4 μM final protein concentration, pH 4.4, 1 day at 37 °C) for 2 minutes. Excess sample was removed and the grids immediately placed briefly on a droplet of 0.1% ammonium acetate followed by 2% uranyl acetate solution for 2 minutes. Excess stain was removed and the grids allowed to thoroughly dry. Grids were then examined on a Philips CM100 electron microscope (FEI, Hillsbrough OR) at 80kV and images collected using a Megaview III CCD camera (Olympus Soft Imaging Solutions, Lakewood CO).
Urea Stability
TTR denaturation curves were prepared by incubating TTR at 0.1 mg/ml (~2 μM) in various concentrations of urea (range 0–8 M) in sodium phosphate buffer (50 mM sodium phosphate, 100 mM KCl, 1 mM EDTA, pH 7.6) for 4 days at r.t. For the renaturation curves, TTR was first denatured in 6.5 M GndCl overnight at r.t., then buffer exchanged into 8M urea and concentrated to 1 mg/ml. Dilutions were prepared to yield a final TTR concentration of 0.1 mg/ml at a wide range of urea concentrations (1–8 M) and were incubated for 24 h at r.t. before measurement of the tertiary and quaternary structures.
Tertiary structure determination by tryptophan fluorescence
The stability of the TTR tertiary structure was determined by measuring the intrinsic tryptophan fluorescence of the protein in the presence of urea, as described in (37). At the time of measurement the samples were vortexed and transferred into ½ area black 96-well plates (Corning) in triplicate (50 μL/well) and the tryptophan fluorescence was measured with Exc 295 nm and Emission at 335 and 355 nm, with 10 nm bandwidth. The percentage of folded protein at each urea concentration was calculated from the fluorescent ratio values (355/335nm) with respect to TTR samples that had not been denatured (100% folded) and those that were completely unfolded (0% folded). The data were fitted to sigmoidal curves with variable slope using Graph Prism software (GraphPrism, San Diego CA) and the concentration of urea at which 50% of TTR was folded (Cm) was calculated. For kinetic studies TTRs were incubated in 6 M urea; a concentration in the post transition region for tertiary structural changes. Tryptophan fluorescence was measured over time as above. To assess tertiary structural changes of the oxidized proteins with respect to their non-oxidized counterparts tryptophan emission spectra from non denatured TTRs (4 μM in GF buffer) were also acquired using Exc 295 nm and a emission wavelength range from 320 to 400 nm with 1 nm steps.
Quaternary structure determination by resveratrol binding fluorescence
The small molecule resveratrol binds in the T4 binding pocket of tetrameric TTR resulting in an increase in the fluorescence quantum yield. The intensity of resveratrol fluorescence is proportional to the concentration of tetrameric TTR in solution (37). To determine the amount of tetrameric TTR present in each of the urea solutions, 4 μL of resveratrol (1 mM in DMSO) were added to 200 μL of the TTR samples in urea. The resveratrol/TTR mixtures were vortexed briefly and incubated for 30 min at r.t. Aliquots of 50 μL were then transferred into ½ area black 96-well plates in triplicate and the resveratrol fluorescence was recorded (Exc 320/Em 394 nm, with 10 nm bandwidth). The proportion of tetrameric protein at each urea concentration was calculated using the average fluorescence values of TTR at very low urea concentrations (0–2 M) (100% tetramer) and the average fluorescent values of completely denatured TTR (0% tetramer). The data were fitted to sigmoidal curves with variable slope using Graph Prism software, and the concentration of urea at which 50% of TTR is tetrameric (Cm) was calculated.
Cell culture
The human cardiomyocyte cell line AC16 (38) was routinely cultured in DMEM/F12 (Mediatech, Manassas VA) supplemented with 10% FBS, 1 mM Hepes, 2 mM L- glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin and incubated at 37 °C in a 5% CO2 environment.
Cytotoxicity assessment
Subconfluent AC16 cells were seeded into black wall clear bottom 96-well plates (Corning) in Opti-MEM (Invitrogen, San Diego CA) supplemented with 5% FBS, 1 mM Hepes, 2 mM L-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin and incubated overnight. The next day, the culture medium was removed and the cells were exposed to different concentrations of TTR (100 μL) in 1:1 Opti-MEM:HBSS supplemented with 0.4 mg/ml BSA, 1 mM Hepes, 2 mM L-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin and 4.5 mM CaCl2. Cytotoxicity was determined by resazurin assay after 24 hours of treatment as previously described (32). Briefly, 10 μL/well of resazurin solution (500 μM in PBS) was added and the cells were incubated for 2 ½ h at 37 °C. Fluorescence resulting from the reduction of resazurin to resorufin by metabolically active cells was measured using Exc/Em 530/590 nm with a 10 nm bandwidth. Cell viability was calculated as the percentage of fluorescence of treated cells with respect to the control cells (vehicle treated cells), after subtraction of the blank (wells with no cells). All experiments were repeated at least twice in triplicate. Averages and standard error of the mean are presented.
RESULTS
TTR methionine and cysteine residues are oxidized by H2O2
WT TTR and V122I TTR were treated with 3% H2O2 as detailed in the Experimental Procedures section. LC-MS analysis showed 100% conversion to a single product with a molecular weight 80 Da above that of the non-oxidized proteins, indicating that each TTR polypeptide chain had incorporated 5 oxygen atoms (Table 1).
Table 1.
Mass spectrometry analysis of several TTR variants subjected to H2O2 oxidation. Shown is the number of Met and Cys in the sequences, the increase in MW observed after H2O2 treatment with respect to native (non-oxidized) TTR and the number oxygen atoms equivalent to the observed MW change. The TTR variants differ only by the number of Met and Cys in the sequence.
| TTR variant | No. of Met | No. of Cys | increase in MW | Oxygen atoms |
|---|---|---|---|---|
| WT | 2 | 1 | 80 | 5 |
| V122I | 2 | 1 | 80 | 5 |
| C10A | 2 | 0 | 32 | 2 |
| M13I | 1 | 1 | 64 | 4 |
| T119M | 3 | 1 | 96 | 6 |
| F87M/L110M | 4 | 1 | 112 | 7 |
| rTTRa | 1 | 1 | 64 | 4 |
| M13I rTTR | 0 | 1 | 48 | 3 |
| C10A rTTR | 1 | 0 | 16 | 1 |
rTTR is recombinant protein lacking Met at position −1 (34)
The sulfur atoms of Met and Cys are the most susceptible to oxidation by ROS (39). Met oxidation can generate two oxidative products, MetO or Met sulfone, by addition of one or two oxygen atoms, respectively (16). Cys oxidation can result in Cys sulfenic, sulfinic or sulfonic acid derivatives by incorporation of one, two or three oxygen atoms, respectively (16;17).
LC-tandem mass spectrometry (MS/MS) analysis of native WT TTR showed oxidation at Met13 due to sample manipulation before analysis (alkylation and trypsinization); therefore, we could not use this technique to determine where oxidation took place in the H2O2-treated TTRs (not shown). Thus, we prepared and purified several recombinant TTR variants containing different numbers of Met or Cys residues in the sequence. These variants were treated with H2O2 and analyzed by LC-MS as above (Table 1). H2O2-treated WT TTR incorporated 5 oxygen atoms, whereas the mutant C10A TTR incorporated 2 oxygen atoms. These results suggest that upon H2O2 treatment Cys is oxidized to Cys-SO3H. Substitution of a Met residue in the sequence (M13I TTR) resulted in the incorporation of 4 oxygen atoms (1 oxygen atom less than WT TTR), whereas addition of one and two Met residues (T119M TTR and F87M/L110M TTR) resulted in the incorporation of 6 and 7 oxygen atoms, respectively. These results indicate that Met is oxidized to MetO (i.e. 1 oxygen atom/Met).
To further confirm our results, we used a plasmid which after expression and purification produces TTR lacking methionine in position −1, named rTTR (34). We used site directed mutagenesis to prepare variants of this rTTR and study their susceptibility to H2O2 oxidation as above (Table 1). Treatment of rTTR and M13I rTTR with H2O2 resulted in the incorporation of 4 and 3 oxygen atoms, respectively, whereas C10A rTTR incorporated only 1 oxygen atom. Taken together these results indicate that under our experimental conditions, H2O2 treatment of TTR results in the oxidation of all Met to MetO and Cys to Cys-SO3H. These Met/Cys oxidized TTRs are referred in the text as oxi-TTRs.
TTR carbonylation in vitro results in multiple oxidative products
WT and V122I TTR oxidative carbonylation was performed as described in (36). The presence of TTR carbonyls was confirmed by Oxyblot technology (Supplemental Figure S1). Carbonylated WT and V122I TTR were further analyzed by LC-MS. Thirty four per cent of WT and 18% of V122I TTR were carbonylated after 5h reaction time, whereas 100% of the proteins were carbonylated after overnight reaction. In all cases a large number of oxidative products was detected consistent with the fact that there are 32 amino acid residues in the TTR sequence that are susceptible to direct oxidative carbonylation (36). The carbonylated TTRs were named car-TTRs. For the aggregation and stability studies described below, we used the 5h carbonylation products of WT and V122I TTRs as mixtures without trying to purify any specific carbonylation species because, when TTR carbonylation occurs in vivo, it is very likely that complex mixtures -and not a single carbonylation product, are generated.
The secondary and tertiary structures of TTR and its oxidized isoforms are similar
Far UV-CD (secondary structure) and near UV-CD as well as tryptophan fluorescence spectra (tertiary structure) were recorded for WT and its oxidized isoforms (oxi-WT and car-WT TTR) as well as for V122I TTR and its oxidized isoforms (oxi-V122I and car-V22I TTR). The far and near UV-CD data show no major differences in the spectra (Supplemental Figure S2) of the oxidized TTRs (oxi- and car-TTRs) with respect to their non-oxidized counterparts. The tryptophan emission spectra are identical for WT and oxi-WT as well as for V122I and oxi-V122I TTR. The carbonylated proteins however, have lower maximum fluorescence intensity than non-oxidized or oxi-TTRs. These differences are not unexpected because of the multiple amino acid residues susceptible to carbonylation in the TTR sequence (32), some of which may be spatially close to the two tryptophan residues (Trp41 and Trp79) of the molecule. For TTR a shift in the maximum emission wavelength from 335 to 355 nm is indicative of TTR unfolding (40). The maximum emission wavelength for the 3 isoforms (non-oxidized, oxi-TTR and car-TTR) is the same (335 nm) suggesting that they are all properly folded (Supplemental Figure S2).
What is the relationship of sample turbidity to TTR aggregation and Thioflavin T binding?
TTR aggregation is inducible in vitro under mildly acidic conditions and its extent can be monitored by recording turbidity at 330–400 nm using UV-Vis spectrophotometry (12;41). Native WT and V122I TTR and their oxidized variants (oxi-TTRs, car-TTRs) were incubated at pH ranging from 4.04 to 5.88 for up to 7 days at 37 °C without agitation, and the extent of aggregation was measured by turbidity at 400 nm (Figure 1). To determine whether turbidity was a measure of aggregation in these highly modified proteins, we also measured the total aggregated (insoluble) protein and the amount of protein remaining in the supernatant. Similar aggregation studies were also performed with shorter (hours) and longer (up to 21 days) incubation times at pH 4.4 – the pH of maximum turbidity for WT and V122I TTR (42) (Supplemental Figure S3). The results show that turbidity does not quantitatively represent aggregated protein (insoluble). At pH 4.04 for example, all the samples have very low turbidity (Figure 1a and b) while it is at this pH where we found the most aggregated (insoluble) TTR (Figure 1c and d). This inconsistency is also observed in the longer kinetic experiments at pH 4.4 where the turbidity of WT TTR and car-WT TTR increases steadily from 3 to 21 days, but the amount of insoluble protein recovered remains constant (Supplemental Figure S3a and S3b). Moreover, for oxi-WT TTR and oxi-V122I TTR the maximum turbidity values (pH 4.37–4.56) are about half the values obtained for their non-oxidized counterparts (WT and V122I TTR) (Figure 1a and 1b), whereas the amount of insoluble protein is slightly lower in oxi-WT compared to WT TTR (Figure 1c) and is the same in V122I and oxi-V122I TTR (Figure 1d). The percentages of protein remaining in the supernatants after centrifugation are complementary to those found in the pellets (not shown). Similar studies to those reported in Figure 1 in which the TTR aggregation reactions were analyzed after 1 and 3 days of incubation at 37 °C gave the same relative patterns of aggregation with respect to turbidity and amount of insoluble protein recovered as those shown in Figure 1 (pH dependent aggregation, 7 days) and Supplemental Figure S3 (time dependent aggregation, pH 4.4).
FIGURE 1.

pH-dependent aggregation of native and oxidized TTRs. WT (a, c) and V122I TTR (b, d) and their oxidized isoforms (oxi-TTRs and car-TTRs) were incubated for 7 days at the designated pH at 37 °C. The extent of aggregation was measured by turbidity at 400 nm (a, b) and percentage of insoluble protein with respect to initial protein content (c, d). Native TTR (solid line, squares), oxi-TTR (broken line, circles), car-TTR (dotted line, triangles). WT TTR and its isoforms are represented by solid symbols; V122I TTR and its oxidized isoforms are represented by open symbols. Data shown for one representative experiment performed in triplicate (mean ± SD).
We used Thioflavin T (ThT) binding fluorescence as an additional method to quantify TTR aggregation and fibril formation in vitro (43). Native and oxidized TTRs were incubated with acidic buffer (pH 4.4) at 37 °C for 1 and 3 days without agitation. ThT fluorescence, the percentage of precipitated protein and turbidity at 400 nm were measured as detailed in the Experimental section. The results shown in Figure 2 (3 days incubation) are presented as percentages relative to native TTRs. The data show that the percentage of ThT fluorescence (white bars) is similar to that of precipitated protein (black bars) for each TTR variant. The same correlation was seen for samples incubated for 1 day at 37 °C, as well as for samples incubated at pH 4.04 for up to 7 days (not shown).
FIGURE 2.

Relative aggregation of WT TTR (a) and V122I TTR (b) and their oxidized isoforms. TTR aggregates and fibrils were generated by incubation at pH 4.4 for 3 days at 37 °C. The extent of aggregation was measured by Thioflavin T fluorescence (white bars), percentage of insoluble protein with respect to initial protein content (black bars) and by turbidity at 400 nm (hatched bars). All the values were normalized to those resulting from the non-oxidized WT TTR and V122I TTR, respectively. The results shown represent the average of three independent experiments (mean ± SD).
Given that turbidity does not reflect the amount of aggregated (insoluble) protein under all conditions or with all the TTR variants, we will refer to protein aggregation as the percentage of precipitated protein after the designated incubation times.
Oxidation modifies TTR aggregation propensity
Figure 1 shows that oxi-WT TTR aggregates slightly less than WT TTR whereas oxi-V122I TTR aggregates to the same extent as V122I TTR. The kinetic aggregation data show that at pH 4.4 both oxi-WT TTR and oxi-V122I TTR aggregate more slowly than WT or V122I TTR, respectively, although the end points of aggregation are similar (Supplemental Figure S3). More interestingly, at pH 4.88 and 5.13, car-WT and car-V122I TTR form more aggregates than non-oxidized WT or V122I TTR, respectively. The differences are particularly significant for car-WT TTR (~21% aggregated protein at pH 4.88) relative to WT TTR (~ 9% aggregated protein at pH 4.88). These findings are relevant because they indicate that in vivo car-WT and car-V122I TTR can aggregate at a pH closer to physiological conditions, hence potentially initiate the senile forms of the TTR amyloid diseases.
Monomeric oxi-TTR aggregates more slowly than monomeric TTR
The aggregation experiments show that oxi-TTRs aggregate at a slower rate than their non-oxidized counterparts (Supplemental Figure S3). To assess whether the decreased rate of aggregation comes exclusively from the tetramers or whether the monomeric subunits are also less aggregation prone, we performed fibril formation kinetic studies at pH 4.4 using the well characterized monomeric TTR variant designated M-TTR (F87M/L110M TTR) (44), and its oxidized isoforms (named oxi-M-TTR and car-M-TTR). M-TTR has the same tertiary structure as each of the subunits of native TTR (WT TTR), but the Met residues introduced at the dimer-dimer interfaces (positions 87 and 110) prevent its tetramerization by steric hindrance. LC-MS analysis of H2O2 treated M-TTR, indicated that all the Met residues in the sequence had gained one oxygen atom (Met to MetO transformation), and Cys10 became Cys-SO3H (Table 1). Carbonylation of M- TTR (5h reaction) was confirmed by Oxyblot (Millipore) and LC-MS. The yield of carbonylation was 42% (not shown). The aggregation and fibril formation data show that oxi-M-TTR aggregates more slowly (measured as insoluble protein) than M-TTR (Figure 3), whereas car-M-TTR and M-TTR aggregate at a similar rate. After 2 days of incubation at 37 °C, both oxi-M-TTR and car-M-TTR aggregated almost to the same extent as M-TTR did.
FIGURE 3.

Kinetics of fibril formation of monomeric M-TTR and its oxidized isoforms. Aggregation of native M-TTR (solid line, squares), oxi-M-TTR (broken line, circles), car-M-TTR (dotted line, triangles) was induced by incubation at pH 4.4 at 37 °C without agitation. Percentage of insoluble TTR was measured over time. Data shown for one representative experiment performed in triplicate (mean ± SD).
Inhibition of aggregation by resveratrol is less effective for carbonylated WT TTR than for non oxidized WT TTR
Resveratrol is a polyphenolic compound known to kinetically stabilize tetrameric TTR by binding in its T4 pocket and thus preventing TTR aggregation and fibril formation in vitro, and TTR-induced cell cytotoxicity in tissue culture (32;33;45). Native and oxidized WT samples were incubated for 3 days at pH 4.4 with or without 2 equivalents of resveratrol. Aggregation was measured as the percentage of insoluble protein formation (Figure 4). The data show that WT and oxi-WT TTR aggregation and fibril formation can be effectively inhibited by resveratrol (>94 ± 0.8% aggregation inhibition with respect to TTR without resveratrol), whereas car-WT TTR is less amenable to resveratrol stabilization (88 ± 0.5 % aggregation inhibition with respect to car-TTR without resveratrol).
FIGURE 4.
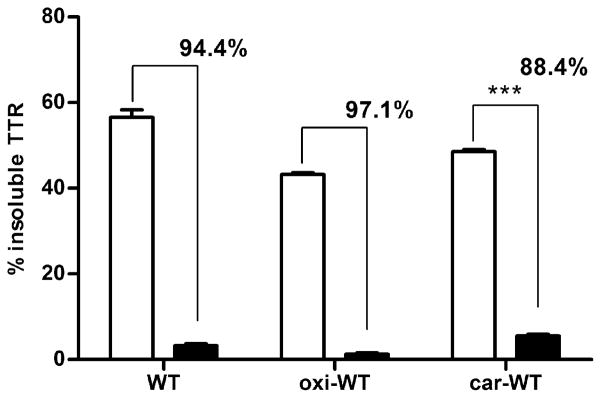
Resveratrol-mediated inhibition of aggregation and fibril formation. WT TTR, oxi-WT and car-WT TTR were incubated at pH 4.4 for 3 days at 37 °C in the presence (black bars) or absence (white bars) of resveratrol. The percentage of insoluble protein was measured with respect to the initial protein content. The numbers over the bars represent the percentage of aggregation inhibition of resveratrol containing samples with respect to vehicle containing samples. The data show that car-WT TTR is less amenable to resveratrol mediated inhibition of aggregation and fibril formation than native WT TTR (88% vs 94%, p ≤ 0.001, unpaired t-test).
Oxi-TTRs aggregate morphology is different than that of non-oxidized TTRs
Transmission electron microscopy (TEM) was used to evaluate the aggregate morphology of the different TTR isoforms under acid-mediated aggregation conditions (pH 4.4, 1 day at 37 °C). Non-oxidized WT and V122I TTR samples were characterized by the presence of elongated material (Fig 5a and d, arrows) as well as abundant small spherical aggregates (Fig 5a and d, circles). In contrast, samples from the oxi-TTR variants (oxi-WT and oxi-V122I TTR) were consistently richer in longer aggregates (Fig 5b and e, arrows) than those found in the non-oxidized proteins. In oxi-TTR samples very few of the spherical aggregates were detected. Carbonylated TTR isoforms were characterized by the presence of elongated material, usually shorter than that found in non-oxidized TTRs samples (Fig 5c and f, arrows) with abundant spherical amorphous aggregates (Fig 5c and f, circles). In some of the regions of the copper grids there were larger amorphous deposits which appeared to be made up of very short, entangled, elongated structures (not shown). These structures were observed in all the samples although they were less abundant in the oxi-TTR isoforms samples than in the native or car-TTRs samples. With longer incubation times (3 and 7 days), particularly with the faster aggregating V122I TTR isoforms, most of the material detected was in the shape of large amorphous aggregates made up of the same short elongated structures mentioned above, or dense deposits opaque to TEM (not shown).
FIGURE 5.

Negative stain TEM images of WT and V122I TTR and their oxidized isoforms subjected to acid-mediated aggregation conditions for 24h at 37 °C. The labeling of the panels corresponds to a) WT TTR, b) oxi-WT TTR, c) car-WT TTR, d) V122I TTR, e) oxi-V122I TTR, f) car-V122I TTR. All images were taken at 92 000 × magnification. Scale bars represent 200 nm.
Oxidative modifications of TTR decrease its thermodynamic stability
The thermodynamic stability of oxidized and non-oxidized TTRs was studied by urea-mediated denaturation and renaturation curves at pH 7.6. As in the process of aggregation and fibril formation, the rate limiting step for TTR unfolding in urea is the dissociation of the tetramer into its constituent monomers (46).
The quaternary structural changes induced by urea (i.e. tetramer dissociation/tetramer reassembly), were measured by resveratrol fluorescence. The binding of resveratrol in the TTR T4 pocket results in a sizable increase in resveratrol fluorescence which is linear with tetrameric TTR concentration (37). Resveratrol does not bind to or fluoresce with monomeric TTR subunits, nor does it shift the monomer-tetramer equilibrium (12); therefore, it is a useful probe to quantify the amount of tetrameric TTR in a given solution. Standard curves using oxidized (oxi-TTRs and car-TTRs) and non-oxidized TTRs in the ranges of concentrations used for these studies (0–0.15 mg/ml) demonstrated that resveratrol fluorescence is linear with tetrameric TTR concentration (not shown).
The tertiary structural changes induced by urea (i.e. unfolding/refolding) were monitored by intrinsic tryptophan fluorescence (37), which allows the quantification of the fraction of unfolded TTR versus folded TTR at any given urea concentration. Figure 6 shows the denaturation and renaturation curves of WT (a–d) and V122I TTR (e–h) and their oxidized isoforms. The data were fitted to a sigmoidal curve and the concentration of urea at the midpoint of denaturation/renaturation (Cm) was determined (Table 2).
FIGURE 6.

Stability curves of native and oxidized TTR variants in urea. WT TTR and its oxidized isoforms (a–d) and V122I and its oxidized isoforms (e–h) were incubated in the presence of various concentrations of urea. The curves were generated in the denaturation direction (a, c, e, g) and in the renaturation direction (b, d, f, h). The amount of folded TTR at any given urea concentration was measured by tryptophan fluorescence (a, b, e, f) and the amount of tetrameric TTR at any given urea concentration was measured by resveratrol fluorescence (c, d, g, h). Non-oxidized TTR (solid line, squares), oxi-TTRs (broken line, circles), car-TTR (dotted line, triangles). WT TTR and its isoforms are represented by solid symbols, V122I TTR and its oxidized isoforms are represented by open symbols. Data shown represents the average of at least 3 independent experiments performed in triplicate (mean ± SD).
Table 2.
Midpoint transition values (Cm) of TTR denaturation/renaturation curves in urea. All Cm values are presented in molar units. TTR unfolding and refolding was measured by tryptophan fluorescence; tetramer disassembly and reassembly was measured by resveratrol binding fluorescence.
| TTR variant | Unfolding | Refolding | Disassembly | Reassembly | ||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Cm | p | Cm | p | Cm | p | Cm | p | |
| WT TTR | 3.5 ±0.06 | 3.5 ±0.12 | 3.4 ±0.09 | 3.4 ± 0.08 | ||||
| oxi-WT | 3.0 ±0.05 | **** | 2.8 ±0.12 | **** | 2.8 ±0.19 | ** | 2.8 ± 0.03 | *** |
| car-WT | 3.4 ±0.15 | ns | 3.3 ±0.05 | ** | 2.8 ±0.27 | ** | 3.1 ±0.00 | ** |
|
| ||||||||
| V122I TTR | 3.4 ±0.06 | 3.4 ±0.07 | 2.8 ±0.07 | 2.9 ±0.08 | ||||
| oxi-V122I | 2.8 ±0.04 | **** | 2.8 ±0.05 | **** | 2.4 ±0.09 | **** | 2.5 ±0.05 | **** |
| car-V122I | 3.4 ±0.03 | ns | 3.3 ±0.06 | ns | 2.5 ±0.04 | *** | 2.5 ±0.02 | **** |
p values measure significant changes in Cm for the oxidized proteins with respect to their non-oxidized counterparts. Unpaired T-test: ns (no significant), p > 0.05;
p ≤0.01;
p ≤0.001;
p ≤0.0001.
For each protein the Cm values for tetramer disassembly and reassembly (measured by resveratrol binding) are equal or smaller than the Cm values for unfolding and refolding (measured by tryptophan fluorescence), respectively. These results are consistent with the notion that the tetramer has to first disassemble in order to unfold. TTR tetramer disassembly and monomer unfolding in urea is a process that take days to reach equilibrium, and is much slower than the refolding and reassembly process which take only few seconds (37). In the disassembly/unfolding direction at urea concentrations > 4 M there is still a considerable amount of tetrameric TTR (>20%), which is not present in the refolding/reassembly direction (Figure 6), indicating that for the denaturation solutions, at the time of measurement (96 h), thermodynamic equilibrium had not been reached. Longer incubation times in urea are not advisable because urea covalently modifies the proteins in a time-dependent manner (47). Although the calculated Cms for the denaturation and the renaturation processes were very similar, for clarity we focus on the refolding/reassembly curves where thermodynamic equilibrium has been reached for all the TTR variants studied.
The data show that for both oxi-TTRs and car-TTRs the Cms for refolding and reassembly are lower than those of their non-oxidized counterparts; these results suggest that oxi-TTRs and car-TTRs are thermodynamically less stable than native TTRs (Table 2).
For WT TTR and its oxidized variants the Cm for refolding and the Cm for tetramer reassembly are very similar, indicating that as soon as the polypeptides refold, they assemble into tetramers. In contrast, for V122I TTR there is a substantial difference between the Cms for refolding and the Cms for tetramer reassembly (0.5, 0.3 and 0.8 molar units for V122I TTR, oxi-V122I TTR and car-V122I TTR, respectively), with the Cms for refolding being higher than those for reassembly. The Cms for refolding of V122I and its oxidized forms are similar to the Cms for refolding of WT and its oxidized products (~3.5, 2.8 and 3.3 M for native, oxi-TTRs and car-TTRs, respectively). Together these results suggest that the monomers of WT and their corresponding V122I variants have similar thermodynamic stability, whereas the tetramers for the WT TTR isoforms have higher thermodynamic stability than those of their respective V122I TTR isoforms. TTR carbonylation has a stronger effect in destabilizing the tetramer as seen by the increased differences in Cm between refolding and reassembly (0.2 M and 0.8 M for car-WT and car-V122I, respectively compared to 0.1 and 0.5 M for WT and V122I TTR, respectively).
Oxidative modifications of TTR change its kinetic stability
We next studied the kinetic stability of the oxidized TTRs in 6 M urea -a concentration at which TTR remains irreversibly unfolded. The percentage of folded protein in solution was measured over time by tryptophan fluorescence. Consistent with published results, the data show that V122I TTR in urea unfolds much faster (75% unfolded in less than 24h) than WT TTR (75% unfolded in near 200h) in urea (Figure 7), indicating a decreased kinetic stability of V122I TTR with respect to WT TTR (42).
FIGURE 7.

Kinetic stability of native and oxidized TTR variants in urea. TTR samples were incubated in 6 M urea at room temperature over a 480 h time course. The degree of folded protein was measured by tryptophan fluorescence. a) WT TTR, black square; oxi-WT TTR, black circles; car-WT TTR, black triangles;. b) V122I TTR, open squares; oxi-V122I TTR, open circles; car-V122I TTR, open triangles. The data shown represents the average of 2 independent experiments performed in triplicate (mean ± SD). To appreciate the relative kinetic stabilities of V122I TTR and its oxidized isoforms the time scale is reduced to 60h.
Oxi-WT TTR unfolds more slowly than native or car-WT TTRs (Figure 7). At 96 hours only ~25% of WT and car-WT remained folded, whereas more than 50% of the oxi-WT TTR was still folded. These results are consistent with an increased kinetic stability of oxi-WT TTR with respect to WT TTR or car-WT TTR. For V122I TTR however, oxi-V122I unfolds at a slightly lower rate than native V122I TTR (i.e. have similar kinetic stability). Car-V122I TTR unfolds at a lower rate than V122I TTR, indicating that it has increased kinetic stability.
Oxidized TTRs have similar cytotoxic potency as native TTRs
We have previously established tissue culture model systems to study the TTR amyloidoses in a physiologic setting (32;33). In these systems we showed that amyloidogenic TTR variants such as V122I TTR or V30M TTR are cytotoxic to cells of human cardiac and neuronal origin in a dose responsive manner (32;33) whereas non amyloidogenic TTR (T119M) is not.
We used the human cardiac tissue culture system to determine the cytotoxic potential of the oxidized forms of TTR with respect to their non-oxidized counterparts. AC16 cells were treated with different concentrations of WT and V122I TTR and its oxidized isoforms for 24h at 37 °C. Cell viability was measured by the resazurin reduction assay. The results show that all oxidized TTR isoforms are cytotoxic to the human cardiac cell line AC16 in a dose-responsive manner (Figure 8); consequently they have the potential to induce tissue damage in vivo.
FIGURE 8.

Cytotoxicity of WT TTR and its oxidized isoforms (a) and V122I TTR and its oxidized isoforms (b) to human cardiomyocytes. AC 16 cells were exposed to several concentrations of native and oxidized TTRs for 24 hours. Cell viability was measured by resazurin reduction assay. Percentage of cell viability was determined with respect to vehicle treated samples (100% viability). Native TTR, black bars; oxi-TTR white bars, car-TTR, hatched bars. Results are representative of 2 independent experiments performed in triplicate (mean ± SD).
DISCUSSION
One of the hallmarks of aging is the increased abundance of oxidized proteins in plasma and tissues. In this paper we explore how oxidation of the plasma protein TTR could result in isoforms with increased aggregation and fibril formation propensity, contributing to the onset of the senile forms of the TTR amyloidoses. We focused on WT and the V122I TTR variant because both proteins are related to late onset TTR aggregation diseases affecting the heart (SSA and FAC).
We have chosen two types of protein oxidative modifications that are relevant to the process of aging, Met oxidation and carbonylation. Far and near UV-CD studies demonstrated that the oxidative modifications created did not alter the secondary or tertiary structure of the protein (Supplemental Figure S2). The tryptophan fluorescence spectra show identical traces for non-oxidized and oxi-TTR and a lower intensity trace -albeit with the same maximum emission-for car-TTR. These results are not unexpected because each TTR polypeptide has 32 amino acid residues susceptible to metal catalyzed carbonylation; some of these carbonylated residues might be spatially close to one or both Trp residues (Trp41 and Trp79) and partially quench their fluorescence intensity. The fact that all the proteins have the same Trp fluorescence maximum emission wavelength at 335 nm indicates that the proteins are folded.
In our extensive aggregation studies at pH ranging from 4.0 to 5.88, we determined that turbidity measurements may not be a reliable measure of aggregated (insoluble) protein content. The differences are particularly striking at the lowest pH studied (pH 4.0) where very little turbidity is detected while there was a very high proportion of insoluble protein (Figure 1). Remarkably, very significant differences were also found for oxi-TTRs at the pH of maximum turbidity (4.37–4.56). At these pH’s the levels of turbidity of oxi-WT and oxi-V122I TTR are less than half of those of WT and V122I TTR respectively, whereas oxi-WT and WT TTR, and oxi-V122I and V122I TTR form very similar amount of insoluble aggregates (Figure 1). TEM analysis of these TTR isoforms with different behavior at pH 4.4 can help to rationalize the discrepancy between the turbidity levels and the amount of insoluble protein detected (Figure 5). WT and V122I TTR at pH 4.4 form short fibrillar structures and small amorphous spherical aggregates, whereas in the preparations of oxi-WT and oxi-V122I TTR we observed essentially longer fibrillar structures and almost none of the spherical aggregates (Figure 5). The concentration of particles for non oxidized WT and V122I TTR, where high levels of turbidity are detected, is greater than for the oxi-TTR isoforms because in the former the aggregates are smaller than in the latter. Thus, turbidity appears to report on the concentration of large particles (large enough to interfere with 330–400 nm light waves) regardless of their particular size. These results are consistent with those reported by Lundberg et al (48) where WT TTR was aggregated at a wide range of pH (2.0–5.5). The turbidity of the samples was measured and atomic force microscopy images were recorded. The images show that at lower pH (2–3.5) where no turbidity is detected, there are large filaments; whereas at higher pH (>4.0) where there is turbidity, the aggregates are small and mainly spherical.
ThT fluorescence signal correlates quite well with the amount of insoluble protein detected (Figure 2). The TEM images show that for non-oxidized TTR both short fibrillar and amorphous aggregates are present in these solutions (Figure 5). Thus, it appears that ThT fluorescence might not be specific for amyloid fibrils, but that it is sensitive to smaller amorphous aggregates as well (11;49). It should be noted however, that ThT fluorescence for TTR is relatively weak compared to that of other amyloidogenic peptides and proteins; therefore, there is the possibility that the apparent correlation between ThT fluorescence and insoluble aggregate formation coincides with the amount of bona fide amyloid fibrils that bind ThT. Given that the turbidity levels do not appear to represent aggregated protein under all conditions and for all TTR isoforms, we relied on insoluble protein formation for the interpretation of the results.
Consistent with our former observations (30), oxi-TTRs form the lowest quantity of aggregates and fibrils within the entire pH range tested at earlier time points (up to 3 days for oxi-WT and up to 8 h for oxi-V122I TTR). The formation of insoluble aggregates becomes similar to those of non-oxidized TTRs and car-TTRs later in the aggregation process (Figure 1 and Supplemental Figure S3). These results suggest that oxi-TTRs might be kinetically stable with respect to native TTRs because they have slower rates of insoluble aggregate formation. The TTR denaturation studies in 6 M urea clearly show a slower rate of unfolding for oxi-WT TTR than for WT TTR. These results demonstrate that oxi-WT TTR is kinetically more stable than non oxidized WT TTR (Figure 7). The V122I TTR isoforms are kinetically much less stable than the WT TTR isoforms, as seen by their faster unfolding rates in 6 M urea (Figure 7). In this case we detected a slight increase in the kinetic stability of oxi-V122I with respect to V122I TTR. These observations are consistent with the fact that for oxi-V122I TTR, aggregation and fibril formation occur at slightly slower rate than for V122I TTR and can only be detected during the first 24h of the aggregation process (Supplemental Figure S3).
With respect to thermodynamic stability, in the presence of urea the calculated midpoints for tetramer reassembly (Cm) are lower for oxi-WT TTR and car-WT TTR than for WT TTR (3.1 M vs 3.4 M), indicating that the oxi- and car-WT TTR are thermodynamically less stable than WT TTR. The same is true for oxi-V122I TTR and car-V122I TTR with respect to V122I TTR (2.5 M vs 2.9 M) (Figure 6 and Table 2). These observations imply that at equilibrium the fraction of monomeric TTR in solution will be higher for the oxi-TTRs and car-TTRs than for their non oxidized counterparts; thus the oxidized proteins will be more prone to aggregation and deposition in vivo.
The TTR aggregation propensity depends on a combination of both, thermodynamic and kinetic stability parameters (13). Oxi-TTRs have the potential to be more amyloidogenic than native TTRs as seen by the lower thermodynamic stability of the tetramer. It is possible however, that their enhanced kinetic stability slows down the dissociation of the tetramer in vivo and thus, decreases the overall TTR aggregation process.
Carbonylated WT and V122I TTR have higher aggregation propensities than their non-oxidized counterparts at pH closer to physiological (4.88 and 5.13). Car-TTRs are also thermodynamically less stable than native TTRs as seen by the lower Cms for refolding and particularly those for tetramer reassembly in urea (Figure 6 and Table 2). Moreover, it appears that car-TTRs have similar kinetic stability to non-oxidized TTRs (Figure 7). Together, these results suggest that in vivo, car-TTRs will have a higher tendency to aggregate and deposit than non-oxidized TTRs and contribute to the onset of the senile forms of the TTR amyloidoses. We hypothesize that a decrease in the thermodynamic stability of TTR (in the absence of a change in kinetic stability) results in aggregation and fibril formation at higher pH in vitro and the development of amyloid deposition in vivo. This notion is supported by other examples in the literature. For example, the V30M TTR variant related to FAP, is thermodynamically unstable (Cm in urea is 2.0 M; compare to 3.4 M for WT TTR), while its pH of maximum fibril formation is 5.2 (compare to 4.4 for WT TTR) (50). Other TTR variants such as L58H or I84S TTR for which both thermodynamic stability parameters (both have Cm =2.4 M in urea) and pH of maximum fibril formation (pH 4.8–4.9) have been reported, follow the same trend (50). Only the T60A TTR variant appears to be an exception to this trend (Cm 2.8 M, and pH of max. fibril formation 4.5). Further support of this hypothesis is found in Zhang et al (51) where the stability and fibril formation propensity of Cys10-modified TTR variants were characterized. They report that TTR variants that are thermodynamically less stable than WT TTR form fibrils at a higher pH range, whereas one of the variants that forms the maximum amount of fibrils and aggregates at the same pH as WT TTR had the same thermodynamic stability (51). It is not known however, whether these variants are involved in early onset amyloid disease.
Aggregation studies performed using the well characterized engineered monomeric TTR (M-TTR) (44) and its oxidized isoforms show that oxi-M-TTR aggregates at slower rate than non-oxidized M-TTR, suggesting that it might also be kinetically more stable (Figure 3). Yet, the rates of aggregation of tetramer TTRs are much slower than those of monomeric TTRs, indicating that even with oxidized proteins the rate limiting step in the aggregation process is the dissociation of the tetramer. The slower aggregation rate of oxi-M-TTR with respect to M-TTR could also be explained by the fact that the former is more hydrophilic than the latter, as seen by faster elution times in an HPLC column in reverse phase (not shown). It is known that protein aggregation is driven by hydrophobic interactions; thus, the decrease in hydrophobicity of oxi-M-TTR with respect to M-TTR could also contribute to the slower aggregation rate observed. These results are consistent with the decreased fibril formation propensities of the MetO Aβ peptide (52), prion protein (53), α-synuclein (54), and apolipoprotein C-II (55) with respect to their non-oxidized counterparts.
In conclusion, the age-related oxidative modifications of TTR studied decrease the protein’s thermodynamic stability and can therefore impact on the onset of the senile forms of the TTR amyloidoses in vivo. Although oxi-WT TTR is kinetically more stable than non-oxidized TTR, it still forms the same amount of aggregates at sufficiently long time points (> 7 days). It is unknown how these two parameters, thermodynamic and kinetic stability, will play in vivo. However, other TTR variants with extremely high kinetic stability such as Y69H or F64S TTR and low thermodynamic stability are known to be amyloidogenic in vivo (13).
The current treatment for TTR amyloidosis related to deposition of mutant TTR is liver transplantation. In this procedure the mutant TTR-producing liver is replaced by a wild type TTR-producing liver (56). More recently, a new pharmacologic-based therapeutic strategy is undergoing human clinical trials and is already approved in Europe to treat early stage TTR amyloidosis (57;58). This strategy consists in the administration of small molecules which by binding in the TTR T4 pocket kinetically stabilize the native tetramer and prevent aggregation and fibril formation (57;59). Resveratrol is one such molecule with high capacity to kinetically stabilize tetrameric TTR and prevent aggregation in vitro (45) and TTR-induced toxicity in tissue culture (32;33). By binding in the TTR T4 pocket, resveratrol precludes the dissociation of the TTR tetramer into its corresponding monomers -the precursors of the fibrils and aggregates. Our studies show that car-WT TTR was less amenable to resveratrol-mediated tetramer stabilization than non-oxidized WT TTR (Figure 4). The TTR T4 binding pocket contains residues which are susceptible to carbonyl modification (Lys15 and Thr119). It is possible that such modification results in a decreased affinity of resveratrol for the TTR T4 binding pocket. These results suggest that for the senile forms of the TTR amyloidoses, where an increased amount of carbonylated TTR might be present, kinetic stabilization of the tetramer by small molecules might be less effective than in cases of early onset disease associated with more aggressive, but non-oxidized TTR mutants. Thus, a therapeutic strategy to delay the onset of the senile forms of the TTR amyloidoses should consider the prevention of oxidative carbonylation of TTR.
In mouse models of the TTR amyloidoses and in human biopsies of asymptomatic mutant TTR carriers there is evidence of tissue damage and cell death well before there is detectable TTR deposition (31). We have previously established a tissue culture model system for the TTR amyloidoses using human-derived cardiomyocytes of the ventricle – the site of TTR deposition in the heart (32). In this system, amyloidogenic TTR variants are toxic to the AC16 human cardiomyocytes, whereas the stable and non-amyloidogenic T119M TTR is not. Herein we show that all the oxidized forms of TTR are toxic to AC16 human cardiomyocytes in a dose-responsive manner (Figure 8) indicating that, like their non-oxidized counterparts, they have potential for tissue damage, before deposition occurs.
In summary, our studies show that an increase in the oxidative state of TTR due to aging might be a factor contributing to the onset of the TTR amyloidoses. The decreased capacity of resveratrol to stabilize carbonylated WT TTR suggests that additional therapeutic strategies to tetramer stabilization, such as anti-oxidant therapy, might be required to delay or prevent the onset of the senile forms of the TTR systemic amyloidoses.
Supplementary Material
Figure S1: Analysis of carbonyl TTRs by western blot.
Figure S2: Far and near UV-CD spectra and tryptophan fluorescence spectra of WT TTR and V122I TTR and their oxidized isoforms.
Figure S3: Kinetics of aggregation and fibril formation of WT and V122I TTR and their oxidized isoforms at pH 4.4 measured by turbidity and insoluble protein content. This material is available free of charge via the internet at http://pubs.acs.org.
Acknowledgments
Funding Source Statement: This research was supported by the National Institutes of Health (NIA, AG032285 to N.R.) and the American Heart Association (award 0865061F to N.R.)
We thank Dr Malcolm R Wood (TSRI) for the acquisition of the TEM images, Jeanine Witkowski for preparing and purifying some of the recombinant protein used for these studies, Dr Mercy Davidson at Columbia University for providing the AC16 cells (38) and Dr Lawreen H Connors at Boston University for generously providing the rTTR plasmid (34). We also thank Dr Jaime Pascual and Dr R Luke Wiseman for carefully reading the manuscript.
ABBREVIATIONS
- CD
circular dichroism
- CSF
cerebrospinal fluid
- Cys
cysteine
- Cys-SO3H
Cys sulfonic acid
- FAC
familial amyloid cardiomyopathy
- GndCl
guanidinium chloride
- GF buffer
10 mM sodium phosphate pH 7.6, 100 mM KCl, 1 mM EDTA buffer
- H2O2
hydrogen peroxide
- HBSS
Hank’s Balanced Salt Solution
- LC-MS
liquid chromatography-Mass spectrometry
- Met
methionine
- MetO
methionine sulfoxide
- rTTR
recombinant transthyretin lacking Met in position −1
- SSA
senile systemic amyloidosis
- T4
thyroxine
- TEM
transmission electron microscopy
- ThT
Thioflavin T
- TTR
transthyretin
- V122I TTR
Val122IIe TTR
- WT TTR
Wild-type TTR
References
- 1.Buxbaum JN. The systemic amyloidoses. Curr Opin Rheumatol. 2004;16:67–75. doi: 10.1097/00002281-200401000-00013. [DOI] [PubMed] [Google Scholar]
- 2.Sipe JD, Benson MD, Buxbaum JN, Ikeda S, Merlini G, Saraiva MJ, Westermark P. Amyloid fibril protein nomenclature: 2010 recommendations from the nomenclature committee of the International Society of Amyloidosis. Amyloid. 2010;17:101–104. doi: 10.3109/13506129.2010.526812. [DOI] [PubMed] [Google Scholar]
- 3.Li X, Masliah E, Reixach N, Buxbaum JN. Neuronal production of transthyretin in human and murine Alzheimer’s disease: is it protective? J Neurosci. 2011;31:12483–12490. doi: 10.1523/JNEUROSCI.2417-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buxbaum JN, Reixach N. Transthyretin: the servant of many masters. Cell Mol Life Sci. 2009;66:3095–3101. doi: 10.1007/s00018-009-0109-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cornwell GG, III, Murdoch WL, Kyle RA, Westermark P, Pitkanen P. Frequency and distribution of senile cardiovascular amyloid. A clinicopathologic correlation. Am J Med. 1983;75:618–623. doi: 10.1016/0002-9343(83)90443-6. [DOI] [PubMed] [Google Scholar]
- 6.Tanskanen M, Peuralinna T, Polvikoski T, Notkola IL, Sulkava R, Hardy J, Singleton A, Kiuru-Enari S, Paetau A, Tienari PJ, Myllykangas L. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: a population-based autopsy study. Ann Med. 2008;40:232–239. doi: 10.1080/07853890701842988. [DOI] [PubMed] [Google Scholar]
- 7.Jacobson DR, Pastore R, Pool S, Malendowicz S, Kane I, Shivji A, Embury SH, Ballas SK, Buxbaum JN. Revised transthyretin Ile 122 allele frequency in African-Americans. Hum Genet. 1996;98:236–238. doi: 10.1007/s004390050199. [DOI] [PubMed] [Google Scholar]
- 8.Buxbaum J, Jacobson DR, Tagoe C, Alexander A, Kitzman DW, Greenberg B, Thaneemit-Chen S, Lavori P. Transthyretin V122I in African Americans with congestive heart failure. J Am Coll Cardiol. 2006;47:1724–1725. doi: 10.1016/j.jacc.2006.01.042. [DOI] [PubMed] [Google Scholar]
- 9.Jacobson DR, Pastore RD, Yaghoubian R, Kane I, Gallo G, Buck FS, Buxbaum JN. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N Eng J Med. 1997;336:466–473. doi: 10.1056/NEJM199702133360703. [DOI] [PubMed] [Google Scholar]
- 10.Kelly JW. The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr Opin Struct Biol. 1998;8:101–106. doi: 10.1016/s0959-440x(98)80016-x. [DOI] [PubMed] [Google Scholar]
- 11.Hurshman AR, White JT, Powers ET, Kelly JW. Transthyretin aggregation under partially denaturing conditions is a downhill polymerization. Biochemistry. 2004;43:7365–7381. doi: 10.1021/bi049621l. [DOI] [PubMed] [Google Scholar]
- 12.Hammarstrom P, Jiang X, Hurshman AR, Powers ET, Kelly JW. Sequence-dependent denaturation energetics: A major determinant in amyloid disease diversity. Proc Natl Acad Sci U S A. 2002;99(Suppl 4):16427–16432. doi: 10.1073/pnas.202495199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sekijima Y, Wiseman RL, Matteson J, Hammarstrom P, Miller SR, Sawkar AR, Balch WE, Kelly JW. The biological and chemical basis for tissue-selective amyloid disease. Cell. 2005;121:73–85. doi: 10.1016/j.cell.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 14.Stadtman ER. Protein oxidation and aging. Free Radic Res. 2006;40:1250–1258. doi: 10.1080/10715760600918142. [DOI] [PubMed] [Google Scholar]
- 15.Stadtman ER, Levine RL. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids. 2003;25:207–218. doi: 10.1007/s00726-003-0011-2. [DOI] [PubMed] [Google Scholar]
- 16.Fedorova M, Kuleva N, Hoffmann R. Identification of cysteine, methionine and tryptophan residues of actin oxidized in vivo during oxidative stress. J Proteome Res. 2010;9:1598–1609. doi: 10.1021/pr901099e. [DOI] [PubMed] [Google Scholar]
- 17.Poole LB. Formation and functions of protein sulfenic acids. Curr Protoc Toxicol Chapter. 2004;17(Unit17) doi: 10.1002/0471140856.tx1701s18. [DOI] [PubMed] [Google Scholar]
- 18.Hoshi T, Heinemann S. Regulation of cell function by methionine oxidation and reduction. J Physiol. 2001;531:1–11. doi: 10.1111/j.1469-7793.2001.0001j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shacter E. Quantification and significance of protein oxidation in biological samples. Drug Metab Rev. 2000;32:307–326. doi: 10.1081/dmr-100102336. [DOI] [PubMed] [Google Scholar]
- 20.Amici A, Levine RL, Tsai L, Stadtman ER. Conversion of amino acid residues in proteins and amino acid homopolymers to carbonyl derivatives by metal-catalyzed oxidation reactions. J Biol Chem. 1989;264:3341–3346. [PubMed] [Google Scholar]
- 21.Dalle-Donne I, Rossi R, Giustarini D, Milzani A, Colombo R. Protein carbonyl groups as biomarkers of oxidative stress. Clin Chim Acta. 2003;329:23–38. doi: 10.1016/s0009-8981(03)00003-2. [DOI] [PubMed] [Google Scholar]
- 22.Levine RL. Carbonyl modified proteins in cellular regulation, aging, and disease. Free Radic Biol Med. 2002;32:790–796. doi: 10.1016/s0891-5849(02)00765-7. [DOI] [PubMed] [Google Scholar]
- 23.Conrad CC, Marshall PL, Talent JM, Malakowsky CA, Choi J, Gracy RW. Oxidized proteins in Alzheimer’s plasma. Biochem Biophys Res Commun. 2000;275:678–681. doi: 10.1006/bbrc.2000.3356. [DOI] [PubMed] [Google Scholar]
- 24.Choi J, Malakowsky CA, Talent JM, Conrad CC, Gracy RW. Identification of oxidized plasma proteins in Alzheimer’s disease. Biochem Biophys Res Commun. 2002;293:1566–1570. doi: 10.1016/S0006-291X(02)00420-5. [DOI] [PubMed] [Google Scholar]
- 25.Floor E, Wetzel MG. Increased protein oxidation in human substantia nigra pars compacta in comparison with basal ganglia and prefrontal cortex measured with an improved dinitrophenylhydrazine assay. J Neurochem. 1998;70:268–275. doi: 10.1046/j.1471-4159.1998.70010268.x. [DOI] [PubMed] [Google Scholar]
- 26.Madian AG, Regnier FE. Profiling carbonylated proteins in human plasma. J Proteome Res. 2010;9:1330–1343. doi: 10.1021/pr900890k. [DOI] [PubMed] [Google Scholar]
- 27.Guidi F, Magherini F, Gamberi T, Bini L, Puglia M, Marzocchini R, Ranaldi F, Modesti PA, Gulisano M, Modesti A. Plasma protein carbonylation and physical exercise. Mol Biosyst. 2011;7:640–650. doi: 10.1039/c0mb00106f. [DOI] [PubMed] [Google Scholar]
- 28.D’Aguanno S, Franciotta D, Lupisella S, Barassi A, Pieragostino D, Lugaresi A, Centonze D, D’Eril GM, Bernardini S, Federici G, Urbani A. Protein profiling of Guillain-Barre syndrome cerebrospinal fluid by two-dimensional electrophoresis and mass spectrometry. Neurosci Lett. 2010;485:49–54. doi: 10.1016/j.neulet.2010.08.060. [DOI] [PubMed] [Google Scholar]
- 29.Ando Y, Nyhlin N, Suhr O, Holmgren G, Uchida K, el SM, Yamashita T, Terasaki H, Nakamura M, Uchino M, Ando M. Oxidative stress is found in amyloid deposits in systemic amyloidosis. Biochem Biophys Res Commun. 1997;232:497–502. doi: 10.1006/bbrc.1996.5997. [DOI] [PubMed] [Google Scholar]
- 30.Maleknia SD, Reixach N, Buxbaum JN. Oxidation inhibits amyloid fibril formation of transthyretin. FEBS J. 2006;273:5400–5406. doi: 10.1111/j.1742-4658.2006.05532.x. [DOI] [PubMed] [Google Scholar]
- 31.Sousa MM, Fernandes R, Palha JA, Taboada A, Vieira P, Saraiva MJ. Evidence for early cytotoxic aggregates in transgenic mice for human transthyretin Leu55Pro. Am J Pathol. 2002;161:1935–1948. doi: 10.1016/S0002-9440(10)64469-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bourgault S, Choi S, Buxbaum JN, Kelly JW, Price JL, Reixach N. Mechanisms of transthyretin cardiomyocyte toxicity inhibition by resveratrol analogs. Biochem Biophys Res Commun. 2011;410:707–713. doi: 10.1016/j.bbrc.2011.04.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reixach N, Deechongkit S, Jiang X, Kelly JW, Buxbaum JN. Tissue damage in the amyloidoses: Transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. Proc Natl Acad Sci U S A. 2004;101:2817–2822. doi: 10.1073/pnas.0400062101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kingsbury JS, Klimtchuk ES, Theberge R, Costello CE, Connors LH. Expression, purification, and in vitro cysteine-10 modification of native sequence recombinant human transthyretin. Protein Expr Purif. 2007;53:370–377. doi: 10.1016/j.pep.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Noble RW, Gibson QH. The reaction of ferrous horseradish peroxidase with hydrogen peroxide. J Biol Chem. 1970;245:2409–2413. [PubMed] [Google Scholar]
- 36.Maisonneuve E, Ducret A, Khoueiry P, Lignon S, Longhi S, Talla E, Dukan S. Rules governing selective protein carbonylation. PLoS One. 2009;4:e7269. doi: 10.1371/journal.pone.0007269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reixach N, Foss TR, Santelli E, Pascual J, Kelly JW, Buxbaum JN. Human-murine transthyretin heterotetramers are kinetically stable and non-amyloidogenic: A lesson in the generation of transgenic models of diseases involving oligomeric proteins. J Biol Chem. 2007;283:2098–2107. doi: 10.1074/jbc.M708028200. [DOI] [PubMed] [Google Scholar]
- 38.Davidson MM, Nesti C, Palenzuela L, Walker WF, Hernandez E, Protas L, Hirano M, Isaac ND. Novel cell lines derived from adult human ventricular cardiomyocytes. J Mol Cell Cardiol. 2005;39:133–147. doi: 10.1016/j.yjmcc.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 39.Stadtman ER, Van RH, Richardson A, Wehr NB, Levine RL. Methionine oxidation and aging. Biochim Biophys Acta. 2005;1703:135–140. doi: 10.1016/j.bbapap.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 40.Lai Z, McCulloch J, Lashuel HA, Kelly JW. Guanidine hydrochloride-induced denaturation and refolding of transthyretin exhibits a marked hysteresis: equilibria with high kinetic barriers. Biochemistry. 1997;36:10230–10239. doi: 10.1021/bi963195p. [DOI] [PubMed] [Google Scholar]
- 41.Colon W, Kelly JW. Partial denaturation of transthyretin is sufficient for amyloid fibril formation in vitro. Biochemistry. 1992;31:8654–8660. doi: 10.1021/bi00151a036. [DOI] [PubMed] [Google Scholar]
- 42.Jiang X, Buxbaum JN, Kelly JW. The V122I cardiomyopathy variant of transthyretin increases the velocity of rate-limiting tetramer dissociation, resulting in accelerated amyloidosis. Proc Natl Acad Sci U S A. 2001;98:14943–14948. doi: 10.1073/pnas.261419998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.LeVine H., III Quantification of beta-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 1999;309:274–284. doi: 10.1016/s0076-6879(99)09020-5. [DOI] [PubMed] [Google Scholar]
- 44.Jiang X, Smith CS, Petrassi HM, Hammarstrom P, White JT, Sacchettini JC, Kelly JW. An engineered transthyretin monomer that is nonamyloidogenic, unless it is partially denatured. Biochemistry. 2001;40:11442–11452. doi: 10.1021/bi011194d. [DOI] [PubMed] [Google Scholar]
- 45.Klabunde T, Petrassi HM, Oza VB, Raman P, Kelly JW, Sacchettini JC. Rational design of potent human transthyretin amyloid disease inhibitors. Nat Struct Biol. 2000;7:312–321. doi: 10.1038/74082. [DOI] [PubMed] [Google Scholar]
- 46.Hammarstrom P, Jiang X, Deechongkit S, Kelly JW. Anion shielding of electrostatic repulsions in transthyretin modulates stability and amyloidosis: insight into the chaotrope unfolding dichotomy. Biochemistry. 2001;40:11453–11459. doi: 10.1021/bi010673+. [DOI] [PubMed] [Google Scholar]
- 47.Bennion BJ, Daggett V. The molecular basis for the chemical denaturation of proteins by urea. Proc Natl Acad Sci U S A. 2003;100:5142–5147. doi: 10.1073/pnas.0930122100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lundberg E, Olofsson A, Westermark GT, Sauer-Eriksson AE. Stability and fibril formation properties of human and fish transthyretin, and of the Escherichia coli transthyretin-related protein. FEBS J. 2009;276:1999–2011. doi: 10.1111/j.1742-4658.2009.06936.x. [DOI] [PubMed] [Google Scholar]
- 49.Lindgren M, Sorgjerd K, Hammarstrom P. Detection and characterization of aggregates, prefibrillar amyloidogenic oligomers, and protofibrils using fluorescence spectroscopy. Biophys J. 2005;88:4200–4212. doi: 10.1529/biophysj.104.049700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miller SR, Sekijima Y, Kelly JW. Native state stabilization by NSAIDs inhibits transthyretin amyloidogenesis from the most common familial disease variants. Lab Invest. 2004;84:545–552. doi: 10.1038/labinvest.3700059. [DOI] [PubMed] [Google Scholar]
- 51.Zhang Q, Kelly JW. Cys10 mixed disulfides make transthyretin more amyloidogenic under mildly acidic conditions. Biochemistry. 2003;42:8756–8761. doi: 10.1021/bi030077a. [DOI] [PubMed] [Google Scholar]
- 52.Hou L, Kang I, Marchant RE, Zagorski MG. Methionine 35 oxidation reduces fibril assembly of the amyloid abeta-(1–42) peptide of Alzheimer’s disease. J Biol Chem. 2002;277:40173–40176. doi: 10.1074/jbc.C200338200. [DOI] [PubMed] [Google Scholar]
- 53.Breydo L, Bocharova OV, Makarava N, Salnikov VV, Anderson M, Baskakov IV. Methionine oxidation interferes with conversion of the prion protein into the fibrillar proteinase K-resistant conformation. Biochemistry. 2005;44:15534–15543. doi: 10.1021/bi051369+. [DOI] [PubMed] [Google Scholar]
- 54.Uversky VN, Yamin G, Souillac PO, Goers J, Glaser CB, Fink AL. Methionine oxidation inhibits fibrillation of human alpha-synuclein in vitro. FEBS Lett. 2002;517:239–244. doi: 10.1016/s0014-5793(02)02638-8. [DOI] [PubMed] [Google Scholar]
- 55.Binger KJ, Griffin MD, Howlett GJ. Methionine oxidation inhibits assembly and promotes disassembly of apolipoprotein C-II amyloid fibrils. Biochemistry. 2008;47:10208–10217. doi: 10.1021/bi8009339. [DOI] [PubMed] [Google Scholar]
- 56.Herlenius G, Wilczek HE, Larsson M, Ericzon BG. Ten years of international experience with liver transplantation for familial amyloidotic polyneuropathy: results from the Familial Amyloidotic Polyneuropathy World Transplant Registry. Transplantation. 2004;77:64–71. doi: 10.1097/01.TP.0000092307.98347.CB. [DOI] [PubMed] [Google Scholar]
- 57.Coelho T, Maia LF, Martins da SA, Waddington CM, Plante-Bordeneuve V, Lozeron P, Suhr OB, Campistol JM, Conceicao IM, Schmidt HH, Trigo P, Kelly JW, Labaudiniere R, Chan J, Packman J, Wilson A, Grogan DR. Tafamidis for transthyretin familial amyloid polyneuropathy: A randomized, controlled trial. Neurol. 2012;79:785–792. doi: 10.1212/WNL.0b013e3182661eb1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bulawa CE, Connelly S, Devit M, Wang L, Weigel C, Fleming JA, Packman J, Powers ET, Wiseman RL, Foss TR, Wilson IA, Kelly JW, Labaudiniere R. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci U S A. 2012;109:9629–9634. doi: 10.1073/pnas.1121005109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berk JL, Suhr OB, Sekijima Y, Yamashita T, Heneghan M, Zeldenrust SR, Ando Y, Ikeda S, Gorevic P, Merlini G, Kelly JW, Skinner M, Bisbee AB, Dyck PJ, Obici L. The Diflunisal Trial: study accrual and drug tolerance. Amyloid. 2012;19(Suppl 1):37–38. doi: 10.3109/13506129.2012.678509. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Analysis of carbonyl TTRs by western blot.
Figure S2: Far and near UV-CD spectra and tryptophan fluorescence spectra of WT TTR and V122I TTR and their oxidized isoforms.
Figure S3: Kinetics of aggregation and fibril formation of WT and V122I TTR and their oxidized isoforms at pH 4.4 measured by turbidity and insoluble protein content. This material is available free of charge via the internet at http://pubs.acs.org.