

Abstract
Background
Soluble oligomers of amyloid β-protein (Aβ) have been increasingly linked to synaptic dysfunction, tau alteration and neuritic dystrophy in Alzheimer’s disease (AD) and mouse models. There is a great need for assays that quantify Aβ oligomers with high specificity and sensitivity.
Methods
We designed and validated two oligomer-specific (o-) ELISAs using either an Aβ aggregate-selective monoclonal for capture and a monoclonal to the free N-terminus for detection or the latter antibody for both capture and detection.
Results
The o-ELISAs specifically quantified pure oligomers of synthetic Aβ with sizes from dimers up to much larger assemblies and over a wide dynamic range of concentrations, whereas Aβ monomers were undetectable. Natural Aβ oligomers of similarly wide size and concentration ranges were measured in extracts of AD and control brains, revealing >1,000-fold higher concentrations of Aβ oligomers than monomers in the soluble fraction of AD cortex. The assays quantified the age-related rise in oligomers in hAPP transgenic mice. Unexpectedly, none of 90 human CSF samples gave a specific signal in either o-ELISA.
Conclusions
These new o-ELISAs with rigorously confirmed specificity can quantify oligomer burden in human and mouse brains for diagnostic and mechanistic studies and for AD biomarker development. However, our data raise the likelihood that the hydrophobicity of Aβ oligomers makes them very low or absent in aqueous CSF.
Keywords: Alzheimer’s disease, amyloid β-peptide, oligomers, cerebrospinal fluid, brain extracts, ELISAs
1. Introduction
The growing recognition of diffusible Aβ oligomers as early neurotoxic species has made these assemblies a topic of special interest in the search for biomarkers of AD. A lowering of Aβ42 monomer levels in human CSF has been widely validated as a robust biomarker for the diagnosis of AD, including in its earliest clinical stages (e.g., [1–4]). Mechanistically, the progressive accumulation of insoluble β-amyloid deposits in brain parenchyma has been postulated to explain the decline in the highly self-aggregating Aβ42 monomer in both CSF and brain interstitial fluid[2, 5, 6]. But beyond monomers, the pathogenic activity of soluble oligomers makes it particularly desirable to measure their levels in brain tissues and biological fluids using assays with high specificity and sensitivity. Oligomer-specific ELISAs performed on CSF or plasma could prove useful for diagnosing AD, following disease progression, and monitoring responses to amyloid-lowering therapeutics. Moreover, such assays would be useful in analyses of biochemical pathology, e.g., to quantify the regional contributions of Aβ oligomers to pathogenic mechanisms in AD and other late-life dementias.
Assays with apparent selectivity for soluble oligomers over monomers and large insoluble assemblies of synthetic Aβ have been reported, but most have not been rigorously validated on natural human oligomers in biological samples[7–15]. Here, we describe and extensively validate two sandwich ELISAs with very high specificity for oligomeric Aβ and sensitivity sufficient to allow detection of low concentrations of natural Aβ oligomers in human brain samples. We demonstrate their utility for quantifying such species in extracts of AD and non-AD cerebral cortex and APP transgenic mouse brains. Unexpectedly, we detected no specific Aβ oligomer signals in 90 human CSF samples from AD and age-matched control subjects. The implications of the latter finding and the potential advantages of these ELISAs over earlier oligomer assays are discussed in detail.
2. Methods
2.1. Antibodies and ELISAs
Sandwich ELISAs were performed using the MSD MULTI-ARRAY® 96-Well Plate platform (Meso-Scale Discovery, Gaithersburg, MD) following the manufacturer’s protocol. For the standard Aβ 1-x ELISA, samples were loaded in duplicate onto plates pre-coated with 30 ul of mAb 266 to the Aβ mid-region (residues 13–28) or mAb 3D6 (to residues 1–5), then detected respectively with biotinylated 3D6 or biotinylated mAb 4G8 (to residues 18–22; Covance) and SULFO-TAG Streptavidin (MSD). For the Aβ o-ELISAs, we used 3D6 or mAb NAB61[14] as capture antibodies and biotinylated 3D6 for detection. Light emitted from the SulfoTAG at the electrode surface was quantified with SECTOR® Imager 2400A. 266 and 3D6 were kindly provided by Elan, plc (South San Francisco, CA), and NAB61 was a gift of Dr. V.M. Lee (U. Penn.). Standard curves were generated with wt synthetic Aβ1-40 for the Aβ 1-x ELISA, and with synthetic Aβ1-40-S26C crosslinked dimers ((Aβ S26C)2) [16] purified by size-exclusion chromatography (SEC) for the o-ELISAs.
2.2 Preparation of Aβ monomers, S26C dimers and aggregates thereof
Oxidized (disulfide crosslinked) Aβ S26C dimers were separated by SEC from higher aggregates and residual S26C monomers immediately before use[17]. Wt Aβ1-40 monomers were also isolated by SEC and used directly. For certain experiments, each of the peptides was aggregated at 10 μM concentration by shaking in 25 mM ammonium acetate, pH 8.5, at 37°C (Labnet Vortemp 56, 700 rpm) for 18 hr.
2.3. Size exclusion chromatography
One ml aliquots of TBS-soluble extracts of human (AD or control) cerebral cortex were injected onto a Superdex 75 10/30 high-resolution SEC column (GE Healthcare) and eluted with 50 mM ammonium acetate, pH 8.5, at 0.6 ml/min into 1 ml fractions. We lyophilized 500 ul of each fraction, reconstituted it in 15 ul of 2X LDS sample buffer (i.e., 4% Lithium dodecyl sulfate; Sigma) and heated it at 65°C for 5 min. Samples were subjected to Western blotting as described[18] using a mixture of 1 ug/ml of mAbs 2G3 (to Aβ40) and 21F12 (to Aβ42) (gifts of Elan, plc) and 0.5 ug/ml of mAb 6E10 (to Aβ3-8; Covance) and imaged on a LiCor Odyssey Infrared System. The remaining 500 ul of each SEC fraction was used for the ELISAs.
2.4. Human and mouse brain samples
Homogenates of human brains (n=28; demographics and diagnoses in Supp. Table S3) were prepared as described[16, 18]. Frozen cortices were provided by Dr. M. Frosch (MGH/Harvard NeuroDiscovery Center) under IRB-approved protocols and by Dr. M. Farrell (Beaumont Hospital, Dublin, Ireland) under Ethical Review Committee/IRB approval. Frozen temporal or frontal cortices were Dounce-homogenized (setting 10, 25 strokes) in ice-cold TBS (20 mM Tris HCl, 150 mM NaCl, pH 7.4) at 4:1 TBS volume/brain wet weight. Homogenates were spun at 175,000g in a TLA100.2 rotor on a Beckman TL100 ultracentrifuge and supernatants (called TBS extracts) aliquoted and stored at −80°C. TBS extracts of wt or J20 hAPP tg mice[19] were prepared identically.
2.5. CSF sample preparation
For lumbar puncture (LP), an IRB-approved published protocol was used[20]. Tubes were pre-cooled, and LP used an atraumatic technique with needle bevel in parallel to dura fibers. After collecting CSF into one 14 ml polypropylene tube and mixing gently by inverting 3–4 times, 500 μl were aliquoted for cell count and 500 μl for glucose and protein levels. CSF was spun at 400g for 10 min; aliquots of the crystal-clear sup were stored at −80°C.
2.6. Immunoprecipitation (IP) and western blotting (WB)
Our IP/WB protocol was described previously [21]. To detect Aβ in TBS extracts of human brain and in CSF, we IPed them with one of our polyclonal Aβ antisera AW8, AW7 or R1282 (1:75) and Protein A Sepharose (PAS; Sigma) or else mAbs 3D6, NAB61 or, as a negative control, 3F3 to the APP C-terminus (each at 3 ug/ml) and Protein G agarose (PGA; Roche) plus PAS. After washing, precipitates were eluted with 10 uL 4% LDS sample buffer, heated at 65°C for 5 min and spun at 8,500g for 5 min. Sups were electrophoresed on 26-well 4–12% bis-Tris gels using MES running buffer (Invitrogen)[18]. Proteins were transferred to 0.2 mm nitrocellulose, and filters were boiled and reacted with a mixture of 1 mg/ml each of 2G3 and 21F12 and 0.5 mg/ml of 6E10 and visualized by LiCor. Some gels were visualized by silver staining[22].
2.7. Aβ immunodepletion of human brain samples and spike-in assays on human fluids
We performed IP of Aβ species in brain TBS extracts or CSF with the indicated antibodies and transferred the immunodepleted sups to new tubes for further experiments. We also prepared serial dilutions of our Aβ peptide standards spiked into human CSF or plasma to search for interference of these fluids with Aβ detection by the ELISAs. Simultaneous serial dilutions of the Aβ standards in regular dilution buffer (1% Blocker A for the MSD® ELISA) served as the control.
2.8. Statistical Analyses
ELISA data on human brain samples were analyzed by nonparametric tests without assuming Gaussian distributions (Mann-Whitney test), followed by two-tailed P values to determine significance.
3. Results
3.1. Design of the 3D6/3D6B and NAB61/3D6B ELISAs and validation of specificity and sensitivity for Aβ oligomers
We first tested several combinations of certain anti-Aβ antibodies to develop sandwich ELISAs specific for Aβ oligomers. For some of the combinations tested, we utilized the same antibody in a sandwich for both antigen capture and detection, a format that should quantify Aβ assemblies containing at least two exposed copies of the same epitope[7, 10]. For all ELISAs, we employed a highly sensitive electrochemiluminescence detection system (MesoScale Discovery), which uses “SULFO-TAG” [(Ru(sulfo-2,2′bipyridine)3)-Streptavidin] for detection of the primary biotinylated detection antibody by emitting light upon electrochemical stimulation initiated at the electrode surfaces of 96-well plates.
To establish the specificity of the ELISAs for oligomeric forms of soluble Aβ (oAβ), we used a synthetic human Aβ1-40 peptide in which Ser26 was changed to cysteine (Aβ40-S26C)[18]. Aβ40-S26C forms disulfide cross-linked dimers when oxidized. Using SEC on a Superdex 75 column, we sized the Aβ40-S26C assemblies and separated Aβ40-S26C dimers from monomers under non-reducing conditions. SDS-PAGE of SEC fractions showed that Fraction 9 contained almost exclusively dimers (hereafter called F9, or (Aβ S26C)2), and Fraction 11 contained almost exclusively monomers (called F11, or Aβ S26C monomers) (Supp. Fig. S1). The Aβ concentration in F9 was estimated to be 90 uM, as judged from quantitative WBs of increasing amounts of synthetic wt Aβ40. F9 was then serially diluted to serve as a standard to develop the oligomer-specific ELISAs. We documented that (Aβ S26C)2 did not aggregate to produce Thioflavin T positive material at high concentrations (12 uM) and at the temperatures and conditions used in our ELISAs; 12 uM is higher than the maximal concentration of (Aβ S26C)2 we routinely used for our o-ELISA standard.
All of the five mAb combinations we initially tested displayed large dynamic ranges, with detection limits as low as ~39 pg/ml and as high as 2,133,000 pg/ml of the F9 synthetic dimers (Supp. Table S1). Among the different antibodies and combinations tested, the two most sensitive combinations were NAB61/3D6B (with a lower detection limit of ~39 pg/ml of the dimeric peptide) and 3D6/3D6B (with a lower detection limit of ~197 pg/ml) (Supp. Table S1). The 3D6/3D6B ELISA employs 3D6 for both capture and biotinylated detector, and the NAB61/3D6B ELISA uses NAB61 for capture and 3D6 as biotinylated detector. 3D6 recognizes a linear epitope of residues 1–5 (DAEFR) having an intact N-terminal aspartate. Importantly, it does not recognize holoAPP and its secreted APPs-α or -β ectodomains [23]. The NAB61 mAb is reported to recognize preferentially a conformational epitope within the N-terminal region of Aβ oligomers; it is not specific for the free N-terminal aspartate[14]. We also tested and rejected (due to less sensitivity) sandwiches of monoclonal antibodies 82E1 (to the free Asp-1 at the Aβ N-terminus; Immuno-Biological Labs, Inc.) and 6E10 (to an Aβ epitope within residues 3–8 (EFRHDS); Covance) (Supp. Table S1).
Based on their sensitivity for the synthetic (Aβ S26C)2 oligomers, the 3D6/3D6B and the NAB61/3D6B ELISAs were selected for further validation; henceforth, we will refer only to these two as the oligomer-ELISAs (o-ELISAs). We found that these o-ELISAs using the MSD ELISA platform showed excellent reproducibility across many runs, with consistently low standard errors of the mean (SEMs) in each run (SEMs are included in all graphs herein, but most are too small to be visible at the wide signal ranges obtained).
Two lines of evidence suggested that the signals in the two o-ELISAs came from oligomeric, not monomeric, Aβ. First, the NAB61/3D6B and the 3D6/3D6B o-ELISAs gave markedly lower signals for the F11 (Aβ S26C monomer) than the F9 ((Aβ S26C)2) SEC fractions (Figs. 1A, B: black and blue traces, respectively; all graphs are plotted on log-log scales because of the very large dynamic range of the assays). Any oxidation of the abundant Aβ40-S26C monomers in F11 will yield small amounts of dimers, especially detectable at high concentrations, and we could indeed detect these by WB, even in the presence of a reducing agent (βME) (Fig. 1E: compare F11 50 ng vs. 10 ng). Accordingly, very high concentrations (i.e., 10 ng/ml or higher) of F11 (S26C monomers) gave detectable, dose-dependent signals in the two o-ELISAs (Fig. 1A, B: blue traces); however, these signals were just 1–3% of the o-ELISA signals that F9 (Aβ S26C)2 gave at the same concentrations (Supp. Table S2). Second, dissociation of the disulfide bond of (Aβ S26C)2 into monomers almost abolished its signal in the two o-ELISAs. Here, we first verified that almost all of the peptide from F9 (Aβ S26C)2 ran as SDS-stable dimers on WB, and that treatment with 3% βME quantitatively reduced the dimers into monomers, although a small amount of residual dimer remained (Fig. 1E: WB with 3D6; Fig. 1F: silver-stained gel). We then serially diluted F9, either with or without βME treatment, and assayed it by the two o-ELISAs (Figs. 1A, B: black and red traces). βME treatment markedly reduced the signals of F9 in both o-ELISAs by more than 97% at most concentrations tested (Supp. Table S2). To exclude the possibility that βME itself interfered with the ELISA method, we applied the same βME treatment to a F11 (S26C monomer) fraction and to wt synthetic Aβ1-40 monomers. βME (which would be present at 0.1% or less final concentration in our ELISAs) had no effect on the measurement of these monomers as detected by two different Aβ 1-x ELISAs: we observed no significant difference between the signal curves of F11 (monomers) with and without βME, and these two curves completely overlapped those of the wt Aβ1-40 peptide with and without βME (Figs. 1C, D; and Supp. Figs. S2A, B).
Figure 1. 3D6/3D6B and NAB61/3D6B ELISAs specifically recognize Aβ dimers but not monomers.

Both NAB61/3D6B (A) and 3D6/3D6B (B) show wide dynamic ranges for quantifying SEC-purified Aβ S26C dimer (F9; black squares), but do not detect F9 reduced to monomers by βME (red triangles) or Aβ S26C monomers (SEC F11; blue inverse triangle). Conventional Aβ ELISAs 266/3D6B (C) and 3D6/4G8B (D) detect F11 (blue inverse triangles), β ME-treated F11 (purple diamond), and the wt synthetic Aβ40 (green circles) equally well, indicating that β ME does not interfere. Data are means ± SEMs (error bars too small to see on this log scale). (E, F) βME effectively dissociates the SDS-stable S26C dimers of F9 into monomers, with small amounts of residual dimers (<10%). At high concentrations, the F11 monomer fraction can be seen to have small amounts of dimers. (E: 3D6 WB; F: silver stain).
To confirm that the small signals detected by our o-ELISAs at high concentrations (0.01 ug/ml – 2.13 ug/ml) of the F11 (Aβ40-S26C monomer) fraction came from the small amount of oxidized dimers remaining in that fraction and not from the monomers themselves, we tested a SEC fraction of wt synthetic Aβ1-40 monomers in the two o-ELISAs. Neither the 3D6/3D6B nor the NAB61/3D6B o-ELISAs gave a signal above background for the wt Aβ1-40 monomers, even at a very high concentration of 2.13 ug/ml. (Supp. Figs. S2C, D). These results strongly suggest that the two o-ELISAs are specific for Aβ oligomers (and see also below). The same wt synthetic Aβ1-40 peptide gave a large dynamic range of signal when assayed by the 266/3D6B (1-x) ELISA or by another conventional ELISA that detects monomers (3D6/4G8B), and βME had no effect whatsoever on the wt Aβ1-40 monomer signals read by these two conventional ELISAs (Supp. Figs. S2A, B). Finally, we tested the o-ELISAs with the SEC dimer fraction (F9) of a tyrosine-10 synthetic Aβ dimer, which is covalently crosslinked and can never dissociate. This dityrosine Aβ dimer only aggregates into species higher than dimers at high uM concentrations. Our two o-ELISAs showed comparable standard curves using the di-tyrosine dimer (data not shown). Taken together, all of the above results strongly indicate that the 3D6/3D6B and the NAB61/3D6B ELISAs specifically measure the levels of bona fide, SEC-purified Aβ dimers in a linear fashion but are insensitive to monomers.
Next, we asked whether the two o-ELISAs can recognize oligomeric Aβ assemblies larger than dimers. To this end, SEC fractions F9 and F11 of Aβ40-S26C and the wt Aβ1-40 monomer SEC fraction were each incubated by shaking at 10 uM concentrations at 37°C overnight. SEC and WB of these peptides post-incubation showed that most of the Aβ had now shifted to higher MW aggregates, i.e. they now eluted principally in the void volume (F4 and F5) of the Superdex 75 column (Fig. 2A–C). When we tested the incubated Aβ peptides in the NAB61/3D6B and 3D6/3D6B o-ELISAs, we found that all three Aβ preparations now gave markedly increased signals (>2 log units higher than the original signals before incubation) (Figs. 2D, E). We conclude that the two o-ELISAs recognize multiple assembly sizes of still water-soluble Aβ oligomers, ranging from dimers to much higher MW pre-fibrillar (non-pelletable) oligomers.
Figure 2. The o-ELISAs recognize a large size range of synthetic Aβ oligomers, including dimers.
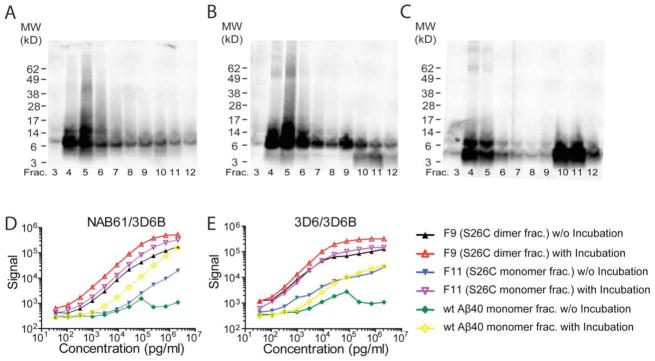
(A–C) WB (6E10+2G3+21F12) of the SEC elution profile of F9 (A), F11 (B) and wt synthetic Aβ40 monomer (C) that had each been incubated at 37°C overnight and then subjected to SEC (WB: 6E10+2G3+21F12). (D, E) Samples before and after 37°C incubation were serially diluted and assayed by NAB61/3D6B (D) or 3D6/3D6B (E); data are means ± SEM.
3.2. The o-ELISAs do not detect APPs-α or APPs-β
As APPs is a prominent secretory product in biological fluids, we asked whether our o-ELISAs show any cross-reactivity with APPs-α and -β. 3D6 antibody was previously shown not to recognize holoAPP or APPs but to sensitively recognize Aβ species with a free N-terminal aspartic acid[23]. Likewise, NAB61 antibody has been reported not to recognize either holoAPP or its C99 fragment[14]. We confirmed that neither o-ELISA detected recombinant APPs-α and APPs-β, even at highly supraphysiological concentrations of ~2.13 ug/ml (Supp. Figs. S3A, B). As a positive control, (Aβ S26C)2 (F9) was assayed simultaneously and again gave an excellent dynamic range in each o-ELISA (Supp. Figs. S3A, B).
3.3. The o-ELISAs detect multiple oligomeric forms of Aβ, including dimers, in soluble extracts of human brain
To assess whether the o-ELISAs can accurately quantify natural Aβ species from human tissue samples, we first prepared TBS-soluble (post-175,000g) extracts of confirmed AD brains (called AD-TBS; see Methods), subjected them to SEC on the Superdex 75 SEC column, and assayed the resultant fractions for Aβ using WB and three ELISAs: the conventional Aβ1-x ELISA (266/3D6B) and the NAB61/3D6B and 3D6/3D6B o-ELISAs. When we analyzed the SEC fractions by WB, we observed that >90% of the Aβ-immunoreactive signal eluted in the void volume fractions (i.e., F4–5) that size at >70 kDa (designated HMW Aβ hereafter) (Figs. 3A–C), which then ran as Aβ monomers and dimers on denaturing SDS-PAGE gels of the SEC fractions (Fig. 3D), just as reported previously using these methods[18]. (NB: The MW sizing of our Superdex 75 Aβ-rich fractions had been established previously using linear polydextran standards, not inappropriate globular protein standards[24]). There was very little Aβ immunoreactivity in the SEC fractions whose elution corresponds to the sizes of dimers (~8 kDa) and monomers (~4 kDa) (Fig. 3A–D). With the goal of attempting to disassemble these HMW Aβ species, we subjected the major void volume fraction (F4) to incubation at 37°C for 2 days in 50 mM ammonium acetate (pH 8.5). A WB showed that this 2-day incubation of the HMW Aβ had induced its dissociation into dimers and monomers; this was proven by a second SEC run that showed a dramatic decrease in the Aβ WB signals in the void volume fractions and a parallel increase in the dimer and monomer fractions (data not shown). ELISA analyses of the first-run SEC fractions by our o-ELISAs showed that the void volume fraction (F4) from the first run (i.e. before the 2-day incubation in buffer) produced robust signals in both o-ELISAs. However, after the 2-day incubation, the Aβ signals detectable by the two o-ELISAs in this void volume fraction from the first SEC run were dramatically decreased, while the monomeric Aβ signals detectable by the conventional 1-x monomer ELISA were correspondingly increased (Table 1). These experiments in which buffer-soluble Aβ species from AD cortex were separated and measured by ELISA confirm that a) the o-ELISAs are specific for oligomeric, not monomeric, Aβ species in human brain extracts, and b) that the o-ELISAs and a conventional Aβ ELISA can be used together to detect their inter-conversion.
Figure 3. The o-ELISAs detect multiple oligomeric forms of Aβ, including dimers, in soluble extracts of human brain tissue.

(A) NAB61/3D6B and (B) 3D6/3D6B o-ELISAs, and (C) 266/3D6B 1-x ELISA. By each method, ~80% of soluble Aβ species elute in the void volume of the Superdex 75 SEC column (i.e., F3–F5, corresponding to >70 kDa). (D) SEC elution profiles of AD-TBS extracts analyzed by WB (6E10+2G3+21F12); synthetic wt Aβ40 (2 ng) is a WB control.
Table 1.
Aβ amount detected by the ELISAs before vs. after incubation
| F4 before incubation (pg/ml) | F4 after incubation (pg/ml) | |
|---|---|---|
| 266/3D6B | 176 ± 16 | 2,578 ± 19 |
| NAB61/3D6B | 94,424 ± 4 | 58,740 ± 5 |
| 3D6/3D6B | 97,926 ± 6 | 18,264 ± 12 |
F4, void volume of SEC with 200 ul of AD-TBS.
SEC F4 was incubated at 37° overnight
We next assayed serial dilutions of AD TBS cortical extracts with the two o-ELISAs and the Aβ1-x ELISA. All three ELISAs showed highly linear dose-response curves (Figs. 4A, B). To confirm that the ELISA signals derived specifically from the Aβ species in the human brain extracts, we performed immunodepletion assays (Fig. 4C). An AD-TBS extract was first subjected to two sequential pre-clearing steps using protein A plus protein G agarose beads. Then, the supernatants of this pre-cleared AD-TBS were subjected to immunodepletions by the Aβ antibodies 3D6 or NAB61 or, as a negative control, by 3F3 (to the APP C-terminus). After removal of the antibody-bead complex by centrifugation, the resultant sups were assayed with the two o-ELISAs. We observed no significant decrease in the o-ELISA signals between the start material and the sups from the first and second pre-clearing steps (Fig. 4C). However, there were marked decreases in signal after immunodepletion with either 3D6 or NAB61; in contrast, there was a negligible decrease with the control 3F3 antibody. A single immunodepletion of the AD-TBS extract with the 3D6 antibody left just 8–9% of the starting Aβ signal as measured by either o-ELISA, whereas a single immunodepletion with the NAB61 antibody left ~28% of the starting signal (Fig. 4C). We checked these ELISA results by performing IP-WB on the same sups and their corresponding pellets. The IP-WB analysis agreed with the ELISA results: Aβ dimers and monomers were markedly decreased after pulling down with 3D6 or NAB61 and were accordingly recovered in the pellets, whereas no significant changes were seen after pre-clearing with protein A+G agarose beads or after precipitation with the control antibody 3F3 (Fig. 4C, bottom). As expected, immunodepletion of AD-TBS extracts with anti-human-Tau or anti-a synuclein antibodies did not decrease the o-ELISA signals, even though these IPs successfully precipitated their respective proteins (data not shown). Therefore, our o-ELISAs cannot detect oligomers of tau or α-synuclein in AD-TBS homogenates. We systematically performed Aβ immunodepletion assays on TBS extracts of 27 of 28 AD brain samples we acquired, i.e., all except the brain of a carrier of the APP D694N (“Iowa”) mutation, which was in low supply (Supp Table S3 lists clinical information on the 28 brains). The AD-TBS extracts were IPed with Aβ antiserum AW8 and the sups measured with the o-ELISAs and the 1-x ELISA; the mean signals were dramatically (97 ± 3%) decreased in all three ELISAs. Taken together, all of the above results indicate that the two o-ELISAs can detect natural oligomers present at pathophysiologically relevant concentrations in AD brain samples and that the signals are derived specifically from Aβ.
Figure 4. The o-ELISAs specifically detect Aβ species in AD TBS brain extracts.

(A–B) AD-TBS was serially diluted and assayed with the two o-ELISAs (A) and the conventional Aβ1-x ELISA (B). All three ELISAs showed highly linear concentration curves. (C) After two sequential pre-clearing steps, AD-TBS was subjected to IP by antibodies 3D6 or 3F3 or NAB61. As numbered, the various supernatants were assayed by o-ELISAs (bar graph) and the corresponding immunoprecipitates by WB. (WB: 6E10+2G3+21F12).
3.4. The o-ELISAs detect rising signals with age in APP transgenic mouse brains
To assess the utility of our assays in an AD mouse model, we applied the o-ELISAs to an analysis of thirty hAPP transgenic mice of increasing age (2–28 months) (J20 line: FAD mutations KM670/671NL + V717F)[6, 19]. The o-ELISA signals in the TBS brain extracts rose in an age-dependent fashion in the NAB61/3D6B and 3D6/3D6B assays (Supp. Fig. S4A, B). Wild-type mouse brain TBS extracts gave a very low signal in the NAB61/3D6B assay and no signal at all in the 3D6/3D6B assay (Supp. Fig. S4A, B). This age-related rise in o-ELISA signals in the J20 mice is consistent with our recently published IP/WB and conventional Aβ ELISA analyses of TBS extracts from the same mouse line[6, 25].
3.5. Quantifying the levels of soluble Aβ oligomers in brain tissues of AD patients vs. age-matched non-AD humans
Once the specificity of the o-ELISAs for natural and synthetic Aβ oligomers had been validated by all of the studies described above, we asked whether these assays could be used routinely to quantify such species in many human brain and CSF samples. We used the two o-ELISAs and the conventional Aβ1-x ELISA to quantify relative amounts of Aβ species in TBS-soluble extracts of cortex from 28 subjects, 13 of whom were clinically and neuropathologically diagnosed with AD (mean age 78, range 55–92 years) and 15 of whom were non-AD controls whose ages spanned the human lifespan (mean age 57, range 0.8–93 years) (Supp. Table S3). Notably, one of the 13 patients had AD caused by the APP D694N (“Iowa”) mutation and the other 12 were “sporadic” late-onset cases. We focused on TBS-soluble extracts, because the presence of dementia in AD has been documented to correlate best with the levels of aqueously soluble Aβ species[26–29]. The mean Aβ levels measured by the three ELISAs for the group of 15 controls and the group of 13 AD subjects are provided in Supp. Table S4.
Using the conventional Aβ1-x ELISA (266/3D6B), we obtained mean total Aβ levels of 165 ± 48 pg/g wet tissue in the 9 controls whose ages matched those of our AD patients (i.e., >55 years old) vs. 1,995 ± 1,217 pg/g in the 13 AD patients (Fig. 5B; see Supp. Table S3 for individual values). Despite a ~13-fold difference in these average Aβ levels, statistical significance was p=0.159 due to the large variability in the AD group and the relatively small sample sizes. We also assayed Aβ1-x as a function of age across all 28 subjects but observed no significant age-dependent trend in this mixed population (not shown).
Figure 5. Aqueously soluble Aβ oligomers are significantly increased in AD vs. control brains.

(A) IP/WB of TBS extracts (IP: AW8; WB: 6E10+2G3+21F12). Each lane represents an individual subject (black: control; red: AD). (B–D) Scatterplots for total Aβ (1-x) levels (B) and Aβ oligomer levels by NAB61/3D6B (C) or 3D6/3D6B (D) in human brain TBS extracts. Each dot represents data from 1 subject. Horizontal lines: means ± SEMs. p values from two-tailed Mann-Whitney t-tests.
We next assayed the 28 TBS-soluble cortical extracts by the two o-ELISAs. In the 13 AD brains, the NAB61/3D6B assay had a mean ± SEM of 4,555,869 ± 1,617,730 pg/g wet tissue, and the corresponding 3D6/3D6B assay values were 2,251,638 ± 863,175 pg/g wet tissue. In the 9 age-matched control cases, the respective values were 89,195 ± 37,691 pg/g and 57,425 ± 24,017 pg/g (Fig. 5C, D; see Supp. Table S3 for individual values). On average, the levels of o-Aβ in the 13 AD cases were ~50 times higher than those of the 9 age-matched controls using the NAB61/3D6B assay (p=0.0173), and ~39 times higher using the 3D6/3D6B assay (p=0.0259).
We correlated the o-ELISA results with those obtained by IP-WB. TBS extracts were IPed with antiserum AW8 to Aβ and blotted with combined mAbs 6E10 (to Aβ3-8), 2G3 (Aβ40 end-specific) and 21F12 (Aβ42 end-specific). This IP-WB analysis revealed Aβ monomers and SDS-stable dimers in the TBS-soluble cortical extracts from all AD subjects (Fig. 5A). Most TBS extracts from controls showed very little or no monomers and negligible amounts of dimers at the level of detection of our IP/WB assay. In general, the individual IP/WB results correlated well with those of the ELISAs (compare Fig. 5A with Supp. Table S3).
3.6. Assaying cerebrospinal fluid for the presence of Aβ oligomers
CSF samples from 89 humans [plus a commercially obtained CSF pool from numerous humans with unknown diagnoses (CSF#12)] were measured using the two o-ELISAs and the conventional Aβ1-x, 1-40 and 1-42 ELISAs (Supp. Table S5). The 266/3D6B, 2G3/3D6B and 21F12/3D6B ELISAs readily detected Aβ 1-x, 1-40 and 1-42 peptide levels, respectively, in all CSF samples. In sharp contrast, both the 3D6/3D6B and the NAB61/3D6B o-ELISAs did not detect any specific Aβ signal in all of the 90 CSF samples, even when certain CSF samples (#6 and #12) were concentrated 5-fold, except in one subject (CSF#27). However, the apparent signal from CSF#27 could not be diluted when diluting the sample two-fold, indicating we were not detecting true oAβ species (Supp. Table S5). This result nicely confirms the oligomer-specific nature of our o-ELISAa, since all CSF samples contained abundant monomers.
The lack o-Aβ signals by both o-ELISAs in all 90 CSF samples suggested several possibilities: 1) that Aβ oligomers are absent in these samples; 2) that the levels of oligomers are below the limit of detection of the o-ELISAs; or 3) that other proteins in CSF interfere with the o-ELISAs, thereby masking any oligomers from detection. To clarify this issue, we IPed 2 ml of CSF from each of 6 of our subjects with Aβ antiserum 1282 (Fig. 6A) and from each of 18 with Aβ antiserum AW7 (not shown) and blotted the precipitates with Aβ mAbs 6E10+2G3+21F12. This IP-WB method revealed that all 24 samples contained robust Aβ monomers but had no detectable SDS-stable dimers. Both the lack of Aβ signal by the o-ELISAs and the lack of Aβ dimers upon IP-WB suggest that there are very low (< 39 pg/ml) or no Aβ oligmers in human CSF. To make low sensitivity a less likely explanation, we increased the CSF volume from 2 ml to 5 ml and performed a first IP with 3D6. Even then, we failed to see any dimers by IP/WB (Fig. 6B). Note that in contrast to the absence of dimers, the Aβ monomers present in the 5 ml CSF samples gave a robust signal even in a second IP with an Aβ antiserum (Fig. 6B). We likewise failed to detect dimers or monomers in 5 ml of CSF IPed with NAB61, and a strong Aβ monomer band was brought down by a second IP with the Aβ antiserum (not shown). This result confirms the oligomer specificity of NAB61. To rule out the possibility that that the failure to detect Aβ oligomers by the o-ELISAs was due to other molecules in CSF interfering with the ELISA method, we spiked the (Aβ S26C)2 SEC fraction (F9) into human CSF, performed serial dilutions and assayed them by the two o-ELISAs (Figs. 6C, D). The ELISA curves generated from these spiked-in CSF samples overlapped perfectly with the curves from the same F9 fraction in standard specimen diluent in both o-ELISAs. An amount of (Aβ S26C)2 as low as ~39 pg per ml of human CSF could be detected by the assays. Finally, aliquots of AD-TBS extracts containing natural human Aβ dimers and monomers were spiked into 1 ml of either artificial CSF or human CSF (#12). IP/WB analysis on these spiked-in samples revealed indistinguishable dimer and monomer bands when spiked into human CSF or into artificial CSF (Supp. Fig. S5). Taken together, these results argue strongly against the possibility of other proteins in human CSF interfering with Aβ oligomer detection in our o-ELISAs.
Figure 6. Lack of Aβ oligomer signals in CSF of representative control and AD subjects by IP/WB and o-ELISAs.

IP/WB analysis on (A) 2 ml CSF (IP: Aβ antiserum R1282; WB: 6E10+2G3+21F12), and (B) 5 ml CSF (1st IP: 3D6 (3ug/ml), 2nd IP: Aβ antiserum R1282; WB: 6E10+2G3+21F12). Clinical information is in Table 3. (C–D) S26C dimer fraction (F9) was spiked into a human CSF, serially diluted, and assayed by NAB61/3D6B (C) and 3D6/3D6B (D) o-ELISAs. Readings generated from F9 diluted in CSF (red squares) show a perfect overlap with those of F9 diluted in specimen diluent (black circles). Data are means ± SEMs.
3.7. Assaying plasmas for the presence of Aβ oligomers
We searched for Aβ oligomers in human plasma. Samples from 6 patients with clinical AD and 5 normal subjects, plus three pooled plasma samples from numerous humans with unknown diagnoses (Supp. Table S6), were assayed by the o-ELISAs and the conventional Aβ1-x ELISA. The Aβ1-x ELISA suggested that all 11 plasma samples contained low amounts of Aβ1-x peptides, with no significant differences between AD and controls (not shown). The 3D6/3D6B o-ELISA gave plasma readings that were on average much lower than those of the 1-x ELISA, and the NAB61/3D6B o-ELISA gave mostly zero signals, with the exception of two samples (one from subject #5 (AD) and one from subject #13 (non-AD control). Importantly, diluting each of the two positive plasmas just two-fold caused a very marked (many-fold) decrease in the oELISA readings, indicating the assays were not detecting true oAβ species (data not shown). To determine whether interfering molecules exist in plasma that preclude reliable detection of oligomers by the o-ELISAs, we spiked the (Aβ S26C)2 SEC fraction (F9) into two human plasmas -- one from an AD patient and one from a control -- and performed serial dilutions (Supp. Figs. S6A, B). Compared to the standard curves generated from the same F9 fraction diluted into plain specimen diluent, the ELISA curves generated from the spiked-in plasmas showed a significant reduction in signal (note the log scale). The NAB61/3D6B o-ELISA showed up to a 93% reduction in the signal of F9 in AD plasma and a 78% reduction in control plasma, with great variability within any one concentration (Supp. Fig. S6A), and the 3D6/3D6B o-ELISA failed to display a dose-dependent linear curve (Supp. Fig. S6B). Repeating this experiment with 3 additional human plasmas again indicated that molecules in human plasma interfere with the o-ELISAs. To confirm furher this interference in plasma, we spiked aliquots of AD-TBS extracts containing natural human Aβ dimers and monomers into human plasma. IP/WB analysis on these spiked-in samples revealed decreased detection of the dimer and monomer bands compared to spiking into TBS buffer. We conclude that any Aβ oligomers that might exist in human plasma cannot be detected by these o-ELISAs due to interfering substances.
4. Discussion
Oligomeric assemblies of Aβ induce altered neuronal structure and function and appear to play an important role early in the development of AD [30]. SDS-stable Aβ oligomers, including dimers, trimers, dodacamers and other relatively low MW assemblies, have been observed by western blotting in AD brains[18, 26, 27, 29, 31–33]. SDS-stable Aβ oligomers can be detected in the brains of APP transgenic mouse models at or before the age at which deficits in learning and memory begin to emerge [25, 34, 35]. These and other studies encourage the inclusion of Aβ oligomers in the search for useful biomarkers of AD. Most Aβ ELISAs reported to date were designed to detect monomers, and their sensitivity for detecting natural oligomers has not been established. Methods that allow one to observe the quality (type) of Aβ species in a sample, such as Western blotting, usually require denaturation to see the peptides, and this could disassemble both large and small oligomers to monomers, since most Aβ oligomers in brain are not known to be covalently bonded. Further, Western blotting is not amenable to high-throughput measurement of many samples. Therefore, we sought to develop an assay that selectively measures natural Aβ oligomers of various sizes. Here, we report two o-ELISAs in which we fully validated specificity for oligomeric Aβ and then showed their utility when applied to human and mouse brain tissues.
Two general approaches have been used to develop ELISAs that could be specific for Aβ oligomers. One uses putatively aggregate-specific antibodies, i.e., antibodies directed against non-linear (non-contiguous) epitopes of Aβ that can selectively recognize assemblies of two or more monomers. The other uses a single monoclonal antibody for both antigen capture and detection. In the first category, a literature screen revealed several reports of antibodies believed to be specific for aggregated Aβ that were used in ELISAs[8, 12–15, 36]. However, only three such ELISAs[8, 15, 36] were tested on biological samples. Englund et al[8] used a mAb158 sandwich ELISA to analyze the media of transfected cells and transgenic mouse brains, and Lord et al[36] then reported that this ELISA detected Aβ protofibrils in brains of tg mice expressing the “Arctic” and “Swedish” hAPP mutations. Another study focused on highly aggregated forms of Aβ present in SDS-solubilized extracts of AD brain[15]. We are unaware that these or other ELISAs relying on aggregate-specific antibodies have been reported to measure aqueously soluble forms of Aβ (e.g., in simple buffer extracts of brain homogenates), which are thought to contain the most bioactive forms of Aβ, or in human fluids.
Regarding the second approach of using a single antibody for both capture and detection, we found only 4 published studies to date[8, 9, 11, 36]. Fukumoto et al.[9] used BAN50 (to Aβ residues 1–16) in a sandwich ELISA, but they reported that their assay could only detect very small amounts of high-molecular weight (HMW) oligomers of synthetic Aβ sized at ~40–200 kDa by SEC, the identities of which were not independently confirmed (e.g., by light scattering or native PAGE). Low MW oligomers (dimers through hexamers) were much more abundant in the synthetic oligomer mixture used as their ELISA standard, but these by themselves gave no signal in the assay. This ELISA was used to assay human CSF, and the levels of the putative HMW Aβ oligomers in each subject were expressed in arbitrary units based on monomer-equivalents in the oligomer standard rather than in actual units of defined Aβ oligomers, making comparisons of oligomer content among CSF samples problematic. This BAN50-BAN50 ELISA was not validated on AD and control brain extracts, nor was the Aβ specificity of the oligomer signal in CSF confirmed by immunodepletion studies.
As mentioned above, Englund et al[8] developed an ELISA using the same conformational antibody for capture and detection, i.e., mAb158 generated against synthetic protofibrils composed of the “Arctic” (E22G) Aβ peptide. The authors stated that the synthetic protofibrils serially diluted to establish a standard for the ELISA were of uncertain size and molarity, so they expressed their protofibril concentrations based on the MW of an Aβ monomer. This 158-158 ELISA detected synthetic protofibrils (estimated to be >100 kDa) much better than it did low MW Aβ mixtures (estimated to be 4–20 kDa). Englund et al used this ELISA to analyze the media of transfected cells and transgenic mouse brains expressing the Swedish APP mutation or the Swedish plus Arctic APP mutations together. The ELISA detected the presence of Aβ protofibrils in both the cell media and mouse brains, and higher protofibril levels were seen in the Arctic-Swedish than Swedish samples. The authors showed that mAb158 does not bind to APP or to other types of amyloids. They did not report immunodepletions to confirm the specificity of the ELISA signals nor assays on human brain samples. A preliminary study from our group[11] described sandwich ELISAs using monoclonal antibodies 82E1 or 3D6. These assays detected signals in human brain homogenates, with higher levels seen in AD than control brains. Human plasmas were also assayed, but many were near or at the lower limit of detection. At that time, we derived only relative oligomer values, as our standard curve employed a synthetic Aβ1-40 peptide that had been quantified by conventional monomer-directed ELISAs. Again, immunodepletion was not used to confirm the specificity of the signals.
To address these drawbacks of published o-ELISAs, we undertook the current experiments to obtain ELISAs whose specificity and sensitivity on both synthetic and natural oligomers were validated in several ways. The o-ELISAs we report here can selectively quantify synthetic and natural oligomers of human Aβ over a wide analytical range. The following observations validate their oligomer specificity. First, the o -ELISAs detected increasing concentrations of pure synthetic (Aβ S26C)2 in a linear and highly reproducible fashion. Treating these cysteine-bonded dimers with βME quantitatively dissociated them to monomers and resulted in a marked (>97%) loss of the o-ELISA signal. The very small residual signal was shown to be due to trace remaining amounts of disulfide-bonded dimers. Second, the o-ELISAs give no signal with wt Aβ40 monomers and gave 1–3% of the signal of (Aβ S26C)2 with the S26C monomer fraction. Third, overnight incubation of (Aβ S26C)2 or wt Aβ40 peptides at 37°C, which polymerized them into higher aggregates (confirmed by SEC), markedly increased the o-ELISA signals, indicating that the assays detect large but still buffer-soluble aggregates far greater than dimers. Fourth, we confirmed that the o-ELISA signals emanate from Aβ and not other soluble cleavage products of APP that contain part or all of the Aβ sequence (APPsa, APPsβ). Fifth, the o-ELISAs specifically detected natural Aβ oligomers in soluble human brain extracts. One round of immunodepletion of Aβ from the TBS extracts (confirmed by WB) reduced the o-ELISA signals to ~8% (3D6) or ~28% (NAB61) of their original values. Sixth, hAPP transgenic mouse brain extracts showed increasing signals over an age range of 2–28 months as brain oligomer levels rise, whereas brain extracts of wt littermates had no detectable signal. We conclude that the two o-ELISAs can specifically quantify a wide size range of oligomeric assemblies of soluble Aβ, even in complex biological samples. The absolute sensitivities of the assays for oAβ were as low as ~6 pg/ml for 3D6/3D6B and ~12 pg/ml for NAB61/3D6B after optimization, based on quantifying the synthetic Aβ dimer standard by amino acid analysis, a preferred method to determine rigorously an absolute protein concentration, allowing us to establish accurate oligomer sensitivities of our ELISAs. It should be noted that our o-ELISAs, in which 3D6B detects Aβ species having a free N-terminal aspartic acid, cannot quantify N-terminally modified Aβ species such as pE3-42, which potentially have a pathogenic role in AD.
Having validated the specificity and sensitivity of the o-ELISAs, we applied them to samples of human brain tissue and biological fluids. Previously, dot blot assays suggested that the levels of soluble Aβ oligomers were on average up to ~70-fold higher in extracts of AD brains than age-matched controls[31]. We measured Aβ oligomer levels in TBS-soluble extracts of 13 AD and 15 non-AD control subjects (9 of the 15 were age-matched). Soluble oligomer levels were ~50-fold higher in our AD than control brains by NAB61/3D6B (p=0.0173) and ~39-fold higher by 3D6/3D6B (p=0.0295) (Fig. 5). One previous study [15] used a sandwich ELISA (either of two conformational antibodies were combined with a polyclonal Aβ1-42 antibody) to assay Aβ oligomers in SDS-solubilized brain homogenates and found that oligomer levels were significantly higher in AD than control cases. Semi-quantitative Western blotting has suggested that the levels of relatively low MW oligomers in buffer-soluble extracts of human brain correlate with the presence of AD dementia [18, 29, 37, 38]. Oligomer-selective ELISAs such as those we report could be useful for quantitative neuropathological analyses relevant to both diagnosis and mechanistic studies of AD.
We applied the two o-ELISAs to search for Aβ oligomers in human CSF but failed to detect any specific signals in >90 subjects. We verified in some of the samples that there were no detectable SDS-stable Aβ assemblies by IP-WB. Importantly, we excluded via spiking experiments that these negative results were due to molecules in CSF that interfere with the assays. Our finding of no measurable levels (i.e., lower than 6 pg/ml) of oligomeric Aβ in human CSF is in contrast to a few prior reports of higher Aβ oligomer levels in the CSF of AD than control subjects[9, 21, 39–43]. Vigo-Pelfrey et al[39] identified three Aβ oligomer forms, stable trimers of (Asp1-Met35)3 and (His6-Ala42)3 and a stable dimer of (Asp1- Val40)2, by purifying Aβ by HPLC from 3 liters of pooled human CSF and performing laser atomic desorption mass spectrometry. Pitschke et al.[41] reported apparent Aβ aggregates in CSF by fluorescence correlation spectroscopy (FCS) that used labeled synthetic Aβ peptides as “seeds” for polymerization; they stated that aggregate formation could be detected at concentrations down to 100 nM. Georganopoulou et al.[42] used a nanoparticle-based biobarcode assay to detect oligomers, using as a calibration curve assemblies of synthetic Aβ1-42 incubated at 7°C for 24 h (“ADDLs”). They observed very low amounts of ADDL signal in CSF (<4.3 pg/ml). The biobarcode assay they reported requires specialized equipment not available in most labs, and it used a polyclonal antiserum not available in quantities needed for large-scale, multicenter validation studies. Also, this assay used polyclonal and monoclonal antibodies raised against synthetic ADDLs that were not proven to be entirely specific to oligomers of Aβ. Santos et al.[43] reported an assay for detection of oligomeric and fibrillar structures based on multiparametric analysis of data obtained by flow cytometry and florescence resonance energy transfer (FRET). Its sensitivity was determined by titration of in vitro-assembled synthetic Aβ fibrils, and the units were FRET events. The authors reported detection of Aβ oligomers in CSFs from 174 non-demented individuals with various neurological disorders. The data showed a large variation in Aβ oligomers and a weak correlation between age and Aβ oligomer levels. Walsh et al.[21] found that 15 of 56 human CSF samples showed small amounts of SDS-stable dimers (~8 kDa) by IP-WB. Klyubin et al[44] reported that 6 CSF samples containing SDS-stable Aβ dimers by IP/WB inhibited hippocampal LTP in living rats and that acute systemic infusion of a mAb to Aβ prevented this.
In summary, we have established two novel o-ELISAs that selectively quantify soluble human Aβ oligomers over a wide range of sizes. The mean levels of oligomeric Aβ were markedly (>1,000-fold) higher than those of monomers in buffer-soluble extracts of AD cortex, signifying that the vast majority of soluble Aβ in AD brain is in oligomeric form. Soluble oligomer levels were significantly higher in AD than age-matched non-AD brains. We obtained no evidence of soluble Aβ oligomers in 90 human CSF samples, despite the fact that the o-ELISAs detected synthetic and natural (AD brain) human oligomers spiked into CSF at very low picogram concentrations. This unexpected finding provides a new biological insight that the relatively hydrophobic Aβ oligomers may have very brief or no dwell times in the aqueous environment of the CSF and may be rapidly adsorbed onto cellular membranes. As detailed above, we believe these o-ELISAs have significant advantages over previous assays for oligomeric Aβ and provide a useful and sensitive method to quantify it in multiple biological samples.
Supplementary Material
Figure S1. Identification of dimer and monomer fractions used to develop o-ELISAs. (A) Chromatography of oxidized Aβ S26C (black curve), reduced Aβ S26C (red curve) or wt Aβ40 (blue dashed curve). (B) Silver stained gels of the indicated SEC fractions.
Figure S2. 3D6/3D6B and NAB61/3D6B o-ELISAs specifically recognize Aβ dimers, not monomers. (A, B) βME does not interfere with the ELISAs. Synthetic wt Aβ40 monomer SEC fractions were treated (or not) with 3% βME and serially diluted. Readings from untreated Aβ (black squares) and βME-treated Aβ (red triangles) overlap perfectly in both 266/3D6B (C) and 3D6/4G8B (D) o-ELISAs. Data are means ± SEMs. (C, D) Neither NAB61/3D6B (C) nor 3D6/3D6B (D) ELISAs detect wt Aβ40 monomers (black squares and red triangles), even at the highest concentrations (2.13 ug/ml). Aβ S26C dimer fraction (F9) was assayed simultaneously as a positive control (blue inverse triangles).
Figure S3. The o-ELISAs do not detect APPs-a and -β. Purified recombinant APPs-a and -β combined were assayed at increasing concentrations by NAB61/3D6B (A) and 3D6/3D6B (B). Neither o-ELISA gave a signal above background for the APPs-a and -β, even at high concentrations (2.13ug/ml). SEC-purified F9 (Aβ S26C dimer SEC fraction) was run in parallel as a positive control. Data are means ± SEMs.
Figure S4. The o-ELISAs detect rising levels of TBS-soluble brain Aβ in hAPP transgenic mice of increasing age. TBS extracts of the brains of J20 transgenic mice were prepared at the indicated ages. Cortical levels of soluble Aβ oligomers were quantified by NAB61/3D6B (A) or 3D6/3D6B (B). Each data point represents mean level ± SEM of duplicate samples from one mouse.
Figure S5. IP/WB of spiked-in AD brain dimers confirms lack of interfering substances in human CSF. AD-TBS extract (at various indicated volumes (ul)) were spiked into 1 ml of artificial CSF or human CSF #12, and IP/WB performed (IP: Aβ antiserum R1280 (1:75); WB: 2G3+21F12+6E10). Natural dimer and monomer signals spiked into human CSF are equal to those spiked into artificial CSF.
Figure S6. Interfering molecules in human plasma preclude reliable detection of Aβ by the o-ELISAs. Aβ S26C dimer SEC fraction (F9) was spiked into plasma from an AD patient (red squares) or a non-AD control subject (green triangles) or into regular specimen diluent (black circles), serially diluted, and assayed with NAB61/3D6B (A) or 3D6/3D6B (B). Data are means ± SEMs.
Acknowledgments
We thank members of the Selkoe and Walsh labs for helpful advice and comments. Supported by NIH grants AG027443 (DJS and DW) and AG012749 (DJS). DJS is a consultant and director of Elan and a consultant to Satori.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Motter R, Vigo-Pelfrey C, Kholodenko D, Barbour R, Johnson-Wood K, Galasko D, et al. Reduction of beta-amyloid peptide 42 in the cerebrospinal fluid of patients with Alzheimer’s disease. Ann Neurol. 1995;38:643–648. doi: 10.1002/ana.410380413. [DOI] [PubMed] [Google Scholar]
- 2.Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006;59:512–519. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- 3.Fagan AM, Head D, Shah AR, Marcus D, Mintun M, Morris JC, et al. Decreased cerebrospinal fluid Abeta(42) correlates with brain atrophy in cognitively normal elderly. Ann Neurol. 2009;65:176–183. doi: 10.1002/ana.21559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, et al. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol. 2009;65:403–413. doi: 10.1002/ana.21610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol. 2007;64:343–349. doi: 10.1001/archneur.64.3.noc60123. [DOI] [PubMed] [Google Scholar]
- 6.Hong S, Quintero-Monzon O, Ostaszewski BL, Podlisny DR, Cavanaugh WT, Yang T, et al. Dynamic analysis of amyloid beta-protein in behaving mice reveals opposing changes in ISF versus parenchymal Abeta during age-related plaque formation. J Neurosci. 2011;31:15861–15869. doi: 10.1523/JNEUROSCI.3272-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.El-Agnaf OM, Mahil DS, Patel BP, Austen BM. Oligomerization and toxicity of beta-amyloid-42 implicated in Alzheimer’s disease. Biochem Biophys Res Commun. 2000;273:1003–1007. doi: 10.1006/bbrc.2000.3051. [DOI] [PubMed] [Google Scholar]
- 8.Englund H, Sehlin D, Johansson AS, Nilsson LN, Gellerfors P, Paulie S, et al. Sensitive ELISA detection of amyloid-beta protofibrils in biological samples. J Neurochem. 2007;103:334–345. doi: 10.1111/j.1471-4159.2007.04759.x. [DOI] [PubMed] [Google Scholar]
- 9.Fukumoto H, Tokuda T, Kasai T, Ishigami N, Hidaka H, Kondo M, et al. High-molecular-weight beta-amyloid oligomers are elevated in cerebrospinal fluid of Alzheimer patients. FASEB J. 2010;24:2716–2726. doi: 10.1096/fj.09-150359. [DOI] [PubMed] [Google Scholar]
- 10.Howlett DR, Perry AE, Godfrey F, Swatton JE, Jennings KH, Spitzfaden C, et al. Inhibition of fibril formation in beta-amyloid peptide by a novel series of benzofurans. Biochem J. 1999;340 (Pt 1):283–289. [PMC free article] [PubMed] [Google Scholar]
- 11.Xia W, Yang T, Shankar G, Smith IM, Shen Y, Walsh DM, et al. A specific enzyme-linked immunosorbent assay for measuring beta-amyloid protein oligomers in human plasma and brain tissue of patients with Alzheimer disease. Arch Neurol. 2009;66:190–199. doi: 10.1001/archneurol.2008.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 13.Barghorn S, Nimmrich V, Striebinger A, Krantz C, Keller P, Janson B, et al. Globular amyloid beta-peptide oligomer - a homogenous and stable neuropathological protein in Alzheimer’s disease. J Neurochem. 2005;95:834–847. doi: 10.1111/j.1471-4159.2005.03407.x. [DOI] [PubMed] [Google Scholar]
- 14.Lee EB, Leng LZ, Zhang B, Kwong L, Trojanowski JQ, Abel T, et al. Targeting amyloid-beta peptide (Abeta) oligomers by passive immunization with a conformation-selective monoclonal antibody improves learning and memory in Abeta precursor protein (APP) transgenic mice. J Biol Chem. 2006;281:4292–4299. doi: 10.1074/jbc.M511018200. [DOI] [PubMed] [Google Scholar]
- 15.van Helmond Z, Heesom K, Love S. Characterisation of two antibodies to oligomeric Abeta and their use in ELISAs on human brain tissue homogenates. J Neurosci Methods. 2009;176:206–212. doi: 10.1016/j.jneumeth.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 16.Hu NW, Smith IM, Walsh DM, Rowan MJ. Soluble amyloid-beta peptides potently disrupt hippocampal synaptic plasticity in the absence of cerebrovascular dysfunction in vivo. Brain. 2008;131:2414–2424. doi: 10.1093/brain/awn174. [DOI] [PubMed] [Google Scholar]
- 17.O’Nuallain B, Freir DB, Nicoll AJ, Risse E, Ferguson N, Herron CE, et al. Amyloid beta-protein dimers rapidly form stable synaptotoxic protofibrils. J Neurosci. 2010;30:14411–14419. doi: 10.1523/JNEUROSCI.3537-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, et al. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teunissen CE, Petzold A, Bennett JL, Berven FS, Brundin L, Comabella M, et al. A consensus protocol for the standardization of cerebrospinal fluid collection and biobanking. Neurology. 2009;73:1914–1922. doi: 10.1212/WNL.0b013e3181c47cc2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walsh DM, Tseng BP, Rydel RE, Podlisny MB, Selkoe DJ. Detection of intracellular oligomers of amyloid β-protein in cells derived from human brain. Biochemistry. 2000;39:10831–10839. doi: 10.1021/bi001048s. [DOI] [PubMed] [Google Scholar]
- 22.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 23.Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, et al. Amyloid precursor protein processing and A beta42 deposition in a transgenic mouse model of Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walsh DM, Townsend TM, Podlisny MB, Shankar GM, Fadeeva J, El-Agnaf O, et al. Certain inhibitors of synthetic Aβ fibrillogenesis block oligomerization of natural Aβ and thereby rescue long term potentiation. J Neurosci. 2005;25:2455–2462. doi: 10.1523/JNEUROSCI.4391-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shankar GM, Leissring MA, Adame A, Sun X, Spooner E, Masliah E, et al. Biochemical and immunohistochemical analysis of an Alzheimer’s disease mouse model reveals the presence of multiple cerebral Abeta assembly forms throughout life. Neurobiol Dis. 2009;36:293–302. doi: 10.1016/j.nbd.2009.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuo Y-M, Emmerling MR, Vigo-Pelfrey C, Kasunic TC, Kirkpatrick JB, Murdoch GH, et al. Water-soluble Aβ (N-40, N-42) oligomers in normal and Alzheimer disease brains. J Biol Chem. 1996;271:4077–4081. doi: 10.1074/jbc.271.8.4077. [DOI] [PubMed] [Google Scholar]
- 27.McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, et al. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 28.Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, et al. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McDonald JM, Savva GM, Brayne C, Welzel AT, Forster G, Shankar GM, et al. The presence of sodium dodecyl sulphate-stable Abeta dimers is strongly associated with Alzheimer-type dementia. Brain. 2010;133:1328–1341. doi: 10.1093/brain/awq065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Selkoe DJ. Resolving controversies on the path to Alzheimer’s therapeutics. Nat Med. 2011;17:1060–1065. doi: 10.1038/nm.2460. [DOI] [PubMed] [Google Scholar]
- 31.Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, et al. Alzheimer’s disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci U S A. 2003;100:10417–10422. doi: 10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, et al. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A. 2009;106:4012–4017. doi: 10.1073/pnas.0811698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pham E, Crews L, Ubhi K, Hansen L, Adame A, Cartier A, et al. Progressive accumulation of amyloid-beta oligomers in Alzheimer’s disease and in amyloid precursor protein transgenic mice is accompanied by selective alterations in synaptic scaffold proteins. Febs J. 2010;277:3051–3067. doi: 10.1111/j.1742-4658.2010.07719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci. 2001;21:372–381. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 36.Lord A, Gumucio A, Englund H, Sehlin D, Sundquist VS, Soderberg L, et al. An amyloid-beta protofibril-selective antibody prevents amyloid formation in a mouse model of Alzheimer’s disease. Neurobiol Dis. 2009;36:425–434. doi: 10.1016/j.nbd.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 37.Bao F, Wicklund L, Lacor PN, Klein WL, Nordberg A, Marutle A. Different beta-amyloid oligomer assemblies in Alzheimer brains correlate with age of disease onset and impaired cholinergic activity. Neurobiol Aging. 2012;33:825 e821–813. doi: 10.1016/j.neurobiolaging.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 38.Sokolow S, Henkins KM, Bilousova T, Miller CA, Vinters HV, Poon W, et al. AD synapses contain abundant Abeta monomer and multiple soluble oligomers, including a 56-kDa assembly. Neurobiol Aging. 2011 doi: 10.1016/j.neurobiolaging.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vigo-Pelfrey C, Lee D, Keim P, Lieberburg I, Schenk DB. Characterization of beta-amyloid peptide from human cerebrospinal fluid. Journal of neurochemistry. 1993;61:1965–1968. doi: 10.1111/j.1471-4159.1993.tb09841.x. [DOI] [PubMed] [Google Scholar]
- 40.Ida N, Hartmann T, Pantel J, Schroder J, Zerfass R, Forstl H, et al. Analysis of heterogeneous A4 peptides in human cerebrospinal fluid and blood by a newly developed sensitive Western blot assay. J Biol Chem. 1996;271:22908–22914. doi: 10.1074/jbc.271.37.22908. [DOI] [PubMed] [Google Scholar]
- 41.Pitschke M, Prior R, Haupt M, Riesner D. Detection of single amyloid beta-protein aggregates in the cerebrospinal fluid of Alzheimer’s patients by fluorescence correlation spectroscopy. Nature medicine. 1998;4:832–834. doi: 10.1038/nm0798-832. [DOI] [PubMed] [Google Scholar]
- 42.Georganopoulou DG, Chang L, Nam JM, Thaxton CS, Mufson EJ, Klein WL, et al. Nanoparticle-based detection in cerebral spinal fluid of a soluble pathogenic biomarker for Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:2273–2276. doi: 10.1073/pnas.0409336102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Santos AN, Torkler S, Nowak D, Schlittig C, Goerdes M, Lauber T, et al. Detection of amyloid-beta oligomers in human cerebrospinal fluid by flow cytometry and fluorescence resonance energy transfer. Journal of Alzheimer’s disease: JAD. 2007;11:117–125. doi: 10.3233/jad-2007-11114. [DOI] [PubMed] [Google Scholar]
- 44.Klyubin I, Betts V, Welzel AT, Blennow K, Zetterberg H, Wallin A, et al. Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J Neurosci. 2008;28:4231–4237. doi: 10.1523/JNEUROSCI.5161-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Identification of dimer and monomer fractions used to develop o-ELISAs. (A) Chromatography of oxidized Aβ S26C (black curve), reduced Aβ S26C (red curve) or wt Aβ40 (blue dashed curve). (B) Silver stained gels of the indicated SEC fractions.
Figure S2. 3D6/3D6B and NAB61/3D6B o-ELISAs specifically recognize Aβ dimers, not monomers. (A, B) βME does not interfere with the ELISAs. Synthetic wt Aβ40 monomer SEC fractions were treated (or not) with 3% βME and serially diluted. Readings from untreated Aβ (black squares) and βME-treated Aβ (red triangles) overlap perfectly in both 266/3D6B (C) and 3D6/4G8B (D) o-ELISAs. Data are means ± SEMs. (C, D) Neither NAB61/3D6B (C) nor 3D6/3D6B (D) ELISAs detect wt Aβ40 monomers (black squares and red triangles), even at the highest concentrations (2.13 ug/ml). Aβ S26C dimer fraction (F9) was assayed simultaneously as a positive control (blue inverse triangles).
Figure S3. The o-ELISAs do not detect APPs-a and -β. Purified recombinant APPs-a and -β combined were assayed at increasing concentrations by NAB61/3D6B (A) and 3D6/3D6B (B). Neither o-ELISA gave a signal above background for the APPs-a and -β, even at high concentrations (2.13ug/ml). SEC-purified F9 (Aβ S26C dimer SEC fraction) was run in parallel as a positive control. Data are means ± SEMs.
Figure S4. The o-ELISAs detect rising levels of TBS-soluble brain Aβ in hAPP transgenic mice of increasing age. TBS extracts of the brains of J20 transgenic mice were prepared at the indicated ages. Cortical levels of soluble Aβ oligomers were quantified by NAB61/3D6B (A) or 3D6/3D6B (B). Each data point represents mean level ± SEM of duplicate samples from one mouse.
Figure S5. IP/WB of spiked-in AD brain dimers confirms lack of interfering substances in human CSF. AD-TBS extract (at various indicated volumes (ul)) were spiked into 1 ml of artificial CSF or human CSF #12, and IP/WB performed (IP: Aβ antiserum R1280 (1:75); WB: 2G3+21F12+6E10). Natural dimer and monomer signals spiked into human CSF are equal to those spiked into artificial CSF.
Figure S6. Interfering molecules in human plasma preclude reliable detection of Aβ by the o-ELISAs. Aβ S26C dimer SEC fraction (F9) was spiked into plasma from an AD patient (red squares) or a non-AD control subject (green triangles) or into regular specimen diluent (black circles), serially diluted, and assayed with NAB61/3D6B (A) or 3D6/3D6B (B). Data are means ± SEMs.