

Abstract
Large scale dynamics within the Michaelis complex mimic of Bacillus stearothermophilus thermophylic lactate dehydrogenase, bsLDH•NADH•oxamate, were studied with site specific resolution by laser induced temperature jump relaxation spectroscopy having a time resolution of 20 ns. NADH emission and Trp emission from the wild type and a series of single-tryptophan bsLDH mutants, with the tryptophan positions at different distances from the active site, were used as reporters of evolving structure in response to the rapid change in temperature. Several distinct dynamical events were observed on the ms - μs time-scale involving motion of atoms spread over the protein, some occurring concomitantly or nearly concomitantly with structural changes at the active site. This suggests that a large portion of the protein-substrate complex moves in a rather concerted fashion to bring about catalysis. The catalytically important surface loop undergoes two distinct movements, both needed for a competent enzyme. Our results also suggest that what is called `loop motion' is not just localized to the loop and active site residues. Rather, it involves the motion of atoms spread over the protein, even some quite distal from the active site. How these results bear on catalytic mechanism of bsLDH is discussed.
INTRODUCTION
The process of formation of a productive enzymic Michaelis complex is one of narrowing the conformational states of the enzyme-substrate system so that, in its search through all the accessible conformations, the system finds the transition state of the on-enzyme chemical reaction in a timely manner. The process involves numerous dynamical events, such as formation of an encounter complex between the substrate and enzyme with re-orientation of the ligand to fit the binding pocket, desolvation, and structural re-arrangements in and around the active site to accommodate the ligand and to establish proper contacts necessary for catalysis. These steps include atomic motions and conformational changes on various scales, occurring in a very wide time range, from femtoseconds to milliseconds.1–4 The details of these changes and motions are essential for understanding the mechanisms of catalysis, but in general, our knowledge about these dynamics within enzyme-substrate systems is very limited. It is clear from earlier studies that enzymes (proteins) exist in an ensemble of conformations, some of which are competent to bind their ligands while others bind poorly or not at all (e.g. for LDH5–7). It has been shown directly that conformational changes occur within the ensemble of enzyme/substrate Michaelis complexes on various timescale from femtoseconds and picoseconds through milliseconds and slower.8–10 Here we investigate, using laser-induced temperature-jump spectroscopy, the dynamics of NADH and oxamate binding to thermophylic Bacillus stearothermophilus lactate dehydrogenase (bsLDH) and the dynamics within the bsLDH•NADH•oxamate complex.
L-lactate dehydrogenase, EC 1.1.1.27, (LDH) catalyzes oxidation of lactate by NAD+ to produce pyruvate and NADH. In LDH, the substrate binding pocket is sequestered inside the protein about 10 Å from the surface.11,12 Based on several X-ray crystallographic data, oxamate is placed near the nicotinamide ring of the NADH and the following key protein residues, His195, Arg106 and Arg171 (Scheme 1). The C2=O bond of oxamate forms hydrogen bonds with His195 and Arg106 while the C1OO− forms a salt bridge with Arg171,13–15 helping to position the substrate. Clarke et al.16 have also shown that Arg173, which lies on the same helix as Arg171, binds the allosteric effector, fructose 1,6–biphosphate (FBP). As the substrate approaches the catalytic site, the following events take place: a catalytically key surface loop (residues 98–110) closes over the ligand bringing residue Arg106 in hydrogen bond contact with ligand, water leaves the pocket, and the pocket geometry rearranges to allow for favorable interactions between the cofactor and the ligand that facilitate on-enzyme catalysis.6,17 The bsLDH protein is an interesting variant of LDH family with a wider temperature range of activity than the corresponding mesophiles. It has also been attractive for studies including site-directed mutations that may provide mechanistic information with residue-specific structural resolution.18
Scheme 1.
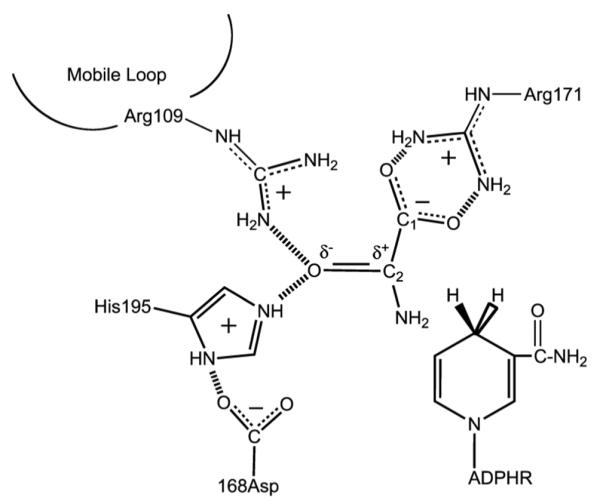
The active site contacts of pyruvate and NADH bound to LDH with key residues as determined by X-ray crystallography. The reaction catalyzed by LDH involves the direct transfer of a hydride ion from C4 of the reduced nicotinamide group of NADH to the C2 carbon of pyruvate accompanied by the protonation of pyruvate's keto-oxygen, the proton being supplied by His195. It is known that either electrostatic stabilization of the transition state in the pyruvate-lactate interconversion, which contains a highly polarized carbonyl moiety, +C-O−, or destabilization of the >C=O ground state (or a combination) is responsible for about half of the rate enhancement brought about by LDH. The other half of the rate enhancement comes about from bringing cofactor and substrate close together in a proper orientation and `activating' cofactor towards catalysis.
In this study, we investigate the atomic motion within the Michaelis complex, bsLDH•NADH•oxamate. To preclude the occurrence of enzyme-catalyzed chemistry that would complicate a kinetic analysis, we used a substrate analog, oxamate, rather than the natural substrate, pyruvate. Oxamate is isoelectric and isosteric to pyruvate and has been shown to have binding kinetics very similar to that of actual substrate, pyruvate. We used T-jump relaxation profiles to monitor the re-equilibration of a chemical system following an instantaneous increase in temperature induced by a laser pulse tuned to an infrared water band. The re-equilibration results in changes in the concentrations of the species involved, and the transient changes are characterized using spectroscopic probes. The chemical conditions have been arranged so that dynamics within the ternary complex are observed.
First, we probed the fluorescence emission of NADH in wild type and mutant bsLDH to report on the time evolution of the changes within the NADH environment over the microseconds to milliseconds time scale. Second, we determined, over the same time scale, the corresponding tryptophan emissions. Several single tryptophan bsLDH mutants were constructed to highlight selected positions within bsLDH. These mutants were created by first replacing all tryptophan residues with tyrosine in wild type bsLDH to create a tryptophan-less template.19 Earlier studies have shown that neither the tryptophan to tyrosine substitutions nor the subsequent single-tryptophan mutants lead to any substantial changes in the biochemical properties of the enzyme.20 To the tryptophan-less template, further mutations were made to add a single tryptophan at strategic sites in the protein. We created four single-tryptophan mutants, one (G106W, designations are based on the tryptophan-less mutant, not the wild type) with a tryptophan on the important surface loop,21 and three (Y190W, Y248W and Y279W) with tryptophan placed at varying distances from the active site (Figure 1). In Y248W, the indole ring is approximately 10.7 Å from the NADH nicotinamide ring, 19.6 Å in Y190W and 22.3 Å in Y279W. Previous time-resolved fluorescence anisotropy studies also suggest that, while the tryptophan in Y248W lies on a very stable helix within the protein, those in Y190W and Y279W do not.19 The tryptophan in G106W reports mainly on the time evolution of the loop motion, and the tryptophans in Y190W, Y248W and Y279W report on conformational changes within the protein distant from the binding pocket.
Figure 1.
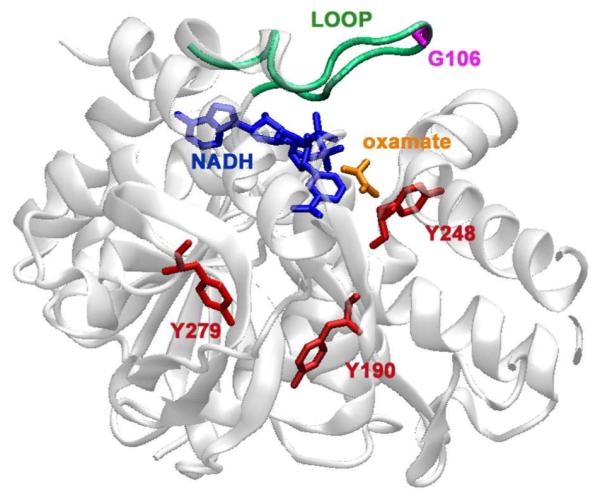
Crystal structure of a bsLDH monomer (PDB file: 1LDN) depicting the sites of mutation. In red are the sites of tyrosine to tryptophan mutations. The indole ring of the tryptophan are placed approximately 10.7 Å from the NADH ring for the Y248W mutant, 19.6 Å in Y190W and 22.3 Å in Y279W. Also highlighted are: the cofactor, NADH, in blue, oxamate in orange, and the catalytically important loop in green. In a fourth mutant, G106W, the tryptophan is placed on the loop (purple). The graphic was generated using VMD.29
MATERIALS AND METHODS
Sample Preparation
All required reagents were purchased from Sigma-Aldrich Co. (St. Louis, MO) except NADH and oxamate, which were purchased from Roche Diagnostic Corp. (Indianapolis, IN). Reagents were used as received. Samples for T-jump measurements were dissolved in 5 mM FBP, 0.1 M triethanolamine (TEA) HCl buffer at pH 6.0 to yield a 250 μL of 50–80 μN solutions (concentration of bsLDH subunits). FBP serves as allosteric activator of the enzyme.22 Different amounts of NADH and oxamate were added as appropriate to form the binary bsLDH•NADH and ternary bsLDH•NADH•oxamate complexes. Fresh solutions of all reagents were prepared for each experiment.
The bsLDH gene was obtained from Genomic DNA from Geobacillus stearothermophilus ATCC 12980D, sub-cloned to pET3a vector, and transformed into C43 (DE3) competent E. coli cells. The growth conditions of the transformed cells and the protein purification followed a published procedure.14
The single-tryptophan mutants were prepared following published protocols.19,21 A tryptophan-less gene, where the three wild-type tryptophan codons (80, 150, and 203) were changed to tyrosine, cloned to pKK223-3 vector and transformed into E. coli TG1 cells. To obtain the mutants, tryptophan replaced glycine at position 106 (G106W), or tyrosine residue at position 190 (Y190W), 248 (Y248W), or 279 (Y279W). The growth conditions of the transformed cells and the protein purification procedures were based on published procedure.19,21 The wild-type protein and all four mutants showed catalytic parameters the same as published values. The values of the mutants are quite close to that of the wild type bsLDH: kcat = 243, 140, 244, 175, 182 s−1 for WT, G106W, Y190W, Y248W, Y279W, respectively (taken from pyruvate side, in 100 mM TEA buffer, pH 6 at 25 °C).19,21 Km of pyruvate remains at 0.06 mM for all proteins except Y279W which is 0.04 mM.
Laser-induced T-Jump
Laser-induced temperature-jump relaxation spectrometers employed in the present study were described previously.7,10 One of these spectrometers was used for NADH fluorescence kinetic measurements, and another one was used for measurements of tryptophan fluorescence kinetics. In the NADH fluorescence measurements, the excitation light, with few milliwatt incident power near 360 nm wavelength (351.1 and 363.8 nm lines), was produced by Innova 70 Ar-ion laser (Coherent, Palo Alto, CA). The excitation beam was focused to ~0.4 mm diameter spot in the center of the heated spot of the sample (~1.5 mm diameter). When tryptophan fluorescence detection was used, the fluorescence was excited by an Innova 200 Ar-ion laser (Coherent, Palo Alto, CA) emitting a group of lines near 300 nm (300.3 and 302.4 nm), with the same ~0.4 mm size of the excitation spot on the sample in the center of ~1.5 mm diameter heated spot. NADH fluorescence passes through a 458 nm narrow-band filter with the bandwidth of 40 nm FWHM before reaching the detector, and to select tryptophan emission, a 340 nm narrow-band filter with 25 nm FWHM bandwidth was used (both from Andover, Salem, NH).
RESULTS AND DISCUSSION
The goal in this study is to concentrate on the dynamics of the conformational ensemble of the bsLDH Michaelis complex. In this work, we employ oxamate as a non-reactive mimic of the substrate pyruvate. Our kinetic studies will then not be influenced by on-enzyme chemistry (i.e., the oxidation of NADH). The conditions are at high protein, NADH, and especially high oxamate concentrations; most studies employed bsLDH•NADH•oxamate at 80/80/1200 μM. This drives the system towards approximately 98% ternary complex, given the Kd's of NADH to bsLDH and oxamate to bsLDH•NADH. Hence, the T-jump relaxation kinetic traces should be dominated by unimolecular interconversions within the ternary complex.23 Fluorescence of NADH is sensitive to its local environment within dehydrogenases and is quite dependent upon local packing interactions.24 Intrinsic tryptophan fluorescence is also sensitive to the local tryptophan local environment and can also be quenched by the nicotinamide ring of bound NADH via Förster resonance energy transfer. The Förster radius for this donor-acceptor pair is about 15–25 Å, depending on actual values of tryptophan quantum yields.25 Oxamate binding leads to strong quenching of the fluorescence of bound NADH, but does not directly affect tryptophan fluorescence.
NADH emission kinetics
Figure 2 shows NADH fluorescence 7°C T-jump kinetics for the wild type bsLDH to a final temperature of 29.5 °C; the initial concentration of the various species was 80 μM bsLDH, 80 μM NADH and 1200 μM oxamate. The relaxation kinetics fit well to a three exponential function with relaxation rates of 327, 1173, and 45800 s−1. This is a very similar kinetic profile to that found in our studies of the pig heart protein by T-jump relaxation studies using NADH emission as probe, a strong intensity rise in the time range from sub-millisecond to few milliseconds due to unimolecular transformations between the so-called lightly populated `encounter complex' between LDH•NADH and oxamate and a substantially populated ternary complex. At 30 °C, the relative NADH quantum yield of emission is: 1.0, 1.5, 0.60 for free NADH, the binary complex, and ternary complex of bsLDH, respectively, very similar to phLDH. In the NADH emission work,23 an accurate kinetic model (along with microscopic rate constants) could be derived based upon extensive studies varying the concentrations of phLDH, NADH, and most importantly oxamate. It was found that the data was well fit by a bi-molecular event (the binding of oxamate to phLDH•NADH) and two unimolecular events amongst three conformations of the ternary complex, phLDH•NADH•oxamate. The NADH emission data could be fit equally well to a `sequential' set of events or to a `branched' kinetic scheme whereby the oxamate binds to the binary complex to form a lightly populated encounter complex and the more populated ternary complexes interconvert via the encounter complex. The IR T-jump only fit the branched model.26 Given the basic similarities between the bsLDH and phLDH enzymes, we expect to obtain a similar kinetic scheme. In fact, IR T-jump studies on the bsLDH protein yielded essentially identical results to that obtained for the phLDH protein.27 We construct kinetic models for the data based on our studies and those of other laboratories below, in the Conclusions section, as a guide for interpreting the data obtained in this study.
Figure 2.

NADH fluorescence T-jump kinetic profile for wild-type bsLDH. Temperature after T-jump is 30 °C after a jump of about 7 °C. The sample contained 80 μM bsLDH, 80 μM NADH and 1200 μM oxamate initial concentrations. The solid line is a three exponential fit to the data; the results of the fit are given in Table 1.
Figure 3 shows NADH fluorescence T-jump kinetics for the wild type bsLDH to a final temperature of 29.5 °C and for the three single-tryptophan mutants, Y279, Y248 and Y190 (total concentrations of all components: 80 μM bsLDH, 80 μM NADH and 1200 μM oxamate). The NADH kinetic profiles for the wild type, Y279 and Y248 mutants are quite similar to each other. At high temperatures, the kinetic traces of NADH within the ternary complex pattern is same as for phLDH:23 Multi-exponential fits of the observed kinetic profiles generally yield three time constants within the observational time; see Table 1. The NADH fluorescence T-jump profiles for the ternary complex of the Y190W mutant differ slightly from the corresponding profiles for wild type bsLDH and other studied mutants, especially at lower post-jump temperatures (Figure 3). The observed exponential rates for the Y190W bsLDH ternary complex fall within the same time ranges as for other mutants, but the percentage contributions of the components are different.
Figure 3.

NADH fluorescence T-jump kinetic profiles for the wild-type bsLDH and three Trp mutant proteins, Y279W, Y248W and Y190W. Temperatures after T-jump of about 7 °C are shown on the graph. All samples contained 80 μM bsLDH, 80 μM NADH and 1200 μM oxamate.
Table 1.
Observed relaxation rates (inverse of relaxation times) for a 23 to 30 °C T-jump for the ternary complexes of bsLDH purified from the wild type and the single-tryptophan mutants.
| Source of bsLDH | NADH fluorescence T-jump trace | Tryptophan fluorescence T-jump trace | |||
|---|---|---|---|---|---|
|
| |||||
| 1 / t1, s−1 | 1 / t2, s−1 | 1 / t3, s−1 | 1 / t1, s−1 | 1 / t2, s−1 | |
|
| |||||
| Wild type | 327 ±20; 78% | 1173 ±500; 8% | 45800 ±5000; 13% | not measured | not measured |
| G106W | not measured | not measured | not measured | 320 ±230; 76% | 2300 ±1000; 24% |
| Y190W | 303 ±11; 56% | 3500 ±1900; 29% | 35500 ±2500; 15% | 913 ± 18; 74% | 16400 ±800; 26% |
| Y248W | 210 ±19; 85% | 2900 ±2400; 5% | 26000 ±7000; 10% | 480 ±40 | |
| Y279W | 460 ±22; 76% | 4100 ±3500; 7% | 19000 ±5000; 17% | 434 ± 13 | |
Most ternary mixtures contain 80 μM bsLDH, 80 μM NADH and 1200 μM oxamate in TEA buffer at pH 6.0. The only exception was for G106W, where similar mixtures were prepared in TEA buffer of pH 7.2. For the G106W, T-jump was from 15 to 24 °C. Percent contribution of each component to the amplitude follows each relaxation rates. For comparison, T-jump IR studies27 measuring the modulation of the C2=O stretch mode of oxamate in the ternary complex of bsLDH found two transients at 490 and 1461 s−1 (at 22 °C).
Tryptophan emission kinetics
The single tryptophan mutants afforded us a probe for monitoring local motions within the protein far from the active site. Earlier studies have shown that replacing all the naturally occurring tryptophans to tyrosine in phLDH, nor subsequent single Trp mutants at specific but strategic locations, does not lead to significant changes in the measured biochemical properties of the enzyme.20 The four single tryptophan mutants studied here include: one with a tryptophan, G106W on the important surface loop, and three (Y248W, Y190W and Y279W) with tryptophan placed at varying distances from the active site (Figure 1). The T-jump kinetic profiles of the tryptophan fluorescence within the four mutants are shown in Figure 4.
Figure 4.

Tryptophan fluorescence T-jump kinetic profiles for the four single-Trp mutant proteins, G106W, Y279W, Y248W and Y190W. For the G106W mutant, temperatures after about 9 °C T-jump were 24, 34 and 44 °C, and measurements were done at pH 7.2. For all other mutants, pH 6.0 buffer was used; temperatures after about 7 °C T-jump are shown on the lower-right graph. All samples contained 80 OM bsLDH, 80 μM NADH and 1200 μM oxamate.
For G106W, wherein the tryptophan is located on the active site loop, the tryptophan fluorescence T-jump profile at 25 °C fits to a double exponential (Table 1). As the post-jump temperature increases, the smaller component of the relaxation kinetics decreases. Due to solubility issues, we were not able to study the G106W mutant as extensively as the other mutants. The G106W bsLDH was previously studied by Holbrook and colleagues,21 who employed stopped flow techniques to determine the kinetic profile of Trp emission upon binding of oxamate to bsLDH•NADH and compared this with the results from single turnover studies of pyruvate to lactate conversion. They found transients for both at 0.125 ms−1 at 25 °C. This is in good agreement with the current work that shows a transient with a relaxation rate of 0.320 ms−1 (at the higher final temperature of 30 °C). However, we additionally find a faster transient at 2.3 ms−1 (presumably simply due to the higher time resolution of the current work). Clarke et al.22 also found two distinct kinetic events on studies of a nitrated phLDH variant by working at low temperatures to bring both events into the range of conventional mixing studies. Their investigation was to look for loop dynamics, focusing on the issue of loop closure upon substrate binding; the mobile surface loop is found to be open in most crystal structures of LDH•NADH and closed in LDH•NADH•oxamate.28 Loop motion was assigned to the fast component in Clarke et al. and to the slower component in Holbrook and colleagues.21 The observed changes in tryptophan emission most likely arise from a modulation of the NADH-Trp radiationless energy transfer; energy transfer is minimal for the loop-open conformation since the Trp and NADH moieties are very far apart but close for closed loop structure(s).22 Taking all the data together, it seems most plausible that two loop motions take place in the bsLDH•NADH•oxamate complex: one at the relatively slow 0.320 ms−1 rate and the other at 2.3 ms−1 (at 30 °C). Single turnover studies revealed two rates associated with oxidizing NADH with similar times as the tryptophan emission found here.22 Loop motion, albeit of different structure composition, appears to be part of both these steps.
The tryptophan fluorescence T-jump profiles obtained for Y248W (Figure 4), wherein the tryptophan indole ring is located approximately 10.7 Å from the NADH nicotinamide ring on a very stable helix,19 fit to a single-exponential regardless of the post-jump temperature. Interestingly, bsLDH from Y279W exhibits similar single-exponential kinetics despite the fact that the indole ring, located 22.3 Å away from the NADH nicotinamide ring, resides on a more flexible helix. Another single-tryptophan bsLDH mutant, Y190W, which has the indole ring located 19.6 Å away from the NADH ring on a different flexible helix, exhibited bi-exponential kinetics (Figure 4; Table 1).
A Förster energy transfer modulation of the Trp emission from the Y190W, Y248W, and Y279W proteins, brought about from NADH release from bsLDH•NADH, can be a complicating factor in interpretating the Trp emission data since we are interested here in discerning protein motions within the ternary complex. Like the modulation of distance between the Trp-NADH moieties brought about by loop motion in the G106W protein, the release of NADH from the binary complex brings about a large change in the Trp-NADH distance. Given the Kd values of NADH with bsLDH and oxamate with bsLDH•NADH, we estimate the steady state concentrations for WT 80/80/1200 sample at 28.8 °C, are: [NADH]free = 1.4 μM; [bsLDH]free = 1.4 μM; [Binary] = 0.3 μM; [oxamate]free = 1122 μM; [ternary] = 78.3 μM. The amount of [bsLDH]free = 1.0 μM at a temperature 7 °C less (i.e., before the T-jump. Supposing that all species of the protein bound have basically the same Trp emission quantum yields the same, then the percentage of the time dependent Trp emission signal that is due to NADH release is given by (χ[1.4 μM] + [78.6 μM])/(χ[1.0 μM] + [79 μM]), where χ is the relative Trp quantum yield between free protein and protein bound with NADH. The release of NADH from the binary complex, at the conditions here, will take place at ca. 1 ms. The T-jump signals should in Figure 4 change by about 3.0% from short time (microsecond time scale) to longer time (millisecond time scale). Although the concentration of the binary complex is very small relative to the ternary, the Trp emission is very strongly modulated by NADH release from the binary complex (i.e., large χ), while the modulation of the Trp emission by local motions within the ternary complex is generally of small amplitude. Using the above relationship, if χ is greater that 8, the reported Trp emissions signals in Figure 4 may arise from NADH release from bsLDH•NADH. Steady state measurements show that only the Y249W mutant (which has the Trp indole ring closest to bound NADH of the Trp mutants studied) has a large, χ=10, relative quantum yield; the others are smaller.
CONCLUSIONS
Several previous studies have revealed a number of features concerning the dynamical nature of LDH and its various complexes. One is that the binary complex exists as an ensemble of substates, some which readily bind substrate and some of the ensemble which do not.6 The conformation of the binding competent LDH•NADH complex appears to be quite remarkable. The derived structural changes show the active site, normally buried about 10 Å from the protein surface, `pushed up' towards the surface; additionally, there are many changes in hydrogen bonding and hydration throughout the protein.17 Once the substrate loosely `docks' to the exposed active site (forming what we have called an `encounter' complex), it is then `dragged' into the heart of the protein to form an ensemble of close packed conformations apparently with varying degrees of catalytic competence.9,23,26,27 There is much in all this that resembles the flavor (if not the physics) of a protein-ligand complex in the process of folding. These results suggest substantial collective motions within and across the enzyme, spread over varying time scales, that will be of importance to catalytic mechanism.
The interest in this work is to probe these motions in more detail. For this purpose, a set of four single Trp mutants were constructed for bsLDH based on the work of Holbrook and colleagues.19 This set of mutant proteins (and others as well) retained basically the same catalytic properties as the wild type protein but are scattered over diverse locations within the protein (see Figure 1). Hence, it is possible to use the Trp residues as reporter of surrounding protein dynamics via its modulation of Trp emission. T-jump relaxation methods are employed to initiate the kinetic event(s), a method that has a 20 ns resolution in our lab. The tryptophan in G106W reports mainly on the time evolution of the motion of the catalytically important surface loop, while the tryptophan in Y248W, Y190W, and Y279W can report on conformational changes within the protein distant from the binding pocket (see Scheme 1 and Figure 1). As shown in Results, the observed Trp emission transients from the Y279W mutant is due, fully or almost fully, to Förster energy transfer modulated by release of NADH from LDH•NADH and so is not of interest to our purposes here. We do not discuss the results from this mutant further. The Trp emission from the Y248W and Y190W mutants contain signatures of local motion of atoms near the Trp reporter.
In stopped-flow emission and absorption studies of pig heart LDH, carried out at −16 to +8 °C, so as to slow the kinetics to a time within the stopped-flow resolution, Holbrook and colleagues observed two rates of on-enzyme NADH oxidation with the slowest corresponding to steady state kcat.22 The kinetic results were fit to a model that has the protein's Michaelis complex occupy two interconverting conformations with only one able to convert LDH•NADH•substrate to LDH•NAD+•product effectively. In Scheme 2, we have graphed Holbrook et al.'s kinetic model, modified by our recent kinetic studies.23,26,27 In our studies, we found that the substrate binds with LDH•NADH to form a `loose' encounter complex. Holbrook et al. had interconversion between the two populated species occurring at the level of the binary complex; substrate binds/releases between two already preformed binary complex structures. In contrast, in our thinking, the encounter complex collapses to form the two more tightly bound complexes. The tightly bound complexes do not interconvert directly, but rather though the encounter complex.9,27 This conclusion is based on kinetic/structural studies of bsLDH that probed the key C=O IR stretch band of bound substrate mimic (oxamate). In that study,27 three Michaelis (i.e., bsLDH•NADH•oxamate) conformations were found, as indicated by a different IR stretching frequencies and the kinetic data, interconverting on times scales with relaxation rates of 490 and 1461 s1 (at 22 °C). Only one of the three conformations would be expected to have any propensity towards catalysis.26,27 The present Trp emission studies of the G106W protein, taken at near physiological temperatures with an approach that can resolve sub-millisecond events, are in close agreement (320 and 2300 s−1 at 30°C; Table 1). Two transients are observed, one near one millisecond, which is the time of the steady state kcat for this enzyme.22 The data from the Y190W mutant reports a very similar result (Table 1). Both events appear to involve the motion of the loop given that the Trp reporter in G106W is very sensitive to loop motion, changes in key hydrogen bond patterns at the active site (from the IR studies), and motion of some parts of the protein far from the active site. This data strongly reinforces the view that the Michaelis complex consists of an ensemble of sub-states interconverting on time scales faster than catalysis, where some are more competent towards on-enzyme catalysis than others.
Scheme 2.
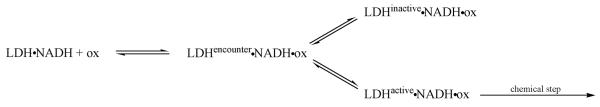
All results combined21,22,27 are in agreement and most simply explained by the above kinetic scheme. The encounter complex presumably has an `open' loop conformation while the two other conformations of the Michaelis complex, labeled inactive and active, are probably `closed' loop with, however, varying loop structure and atomic arrangements at the active site. The two `closed' conformations do not appear to directly interconvert, rather via the encounter complex.
Given that the IR studies probe changes in structure of the active site, specifically the H-bonding patters between bound substrate and adjacent active site residues of Arg109 and His195 (see Scheme 1), and G106W and NADH emission studies probe motion of the surface mobile loop and the structural rigidity of the active site respectively, we ask if there are motions of residues within the LDH•NADH•oxamate Michaelis complex further from the active site on similar time scales? The Y248W and Y190W proteins both show a component of protein motion on the one millisecond time scale, very close to the time associated with loop motion and the rate limiting step to kcat (see Table 1), that shows up in the NADH emission profiles as well as the Trp emission. The Y190W protein relaxation spectra show additionally a much faster transient. This result suggests that what is called `loop motion' is not just localized to the loop. Rather, it involves the motion of atoms spread over the protein, even some quite distal from the active site. This is in close accord with our previous molecular dynamics study and experimental work that `closure' of the key surface loop is not a localized hinge motion (as often supposed) but rather is a sliding motion happening together with a substantial conformation change of the protein involving many changes to internal hydrogen bonding patterns.6,17
Acknowledgments
FUNDING SOURCE This work was supported by the Institute of General Medicine of the National Institutes of Health, program project grant No. 5P01GM068036.
ABBREVIATIONS
- LDH
L-lactate dehydrogenase
- bsLDH
thermophylic B. stearothermophilus lactate dehydrogenase
- phLDH
pig heart lactate dehydrogenase
- FBP
fructose 1,6–biphosphate
- TEA
triethanolamine
- FWHM
full width at half maximum
Footnotes
Author Contributions The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
REFERENCES
- (1).Karplus M, McCammon JA. Dynamics of proteins: elements and function. Ann. Rev. Biochem. 1983;53:263–300. doi: 10.1146/annurev.bi.52.070183.001403. [DOI] [PubMed] [Google Scholar]
- (2).Hammes GG. Multiple Conformational Changes in Enzyme Catalysis. Biochemistry. 2002;41:8221–8228. doi: 10.1021/bi0260839. [DOI] [PubMed] [Google Scholar]
- (3).Frauenfelder H, Chen G, Berendzen J, Fenimore PW, Jansson H, McMahon BH, Stroe IR, Swenson J, Young RD. A unified model of protein dynamics. Proc Natl Acad Sci U S A. 2009;106:5129–5134. doi: 10.1073/pnas.0900336106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Eisenmesser EZ, Bosco DA, Akke M, Kern D. Enzyme dynamics during catalysis. Science. 2002;295:1520–1523. doi: 10.1126/science.1066176. [DOI] [PubMed] [Google Scholar]
- (5).Gulotta M, Qiu L, Rosgen J, Bolen DW, Callender R. Effects of Cell Volume Regulating Osmolytes on Glycerol-3-phosphate Binding to Triosphosphate Isomerase. Biochemistry. 2007;46:10055–10062. doi: 10.1021/bi700990d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Qiu L, Gulotta M, Callender R. Lactate Dehydrogenase Undergoes a Substantial Structural Change to Bind its Substrate. Biophysical J. 2007;93:1677–1686. doi: 10.1529/biophysj.107.109397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zhadin N, Callender R. The Effect of Osmolytes on Protein Dynamics in the LDH-Catalyzed Reaction. Biochemistry. 2011;50:1582–1589. doi: 10.1021/bi1018545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Basner JE, Schwartz SD. Donor-Acceptor Distance and Protein Promoting Vibration Coupling to Hydride Transfer: A possible mechanism for kinetic control in isozymes of human lactate dehydrogenase. J. Phys. Chem. B. 2004;108:444–451. [Google Scholar]
- (9).Deng H, Brewer SH, Vu DV, Clinch K, Callender R, Dyer RB. On the Pathway of Forming Enzymatically Productive Ligand-Protein Complexes in Lactate Dehydrogenase. Biophysical J. 2008;95:804–813. doi: 10.1529/biophysj.108.128884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zhadin N, Gulotta M, Callender R. Probing the Role of Dynamics in Hydride Transfer Catalyzed by Lactate Dehydrogenase. Biophysical J. 2008;95:1974–1984. doi: 10.1529/biophysj.108.132464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Holbrook JJ, Liljas A, Steindel SJ, Rossmann MG. The Enzymes. Vol. XI. Academic Press; New York: 1975. Lactate Dehydrogenase; pp. 191–293. [Google Scholar]
- (12).Griffith JP, Rossmann MG. M4 Lactate Dehydrogenase Ternary Complex with NAD and Oxamate. Brookhaven Data Bank 1LDM. 1987 [Google Scholar]
- (13).Luyten MA, Gold M, Friesen JD, Bryan Jones J. On the effects of replacing the carboxylate-binding arginine-171 by hydrophobic tyrosine or tryptophan residues in the l-lactate dehydrogenase. Biochemistry. 1989;28:6605–6610. doi: 10.1021/bi00442a011. [DOI] [PubMed] [Google Scholar]
- (14).Deng H, Zheng J, Clarke A, Holbrook JJ, Callender R, Burgner JW. Source of Catalysis in the Lactate Dehydrogenase System: Ground State Interactions in the Enzyme•Substrate Complex. Biochemistry. 1994;33:2297–2305. doi: 10.1021/bi00174a042. [DOI] [PubMed] [Google Scholar]
- (15).Hart KW, Clarke AR, Wigley DB, Waldman ADB, Chia WN, Barstow DA, Atkinson T, Jones JB, Holbrook JJ. A strong carboxylate-arginine interaction is important in substrate orientation and recognition in lactate dehydrogenase. Biochim. Biophys. Acta. 1987;914:294–298. doi: 10.1016/0167-4838(87)90289-5. [DOI] [PubMed] [Google Scholar]
- (16).Clarke AR, Wigley DB, Barstow DA, Chia WN, Atkinson T, Holbrook JJ. A single amino acid substitution deregulates a bacterial lactate dehydrogenase and stabilizes its tetrameric structure. Biochim Biophys Acta. 1987;913:72–80. doi: 10.1016/0167-4838(87)90234-2. [DOI] [PubMed] [Google Scholar]
- (17).Pineda JRET, Callender R, Schwartz SD. Ligand Binding and Protein Dynamics in Lactate Dehydrogenase. Biophysical J. 2007;93:1474–1483. doi: 10.1529/biophysj.107.106146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Atkinson T, Barstow D, Chia W, Clarke A, Hart K, Waldman A, Wigley D, Wilks H, Holbrook JJ. Mapping Motion in Large Proteins by Single Tryptophan Probes Inserterd by Site-Directed Mutagenesis: Lactate Dehydrogenase. Biochem. Soc. Trans. 1987;15:991–993. doi: 10.1042/bst0150991. [DOI] [PubMed] [Google Scholar]
- (19).Smith CJ, Clarke AR, Chia WN, Irons L, Atkinson T, Holbrook JJ. Detection and Characterization of Intermediates in the Folding of Large Proteins by the Use of Genetically Inserted Tryptophan Probes. Biochemistry. 1991;30:1028–1036. doi: 10.1021/bi00218a021. [DOI] [PubMed] [Google Scholar]
- (20).Roper DI, Moreton KM, Wigley DB, Holbrook JJ. The structural consequences of exchanging tryptophan and tyrosine residues in B. stearothermophilus lactate dehydrogenase. Protein Eng. 1992;5:611–615. doi: 10.1093/protein/5.7.611. [DOI] [PubMed] [Google Scholar]
- (21).Waldman ADB, W. HK, Clarke AR, Wigley DB, Barstow DA, Atkinson T, Chia WN, Holbrook JJ. The Use of a Genetically Engineered Tryptophan to Identify the Movement of a Domain of B. Stearothermophilus Lactate Dehydrogenase with the Process which Limits the Steady-State Turnover of the Enzyme. Biochem. Biophys. Res. Comm. 1988;150:752–759. doi: 10.1016/0006-291x(88)90455-x. [DOI] [PubMed] [Google Scholar]
- (22).Clarke AR, Waldman ADB, Hart KW, Holbrook JJ. The Rates of Defined Changes in Protein Structure During the Catalytic Cycle of Lactate Dehydrogenase. Biochimica et Biophysica Acta. 1985;829:397–407. doi: 10.1016/0167-4838(85)90250-x. [DOI] [PubMed] [Google Scholar]
- (23).McClendon S, Zhadin N, Callender R. The Approach to the Michaelis Complex in Lactate Dehydrogenase: the substrate binding pathway. Biophysical J. 2005;89:2024–2032. doi: 10.1529/biophysj.105.062604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Velick SF. Fluorescence Spectra and Polarization of Glyceraldehyde-3-phosphate and Lactic Dehydrogenase Coenzyme Complexes. J. Biol. Chem. 1958;233:1455–1467. [PubMed] [Google Scholar]
- (25).Steinberg IZ. Long-range nonradiative transfer of electronic excitation energy in proteins and polypeptides. Annu. Rev. Biochem. 1971;40:83–114. doi: 10.1146/annurev.bi.40.070171.000503. [DOI] [PubMed] [Google Scholar]
- (26).McClendon S, Vu D, Clinch K, Callender R, Dyer RB. Structural Transformations in the Dynamics of Michaelis Complex Formation in Lactate Dehydrogenase. Biophysical J. 2005;89:L07–L09. doi: 10.1529/biophysj.105.064675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Deng H, Vu DV, Clinch K, Desamero R, Dyer RB, Callender R. Conformational heterogeneity within the Michaelis complex of lactate dehydrogenase. J, Phys. Chem. B. 2011;115:7670–6778. doi: 10.1021/jp2015929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Wigley DB, Muirhead H, Gamblin SJ, Holbrook JJ. Crystallization of a Ternary Complex of Lactate Dehydrogenase from Bacillus stearothermophilus. J. Mol. Biol. 1988;204:1041–1043. doi: 10.1016/0022-2836(88)90060-5. [DOI] [PubMed] [Google Scholar]
- (29).Humphrey W, Dalke A, Schulten K. VMD-visual molecular dynamics. J. Molec. Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]