

Abstract
Background
The only therapeutic options that exist for squamous cell lung carcinoma (SCC) are standard radiation and cytotoxic chemotherapy. Cancer stem cells (CSCs) are hypothesized to account for therapeutic resistance, suggesting that CSCs must be specifically targeted. Here, we analyze the transcriptome of CSC and non-CSC subpopulations by RNA-seq to identify new potential therapeutic strategies for SCC.
Methods
We sorted a SCC into CD133− and CD133+ subpopulations and then examined both by copy number analysis (CNA) and whole genome and transcriptome sequencing. We analyzed The Cancer Genome Atlas (TCGA) transcriptome data of 221 SCCs to determine the generality of our observations.
Results
Both subpopulations highly expressed numerous mRNA isoforms whose protein products are active drug targets for other cancers; 31 (25%) correspond to 18 genes under active investigation as mAb targets and an additional 4 (3%) are of therapeutic interest. Moreover, we found evidence that both subpopulations were proliferatively driven by very high levels of c-Myc and the TRAIL long isoform (TRAILL) and that normal apoptotic responses to high expression of these genes was prevented through high levels of Mcl-1L and Bcl-xL and c-FlipL—isoforms for which drugs are now in clinical development. SCC RNA-seq data (n = 221) from TCGA supported our findings. Our analysis is inconsistent with the CSC concept that most cells in a cancer have lost their proliferative potential. Furthermore, our study suggests how to target both the CSC and non-CSC subpopulations with one treatment strategy.
Conclusions
Our study is relevant to SCC in particular for it presents numerous potential options to standard therapy that target the entire tumor. In so doing, it demonstrates how transcriptome sequencing provides insights into the molecular underpinnings of cancer propagating cells that, importantly, can be leveraged to identify new potential therapeutic options for cancers beyond what is possible with DNA sequencing.
Introduction
Lung cancer accounts for 28% of all cancer deaths—the highest percentage of all cancers [1]. Non-small cell lung cancer (NSCLC) accounts for ∼85–90% of lung cancers, of which adenocarcinoma and squamous cell carcinoma are the most common subtypes [1]. Although upwards of 70% of NSCLC patients have advanced disease that is rarely curable when diagnosed, new advances for subsets of lung adenocarcinomas that harbor EGFR mutations or EML4-ALK gene fusions encourage the development of targeted therapies that may alter this dire situation [2]. These genetic alterations primarily occur in adenocarcinomas of patients who never smoked, and are uncommon in SCC which is predominantly associated with smoking [3]–[5]. While FGFR1 [6] and DDR2 [7] have recently emerged as potential therapeutic targets for some SCC patients, inhibitors have yet to reach clinical trials. Recent NSCLC high throughput sequencing studies primarily focused on analyzing DNA have shown that few genes are mutated at a sufficiently high frequency to be useful for targeted therapy; however these studies do predict DNA alterations that are frequently clustered in a limited number of important molecular pathways suggesting that targeting these pathways may be a viable therapeutic strategy [8]–[12]. Deep transcriptome (RNA-seq) profiling of NSCLC to identify genes with deregulated expression that is common between tumors has not yet been reported, although such reports are to be expected given the large RNA-seq datasets being generated by TCGA [13] and other consortia.
Cancer cells within an individual tumor exist in distinct phenotypic states that often exhibit important functional differences. A subpopulation of cells with self-renewing and tumor-initiating capabilities, commonly referred to as cancer-stem-like cells (CSCs), have been identified in a variety of tumor types including NSCLC [14]. Mounting evidence suggests that CSCs are resistant to anticancer therapies and underlie metastasis [15], [16], and hence are the primary cancer cell type responsible for relapse and progression of malignant tumors. The immediate implication is that by targeting CSCs it should be possible to eradicate the drug resistant and metastatic subpopulation of a cancer [14]. However, recent studies have demonstrated that the CSC phenotype is plastic and can be reconstituted by other, non-CSC, tumor cells [17], [18]; thus not just CSCs but all tumor subpopulations that are “potential CSCs” must be targeted. Transcriptome sequencing of CSC and non-CSC subpopulations in NSCLC would provide insights into the molecular basis underlying their phenotypic similarities and differences and facilitate the identification of novel therapeutic targets. Such analysis will be an important and necessary complement to the bulk tumor transcriptome profiling being performed by TCGA and others.
The observations that non-CSCs can reconstitute CSCs, and vice versa, suggest that the phenotypic differences between these subpopulations are due to epigenetic rather than genetic differences. Therefore, exome and genome sequencing experiments aimed at identifying somatic mutations are not expected to reveal differences between sorted CSC and non-CSC subpopulations. On the other hand, transcriptome profiling, which is a readout of the epigenome (i.e. histone marks and DNA methylation that regulate expression), should be an excellent method for profiling CSCs and non-CSCs to reveal mechanistic differences. The advantage of RNA-seq data over microarrays is the ability to analyze isoform expression differences [19]. In cancer cells, alternative mRNA isoforms can produce protein isoforms with dominant negative activity. The pathogenic role of cancer-specific isoforms has been extensively demonstrated across all aspects of cellular physiology, including cellular adhesion and metastasis (CD44 and RON), cell growth and tumorigenesis (PKM2, MDM2, FGFR2, CRK, NUMB), cell cycle (PYK), angiogenesis (VEGF), apoptosis (GS3KB, CD95, Bcl-X, caspase-2, caspase-9), metabolism (PK), and drug resistance (AR and MRP-1) [20]–[24]. These examples underscore the advantage of isoform-level transcriptome information over whole gene expression for gaining insights into the molecular mechanisms underlying CSC and non-CSC phenotypic differences.
Here we report the application of genomics technologies to a SCC xenograft that was sorted into CSC and non-CSC subpopulations based on the CD133 marker (Figure 1). CD133 (PROM1;Prominin-1) is a 5-transmembrane glycoprotein that is considered to be a marker for the subpopulation of CSCs in both subtypes of NSCLC [15], [25]–[27]. In NSCLC the CD133+ subpopulation has been shown to have higher tumorigenic potential in SCID mice, to express higher levels of stemness genes and to be more resistant to conventional chemotherapy than the CD133− subpopulation [15]. Importantly, so that the SCC xenograft would be more representative of primary tumor, it was directly engrafted as minced primary tumor into NSG mice and was never grown in vitro. Whole-genome DNA analysis revealed that the chromosomes of CD133+ and CD133− subpopulations were highly deranged in a very similar manner; however, as expected the tumor did not harbor clinically actionable mutations. Analysis of mRNA splice isoform expression profiles of the CD133+ and CD133− subpopulations resulted in the identification of SCC as a potential new indication for numerous drugs currently in development and suggest several additional new promising targets. Finally, analysis of The Cancer Genome Atlas (TCGA) publicly available transcriptome RNA-seq data of 221 SCCs [28] supports the generality of our transcriptome findings for this disease. Altogether our study demonstrates the capability of transcriptome sequencing of sorted cancer cell subpopulations to inform clinical development in ways that are not possible with DNA sequencing.
Figure 1. Study design.
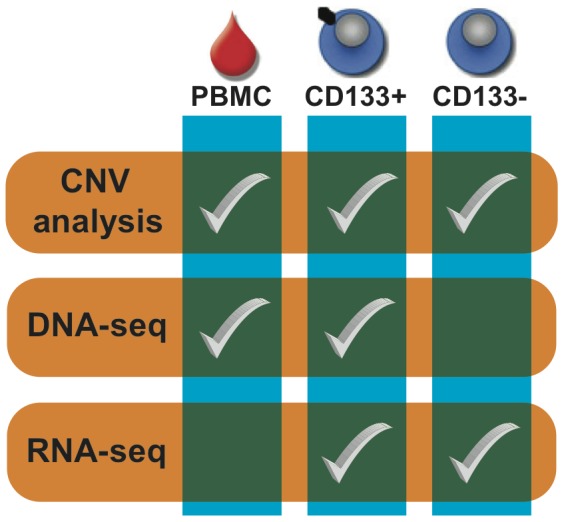
We sorted the human tumor cells of a squamous cell lung cancer xenograft into CD133+ and CD133− subpopulations. We then performed CNA analysis using genotyping arrays on peripheral blood mononuclear cells (PBMC) as well as both the CD133+ and CD133− subpopulations. We compared the genomes of the CD133+ population and PBMC sample to identify somatic mutations. Finally, we performed whole transcriptome sequencing (RNA-seq) of the CD133+ and CD133− subpopulations to evaluate their mRNA isoform expression differences and similarities.
Methods
Xenografting
A UCSD Human Research Protections Program Institutional Review Board (IRB) approved this study prior to any study-related activities. All animal experimental procedures complied with the Guide for the Care and Use of Laboratory Animals (Institute for Laboratory Animal Research, 1996) and were approved by the Pfizer Global Research and Development Institutional Animal Care and Use Committee. A 75-year-old man with a 30-pack-year history of smoking and an incidentally found lung nodule provided written documented informed consent according to this IRB approved protocol prior to surgery. Resection revealed a T1 moderately differentiated squamous cell carcinoma. A portion of his resected fresh tumor was taken for xenografting into NSG (exact strain name NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice (The Jackson Laboratory). Subsequent passages were made in CB17.Cg-PrkdcscidLystbg/Crl mice from Charles River. Minced fresh tumor was mixed with Matrigel (BD Biosciences) and inserted subcutaneously. Mice were observed until palpable tumors grew and these were harvested for serial implantation and study. At two-year follow up the subject remained without clinical or radiographic evidence of relapse.
Isolation of RNA and DNA
Lysates from the CD133−/EpCAM+ and CD133+/EpCAM+ sorted samples were processed for RNA and DNA extraction using Qiagen's All Prep Mini Kit (Valencia, CA). Samples were processed according to manufacturer recommendations. No more than four volumes were loaded on a single RNeasy Mini Spin column. Total RNA samples were evaluated for purity and concentration using the Agilent 2100 Bioanalyzer at the Genechip Microarray Core.
RNA sequencing
Total RNA from both the CD133−/EpCAM+ and CD133+/EpCAM+ tumor subpopulations (∼230 ng each) were used to generate whole transcriptome libraries for sequencing on the SOLiD platform following manufacturer's recommendation (Life Technologies, Carlsbad CA, USA). The RNA samples were fragmented by RNAse III and concentrated using RiboMinus concentration Module (Invitrogen, Carlsbad CA, USA). RNA fragments were ligated to SOLiD adaptor mixes and reverse transcription performed. Purified cDNAs were size selected (150–250 bp) using the MinElute PCR purification kit (Qiagen Inc., Valencia, CA, USA), and then amplified 15 cycles using SOLiD 3′ and SOLID 5′ primers. Amplified cDNA was purified using PureLink PCR Micro kit (Invitrogen, Carlsbad CA, USA), QC'ed by the Bioanalyzer 2100 DNA 1000 kit (Agilent, Santa Clara, CA, USA), and quantified using the Qubit 2.0 Fluorometer (Invitrogen, Carlsbad CA, USA). The whole transcriptome libraries were used for making SOLiD templated beads following the SOLiD Templated Bead Preparation Guide and sequenced using the 50×25 paired-end protocol, for each sample generating more than 500 M read pairs per sample.
Computational analysis of RNA-seq data
Transcriptome sequencing of the CD133+ and CD133− subpopulations produced 529 M and 531 M 50×25 read pairs, respectively. We first aligned the whole transcriptome paired-end reads to rRNA and tRNA genes using version 0.6.4 f of bfast+bwa [29] and retained only those pairs in which neither read was aligned, resulting in data sets comprising 77 M and 79 M read pairs. We then aligned each set of retained read pairs with bfast+bwa to a custom non-redundant database of mRNA isoforms constructed with the “cuffcompare” program from the version 0.9.3 of the Cufflinks software suite(79) and all isoform models in the RefSeq [30], UCSC Known Genes [31], and Ensembl [32] databases. We next converted the read alignment coordinates in the mRNA isoforms database to hg19 genomic coordinates using a custom script. From these alignments we computed expression and differential expression statistics with the “cufflinks” and “cuffdiff” programs of Cufflinks, respectively. When using both programs we used upper quartile normalization and the hg19 human genome reference sequence for bias correction. We computed that 13,612 genes and 43,120 gene isoforms had some non-zero level of expression in at least one of the two subpopulations. Figure S1 in Information S1 shows a histogram of these pre-filtered isoform expression values in both cell subpopulations. A majority of these instances were due to read mapping noise or to “leaky” gene expression that is of unknown physiological significance [33], so we applied minimum expression and read coverage criteria (>1 FPKM and 60% coverage) to obtain an initial isoform expression estimates in the two subpopulations. We observed that 3,844 genes with 7,558 mRNA isoforms met these minimal criteria. To be highly confident in our expression and differential expression results, we applied stringent filtering to the initial isoform expression estimates; we limited our analysis to mRNA isoforms that were expressed at a level of at least 30 FPKM in one of the subpopulations and which were covered over at least 60% of their length by sequencing reads. Additionally, for an isoform to be called differentially expressed, its differential expression had to be called significant at an FDR of 0.01 and to have changed at least four-fold between the two subpopulations. Application of these criteria yielded 572 genes had at least one isoform that was significantly differentially expressed. As a final result, we computed 671 of the 43,120 isoforms (1.6%) to be significantly differentially expressed. Table S1 in Information S1 shows how these numbers change for different FPKM and fold change criteria.
Results
Isolation of CSC and non-CSC subpopulations
We isolated human CSCs (CD133+/EpCAM+) and non-CSCs (CD133−/EpCAM+) from a squamous cell lung cancer xenograft using fluorescence-activated cell sorting (FACS) and respectively collected a total of 9.19×104 (3.4%) and 2.18×106 (96.6%) live cells corresponding to the two subpopulations (Figure 2). To assess the quality of our sort we measured expression levels of CD133 isoforms (Figure S2 in Information S1) in the whole transcriptome data (described below) and observed moderate expression of three isoforms in the CD133+ subpopulation and no detectable level of expression in the CD133− subpopulation. We performed a second FACS for the same xenograft isolated from a different animal and obtained a similar number and proportion of CD133+ and CD133− cells (Figure S3 in Information S1). Our results show that we are able to sort into pure CD133+ and CD133− subpopulations and that the CD133+ subpopulation constitutes a minor fraction of the cancer cells in the squamous cell lung tumor, which is consistent with previously published studies [15], [26].
Figure 2. FACS isolation of CSCs (CD133+) and non-CSCs (CD133−) populations from a human squamous cell lung cancer xenograft.

(A) Live cells displayed distinct populations of either mouse CD45/MHC I or human EpCAM expression. (B) Isolation of human CD133+ cells (red) from human CD133− cells (green) and mouse positive cells (blue). (C) Reanalysis of cell populations in panel B shows that the C133+ cells (red) are 3.4% of the EpCAM+ population.
The mutational landscape of squamous cell lung tumor subpopulations
We performed Copy Number Alteration (CNA) analysis by microarray (see Results and Methods in Information S1) of germline, CD133+, and CD133− DNA and found that approximately half of the entire genome in both the CD133− and CD133+ subpopulations was involved in large CNAs (Figure S4 in Information S1). Overlap analysis revealed the potential existence of CNAs specific to each subpopulation, but upon manual inspection of all CNAs and taking into account the accuracy of microarrays [34], we could not confidently identify differences. Thus we concluded that the chromosomes of the CD133+ and CD133− subpopulations were highly altered in a largely indistinguishable manner. Additionally, we analyzed whole genome sequence data of the CD133+ subpopulation and matching germline DNA and concluded that the tumor studied did not contain any clinically actionable mutations (Tables S1, S2, S3, S4, S5, S6, S7, S8, S9, S10, S11, S12 in Information S2 and Figure S8 in Information S1).
Evaluation of mRNA isoforms of genes encoding cell surface proteins
Deregulated expression of mRNA isoforms can result in the generation of cancer-associated cell surface proteins that are potential antigenic targets for therapeutic monoclonal antibodies. To identify such targets, we measured the expression of 1,191 mRNA isoforms of 426 CD Molecule [35], 354 GPCR [36], and 212 ion channel [36] genes (992 total genes). We limited our analysis to isoforms that were at least moderately expressed (30 FPKM) and covered over at least 60% of their length by sequencing reads in either or both the CD133+ and CD133− subpopulations, yielding an evaluation set of 124 isoforms representing 80 genes (10.4% of the 1,191 isoforms and 8.0% of the 992 genes) (Figure 3; Figures S5A, S5B in Information S1). Of the 124 highly expressed isoforms, 43 (in 25 genes) have at least four-fold expression differences (FDR<0.01), with 24 showing decreased and 19 showing increased expression in CD133+ cells relative to CD133− cells (Figure 3). Interestingly, 31 (25%) of the 124 highly expressed isoforms correspond to 18 genes currently under active investigation as drug targets for numerous cancers (specifically ABCG2 [37], ADAM10 [38], ALCAM [39], [40], BSG [41], CD151 [42], CD44 [43], CD47 [44], [45], CD9 [46]–[50], CEACAM5 [51], [52], CEACAM6 [52], CXCR4 [53], EPCAM [54], ERBB2 [55], ICAM1 [56], IGF1R [57], NRP1 [58], NT5E [59], TRPM7 [60]. Of note, the ABCG2 isoform (16X higher expression in the CD133+ subpopulation) and the CXCR4 isoform (4X higher expression in the CD133+ subpopulation) have previously been identified as biomarkers of lung cancer tumor initiating cells spared by cisplatin treatment [15]. In addition to these 18 genes under active investigation as drug targets two other highly expressed genes—CD97 [61], and IFITM1 [62]—have been proposed but not actively pursued as therapeutic antibody targets for primary or metastatic tumors. We believe that other highly expressed cell-surface genes shown in Figure 3 may also be potential drug targets. For example, an isoform of DDR1, a homolog of DDR2 that is a promising target for SCC [7], is highly expressed in CD133+ cells. ICAM4, which has a demonstrated role in carcinogenesis and is located in the 19p13.2 susceptibility locus for multiple cancers [63], [64], is also highly expressed. As these 22 cell surface genes of therapeutic interest encode for proteins involved in cell-cell interactions, cell adhesion, migration, and chemoresistance, their expression similarities and differences identified here illustrates how isoform-level analyses can provide a new level of information for both understanding the physiology of, and for targeting, specific tumor subpopulations.
Figure 3. Isoform expression of cell surface genes in CD133+ and CD133− subpopulations.

Displayed are the expression levels of 1,191 isoforms of 426 CD Molecule, 354 GPCR and 212 ion channel genes. Grey circles correspond to isoforms that did not meet our minimum expression level criteria and blue circles to isoforms that did but were not significantly differentially expressed. Colored triangles indicate significantly differentially expressed isoforms; green down-pointing triangles indicate decreased expression and red up-pointing triangles indicate increased expression in the CD133+ cells. The diagonal grey lines are fold change clines. Isoforms discussed in the text are named. Note that CD133/PROM1 does not meet our expression criterion, underlying the stringency of our data filtering; but is shown for comparison. Here we use HGNC-approved gene symbols [96] and UCSC isoform designations [31].
To assess the degree to which high expression of the therapeutically attractive cell surface proteins described above for the studied tumor is a common phenomenon in SCC we examined TCGA transcriptome data of 221 squamous cell lung cancers [28], [65]. Importantly, TCGA sequencing is performed on bulk tumor, so the subpopulation composition is expected to be quite different than the CD133+ and CD133− subpopulations of the squamous cell carcinoma in our study. The publicly available TCGA expression data is analyzed at the exon-, splice junction-, and gene-level. Since there is no established way to infer isoform-level expression from exon and splice junction expression, we chose to compare our isoform level expression estimates with TCGA gene level expression estimates. Based on the fact that our analysis focused on isoforms with high expression, we reasoned that if our findings are general to SCC then we would observe high expression of the corresponding whole genes across a majority of the 221 tumor samples. As shown in Figure 4A, 4C, the majority of the 22 cell surface genes of therapeutic interest are robustly expressed across all 221 samples with 12 placing in the top 10% (median expression >31 RPKM) of the most highly expressed genes. Interestingly, ABCG2 and ICAM4, which have the two lowest expression levels of the 22 genes, were predominantly expressed in the CD133+ subpopulation. This situation mirrors that of CD133/PROM1 (Figure 3), so we speculate that the apparently low tumor-wide expression of ABCG2 and ICAM4 is because they are robustly expressed only in CD133+ cells, which constitute a minor fraction of the tumor cells. Overall, these results indicate that our findings are general for SCC and so could impact clinical development because they identify this disease as a potential new indication for numerous drugs currently under development and suggest several additional new promising targets.
Figure 4. Box plots of whole gene expression measurements from TCGA lung squamous cell carcinomas.

A) Cell surface and B) apoptosis-related whole gene expression estimates from 221 lung squamous cell carcinoma samples. C) The distribution of expression levels estimated for 20,533 genes per sample, averaged across all 221 samples. The dotted horizontal line indicates the top 5% of genes, when ranked by expression.
Evaluation of mRNA isoforms of genes involved in apoptosis
Given the extent of genomic alterations observed in the CNA analysis of the studied SCC and the multitude of known cell death mechanisms that respond to DNA damage, we suspected that the cancer cells must have altered expression of apoptosis-related genes in order to survive under such a high mutational load. To gain insights into the survival capacity of the CD133+ and CD133− subpopulations, we performed a focused expression analysis on a set of 106 apoptosis-related genes (Table S2 in Information S2) that altogether had 434 mRNA isoforms. As above, we limited our analysis to isoforms that were expressed at the level of at least 30 FPKM and that were covered over at least 60% of their length by sequencing reads in either or both of the subpopulations. Thirty of the 106 genes (28%) and 37 of the 434 isoforms (9%) met these criteria (Figure 5; Figure S6 in Information S1). Interestingly, only 9 isoforms in 6 genes were significantly differentially expressed with at least a four-fold difference between the two subpopulations (FDR<0.01). Noteworthy is the high expression of an L-Myc isoform (MYCL1,uc001cer) and an XIAP isoform (XIAP, ucoo4etx) in both subpopulations, with four-fold upregulation in the CD133+ and the CD133− subpopulations, respectively. L-Myc is a globally acting transcription factor and of all of the proteins in the Myc family (N-Myc, c-Myc, and L-Myc) it has the strongest and most specific activity in promoting human iPSC generation—presumably through its ability to suppress differentiation-associated genes [66]. XIAP is a well-known suppressor of apoptosis in addition to other physiological roles related to its E3 ubiquitin ligase activity [67]. Also noteworthy are the few isoforms (i.e. BCLAF1 and BFAR isoforms) with >16x differential expression. BCLAF1, which has an isoform highly expressed only in the CD133− subpopulation, plays critical roles in many processes [68], including lung development [69]. BFAR, an E3 ubiquitin ligase that likely mediates crosstalk between the intrinsic and extrinsic apoptotic pathways [70], has one isoform expressed exclusively in CD133+ and another exclusively in CD133−. These results suggest that, although overall the CD133+ and CD133− subpopulations similarly express apoptosis-related genes, expression differences of isoforms of key apoptosis genes may in part contribute to self-renewal and survival differences between these cancer cell types.
Figure 5. Isoform expression of apoptosis-related genes in CD133+ and CD133− subpopulations.

Displayed are the expression levels of 434 isoforms of 106 apoptosis-related genes. Grey circles correspond to isoforms that did not meet our minimum expression level criteria and blue circles to isoforms that did but were not significantly differentially expressed. Colored triangles indicate significantly differentially expressed isoforms; green down-pointing triangles indicate decreased expression and red up-pointing triangles indicate increased expression in the CD133+ cells. The diagonal grey lines are fold change clines. Isoforms discussed in the text are named. Isoforms discussed in the text are named. Here we use HGNC-approved gene symbols [96] and UCSC isoform designations [31].
We next focused on the most highly expressed apoptosis-related genes in both the CD133+ and CD133− subpopulations to gain insights into survival mechanisms common between these cancer cell types. This focused analysis revealed c-Myc (MYC,uc003ysi), and the long isoforms of TRAIL/TNFSF10 (TNFSF10,uc003fid), Mcl-1 (MCL1,uc010pch), and Bcl-x (BCL2L1,uc002wwl) to be among the most highly expressed apoptosis-related genes (Figure 5). We confirmed by RT-qPCR the high expression of c-Myc, Mcl-1L, and Bcl-xL in both the CD133+ and CD133− subpopulations (Figure S7 in Information S1). It is well established that high c-Myc expression potently induces the intrinsic (mitochondria-mediated) cell death pathway; however concurrent high expression of Mcl-1L and Bcl-xL can sequester the mitochondrial outer membrane proteins Bak and Bax [71], [72], and thereby block the apoptotic consequences of c-Myc. Such a role for Mcl-1L and Bcl-xL has been recently demonstrated in 14 c-Myc-driven non-squamous cell lung cancer human cell lines [73], in mouse models of adenocarcinoma lung cancer [74], and in other malignancy contexts [75]. Our data implicate Mcl-1L and Bcl-xL in blocking the apoptotic effects of c-Myc in both the CSC and the non-CSC subpopulation of the studied squamous cell carcinoma. We note that while the long isoforms of Mcl-1 and Bcl-x have demonstrated pro-survival functions, the short isoforms of these genes, which were not expressed, are pro-apoptotic [76]. High expression of the long isoform of TRAIL (TRAILL) in both subpopulations is significant, as this cytokine is under investigation as an anti-cancer agent [77] because it can selectively kill tumor cells via receptor-mediated apoptosis. However, prolonged exposure to high levels of TRAILL can result in TRAILL resistance, after which TRAILL becomes a strong inducer of proliferation and metastasis [78]. Previous studies have shown both Mcl-1L, and Bcl-xL to be responsible for acquired TRAILL resistance in non-squamous lung cancer cell lines [79] and in other cancer cell lines [80]–[83]. Of note, another major determinant of TRAIL resistance, c-FlipL (CFLAR,uc002uxb)–the long isoform of Flip/CFLAR that is a catalytically inactive homolog of caspase-8–is also highly expressed in both subpopulations of our studied SCC (Figure 5) [79], [84]–[87]. The short isoforms of both TRAIL and c-Flip also have antithetical pro-apoptotic roles compared to the long isoforms [79], [88]. Altogether our isoform-level gene expression analysis suggests that in the studied SCC both the CD133+ and CD133− subpopulations were driven by very high levels of c-Myc and TRAILL and that the normal apoptotic response to the high expression of these genes was prevented through very high levels of Mcl-1L and Bcl-xL and c-FlipL.
Again we used TCGA transcriptome data of 221 squamous cell lung cancers to determine the generality of our apoptosis gene isoform analysis results. As above for ABCG2 and ICAM4, we attribute the apparently low tumor-wide expression of L-Myc (MYCL1) (Figure 4B, 4C) to the possibility that in SCC it is robustly expressed only in CD133+ cells, which constitute a minor fraction of the tumor cells. Myc (MYC), TRAIL (TNFSF10) and Mcl-1 (MCL1) are among the top 5% of most highly expressed genes (median expression >63 RPKM) and Bcl-x (BCL2L1) and c-Flip (CFLAR) are among the top 10% of most highly expressed genes across all 221 samples (Figure 4B, 4C). TCGA data does not provide mRNA isoform level expression estimates, though, which is crucial for accurate biological interpretation given the antithetical roles of the long and short isoforms of Mcl-1, Bcl-x, c-Flip, and TRAIL [89], [90]. Thus, our isoform analysis provides insights into the shared biological mechanisms underlying survival of both CSC and non-CSC subpopulations in the studied SCC and TCGA data support the generality of our findings.
Discussion
Our study is an important contribution to the understanding of the potential of CSC-targeted therapy [14]. To our knowledge, it is the first to leverage transcriptome sequencing for the purpose of discovering potentially translatable therapeutic strategies based on CSCs. Indeed, few (if any) genomic investigations that leverage current understanding of the CSCs have been reported. There are two main reasons. First, the CSC concept posits that the difference between CSCs and non-CSCs is epigenetic and not genetic, so comparative DNA sequencing is not likely to yield any results that could not be obtained from bulk tumor sequencing. Comparative RNA sequencing, on the other hand, should yield new insights given the expected physiological differences between CSCs and non-CSCs. RNA-seq, though, is a relatively new technique and the needed informatics methods for transforming raw sequencing output into information that reliably reflects the complexity of the transcriptome (particularly isoform calling) are still under development.
While the use of CD133 as a CSC marker is an unresolved issue, our transcriptome measurements do indeed reveal pronounced gene usage in the CD133+ subpopulation that one would expect in a CSC population (e.g. L-Myc, ABCG2, CXCR4). But the expression differences of these genes' isoforms are a matter of degree and do not support a clear and definitive distinction between the two subpopulations—consistent with recent demonstrations of the plasticity of the CSC phenotype. Furthermore, our transcriptome measurements are inconsistent with the CSC concept tenet that a majority of the cells in a cancer have lost their proliferative potential; because c-Myc and TRAILL are very highly expressed in both CD133+ and CD133− cells, both subpopulations have high tumorigenic potential. Tumorigenic potential is the key consideration for defining a targeted therapy, so our analysis results suggest that essentially all of the cells in the studied tumor would need to be targeted, regardless of whether or not SCC harbors CSCs and whether or not CD133 is the most accurate way to identify them.
It is important to highlight that the extra expense and effort associated with transcriptome sequencing and cell sorting were necessary for obtaining our results. For instance, expression profiling of bulk tumor would have revealed high expression of c-Myc, L-Myc, Mcl-1, Bcl-x, c-Flip, and TRAIL, but (critically) would not have revealed which isoforms of Mcl-1, Bcl-x, c-Flip, and TRAIL were being expressed. Furthermore, if we had not sorted the tumor cells using what is currently the best marker of CSCs in SCC we would not have been able to identify phenotypic overlap between CSCs and non-CSCs and targetable mechanisms and proteins that could potentially eradicate all tumor cells.
For complete eradication of a malignancy, it is often presumed that separate therapies targeting the CSC and the non-CSC subpopulations will be required. Our results, on the other hand, suggest the possibility of targeting both subpopulations with one treatment strategy. Our analysis indicates that blocking the anti-apoptotic effects of Bcl-xL and Mcl-1L may render both the CSC and non-CSC subpopulations susceptible to c-Myc- and TRAILL-induced apoptosis. Such a strategy could potentially be achieved using the BCL2 family pan-inhibitor Sabutoclax [91], currently being investigated for leukemias because of its ability to inhibit both Bcl-xL and Mcl-1L [92]. Our observation of high c-FlipL expression suggests an even more robust strategy in which both subpopulations could be additionally sensitized to TRAILL-induced apoptosis via the extrinsic cell death pathway by treatment with, for instance, triterpenoids [93], [94], troglitazone [95], or flavopiridol(69). While to our knowledge no therapies targeting both the CSCs and non-CSCs in a single type of malignancy have been articulated, others [92] have suggested that such a dual-target therapy might prove the most effective for tumor eradication because all tumor cells would be targeted—which is the safest approach given the distinct possibility that the CSC phenotype is a not a stable trait.
Conclusions
We have used a SCC xenograft to demonstrate the power of transcriptome sequencing of CSC-marker-sorted subpopulations to: 1) identify SCC as a new potential indication for numerous ongoing drug development efforts; 2) illustrate how isoform level expression measurements can be used to inform the targeting of tumor subpopulations; and 3) gain insight into tumor physiology and develop a new therapeutic strategy with high clinical potential. Our study is relevant to SCC in particular for it presents numerous possible options to standard radiation and cytotoxic chemotherapy where none currently exist, and it is relevant to cancers in general because it demonstrates how leveraging the CSC concept and isoform-level transcriptome profiling can provide new insights into cancer therapy beyond what would be possible with DNA sequencing.
Supporting Information
Supplemental Materials.
(DOCX)
Supplemental Table.
(XLSX)
Acknowledgments
We would like to thank Xiaoyun Wang for helping with laboratory procedures; Louise Laurent for enabling the microarray experiments; Brian Coullahan from Life Technologies for support and assistance with library preparation and SOLiD sequencing; and Shawn Yost, Erin Smith and Chris DeBoever for stimulating scientific discussions.
Funding Statement
This work was supported by the Center for Translational Science Award (UL1RR031980 and UL1TR000100) from the National Center for Research Resources, three grants (1R21CA152613-01, 1R21CA155615-01A1, CA69375) from the National Cancer Institute, four grants from the California Institute for Regenerative Medicine (CL1-00502, RT1-01108, TR1-01250, TR2-01789) and a grant from the National Institute of Allergy and Infectious Diseases (P30 AI036214). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Siegel R, Ward E, Brawley O, Jemal A (2011) Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA 61: 212–236 doi:10.3322/caac.20121. [DOI] [PubMed] [Google Scholar]
- 2. Heukamp LC, Wolf J, Büttner R (2011) Molecular diagnostics of lung cancer for treatment stratification. Der Internist 52: 146, 148–150, 152–154 doi:10.1007/s00108-010-2698-y. [DOI] [PubMed] [Google Scholar]
- 3. Johnson DH, Fehrenbacher L, Novotny WF, Herbst RS, Nemunaitis JJ, et al. (2004) Randomized phase II trial comparing bevacizumab plus carboplatin and paclitaxel with carboplatin and paclitaxel alone in previously untreated locally advanced or metastatic non-small-cell lung cancer. J Clin Oncol 22: 2184–2191 doi:10.1200/JCO.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 4. Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, et al. (2006) Paclitaxel–Carboplatin Alone or with Bevacizumab for Non–Small-Cell Lung Cancer. N Engl J Med 355: 2542–2550. [DOI] [PubMed] [Google Scholar]
- 5. Langer CJ, Besse B, Gualberto A, Brambilla E, Soria J-C (2010) The evolving role of histology in the management of advanced non-small-cell lung cancer. J Clin Oncol 28: 5311–5320 doi:10.1200/JCO.2010.28.8126. [DOI] [PubMed] [Google Scholar]
- 6. Weiss J, Sos ML, Seidel D, Peifer M, Zander T, et al. (2010) Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci Transl Med 2: 62ra93 doi:10.1126/scitranslmed.3001451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hammerman PS, Sos ML, Ramos AH, Xu C, Dutt A, et al. (2011) Mutations in the DDR2 Kinase Gene Identify a Novel Therapeutic Target in Squamous Cell Lung Cancer. Cancer Discov 1: 78–89 doi:10.1158/2159-8274.CD-11-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, et al. (2008) Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455: 1069–1075 doi:10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weir BA, Woo MS, Getz G, Perner S, Ding L, et al. (2007) Characterizing the cancer genome in lung adenocarcinoma. Nature 450: 893–898 doi:10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bell D, Berchuck A, Birrer M, Chien J, Cramer DW, et al. (2011) Integrated genomic analyses of ovarian carcinoma. Nature 474: 609–615 doi:10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, et al. (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17: 98–110 doi:10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, et al. (2008) Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455: 1069–1075 doi:10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.The Cancer Genome Atlas website. Available: http://cancergenome.nih.gov.
- 14. Clevers H (2011) The cancer stem cell: premises, promises and challenges. Nature Med 17: 313–319 doi:10.1038/nm.2304. [DOI] [PubMed] [Google Scholar]
- 15. Bertolini G, Roz L, Perego P, Tortoreto M, Fontanella E, et al. (2009) Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc Natl Acad Sci USA 106: 16281–16286 doi:10.1073/pnas.0905653106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Perona R, López-Ayllón BD, Castro Carpeño J, Belda-Iniesta C (2011) A role for cancer stem cells in drug resistance and metastasis in non-small-cell lung cancer. Clin Transl Oncol 13: 289–293 doi:10.1007/s12094-011-0656-3. [DOI] [PubMed] [Google Scholar]
- 17. Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, et al. (2011) Stochastic State Transitions Give Rise to Phenotypic Equilibrium in Populations of Cancer Cells. Cell 146: 633–644 doi:10.1016/j.cell.2011.07.026. [DOI] [PubMed] [Google Scholar]
- 18. Quintana E, Shackleton M, Foster HR, Fullen DR, Sabel MS, et al. (2010) Phenotypic heterogeneity among tumorigenic melanoma cells from patients that is reversible and not hierarchically organized. Cancer Cell 18: 510–523 doi:10.1016/j.ccr.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, et al. (2008) A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 321: 956–960 doi:10.1126/science.1160342. [DOI] [PubMed] [Google Scholar]
- 20. Skotheim RI, Nees M (2007) Alternative splicing in cancer: noise, functional, or systematic? Int J Biochem Cell Biol 39: 1432–1449 doi:10.1016/j.biocel.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 21. He X, Ee PLR, Coon JS, Beck WT (2004) Alternative splicing of the multidrug resistance protein 1/ATP binding cassette transporter subfamily gene in ovarian cancer creates functional splice variants and is associated with increased expression of the splicing factors PTB and SRp20. Clin Cancer Res 10: 4652–4660 doi:10.1158/1078-0432.CCR-03-0439. [DOI] [PubMed] [Google Scholar]
- 22. Parajes M, Ezponda M, Catena R, Calvo A, Pio R, et al. (2007) Alternative splicing: an emerging topic in molecular and clinical oncology. Lancet Oncol 8: 349–357 doi:10.1016/S1470-2045(07)70104-3. [DOI] [PubMed] [Google Scholar]
- 23. Grosso AR, Martins S, Carmo-Fonseca M (2008) The emerging role of splicing factors in cancer. EMBO Reports 9: 1087–1093 doi:10.1038/embor.2008.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Venables JP (2006) Unbalanced alternative splicing and its significance in cancer. BioEssays 28: 378–386 doi:10.1002/bies.20390. [DOI] [PubMed] [Google Scholar]
- 25. Tirino V, Camerlingo R, Franco R, Malanga D, La Rocca A, et al. (2009) The role of CD133 in the identification and characterisation of tumour-initiating cells in non-small-cell lung cancer. Eur J Cardio-thoracic Surgery 36: 446–453 doi:10.1016/j.ejcts.2009.03.063. [DOI] [PubMed] [Google Scholar]
- 26. Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, et al. (2008) Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ 15: 504–514 doi:10.1038/sj.cdd.4402283. [DOI] [PubMed] [Google Scholar]
- 27. Chen Y-C, Hsu H-S, Chen Y-W, Tsai T-H, How C-K, et al. (2008) Oct-4 expression maintained cancer stem-like properties in lung cancer-derived CD133-positive cells. PloS ONE 3: e2637 doi:10.1371/journal.pone.0002637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hammerman PS, Hayes DN, Wilkerson MD, Schultz N, Bose R, et al. (2012) Comprehensive genomic characterization of squamous cell lung cancers. Nature 489: 519–525 doi:10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Homer N, Merriman B, Nelson SF (2009) BFAST: an alignment tool for large scale genome resequencing. PloS ONE 4: e7767 doi:10.1371/journal.pone.0007767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pruitt KD, Tatusova T, Maglott DR (2007) NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 35: D61–5 doi:10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hsu F, Kent WJ, Clawson H, Kuhn RM, Diekhans M, et al. (2006) The UCSC Known Genes. Bioinformatics 22: 1036–1046 doi:10.1093/bioinformatics/btl048. [DOI] [PubMed] [Google Scholar]
- 32. Hubbard T (2002) The Ensembl genome database project. Nucleic Acids Res 30: 38–41 doi:10.1093/nar/30.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hebenstreit D, Fang M, Gu M, Charoensawan V, Van Oudenaarden A, et al. (2011) RNA sequencing reveals two major classes of gene expression levels in metazoan cells. Mol Systems Biol 7: 497 doi:10.1038/msb.2011.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pinto D, Darvishi K, Shi X, Rajan D, Rigler D, et al. (2011) Comprehensive assessment of array-based platforms and calling algorithms for detection of copy number variants. Nat Biotechnol 29: 512–520 doi:10.1038/nbt.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Human Cell Differentiation Molecules Organization website. Available: http://www.hcdm.org.
- 36.The International Union of Basic and Clinical Pharmacology website. Available: http://www.iuphar.org.
- 37. Tiwari AK, Sodani K, Wang S-R, Kuang Y-H, Ashby CR, et al. (2009) Nilotinib (AMN107, Tasigna) reverses multidrug resistance by inhibiting the activity of the ABCB1/Pgp and ABCG2/BCRP/MXR transporters. Biochem Pharm 78: 153–161 doi:10.1016/j.bcp.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 38. Moss ML, Stoeck A, Yan W, Dempsey PJ (2008) ADAM10 as a Target for Anti-Cancer Therapy. Curr Pharm Biotechnol 9: 7. [DOI] [PubMed] [Google Scholar]
- 39. Roth A, Drummond DC, Conrad F, Hayes ME, Kirpotin DB, et al. (2007) Anti-CD166 single chain antibody-mediated intracellular delivery of liposomal drugs to prostate cancer cells. Mol Cancer Ther 6: 2737–2746 doi:10.1158/1535-7163.MCT-07-0140. [DOI] [PubMed] [Google Scholar]
- 40. Wiiger MT, Gehrken HB, Fodstad Ø, Maelandsmo GM, Andersson Y (2010) A novel human recombinant single-chain antibody targeting CD166/ALCAM inhibits cancer cell invasion in vitro and in vivo tumour growth. Cancer Immunol Immunother 59: 1665–1674 doi:10.1007/s00262-010-0892-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kanekura T, Chen X (2010) CD147/basigin promotes progression of malignant melanoma and other cancers. J Dermatol Sci 57: 149–154 doi:10.1016/j.jdermsci.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 42. Yang XH, Flores LM, Li Q, Zhou P, Xu F, et al. (2010) Disruption of laminin-integrin-CD151-focal adhesion kinase axis sensitizes breast cancer cells to ErbB2 antagonists. Cancer Res 70: 2256–2263 doi:10.1158/0008-5472.CAN-09-4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pham PV, Phan NLC, Nguyen NT, Truong NH, Duong TT, et al. (2011) Differentiation of breast cancer stem cells by knockdown of CD44: promising differentiation therapy. J Transl Med 9: 209 doi:10.1186/1479-5876-9-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Uluçkan O, Becker SN, Deng H, Zou W, Prior JL, et al. (2009) CD47 regulates bone mass and tumor metastasis to bone. Cancer Res 69: 3196–3204 doi:10.1158/0008-5472.CAN-08-3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chao MP, Weissman IL, Majeti R (2012) The CD47-SIRPα pathway in cancer immune evasion and potential therapeutic implications. Curr Opin Immunol 24: 225–232 doi:10.1016/j.coi.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hwang JR, Jo K, Lee Y, Sung B-J, Park YW, et al. (2012) Upregulation of CD9 in ovarian cancer is related to the induction of TNF-α gene expression and constitutive NF-κB activation. Carcinogenesis 33: 77–83 doi:10.1093/carcin/bgr257. [DOI] [PubMed] [Google Scholar]
- 47. Ovalle S, Gutiérrez-López MD, Olmo N, Turnay J, Lizarbe MA, et al. (2007) The tetraspanin CD9 inhibits the proliferation and tumorigenicity of human colon carcinoma cells. Int J Cancer 121: 2140–2152 doi:10.1002/ijc.22902. [DOI] [PubMed] [Google Scholar]
- 48. Nakamoto T, Murayama Y, Oritani K, Boucheix C, Rubinstein E, et al. (2009) A novel therapeutic strategy with anti-CD9 antibody in gastric cancers. J Gastroenterol 44: 889–896 doi:10.1007/s00535-009-0081-3. [DOI] [PubMed] [Google Scholar]
- 49. Murayama Y, Miyagawa J, Oritani K, Yoshida H, Yamamoto K, et al. (2004) CD9-mediated activation of the p46 Shc isoform leads to apoptosis in cancer cells. J Cell Sci 117: 3379–3388 doi:10.1242/jcs.01201. [DOI] [PubMed] [Google Scholar]
- 50. Kohmo S, Kijima T, Otani Y, Mori M, Minami T, et al. (2010) Cell surface tetraspanin CD9 mediates chemoresistance in small cell lung cancer. Cancer Res 70: 8025–8035 doi:10.1158/0008-5472.CAN-10-0996. [DOI] [PubMed] [Google Scholar]
- 51. Govindan SV, Cardillo TM, Moon S-J, Hansen HJ, Goldenberg DM (2009) CEACAM5-targeted therapy of human colonic and pancreatic cancer xenografts with potent labetuzumab-SN-38 immunoconjugates. Clin Cancer Res 15: 6052–6061 doi:10.1158/1078-0432.CCR-09-0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Blumenthal RD, Hansen HJ, Goldenberg DM (2005) Inhibition of adhesion, invasion, and metastasis by antibodies targeting CEACAM6 (NCA-90) and CEACAM5 (Carcinoembryonic Antigen). Cancer Res 65: 8809–8817 doi:10.1158/0008-5472.CAN-05-0420. [DOI] [PubMed] [Google Scholar]
- 53. Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, et al. (2007) Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 1: 313–323 doi:10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 54. Linke R, Klein A, Seimetz D (2010) Catumaxomab: clinical development and future directions. mAbs 2: 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yamazaki M, Yamashita Y, Kubo N, Yashiro M, Ohira M, et al. (2012) Concurrent biological targeting therapy of squamous cell carcinoma of the esophagus with cetuximab and trastuzumab. Oncol Rep 28: 49. [DOI] [PubMed] [Google Scholar]
- 56. DiPaola RS, Plante M, Kaufman H, Petrylak DP, Israeli R, et al. (2006) A phase I trial of pox PSA vaccines (PROSTVAC-VF) with B7-1, ICAM-1, and LFA-3 co-stimulatory molecules (TRICOM) in patients with prostate cancer. J Trans Med 4: 1 doi:10.1186/1479-5876-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fagan DH, Uselman RR, Sachdev D, Yee D (2012) Acquired resistance to tamoxifen is associated with loss of the type I insulin-like growth factor receptor (IGF1R): implications for breast cancer treatment. Cancer Res To Appear doi:10.1158/0008-5472.CAN-12-0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ueyama H, Horibe T, Nakajima O, Ohara K, Kohno M, et al. (2011) Semaphorin 3A lytic hybrid peptide binding to neuropilin-1 as a novel anti-cancer agent in pancreatic cancer. Biochem Biophysical Res Comm 414: 60–66 doi:10.1016/j.bbrc.2011.09.021. [DOI] [PubMed] [Google Scholar]
- 59. Zhang B (2010) CD73: a novel target for cancer immunotherapy. Cancer Res 70: 6407–6411 doi:10.1158/0008-5472.CAN-10-1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zierler S, Yao G, Zhang Z, Kuo WC, Pörzgen P, et al. (2011) Waixenicin A inhibits cell proliferation through magnesium-dependent block of transient receptor potential melastatin 7 (TRPM7) channels. J Biol Chem 286: 39328–39335 doi:10.1074/jbc.M111.264341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ward Y, Lake R, Yin JJ, Heger CD, Raffeld M, et al. (2011) LPA receptor heterodimerizes with CD97 to amplify LPA-initiated RHO-dependent signaling and invasion in prostate cancer cells. Cancer Res 71: 7301–7311 doi:10.1158/0008-5472.CAN-11-2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yu F, Ng SSM, Chow BKC, Sze J, Lu G, et al. (2011) Knockdown of interferon-induced transmembrane protein 1 (IFITM1) inhibits proliferation, migration, and invasion of glioma cells. J Neurooncol 103: 187–195 doi:10.1007/s11060-010-0377-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Roland CL, Harken AH, Sarr MG, Barnett CC (2007) ICAM-1 expression determines malignant potential of cancer. Surgery 141: 705–707 doi:10.1016/j.surg.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 64. Kammerer S, Roth RB, Reneland R, Marnellos G, Hoyal CR, et al. (2004) Large-scale association study identifies ICAM gene region as breast and prostate cancer susceptibility locus. Cancer Res 64: 8906–8910 doi:10.1158/0008-5472.CAN-04-1788. [DOI] [PubMed] [Google Scholar]
- 65.The Cancer Genome Atlas website. Available: http://cancergenome.nih.gov.
- 66. Nakagawa M, Takizawa N, Narita M, Ichisaka T, Yamanaka S (2010) Promotion of direct reprogramming by transformation-deficient Myc. Proc Natl Acad Sci USA 107: 14152–14157 doi:10.1073/pnas.1009374107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Galbán S, Duckett CS (2010) XIAP as a ubiquitin ligase in cellular signaling. Cell Death Differ 17: 54–60 doi:10.1038/cdd.2009.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sarras H, Alizadeh Azami S, McPherson JP (2010) In search of a function for BCLAF1. Sci World J 10: 1450–1461 doi:10.1100/tsw.2010.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. McPherson JP, Sarras H, Lemmers B, Tamblyn L, Migon E, et al. (2009) Essential role for Bclaf1 in lung development and immune system function. Cell Death Differ 16: 331–339 doi:10.1038/cdd.2008.167. [DOI] [PubMed] [Google Scholar]
- 70. Zhang H (2000) BAR: An apoptosis regulator at the intersection of caspases and Bcl-2 family proteins. Proc Natl Acad Sci USA 97: 2597–2602 doi:10.1073/pnas.97.6.2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Edlich F, Banerjee S, Suzuki M, Cleland MM, Arnoult D, et al. (2011) Bcl-xL Retrotranslocates Bax from the Mitochondria into the Cytosol. Cell 145: 104–116 doi:10.1016/j.cell.2011.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Dunkle A, Dzhagalov I, He Y-W (2010) Mcl-1 promotes survival of thymocytes by inhibition of Bak in a pathway separate from Bcl-2. Cell Death Differ 17: 994–1002 doi:10.1038/cdd.2009.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhang H, Guttikonda S, Roberts L, Uziel T, Semizarov D, et al. (2011) Mcl-1 is critical for survival in a subgroup of non-small-cell lung cancer cell lines. Oncogene 30: 1963–1968 doi:10.1038/onc.2010.559. [DOI] [PubMed] [Google Scholar]
- 74. Allen TD, Zhu CQ, Jones KD, Yanagawa N, Tsao M-S, et al. (2011) Interaction between MYC and MCL1 in the genesis and outcome of non-small-cell lung cancer. Cancer Res 71: 2212–2221 doi:10.1158/0008-5472.CAN-10-3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Meyer N, Penn LZ (2008) Reflecting on 25 years with MYC. Nat Rev Cancer 8: 976–990 doi:10.1038/nrc2231. [DOI] [PubMed] [Google Scholar]
- 76.Ding W, Yin X (2009) The BCL-2 Family Proteins. In: Dong Z, Yin X-M, editors. Essentials of Apoptosis. Totowa, NJ: Humana Press. doi:10.1007/978-1-60327-381-7.
- 77. Lacour S, Hammann A, Wotawa A, Corcos L, Solary E, et al. (2001) Anticancer Agents Sensitize Tumor Cells to Tumor Necrosis Factor-related Apoptosis-inducing Ligand-mediated Caspase-8 Activation and Apoptosis. Cancer Res 61: 1645–1651. [PubMed] [Google Scholar]
- 78. Malhi H, Gores GJ (2006) TRAIL resistance results in cancer progression: a TRAIL to perdition? Oncogene 25: 7333–7335 doi:10.1038/sj.onc.1209765. [DOI] [PubMed] [Google Scholar]
- 79. Wang X, Chen W, Zeng W, Bai L, Tesfaigzi Y, et al. (2008) Akt-mediated eminent expression of c-FLIP and Mcl-1 confers acquired resistance to TRAIL-induced cytotoxicity to lung cancer cells. Mol Cancer Ther 7: 1156–1163 doi:10.1158/1535-7163.MCT-07-2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Taniai M, Grambihler A, Higuchi H, Werneburg N, Bronk SF, et al. (2004) Mcl-1 Mediates Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand Resistance in Human Cholangiocarcinoma Cells. Cancer Res 64: 3517–3524 doi:10.1158/0008-5472.CAN-03-2770. [DOI] [PubMed] [Google Scholar]
- 81. Zhu H, Guo W, Zhang L, Davis JJ, Wu S, et al. (2005) Enhancing TRAIL-induced apoptosis by Bcl-X(L) siRNA. Cancer Biol Therapy 4: 393–397. [DOI] [PubMed] [Google Scholar]
- 82. Meng XW, Lee S-H, Dai H, Loegering D, Yu C, et al. (2007) Mcl-1 as a buffer for proapoptotic Bcl-2 family members during TRAIL-induced apoptosis: a mechanistic basis for sorafenib (Bay 43-9006)-induced TRAIL sensitization. J Biol Chem 282: 29831–29846 doi:10.1074/jbc.M706110200. [DOI] [PubMed] [Google Scholar]
- 83. Kim S-H, Ricci MS, El-Deiry WS (2008) Mcl-1: a gateway to TRAIL sensitization. Cancer Res 68: 2062–2064 doi:10.1158/0008-5472.CAN-07-6278. [DOI] [PubMed] [Google Scholar]
- 84. Raclaw KA, Heemers HV, Kidd EM, Dehm SM, Tindall DJ (2008) Induction of FLIP expression by androgens protects prostate cancer cells from TRAIL-mediated apoptosis. The Prostate 68: 1696–1706 doi:10.1002/pros.20844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. El-Gazzar A, Wittinger M, Perco P, Anees M, Horvat R, et al. (2010) The role of c-FLIP(L) in ovarian cancer: chaperoning tumor cells from immunosurveillance and increasing their invasive potential. Gynecologic Oncol 117: 451–459 doi:10.1016/j.ygyno.2010.02.024. [DOI] [PubMed] [Google Scholar]
- 86. Haag C, Stadel D, Zhou S, Bachem MG, Möller P, et al. (2011) Identification of c-FLIP(L) and c-FLIP(S) as critical regulators of death receptor-induced apoptosis in pancreatic cancer cells. Gut 60: 225–237 doi:10.1136/gut.2009.202325. [DOI] [PubMed] [Google Scholar]
- 87. Koehler B-C, Urbanik T, Vick B, Boger R-J, Heeger S, et al. (2009) TRAIL-induced apoptosis of hepatocellular carcinoma cells is augmented by targeted therapies. World J Gastroenterol 15: 5924–5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Krieg A, Krieg T, Wenzel M, Schmitt M, Ramp U, et al. (2003) TRAIL-beta and TRAIL-gamma: two novel splice variants of the human TNF-related apoptosis-inducing ligand (TRAIL) without apoptotic potential. Br J Cancer 88: 918–927 doi:10.1038/sj.bjc.6600772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Bae J, Leo CP, Hsu SY, Hsueh AJ (2000) MCL-1S, a splicing variant of the antiapoptotic BCL-2 family member MCL-1, encodes a proapoptotic protein possessing only the BH3 domain. J Biol Chem 275: 25255–25261 doi:10.1074/jbc.M909826199. [DOI] [PubMed] [Google Scholar]
- 90. Yang E, Zha J, Jockel J, Boise LH, Thompson CB, et al. (1995) Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 80: 285–291. [DOI] [PubMed] [Google Scholar]
- 91. Dash R, Azab B, Quinn BA, Shen X, Wang X-Y, et al. (2011) Apogossypol derivative BI-97C1 (Sabutoclax) targeting Mcl-1 sensitizes prostate cancer cells to mda-7/IL-24-mediated toxicity. Proc Natl Acad Sci USA 108: 8785–8790 doi:10.1073/pnas.1100769108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Willis SN, Fletcher JI, Kaufmann T, Van Delft MF, Chen L, et al. (2007) Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 315: 856–859 doi:10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- 93. Hyer ML, Croxton R, Krajewska M, Krajewski S, Kress CL, et al. (2005) Synthetic triterpenoids cooperate with tumor necrosis factor-related apoptosis-inducing ligand to induce apoptosis of breast cancer cells. Cancer Res 65: 4799–4808 doi:10.1158/0008-5472.CAN-04-3319. [DOI] [PubMed] [Google Scholar]
- 94. Zou W, Chen S, Liu X, Yue P, Sporn MB, et al. (2007) c-FLIP downregulation contributes to apoptosis induction by the novel synthetic triterpenoid methyl-2-cyano-3, 12-dioxooleana-1, 9-dien-28-oate (CDDO-Me) in human lung cancer cells. Cancer Biol Therapy 6: 1614–1620. [DOI] [PubMed] [Google Scholar]
- 95. Schultze K, Böck B, Eckert A, Oevermann L, Ramacher D, et al. (2006) Troglitazone sensitizes tumor cells to TRAIL-induced apoptosis via down-regulation of FLIP and Survivin. Apoptosis 11: 1503–1512 doi:10.1007/s10495-006-8896-3. [DOI] [PubMed] [Google Scholar]
- 96. Seal RL, Gordon SM, Lush MJ, Wright MW, Bruford EA (2011) genenames.org: the HGNC resources in 2011. Nucleic Acids Res 39: D514–9 doi:10.1093/nar/gkq892. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Materials.
(DOCX)
Supplemental Table.
(XLSX)