

Abstract
More than 60% of neuroendocrine tumours, also called carcinoids, are localised within the gastrointestinal tract. Small bowel neuroendocrine tumours have been diagnosed with increasing frequency over the past 35 years, being the second most frequent tumours of the small intestine. Ileal neuroendocrine tumours diagnosis is late because patients have non-specific symptoms. We have proposed to illustrate as an example the case of a patient, and on its basis, to make a brief review of the literature on small bowel neuroendocrine tumours, resuming several recent changes in the field, concerning classification criteria of these tumours and new recommendations and current advances in diagnosis and treatment. This patient came to our emergency department with a complete bowel obstruction, along with a 2-year history of peristaltic abdominal pain, vomits and diarrhoea episodes. During emergency laparotomy, an ileal stricture was observed, that showed to be a neuroendocrine tumour of the small bowel.
Background
The neuroendocrine tumours (NETs) of the digestive tract or carcinoid tumours are a diverse group of malignancies considered to be rare, but recent data are indicating a significant increase in both incidence and prevalence.
The purpose of this manuscript is to non-systematically review the subject of small bowel NETs starting from the basis of a rather typical case of gastrointestinal NET; though a rare tumour, in recent years, this pathology has become more and more frequent but the diagnosis remain difficult because of non-specific symptoms. The aim of this review is to report several recent publications in the field concerning new classification criteria of this type of tumour and we try to give some recommendations that take into account current advances in the diagnosis and treatment of NETs.
Case presentation
We report the case of a patient with a long medical history of abdominal cramp, vomit episodes in alternation with diarrhoea (until 10–12 stools a day) and reduction of weight. A total colonoscopy performed, 2 years before, had failed to show a tumour or any lesion on the large bowel or the terminal ileum. The patient was treated symptomatically and an abdominal CT done, 1 year later, was also non-specific; no further investigations were done except for microbiological stool examination and faecal occult blood test, both being negatives.
The patient came at the emergency department for superior abdominal pain rapidly progressive with uncontrollable vomiting and no intestinal transit. Laboratory tests identified a moderate hypochromic microcytic anaemia, otherwise showing normal values. An abdominal CT scan revealed a thickened intestinal wall in the right lower quadrant and upstream-dilated small bowel loops sustaining a mechanical small bowel obstruction (figure 1). The patient underwent exploratory laparotomy, laparoscopy being considered too risky because of bowel dilation. During surgery, we identified an ileal stricture with enlarged nodes in the mesentery (figure 2); a second lesion was observed on another small bowel loop without stenosis. Double segmentary enterectomy and mesenteric lymphadenectomy were performed. The histopathological workup showed a double well-differentiated (G1) NET of the ileum (figures 3 and 4) with mesenteric node metastasis; the surgical margins were free of tumour (R0). The postoperative course was uneventful and the patient was discharged home 7 days after the surgery without any specific treatment. At 1-year follow-up, the patient was free of symptoms and the thoraco-abdominal CT scan showed neither local recurrence nor distant metastasis.
Figure 1.

Abdominal CT scan demonstrating small bowel wall thickening and dilated intestinal loops.
Figure 2.
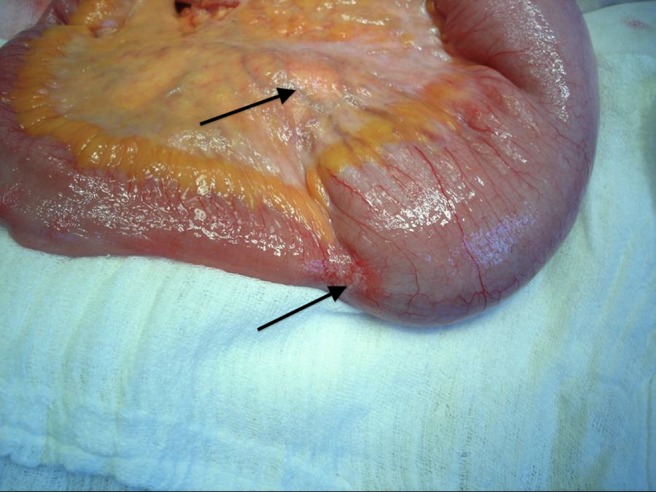
Main carcinoid tumour of the ileum with bowel obstruction and enlarged mesenteric lymph nodes (arrows).
Figure 3.
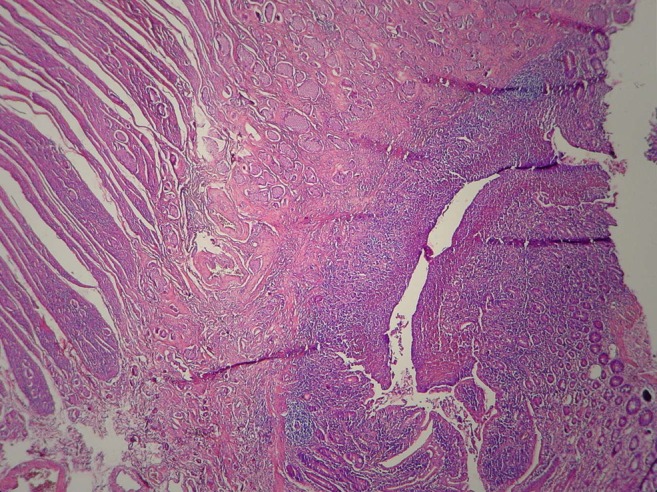
Microscopy sample showing the mucosa and submucosa of ileum invaded by several solid nests of cells, corresponding to type I growth and well-differentiated neuroendocrine tumour (G1).
Figure 4.
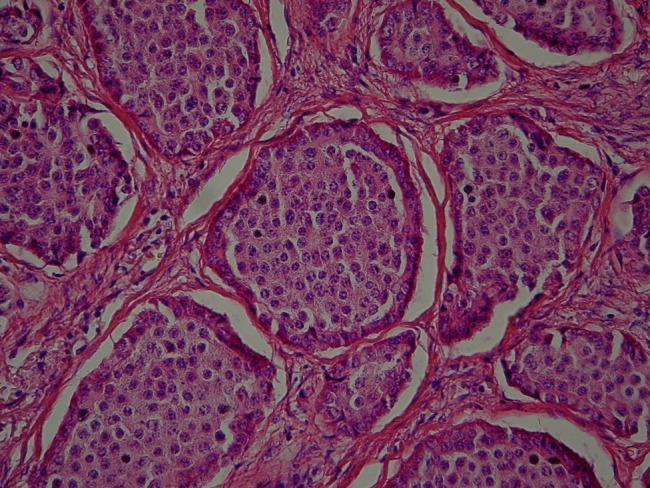
Microscopy sample showing nests of cells of well-differentiated neuroendocrine tumour (G1).
Discussion
Introduction
The endocrine tumours of the digestive tract (gastro-entero-pancreatic neuroendocrine neoplasm, GEP-NETs), formerly called NETs or carcinoid tumours, are a diverse group of malignancies considered to be rare, but recent data are indicating a significant increase in both incidence and prevalence during the last few decades. Current figures indicate an incidence of 5.1/1 00 000/year and a prevalence of 35/1 00 000.1
Epidemiology, nomenclature and classification
The age-adjusted diagnosed incidence rate of malignant NETs arising from jejunum and ileum, in the Surveillance, Epidemiology and End Results (SEER) program database, is 0.67/100 000 per year. Time-trend analyses have shown a rise in the diagnosed incidence of NETs including those of the jejunum, ileum, appendix and caecum. The true incidence rates of NETs at these sites are likely to be substantially higher for several reasons. First, NETs of the small bowel are difficult to diagnose and likely account for a substantial number of unknown primary NETs. Second, data obtained from the SEER registries likely underestimated the total number of patients with NETs. Only patients with malignant NETs are included in the SEER registries.2
Intestinal NETs have been diagnosed with increasing frequency over the past 35 years and presently account for approximately 2% of all gastrointestinal neoplasm, being the second most frequent tumours of the small intestine.3 NETs, arising from entero-chromaffin cells, classified in Apud system, are more often localised in the appendix and the rectum; these are small and benign tumours, discovered fortuitously at the time of appendectomy or colonoscopy removal.4
The incidence rates of appendiceal and caecal NETs were similar between male and female. However, for jejunal and ileal NETs, the incidence rate was slightly higher among males. The median age at diagnosis for midgut NETs was 64 (SD 15.5) years in the SEER registry. Patients in appendiceal subgroup were younger at diagnosis, with a median age of 47 (SD 18) years. However, the true age at diagnosis for appendiceal NETs is likely even lower because most small tumours found incidentally at appendectomy are considered benign and not reported to SEER. Those with jejunal/ileal and caecal NETs had similar age at diagnosis (66 (SD 13) and 68 (SD 14) years, respectively).2
It seems that all GEP-NETs are potentially malignant neoplasm. However, the various entities that are recognised in the gastrointestinal tract and the pancreas differ considerably in their metastasising capacity. It has, therefore, always been difficult to classify the GEP-NETs.5
In recent years, it was felt that the 2000 WHO's classification of the neuroendocrine neoplasm should be supplemented by criteria that may refine the prognostic stratification of GEP-NETs to allow a better stage-adjusted treatment of the patients. Therefore, in 2010, a revised version of the WHO classification of GEP-NETs appeared. This new classification introduced several changes, a carcinoid being now defined as a grade 1 or 2 NET and grade 3, small-cell or large-cell neuroendocrine carcinoma (NEC; table 1).6–8
Table 1.
Classification and nomenclature of gastro-entero-pancreatic neuroendocrine tumours (according to refs. 8 and 9)
| Grade | (WHO 2010)17 |
|---|---|
| Low | Neuroendocrine neoplasm, grade 1 (NET—G1) |
| Intermediate | Neuroendocrine neoplasm, grade 2 (NET—G2) |
| High | Neuroendocrine carcinoma, grade 3 (G3), small cell carcinoma (NEC) |
| Neuroendocrine carcinoma grade 3 (G3), large cell neuroendocrine carcinoma (NEC) |
The grade of the tumour MUST be included in the pathology report, along with a reference to the specific grading system being used. Unqualified terms such as neuroendocrine tumour or neuroendocrine carcinoma without reference to grade do not provide adequate pathology information.
NET, neuroendocrine tumour.
Although NETs at any site can produce hormone(s), well-differentiated NETs of the jejunum, ileum, appendix and caecum are most closely associated with the classic carcinoid syndrome and are often described as midgut carcinoids because of their common embryological origin.2
The highest frequency of small intestinal NETs is in the ileum, and is ∼7 times more frequent than in the duodenum and the jejunum.9 Small intestinal NETs exhibit an overall higher frequency of metastases at the time of diagnosis (∼60% of staged tumours) compared to all GEP-NETs (26% of staged tumours). The ileal NETs are small and frequently multiple, presenting complicated in 30–50% of cases by bowel obstruction, mesenteric invasion or bleeding.4 9 10
Pathogenesis and pathology
The biological basis of small intestinal neuroendocrine pathogenesis, malignancy and metastasis is unknown. NETs are considered to arise from abnormal mucosal precursor cells. It is likely that the cell that accumulates the mutations necessary for the development of NETs is a committed neuroendocrine progenitor, a cell not as yet defined in the human gastrointestinal tract. The precise mechanisms underlying the lineage pathways of neuroendocrine cells and their precursor remains poorly defined but the Notch signalling pathway is implicated in regulation of cell differentiation from stem cells. Typical small bowel NETs display an insular growth pattern (type I), which consists of solid nests or cords of cells with clearly defined boundaries. A trabecular pattern (type II) consists of narrow cell bands forming ribbons, regularly anastomosing along a highly differentiated vascular network. Type III has a glandular pattern, consisting of cells arranged in alveolar, acinar or rosette patterns with glandular cavities or pseudocavities. Types IV and V NETs consist of undifferentiated and mixed cells, respectively. Multifocal lesions are evident in ∼30% of small bowel NETs.7
The tumour cells are characteristically argyrophil and argentaffin, and over 85% of the tumours exhibit positive immunohistochemistry for CgA, Leu-7, neuron-specific enolase (NSE) and 5-HT. The vast majority of these lesions are ‘classical’ ileal carcinoids with production of 5-HT and substance P, but rare tumours producing enteroglucagon, PP or peptide YY may occur. Small bowel NETs exhibit the highest frequency of non-NET tumour association, for example, colorectal cancers (39%). Other associated non-endocrine tumours include adenocarcinomas of the small bowel, stomach, lung, prostate and cervix uteri.7
Clinical aspects and molecular markers
The gastrointestinal NETs have classically a long-lasting clinical silence and slow-evolution to advanced stage, the symptoms and sign being non-specific. The clinical diagnosis is difficult and the importance of complementary investigations is crucial. Another clinical particularity of NET is the carcinoid syndrome. It consists of abdominal pain, flushing, diarrhoea, hypertension, bronchospasm and right-side cardiac vegetations and is caused by the hypersecretion of serotonin into the systemic circulation. One difficulty is that these symptoms occur in less than 10% of cases and are usually associated with the presence of hepatic metastases, being late signs of diagnosis.11
Basic laboratory tests are usually not helpful, being most of the time normal, and neuroendocrine markers could guide the diagnosis of NETs. Determination of 5-hydroxyindoleacetic acid (5-HIAA) in 24 h urine sample and serum chromogranin-A (CgA) measurements are used as biochemical tumour markers for clinical diagnosis, and moreover as monitors of treatment effects and prognostic predictors; CgA and 5-HIAA levels are increased especially in metastatic tumours.11 12
The sensitivity of CgA in diagnosis of GEP-NETs was as high as 82.1%, and the specificity was 96.2% in a recent report. It were also showed significant differences in CgA levels in patients with metastatic and non-metastatic tumours.13
It is considered that CgA is the most important tumour marker for well-differentiated NETs whereas, for poorly differentiated NEC, NSE is a better indicator. Between other markers, cytokeratin fragments (CKfr) was recommended to be used in patients with well-differentiated NET, and both CKfr and progastrin-releasing peptide in patients with poorly differentiated NEC.14
Diagnostic
One of the diagnostic steps that increased in its importance is the need to assess primary tumour location and extent in these patients. Without such information, it is not possible to adequately manage the disease. Conventional imaging studies (CT scan, MRI, ultrasound, angiography and enteroclysis), functional localisation studies measuring hormonal gradients, endoscopic ultrasound and somatostatin receptor scintigraphy (Octreoscan) have all been advocated to localise NETs. It has now been established that for all NETs, except for insulinomas, Octreoscan has the greatest sensitivity and therefore it is proposed that Octreoscan should be the initial tumour imaging study; it is clear that, for small bowel NETs, this imaging study becomes even more important, considering the low sensitivity of the different localisation methods. In one report, the Octreoscan was positive in 94% of patients with metastatic disease, showing clearly better results than CT or MRI.11 15
18F-deoxyglucose (FDG)—positron emission tomography (PET) has also been used to diagnose tumours of neuroendocrine origin, even if the tracer has not demonstrated a significant uptake in well-differentiated neuroendocrine tissues. It is recommended that 18F-FDG-PET should be performed only if somatostatin receptor scintigraphy (alone or with 99mTc-DMSA) is negative. On the contrary, other positron emitter tracers seem to be more promising, as 68Ga-DOTA-NOC (tetraazycyclododecanetetraacetic acid-octreotide) has been used for the detection of NETs in preliminary studies. These reports show higher sensitivity for the whole body PET-CT in detecting more lesions than with CT or octreotide scintigraphy.16 Recently, capsule endoscopy has been reported as a useful method to localise small bowel NET. The final diagnosis of ileal carcinoid tumours, generally, takes place during emergency surgery for intestinal occlusion or, much less frequently, for gastrointestinal bleeding.17 18
Localisation specificity
Most appendiceal carcinoids are located at the tip of the appendix. Approximately 10% of patients will be found to have the base of the appendix involved with tumour. Small (<1 cm) well-differentiated carcinoids confined to the tip of the appendix that are completely excised can be regarded as cured, if there is no evidence of lymphovascular invasion or invasion into the mesoappendix. Careful pathological examination of the specimen is required. There are other factors, however, that make appendiceal carcinoids worrisome.2
Caecal carcinoids are a small subgroup of NETs that are frequently metastatic at the time of diagnosis. They often present as silent large bulky lesions, presenting with gastrointestinal haemorrhage or obstruction. Their biological behaviour is generally more aggressive than appendiceal carcinoid. Surgical excision along standard oncological principles applies, with attention to adequate resection of mesenteric lymph node-bearing tissue. When small lesions are encountered, careful pathological examination is required.2
Carcinoid tumour of the jejunum and ileum are generally thought to have greater malignant potential than appendiceal carcinoid. Even small lesions may be associated with regional or distant metastases. Patients are often diagnosed at the time of an operation undertaken for some other reason. The discovery of these tumours often occurs as a result of surgical exploration for chronic blood loss, intestinal obstruction or in the course of evaluation of metastatic disease. Regardless of how they are found, the discovery of a primary gut-based NET should engender a diligent search for additional tumours by inspection and palpation. This often requires conversion of laparoscopic procedures to open procedures, because these tumours are often small and multiple. In our experience, as many as 70 tumours have been found along a segment of bowel. Tumours larger than 1.5 cm in diameter are usually associated with metastasis at the time of discovery. Resection should proceed along oncological principles, even in the face of metastatic disease, if it is technically feasible. With modern therapies, patients with intestinal NETs may live long enough to develop mesenteric vascular ischaemia or recurrent obstruction from lymph node metastases that are left behind in the mesentery.2
Management and prognosis
A treatment plan is devised on the basis of the pathological-anatomic classification of the tumour. Prospective studies assessing the efficacy of surgical treatment strategies for NETs of the small intestine do not exist. However, retrospective studies have demonstrated that curative as well as palliative resection of the primary tumour improves the prognosis and the quality of life of patients. Besides limited resection of the small bowel, to avoid postoperative short bowel syndrome, an effective clearance of the regional lymph nodes is essential. A primary tumour site in the terminal ileum requires dissection of the lymph nodes on the right side of the ileocolic artery, usually implying an additional resection of the right colon. In cases of a primary tumour site located in the lower ileum up to the distal jejunum, a cone-shaped resection of the mesenterium of the small bowel with extension of lymphadenectomy into adjacent segments with preservation of vascularisation is performed.3
In patients with metastatic disease, the administration of somatostatin analogues improves their quality of life.11 18 Although substantial improvements in the management of carcinoid syndrome have been made, no new agent has been approved for the control of tumour growth over the last three decades. Despite having a reputation for being indolent, advanced midgut NET remains a deadly disease. Several agents have been found to have a varying degree of activity in stabilising tumour growth. Tumour regressions, however, are rare. Novel targeted agents such as vascular endothelial growth factor and mTOR (mammalian target of rapamycin) inhibitors have been found to be promising in NETs and are under development.2
The overall 5-year-survival rate of small intestine NETs is 68.1%. The 5-year-survival rate of patients with hepatic tumour spread is 18–32%. An increased median survival (4.4 years) is evident in patients with jejuno-ileal carcinoids that exhibit a mixed insular/glandular pattern. In contrast, patients with an undifferentiated pattern have a median survival of only 6 months.9 The relatively poor prognosis of small intestinal NETs reflects the inherent clinical difficulty in identifying small bowel malignancies, as well as the intrinsically malignant nature of the tumour with dissemination to both the lymph nodes and the liver.9
Our example is interesting for several reasons. First of all, the 2-year history of diarrhoea and abdominal pain could have determined further investigation (eg, 5-HIAA or CgA) in searching of a carcinoid syndrome, but liver metastases were not found on CT. The presence of a carcinoid syndrome without metastasis is extremely rare. The main hypothesis that the patient's general practitioner had was an inflammatory bowel disease, conducting his differential diagnosis in this direction and infirming it by colonoscopy and negative biopsy; however, Crohn's disease is one of the main differential diagnosis for intestinal carcinoids.
To confirm even more the absence of carcinoid syndrome, at 1 month after surgery, 5-HIAA revealed to be normal, and the 1-year follow-up CT scan did not show liver metastasis. The only explanation we could find for the recurrent diarrhoea syndrome in this case is that mechanically the tumour had caused intermittent bowel obstruction (obstructive diarrhoea). Another particularity was the presence of a multifocal NET of the small bowel without liver metastasis, even if in the literature, these tumours are metastasised at the time of diagnosis in nearly two-thirds of cases.
Learning points.
In the past, these tumours were considered under a common rubric ‘carcinoid’ as indolent and uncommon, but in reality, they exhibit distinct cellular and clinical behaviours; each cell-specific lesion should therefore be considered and examined as a separate entity.
Overall, NETs occur far more frequently than previously considered and therefore should be carefully identified and treated, even if the present diagnostic tools do not always guarantee the results.
The gastrointestinal NETs have classically a long-lasting clinical silence and slow evolution to advanced stage, the symptoms and sign being non-specific. The clinical diagnosis is difficult and the importance of complementary investigations is crucial.
The presence of a carcinoid syndrome without metastasis is extremely rare, symptoms occurring in less than 10% of cases and being late signs of diagnosis.
Molecular imaging will increase in importance in the near future. There is still an unmet need for more sensitive biomarkers for diagnosis and follow-up.
Footnotes
Competing interests: None.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Oberg K. Neuroendocrine tumors: recent progress in diagnosis and treatment. Endocr Relat Cancer 2011;18(Suppl 1):E3–6 [DOI] [PubMed] [Google Scholar]
- 2.Boudreaux JP, Klimstra DS, Hassan MM, et al. The NANETS consensus guideline for the diagnosis and management of neuroendocrine tumors: well-differentiated neuroendocrine tumors of the jejunum, ileum, appendix, and cecum. North American Neuroendocrine Tumor Society (NANETS). Pancreas 2010;39:753–66 [DOI] [PubMed] [Google Scholar]
- 3.Musholt TJ. Extent of resection for neuroendocrine tumors of the small intestine. Chirurg 2011;82:591–7 [DOI] [PubMed] [Google Scholar]
- 4.Kianmanesh R, et al. (Surgical treatment of gastric, enteric, and pancreatic endocrine tumors Part 1. Treatment of primary endocrine tumors). J Chir (Paris) 2005;142:132–49 [DOI] [PubMed] [Google Scholar]
- 5.Kloppel G. Classification and pathology of gastroenteropancreatic neuroendocrine neoplasms. Endocr Relat Cancer 2011;18(Suppl 1):S1–16 [DOI] [PubMed] [Google Scholar]
- 6.Rindi G, Wiedenmann B. Neuroendocrine neoplasms of the gut and pancreas: new insights. Nat Rev Endocrinol 2011;8:54–64 [DOI] [PubMed] [Google Scholar]
- 7.Rindi G, et al. TNM staging of midgut and hindgut (neuro) endocrine tumors: a consensus proposal including a grading system. Virchows Arch 2007;451:757–62 [DOI] [PubMed] [Google Scholar]
- 8.Klimstra DS, et al. The pathologic classification of neuroendocrine tumors: a review of nomenclature, grading, and staging systems. Pancreas 2010;39:707–12 [DOI] [PubMed] [Google Scholar]
- 9.Schimmack S, et al. The diversity and commonalities of gastroenteropancreatic neuroendocrine tumors. Langenbecks Arch Surg 2011;396:273–98 [DOI] [PubMed] [Google Scholar]
- 10.Russo A, et al. Malignant carcinoid of the last ileal ansa. Report on 2 consecutive clinical cases. Minerva Chir 2002;57:203–11 [PubMed] [Google Scholar]
- 11.Nikou GC, et al. Current diagnosis and treatment of gastrointestinal carcinoids in a series of 101 patients: the significance of serum chromogranin-A, somatostatin receptor scintigraphy and somatostatin analogues. Hepatogastroenterology 2005;52:731–41 [PubMed] [Google Scholar]
- 12.Akerstrom G, et al. Management of midgut carcinoids. J Surg Oncol 2005;89:161–9 [DOI] [PubMed] [Google Scholar]
- 13.Yang XO, et al. The diagnostic value of plasma chromogranin A in neuroendocrine tumors. Zhonghua Nei Ke Za Zhi 2011;50:124–7 [PubMed] [Google Scholar]
- 14.Korse CM, et al. Choice of tumour markers in patients with neuroendocrine tumours is dependent on the histological grade. A marker study of Chromogranin A, neuron specific enolase, progastrin-releasing peptide and cytokeratin fragments. Eur J Cancer 2012;48:662–71 [DOI] [PubMed] [Google Scholar]
- 15.Jensen RT, Gibril F, Termanini B. Definition of the role of somatostatin receptor scintigraphy in gastrointestinal neuroendocrine tumor localization. Yale J Biol Med 1997;70:481–500 [PMC free article] [PubMed] [Google Scholar]
- 16.Junik R, et al. The role of positron emission tomography (PET) in diagnostics of gastroenteropancreatic neuroendocrine tumours (GEP NET). Adv Med Sci 2006;51:66–8 [PubMed] [Google Scholar]
- 17.García-Compean D, González JA, Gonzalez Giasi E, et al. Ileal carcinoid tumor manifesting as gastrointestinal hemorrhage and diagnosed with capsule endoscopy: case report. Rev Gastroenterol Mex 2005;70:164–8 [PubMed] [Google Scholar]
- 18.Marzocca G, et al. Intestinal occlusion by ileal carcinoid. Ann Ital Chir 2008;79:457–61 [PubMed] [Google Scholar]