

Abstract
Rathke's cleft cyst is a benign growth found on the pituitary gland in the brain, specifically a fluid-filled cyst in the posterior portion of the anterior pituitary gland. It occurs when the Rathke's pouch does not develop properly, and ranges in size from 2 to 40 mm in diameter. Asymptomatic cysts are common, detected during autopsies of 2–26% of individuals who have died of unrelated causes. Symptomatic cysts are rare and only approximately 150 cases have been reported. Females are twice as likely as males to have a cyst. Symptomatic cysts can trigger visual disturbances, pituitary dysfunction and headaches. Here we present a case of a 40-year-old female patient who presented with complains of visual disturbances, headache and amenorrhoea. On investigations, MRI of brain revealed findings suggestive of Rathke's cleft cyst.
Background
Rathke's cleft cyst (RCC) is a not-so-common pituitary developmental disorder. Most of the cyst are usually asymptomatic and found only on autopsy findings.1 2 Symptomatic cysts are very rare. Only around 150 cases have been documented in the literature. Due to rarity and unusual presentation of the disease we are writing this case report.
Case presentation
A 40-year-old female patient presented with frontotemporoparietal headache 3 months back, throbbing in nature intermittent and not relieved on taking medication. Patient also complained of blurring of vision and giddiness on seeing illuminated objects for a longer time and also occasionally complained of difficulty in viewing surrounding objects while looking straight.
Patient had earlier gone to a neurophysician and was given medications for migraneous headache but did not get relief. CT of brain was done which was found normal. The patient was advised to visit an ophthalmologist where she was checked for refractive error and fundus which were found normal. The patient was reassured and sent home. Despite all medications the patient did not get relief from the symptoms which increased in intensity. The patient visited our hospital on outpatient department and was admitted for further investigations and diagnosis.
No C/O fever, vomiting, weight loss, increased appetite, thirst or urine frequency.
No H/O diabetes mellitus, ischaemic heart disease, chronic obstructive pulmonary disease and drug allergy.
K/C/O hypertension since 5 months.
Operated for umbilical hernia 5 months back.
On general examination—the patient was afebrile, pulse rate 84/min, blood pressure 126/80 mm Hg, height 5 feet 2 inches and weight 56 kg. Arm span was equal to the height. The growth was consistent with the mid-parental height and sibling also had similar height. There was no pallor, icterus, cyanosis, lymphadenopathy, jugular venous pressure not raised, abdomen was soft, no lump or organomegaly, bowel sounds were present. The cardiovascular system and the respiratory system examination were within normal limits. On central nervous system examination no neck rigidity was found; pupil bilateral reacting to light and plantar bilateral flexion. On bedside examination of vision we found that the patient had decreased peripheral visual field for which perimetry was advised. On the basis of symptoms and examination of the patient we suspected lesion in the brain obstructing the optic pathway and therefore suggested MRI. Menstrual history—amenorrhoea since 5 months which was earlier than the normal age (45–50 years). Sleep, bowel, bladder and appetite were normal.
Investigations
On blood investigation: haemoglobin 13.8, total count 7150; differential count: N-50 , L-40, M-3, E-7, B-0; platelet count 3.5 lakh/mm3. Urea 10, creatine 0.71, Na 139 meq/l, K 3.5 meq/l, T3 0.73 ng/ml (0.58–1.59 ng/ml), T4 4.29 μg% (4.87–11.72 μg%), thyroid stimulating hormone 0.79 μIU/ml (0.35–4.94 μIU/ml) (tests done by chemiluminescent assay), luteinising hormone 0.38 μIU/ml (adult female normal values—follicular=1.0–18.0, mid-cycle=24.0–105.0, leutal=0.4–20.0, menopause=15.0–62.0), follicle stimulating hormone 2.32 μIU/ml (adult female normal values—follicular=3.3–8.8, mid-cycle=5.4–20.0, leutal=1.6–8.7, menopause=42.0–126.0). S. prolactin 69.38 ng/ml (1.2–29.93) but galactorrhoea was absent but the patient had amenorrhoea which was earlier than the normal age (45–50 years). Synacthen test (adrenocorticotropic hormone, ACTH stimulation test) was performed and post-ACTH S. cortisol level was 9.8 µg/dl suggestive of hypocortisolaemia. Serial input output charting was done which suggested average intake of about 1.5 litres and output of about 1.1 litre, which ruled out the possibility of diabetes insipidus. Urine routine was normal.
Perimetry revealed constriction of peripheral visual field. On doing radiological investigations—ultrasonography (abd, kub) was normal. Chest x-ray and ECG were normal. CT scan was normal. MRI brain study revealed well-defined altered signal intensity lesion in pituitary fossa which appeared isointense on T1-weighted images (T1WI) and hyperintense on T2-weighted images (T2WI). The lesion measured 16.3×10.9 mm. The lesion shows suprasellar extension with compression over optic chiasma. Findings suggest peripherally enhancing cystic lesion in sella with suprasellar extension suggesting possibility of RCC.
Differential diagnosis
The differential diagnosis for RCCs includes craniopharyngioma, cystic pituitary adenoma or other non-neoplastic cysts (arachnoid cysts or epidermoids).
Craniopharyngiomas are benign, suprasellar cystic masses are derived from Rathke's pouch and arise near the pituitary stalk, commonly extending into the suprasellar cistern. Craniopharyngiomas are often large, cystic and locally invasive. More than half of all patients present before the age of 20, usually with signs of raised intracranial pressure, including headache, vomiting, papilledema and hydrocephalus. Associated symptoms include visual field abnormalities, personality changes and cognitive deterioration, cranial nerve damage and weight gain. Hypopituitarism can be documented in about 90%, and diabetes insipidus occurs in about 10% of patients.
Radiologically, unlike RCCs, craniopharyngiomas typically demonstrate calcification and approximately 90% have nodular, globular or rim enhancement.
The presence of solid enhancing nodules in the cyst wall also favours the diagnosis of craniopharyngioma.
The rare non-calcified cystic non-enhancing craniopharyngioma, a finding more common in adults than in children, may be impossible to distinguish from RCC with imaging findings alone.
The age, symptoms and radiological findings in this patient favour the diagnosis of RCC.
Cystic lesions of the brain
| Common | Uncommon | Rare | Uncommon | Uncommon | More common than dermoid cyst. |
|---|---|---|---|---|---|
| Multiple possible mechanisms including leptomeningitis, trauma and malformation | Probably due to maldevelopment and most investigators favoured a neuroepithelial origin. There are also evidence that favours endoderm origin | Probably due to maldevelopment | Cystic enlargement of remnants of Rathke's pouch | Inclusion of ectodermal elements during closure of neural groove between the third and fifth weeks of embryonic life. Malformation of the adjacent structure can occur. Some of them may remain communicating with the overlying skin through a dermal sinus. Traumatic introduction of skin fragment is also a possible mechanism | Similar to dermoid cyst |
| About half of the cases occur along the Sylvian fissure, other common locations include the cerebellar pontine angle and quadrigerminal area, vermian area, sellar and suprasellar area | Third ventricle | Spinal cord, most common in the cervical and upper thoracic segment. Less common in other spinal levels. Rarely seen as intracranial cyst | Pituitary. The smaller cysts are usually identified as a small cyst at the interface of the anterior and posterior pituitary; most of these small cysts are asymptomatic | Most of them are intracranial and arise in the posterior fossa, usually along the midline in the vermis. About one-tenth of all cases are intraspinal and they are most commonly found in the lumbosacral region; they can be both intra- or extramedullary | Most of them are seen in the cerebellopontine angle and parapituitary area. Less than one-tenth of the cases occur in the spinal cord. Parapituitary tumours are usually embedded in the temporal bone. Similar to dermoid cyst, the pineal area is rarely involved |
| May be clinically silent. Symptomatic cases have manifestations of a gradually expansile mass leading to increased intracranial pressure. May cause hydrocephalus, particular the cerebellar cysts | Clinical presentations are often seen in the third to fifth decades of life. The location of colloid cyst often makes it an effective block of foramen of Monro leading to acute hydrocephalus. Its pedunculated nature, however, allows change in position easily and release of the pressure built up. As a result, they are often associated with intermittent headache. Sudden death has been well documented. Smaller cysts may remain asymptomatic | Clinical manifestation is related to compression and disruption of spinal cord function | Symptoms are due to compression of local structures and may lead to headache, hypopituitarism, hyperprolactinaemia, visual disturbance and, rarely, diabetes insipidus | Most of the spinal cases are discovered in the first and second decades of life; intracranial cases are seen in slightly older patients. Symptoms are usually due to local compression, irritation and to increased intracranial pressure due to hydrocephalus. Leakage of the kertinous content will lead to extensive granulomatous meningitis | They are found over a wide age range but most of them are discovered in the fourth to sixth decades. The clinical manifestations are due to local compression, irritation and hydrocephalus. Leakage of the kertinous content will lead to extensive granulomatous meningitis |
| The size is variable but some can attain a very large size. They are fluid filled cysts with thin, transparent wall. The cyst can be well distincted from the leptomeninges and dura. The fluid is usually clear and colourless | The size varies form several millimetres to 3–4 cm. The cyst is round, unilocular and have a thin wall. After fixation, the cyst content appears as a grey hyaline substance with consistency of soft cartilage | Most of them are intradural or subdural, occasionally intramedullary | Well-defined, small, thin walled cyst with a watery to mucinous, occasionally xanthochromatic due to prior haemorrhage, content | Well-defined round to oval, opaque, ‘pearly’ mass of variable size. The thickness of the wall is variable. The content is greasy material due to secretion of sebaceous glands. A variable amount of hair is present. Solid components may be present. The distinction between dermoid cyst and cystic teratoma is not entirely sharp | Well-defined round mass with an irregularly nodular capsule with a pearly discolouration |
| Most of them have a thin, collageneous cyst wall lined by stratified and compressed meningothelial (arachinoid) cell | The internal lining range from cuboidal, columnar, to ciliated. Mucin-producing cells can be found in some cases. The cyst content is periodic acid–Schiff (+). Rathke's cleft cyst cannot be histologically differentiated from colloid cyst of the third ventricle. Clinical correlation is important | The cyst wall is composed of connective tissue. The lining epithelium varies from low to high columnar cells that may have pseudostratification. Mucous secreting cells can be found. Occasionally, they are ciliated | The cyst is lined by a single layer of cuboidal to columnar, ciliated or mucin-producing epithelium. Squamous metaplasia may occur and raises the possibility of a craniopharyngioima. Occasional cells of anterior pituitary gland may be present. Rathke's cleft cyst cannot be histologically differentiated from colloid cyst of the third ventricle. Clinical correlation is important | The internal lining is very similar to skin and skin appendages are present. Leakage or escape of the content may lead to a wide spread granulomatous meningitis with foreign body giant cell reaction | The histology is similar to that of dermoid cyst except that no skin appendages are found. Rare cases of malignant transformation into invasive squamous cell carcinoma have been documented |
Treatment
Cortisol replacement in the form of Tab Hydrocortisone (10 mg) was started 1–1–1/2, a total of 25 mg/day.
Oestrogen was not given as the patient did not want to have menstruation. However, other preventive measures for osteoporosis in the form of vitamin D, calcium supplements were started.
Tab Cabergoline (0.5 mg) twice a week, every Monday and Thursday was started.
Tab Thyroxin (50 mcg) once a day was started for central hypothyroidism.
Outcome and follow-up
The patient was transferred to the neurosurgical department for further management.
Discussion
RCCs are benign, epithelium-lined intrasellar cysts believed to originate from remnants of the Rathke's pouch. RCCs commonly have a round, ovoid or dumbbell shape.
As Voelker et al3 have stated, the most common theory about the origin of RCCs is that the cysts are derived from true remnants of the embryological Rathke's pouch.
On or about the 24th day of embryonic life, the Rathke's pouch arises as a dorsal diverticulum from the stomodeum; it is lined with epithelial cells of ectodermal origin. At approximately the same time, the infundibulum forms as a downgrowth of the neuroepithelium from the diencephalon. By the fifth week, the Rathke's pouch comes into contact with the infundibulum, and the neck of the pouch becomes occluded at the buccopharyngeal junction. During the sixth week, the Rathke's pouch separates from the oral epithelium. Subsequently, the pars distalis of the pituitary gland develops from the anterior wall of the pouch. The posterior wall does not proliferate and remains as the poorly defined pars intermedia. The residual lumen of the pouch is reduced to a narrow Rathke's cleft, which generally regresses. The persistence and enlargement of this cleft are considered to cause the RCC.
Other authors have different theories regarding the formation of RCCs, suggesting instead that the cells of origin are derived from the neuroepithelium or the endoderm, or that they come from metaplastic anterior pituitary cells.
On pathological examination, the size of the cyst can vary from 2 to 40 mm. The cystic capsule frequently is described as thin and has been reported as being transparent, blue, grey, white, yellow, pink, red, tan or green. The cystic fluid commonly is thick or gelatinous, but it also can be watery, serous or similar in consistency to motor oil. The cystic fluid most often is yellow, but it also has been described as white, clear, grey or green.
At histological examination, the cysts typically are composed of vascularised stroma of connective tissue and three types of epithelial cells: ciliated, non-ciliated epithelial and mucous secreting. Non-ciliated cells appear as a single layer of flat cells or as stratified columnar cells. Fager and Carter concluded that the presence of ciliated epithelial and mucous-secreting cells in a pituitary gland is pathognomonic for RCC.4
In a retrospective analysis, Shin et al4 found that all the RCCs studied were intrasellar in origin at their primary location.4 Because of their cystic nature, most of the RCCs that were studied expanded into the suprasellar cistern through the cleft of the diaphragma sella.
In Voelker's report, a retrospective study of 155 patients with symptomatic RCC, the cyst was found in intrasellar and suprasellar locations in 71% of the patients.3 The sella was enlarged in 80%.
RCCs often produce no symptoms and so are usually discovered incidentally, when radiographic or necropsy findings are reviewed. Most cysts are smaller than 2 cm in diameter. Symptomatic RCCs are uncommon, but cysts can enlarge and cause symptoms secondary to compression of the pituitary gland, pituitary stalk, optic chiasma or hypothalamus.
Symptomatic RCCs vary in presentation. In a study of 11 symptomatic patients by Rao et al5, eight patients initially had visual symptoms. In another study, by Eguchi et al6, visual symptoms occurred in 47% of patients. Signs and symptoms included reduced visual acuity, optic atrophy, visual field defects and a chiasmatic syndrome. Eguchi's study looked at 19 patients, 4 of whom had diabetes insipidus, 3 of whom had amenorrhoea and/or galactorrhoea and 2 of whom had panhypopituitarism. With systematic endocrinological examinations, various degrees of pituitary dysfunction were observed. Other abnormal findings included headaches and, less commonly, epilepsy.
In the study by Voelker et al3, the most common finding was pituitary hypofunction with multiple endocrinopathies. Younger patients, aged 4–22 years, had evidence of hypopituitarism from an early age, with resultant growth retardation.7 The second most common symptom was visual disturbance, which included visual field defects resulting from chiasmatic compression. The next most common symptom was headache, of which 57% were frontal. Shin and co-authors described impotence or low libido as the most common endocrine abnormality in men; in women, the most common was hyperprolactinaemia. Many RCCs are associated with pituitary adenomas.4 In addition to the symptoms described above, endocrine symptoms occur as a result of the adenoma.
In another study by JJ Mukherjee 9 of 12 patients (75%) were symptomatic; visual symptoms were the commonest, and 8 had visual field defects. The median duration of symptoms was 12 months (range 3–24 months). Three patients (25%) had panhypopituitarism, two of whom also had diabetes insipidus (17%). The cysts varied in size from 6 to 50 mm, one being entirely suprasellar.8
Other unique or unusual findings associated with RCC include pituitary apoplexy,9 hypophysitis, giant cysts, large frontal extension with an unusual shape, aseptic meningitis, intracystic abscess, sphenoid sinusitis and empty sella syndrome.
MRI is the modality of choice in the detection of RCCs.10 Thin-section sagittal and coronal MRI scans should be obtained through the sella. CT scans are also useful for evaluating RCCs. They are particularly helpful in delineating associated bony remodelling and in assessing calcification.
MRI is superior to CT scanning for evaluating RCC mass extension. Sagittal and coronal MRI scans provide reliable information concerning the relationship of the mass to the optic nerves, optic chiasma and hypothalamus. Coronal MRI is also helpful in the evaluation of the lateral extension of the sellar cyst and its relationship to the internal carotid arteries and cavernous sinuses. MRI also has superior multiplanar capabilities and contrast resolution compared with those of CT scanning.
The advantage of CT scanning is that it is superior to MRI in depicting small amounts of calcium. This advantage can be important, because the presence of calcification tends to indicate an alternative diagnosis, such as craniopharyngioma, although small calcifications are observed in some cases of RCC.11 CT scanning is also superior to MRI in the evaluation of associated bony remodelling.
CT scan findings
RCCs frequently appear as well-circumscribed, hypoattenuating, cystic sellar masses that may have suprasellar extension. As a result of the different cystic contents, RCCs may appear isoattenuating or hyperattenuating relative to the brain parenchyma. RCCs usually have a thin wall that may enhance.
Variability in CT scan contrast enhancement among individual cysts may reflect squamous metaplasia in the wall or a peripherally displaced rim of pituitary tissue.
Extravasation of cystic contents may inflame nearby structures, resulting in enhancement. Large cysts may cause bony remodelling.
MRI findings
The best imaging clue is a non-enhancing non-calcified intrasellar and/or suprasellar cyst with an intracystic nodule. While this is the typical picture, the imaging characteristics vary widely.
Approximately half are hyperintense on T1WI, while half are hypointense. On T2WI, 70% are hyperintense and 30% are isointense or hypointense.
Although no characteristic MRI features have been identified, many RCCs are in one of the following two groups:
RCCs with low signal intensity on T1WI and high signal intensity on T2WI.
RCCs with high signal intensity on T1WI and variable signal intensity on T2WI.
The cystic contents of the first group resemble those of cerebrospinal fluid (CSF). In the second group, an increase in the signal on T1WI has been related to the high content of mucopolysaccharides, which is believed to result from an increase in the number of mucin-secreting cells in the cyst wall, as well as from an increase in the activity of these cells.
Uncommon cases with high signal intensity on T1WI and low signal intensity on T2WI have been suggested to result from a combination of factors, including the presence of mucopolysaccharides, chronic haemorrhage, a high cholesterol content and cellular debris from the cyst wall.
Figure 1.
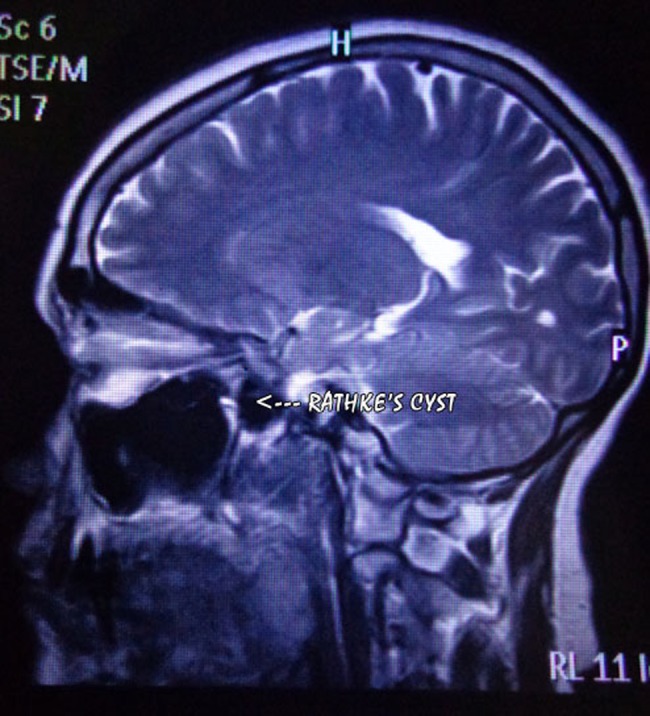
T2W image, sagittal section of brain.
Figure 2.
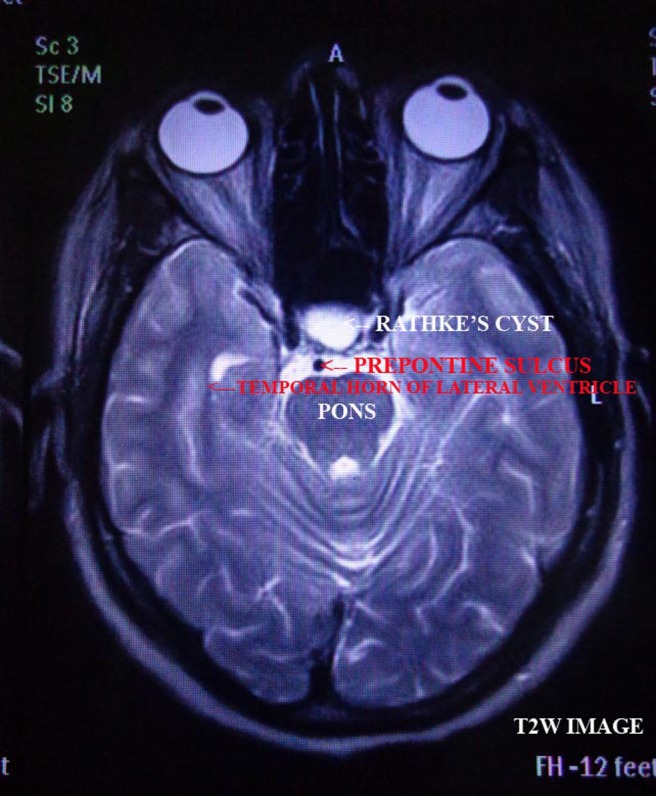
T2W image, axial section of brain.
Figure 3.
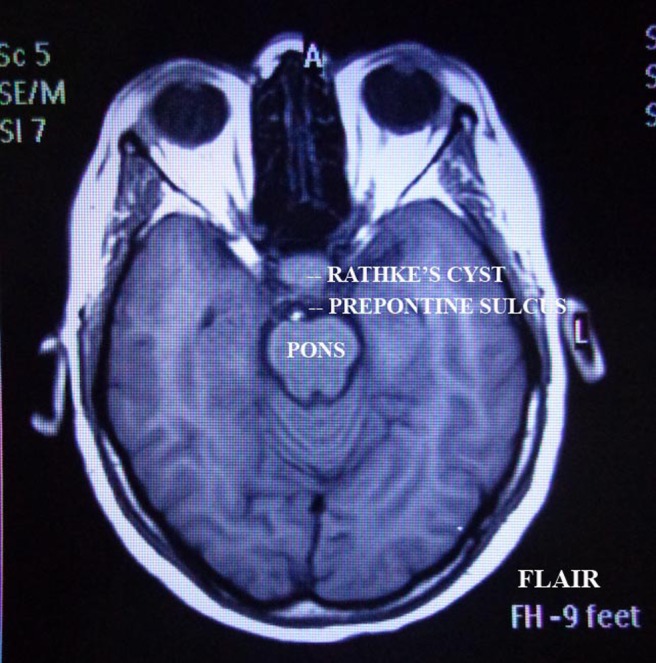
FLAIR image, axial section of brain.
Figure 4.
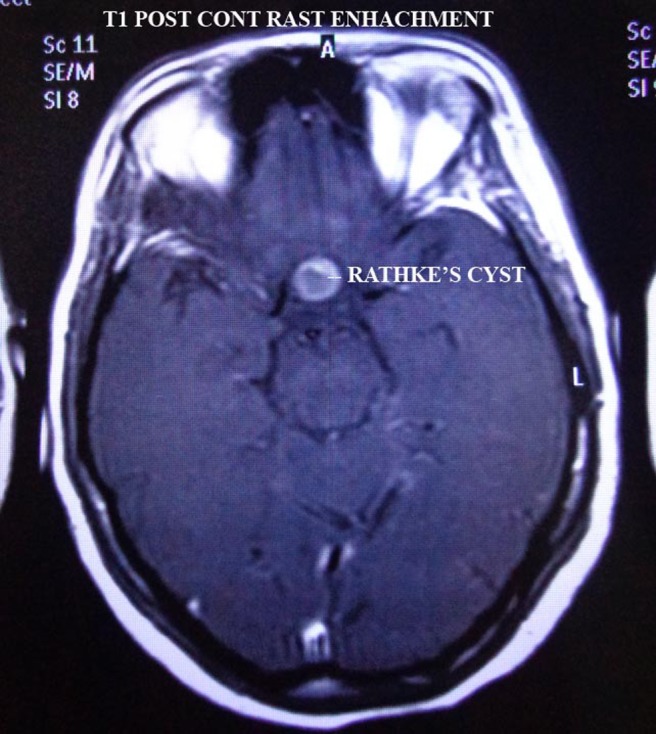
T1W, post contrast image.
Figure 5.

Right eye perimetry.
Figure 6.

Left eye perimetry.
A small non-enhancing intracystic nodule is considered as virtually pathognomonic sign of a RCC. These nodules show high signal intensity on T1WI and low signal intensity on T2WI, and they do not enhance.
RCCs do not enhance after contrast material administration, although an enhancing rim of displaced compressed pituitary gland is present in approximately half of the cases.
The most common approach in the treatment of RCCs is transsphenoidal surgery, in which the cyst is partially excised and drained.12 13 This method is effective and helps to preserve pituitary function. Radical excision can cause additional and unnecessary pituitary damage; therefore, it is not the treatment of choice. In transsphenoidal surgery, the cyst is opened, a biopsy specimen is obtained from the wall and the cyst is drained into the sphenoid sinus. No sellar reconstruction is needed unless a CSF leak is noted during surgery. In patients in whom this approach is not appropriate because of an inaccessible cyst, craniotomy is performed, usually via a right frontal flap.
In some patients, the postoperative follow-up period ranges from 1 week to 26 years (average, 34 months). The postoperative outcome for most patients is resolution of or improvement in symptoms. In patients with hypopituitarism or diabetes insipidus, the RCC usually fails to improve. After surgery, the greatest improvement in symptoms occurs with a resolution of neurological symptoms (71% of patients), followed by a resolution of ophthalmological symptoms (70% of patients). In more than 65% of patients, amenorrhoea, galactorrhoea and oligomenorrhoea improve. Headaches and visual field defects resolve in 82% and 70% of patients, respectively.
Surgical complications include CSF rhinorrhoea (7% of patients), diabetes insipidus (4% of patients) and meningitis (4% of patients). A rare surgical complication noted by Baskin was nasal septal perforation.9 Abscess formation in a cyst is a treatable complication; this complication should be considered even in the absence of fever when radiological features suggest the condition. Treatment with surgery and antibiotic medications is effective in most patients.
The recurrence rate after craniotomy is twice as high as that after transsphenoidal surgery. In addition, leakage of the cystic contents into the subarachnoid space has been reported; this can cause aseptic meningitis. In cases of recurrence, extensive removal of the cyst wall is most appropriate, and some recommend external beam pituitary radiation therapy, although its role in preventing further recurrence remains unclear. Fager and Carter14 recommend full evacuation of the cystic contents and liberal opening of the wall in the treatment of symptomatic RCCs. When this procedure is performed, recurrence is rare.
Learning points.
Consider Rathke's cyst as one of the pituitary lesions causing symptoms of headache, visual disturbances and panhypopituitarism.
Rathke's cyst though rare can be symptomatic.
Headache, visual disturbances and pituitary dysfunction are the most common presenting features of a symptomatic Rathke's cyst.
Footnotes
Patient consent: Obtained.
Competing interests: None.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.‘Rathke's Cleft Cyst’. UCLA. http://neurosurgery.ucla.edu/body.cfm?id=215.
- 2.Islam O, (28 May 2008). ‘Rathke Cleft Cyst: Overview’. http://emedicine.medscape.com/article/343629-overview
- 3.Voelker JL, Campbell RL, Muller J. Clinical, radiographic, and pathological features of symptomatic Rathke's cleft cysts. J Neurosurg 1991;74:535–44 [DOI] [PubMed] [Google Scholar]
- 4.Shin JL, Asa SL, Woodhouse LJ, et al. Cystic lesions of the pituitary: clinicopathological features distinguishing craniopharyngioma, Rathke's cleft cyst, and arachnoid cyst. J Clin Endocrinol Metab 1999;84:3972–82 [DOI] [PubMed] [Google Scholar]
- 5.Rao GP, Blyth CP, Jeffreys RV. Ophthalmic manifestations of Rathke's cleft cysts. Am J Ophthalmol 1995;119:86–91 [DOI] [PubMed] [Google Scholar]
- 6.Eguchi K, Uozumi T, Arita K, et al. Pituitary function in patients with Rathke's cleft cyst: significance of surgical management. Endocr J 1994;41:535–40 [DOI] [PubMed] [Google Scholar]
- 7. Harrison (Medicine) 18th edn, p 2878.
- 8.Mukherjee JJ, Islam N, Kaltsas G, et al. http://jcem.endojournals.org/content/82/7/2357.full. [Google Scholar]
- 9.Binning MJ, Liu JK, Gannon J, et al. Hemorrhagic and nonhemorrhagic Rathke cleft cysts mimicking pituitary apoplexy. J Neurosurg 2008;108:3–8 [DOI] [PubMed] [Google Scholar]
- 10.Nakasu Y, Isozumi T, Nakasu S, et al. Rathke's cleft cyst: computed tomographic scan and magnetic resonance imaging. Acta Neurochir (Wien) 1990;103:99–104 [DOI] [PubMed] [Google Scholar]
- 11.Le BH, Towfighi J, Kapadia SB, et al. Comparative immunohistochemical assessment of craniopharyngioma and related lesions. Endocr Pathol 2007;18:23–30 [DOI] [PubMed] [Google Scholar]
- 12.Frank G, Sciarretta V, Mazzatenta D, et al. Transsphenoidal endoscopic approach in the treatment of Rathke's cleft cyst. Neurosurgery 2005;56:124–8; discussion 129 [DOI] [PubMed] [Google Scholar]
- 13.Cavallo LM, Prevedello D, Esposito F, et al. The role of the endoscope in the transsphenoidal management of cystic lesions of the sellar region. Neurosurg Rev 2008;31:55–64; discussion 64 [DOI] [PubMed] [Google Scholar]
- 14.Fager CA, Carter H. Intrasellar epithelial cysts. J Neurosurg 1966;24:77–81 [DOI] [PubMed] [Google Scholar]