

Abstract
Background/Aims
Acute coronary syndrome (ACS) is characterized by increased inflammatory processes and endothelial activation. We investigated the association between ACS and inflammatory mediators and matrix-degrading enzymes.
Methods
We prospectively enrolled 55 consecutive patients with ACS: 25 with unstable angina (UA) and 30 with non-ST elevated myocardial infarction (NSTEMI). For comparison, 25 age- and sex-matched subjects with no significant coronary artery stenosis were included as the control group. Peripheral serum levels of interleukin (IL)-33, matrix metalloproteinase (MMP)-9, tissue inhibitor of MMP-1, and C-reactive protein (CRP) were measured on admission, and at 12, 24, 48, and 72 hours after the initial evaluation.
Results
Compared to serum levels in the control group, serum levels of IL-33 decreased in the NSTEMI group (p < 0.05), and levels of MMP-9 and tissue inhibitor of matrix metalloproteinase (TIMP)-1 increased in the UA group (p < 0.01, p < 0.05, respectively) and NSTEMI group (p < 0.05, p < 0.05, respectively). IL-33 levels were significantly lower on admission than at 12 hours after the initial evaluation (p < 0.05). IL-33 levels were negatively correlated with MMP-9 levels (r = -0.461, p < 0.05) and CRP levels (r = -0.441, p < 0.05).
Conclusions
Elevated levels of MMP-9, TIMP-1, and decreased levels of IL-33 play a role in the development and progression of ACS.
Keywords: Inflammation, Interleukin-33, Matrix metalloproteinase 9, Myocardial infarction
INTRODUCTION
Coronary artery disease (CAD), a common manifestation of atherosclerosis, is responsible for a high percentage of morbidity and mortality in the Western world [1]. A large body of evidence supports the idea that inflammatory mechanisms play an important role throughout all phases of atherogenesis, from the formation of fatty streaks to the acute coronary event due to vulnerable plaque rupture [1-3]. Smooth muscle cells produce extracellular matrix (ECM), leading to the formation of the fibrous cap that covers the atheromatic plaque [4]. Conversely, proteases with collagenolytic properties counterbalance collagen synthesis. ECM degradation is mainly achieved through the action of matrix metalloproteinases (MMPs), a family of zincand calcium-dependent endopeptidases and macrophages proposed to be the main contributors of MMPs within the atherosclerotic plaque [5,6]. Interleukin (IL)-6, tumor necrosis factor-α, and IL-1β increase the expression of MMPs [7,8], whereas cytokines that exert atheroprotective actions reduce MMP production [9]. Among the MMPs, MMP-9 is highly expressed in the vulnerable regions of the atherosclerotic plaque and has been suggested to be causally involved in plaque rupture [10]. Plasma MMP-9 concentrations are elevated in patients with acute myocardial infarction (MI) [11-13]. Endogenous MMP activity is regulated by the tissue inhibitors of matrix metalloproteinase (TIMP), in particular TIMP-1, which binds to several MMPs with high affinity [14,15].
Several cytokines exert anti-inflammatory and atheroprotective actions. IL-33 is expressed in coronary artery smooth muscle cells [16], coronary artery endothelium [17], nonhigh endothelial venule endothelial cells [18], and cardiac fibroblasts, suggesting that IL-33 may play a role in various cardiovascular disorders [19]. In addition, IL-33 markedly increases levels of IL-4, -5, and -13, but decreases interferon (IFN)-γ levels in serum and lymph node cells. IL-33 may play a protective role in the development of atherosclerosis via the induction of IL-5 and oxidized low density lipoprotein (ox-LDL) antibodies [20]. The immunological response of Th1 type and its mediators (i.e., IFN-γ) is believed to accelerate atherosclerosis, whereas the response of Th2 type and its mediators (i.e., IL-4, IL-5, and IL-13) inhibit the development of atherosclerosis [20-23]. IL-33 reduces MMP release by intentionally decreasing IFN-γ levels having inflammatory effects and inhibits plaque rupture.
The correlation between the time course of serum concentration changes in IL-33, MMP-9, and TIMP-1 and the extent of acute coronary syndrome (ACS) during and after acute MI is unclear. Therefore, in the present study, we evaluated the serum concentrations of these markers at various time points in patients with ACS.
METHODS
Patients
The study population was composed of 55 patients with complaints of sudden chest pain who were admitted to the emergency department within the first 6 hours of pain onset. Exclusion criteria were as follows: a diagnosis of renal failure, collagen tissue disease, infection, vasculitis, depression, or somatization or having undergone angioplasty, bypass surgery, or open-heart surgery. The patient group was divided into two subgroups: patients with unstable angina (UA; n = 25) and those with non-ST elevated myocardial infarction (NSTEMI; n = 30). The leading symptom of ACS is typically chest pain. The working diagnosis of NSTEMI-ACS is a rule-out diagnosis based on the electrocardiogram showing a lack of persistent ST elevation. Biomarkers (troponins) further distinguish NSTEMI and UA [24]. The control group consisted of 25 patients who had no significant coronary artery stenosis (25% of luminal diameter) and were age- and sex-matched with the patient groups. The study was performed in compliance with the Declaration of Helsinki.
Venous blood samples were obtained when patients with NSTEMI were admitted to our hospital. Postadmission blood samples were obtained at 12, 24, and 48 hours after percutaneous coronary intervention. Blood samples were obtained from the control group in the morning. Triglycerides, total cholesterol, high density lipoprotein cholesterol (HDL-C), C-reactive protein (CRP), fibrinogen, troponin I, aspartate aminotransferase (AST), lactate dehydrogenase (LDH), and isoenzymes of phosphocreatine kinase (creatine kinase-MB, CK-MB) were measured immediately. For other measurements, the blood samples were centrifuged at 1,500 ×g and 4℃ for 10 minutes and stored at -86℃ until analysis. Blood samples were drawn on admission and at 12, 24, and 48 hours after the onset of infarction for measuring MMP-9, TIMP-1, and IL-33 levels.
MMP-9 measurement
The concentrations of MMP-9 were determined in duplicate using the RayBio enzyme-linked immunosorbent assay (ELISA) kits (RayBiotech Inc., Norcross, GA, USA) designed to measure their pro and active forms. The detection limit of this assay was 10 pg/mL. Intra and interassay variations were less than 12%.
IL-33 measurement
IL-33 levels were determined using the ELISA Kit (USCN Life Science Inc., Houston, TX, USA). A monoclonal antibody specific for IL-33 was precoated onto a microplate. The detection limit of this assay was less than 6.4 pg/mL.
TIMP-1 measurement
The concentrations of TIMP-1 were determined in duplicate using the RayBio ELISA kits (RayBiotech Inc.). The detection limit of this assay was 40 pg/mL. Intra and interassay variations were less than 12%.
Others assays
For the CRP measurement, the particle-enhanced immunoturbidimetric method was performed using a Behring Nephelometer BN-100 (Behring Diagnostic, Frankfurt, Germany). The sensitivity of the test was 0.1 mg/L.
Serum cholesterol, triglyceride, and HDL-C were measured using enzymatic colorimetric methods with commercially available kits (Cobas 311, Roche Diagnostics GmbH, Mannheim, Germany) and LDL-C was calculated according to the Friedewald formula. Serum glucose levels were determined enzymatically by the hexokinase method (Roche Diagnostics GmbH).
Cardiac troponin T (cTnT) was measured by an electrochemiluminescence immunoassay (Troponin T, Roche Diagnostics GmbH) using an Elecsys E170 analyzer (Roche Diagnostics GmbH). The lower limit of detection for the cTnT assay was 3 ng/L. Serum CK-MB, AST, and LDH levels were determined in a Roche/Cobas 311 automated clinical chemistry analyzer. Fibrinogen levels were determined by the Clauss method using Multifibren U (Dade Behring, Marburg, Germany), and leukocyte levels were measured with an automatic hematology analyzer (Beckman Coulter, Brea, CA, USA).
Statistical analysis
Statistical analyses were performed with a IBM SPSS version 18.0 package (IBM, Armonk, NY, USA). Values were expressed as means ± SD. The significance of the mean differences between groups was assessed by a Mann-Whitney and Student's t test. Relationships between variables were tested using Pearson's correlation analysis. The p values less than 0.05 were considered statistically significant.
RESULTS
No significant differences were observed between patients with ACS and the healthy control group with respect to age. As shown in Table 1, when compared with the UA and control groups, serum MMP-9 levels were significantly higher in the NSTEMI group on admission (p < 0.01, p < 0.05, respectively). Additionally, MMP-9 levels were significantly higher in the UA group compared to the control group (p < 0.05) (Table 1). TIMP-1 levels were significantly higher in the NSTEMI group than either the UA or control groups (p < 0.05). The UA group had significantly lower TIMP-1 levels (p < 0.05) than the MI group (Table 1). IL-33 levels were significantly lower in the NSTEMI group compared to the control group (p < 0.05) (Table 1).
Table 1.
Values and statistical significance of the analyzed parameters in the NSTEMI, UA, and control groups
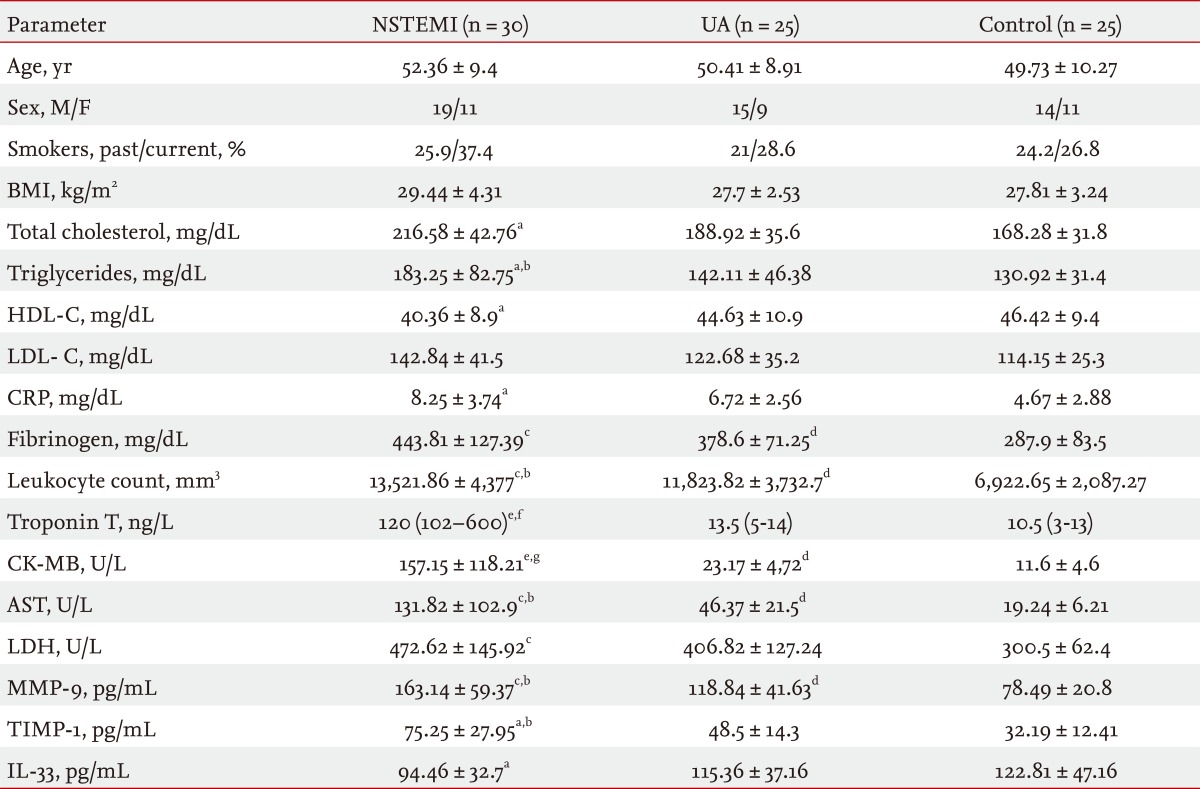
Values are presented as mean ± SD.
NSTEMI, non-ST elevated myocardial infarction; UA, unstable angina; M, male; F, female; BMI, body mass index; HDL-C, high density lipoprotein cholesterol; LDL-C, low density lipoprotein cholesterol; CRP, C-reactive protein; CK-MB, creatine kinase-MB; AST, aspartate aminotransferase; LDH, lactate dehydrogenase; MMP-9, matrix metalloproteinase-9; TIMP-1, tissue inhibitor of matrix metalloproteinase-1; IL-33, interleukin-33.
aBetween NSTEMI and control groups, p < 0.05.
bBetween NSTEMI and UA groups, p < 0.05.
cBetween NSTEMI and control groups, p < 0.01.
dBetween UA and control groups, p < 0.05.
eBetween NSTEMI and control groups, p < 0.001.
fBetween NSTEMI and UA groups, p < 0.001.
gBetween NSTEMI and UA groups, p < 0.01.
As for enzymes indices, the NSTEMI group had significantly higher CK-MB, AST, and LDH levels (p < 0.01, p < 0.001, p < 0.01, respectively) than the control group. A comparison between the NSTEMI and UA groups revealed significantly higher CK-MB and AST levels (p < 0.01, p < 0.05, respectively) in the NSTEMI group (Table 1). In addition, CK-MB and AST levels were significantly higher in the UA group compared to the control group (p < 0.05 for both) (Table 1).
As for inflammatory parameters, the NSTEMI group had significantly higher CRP, fibrinogen, and leukocyte count levels (p < 0.05, p < 0.01, p < 0.05, respectively) than the control group. A comparison between the NSTEMI and UA groups revealed significantly higher leukocyte count levels (p < 0.05) in the NSTEMI group (Table 1). In addition, fibrinogen and leukocyte count levels were significantly higher in the UA group compared to the control group (p < 0.05) (Table 1).
When compared to the control group, the NSTEMI group had significantly higher triglyceride and total cholesterol levels (p < 0.05). In addition, HDL-C levels were significantly lower in the NSTEMI patient group compared to the control group (p < 0.05). A comparison of the NSTEMI and UA patient groups revealed significantly higher triglyceride levels (p < 0.05) in the NSTEMI group (Table 1).
Table 2 shows the time course changes in the serum MMP-9, TIMP-1, and IL-33 concentrations after NSTEMI. The MMP-9 levels on admission were significantly higher than those observed at 24 and 48 hours (p < 0.05). The MMP-9 level at 12 hours was significantly higher than that at 48 hours (p < 0.05). The serum TIMP-1 level on admission was significantly lower than the level at 48 hours (p < 0.05), whereas the level of IL-33 on admission was significantly lower than the level at 12 hours (p < 0.05) (Table 2).
Table 2.
Time course of changes in MMP-9, TIMP-1, and IL-33 after NSTEMI

Values are presented as mean ± SD.
MMP-9, matrix metalloproteinase-9; TIMP-1, tissue inhibitor of matrix metalloproteinase-1; IL-33, interleukin-33; NSTEMI, non-ST elevated myocardial infarction.
a0 to 24 hr, p < 0.05.
b0 to 48 hr, p < 0.05.
c12 to 48 hr, p < 0.05.
d0 to 12 hr, p < 0.05.
Significant positive correlations were observed between MMP-9 and TIMP-1, CRP, and LDH levels (r = 0.307, p < 0.01 and r = 0.321, p < 0.05 and r = 0.387, p < 0.01, respectively). MMP-9 levels were negatively correlated with IL-33 levels (r = -0.461, p < 0.05). TIMP-1 levels were positively correlated with both CRP and leukocyte count levels (r = 0.394, p < 0.05 and r = 0,420, p < 0.05, respectively) and negatively correlated with IL-33 levels (r = -0.288, p < 0.05). IL-33 levels were negatively correlated with CRP levels (r = -0.441, p < 0.05) (Table 3).
Table 3.
Correlations among MMP-9, TIMP-1, and IL-33 in NSTEMI
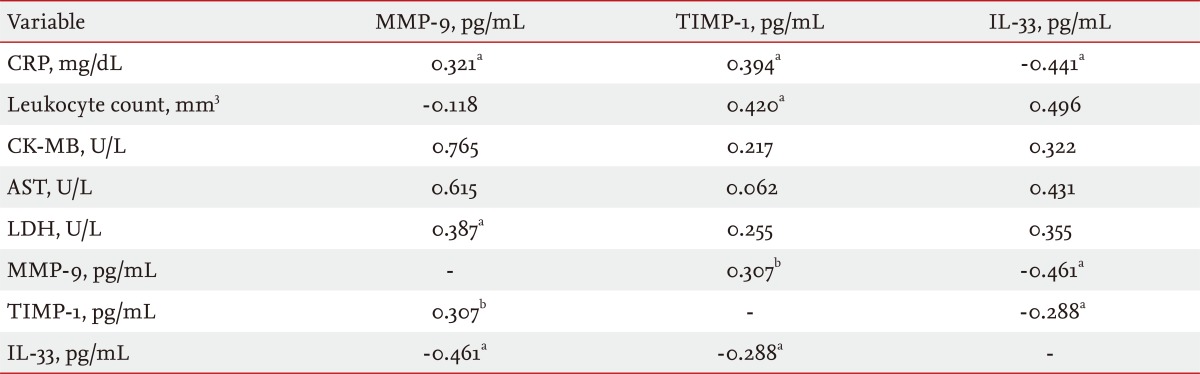
MMP-9, matrix metalloproteinase-9; TIMP-1, tissue inhibitor of matrix metalloproteinase-1; IL-33, interleukin-33; NSTEMI, non-ST elevated myocardial infarction; CRP, C-reactive protein; CK-MB, creatine kinase-MB; AST, aspartate aminotransferase; LDH, lactate dehydrogenase.
ap < 0.05.
bp < 0.01.
DISCUSSION
MMP, an ECM-degrading enzyme, plays a crucial role in the breakdown of the fibrous cap of plaque and subsequent rupture in the pathogenesis of ACS [25]. MMP-9 affects plaque stability in association with various inflammatory cytokines [26].
The results of this study indicate that the serum levels of the inflammatory marker MMP-9, CRP, and leukocyte count were significantly higher at the acute phase of patients with either NSTEMI or UA. Patients with NSTEMI had higher levels of MMP-9 and a higher leukocyte count compared to patients with UA. These findings provide evidence that both NSTEMI and UA are accompanied by an increased inflammatory process. Our observation showing elevated serum levels of MMP-9 in subjects with acute MI is consistent with published data [11,12,27,28]. Several studies have demonstrated that plasma levels of MMP-9 are significantly higher in the coronary artery than in the systemic circulation of patients with acute MI [11,29]. Thus, the atherosclerotic plaque may be the major source of MMP-9 released into the circulation [13].
TIMP-1 is a major inhibitor of MMP-9 [15]. In our study, serum levels of TIMP-1 were increased in both UA and NSTEMI groups. Patients with NSTEMI had higher levels of TIMP-1 compared to patients with UA. Controversial findings are reported in the literature regarding the levels of TIMP-1 in patients with NSTEMI. Some studies have reported a decrease in TIMP-1 levels in patients with acute MI [30], while others have described elevated levels of TIMP-1 in patients with acute MI [29,31,32]. In a report by Inokubo et al. [33], elevated levels of TIMP-1 during acute MI were demonstrated only in blood taken from the coronary sinus, but not in aortic blood; this observation was attributed to the release from the ruptured atherosclerotic plaque.
We examined a time course series of plasma-level changes of MMP-9 and TIMP-1 (on admission, and at 12, 24, 48, and 72 hours after admission). Serum levels of MMP-9 and TIMP-1 were found to be the highest at the onset of NSTEMI. The MMP-9 level gradually decreased and reached its lowest value at 48 hours. The MMP-9 levels on admission were significantly higher when compared to the blood samples collected at 24 and 48 hours. The MMP-9 level at 12 hours was significantly higher than the level at 48 hours. In contrast, the TIMP-1 level on admission gradually increased and reached its highest value at 72 hours. However, our results show that the atherosclerotic plaque is not the only source from which MMP-9 is released: it may be released from the interstitium of the ischemic myocardium in the early period as well after an NSTEMI. TIMP-1 has been shown to be upregulated after NSTEMI and to be released as a response to prevent any damage that might occur in the matrix due to the sudden increase in the MMP-9 levels at the onset of NSTEMI.
Kai et al. [12] found that plasma MMP-9 levels on day 0 were about 3-fold and 2-fold higher in the UA group than in the control and EA groups, respectively. Tziakas et al. [34] found similar results as ours regarding the alterations in the MMP-9 and TIMP-1 levels at 24 and 72 hours. Etoh et al. [13] demonstrated increased expression of soluble interstitial MMP-9 as early as 2 hours after onset of the infarction. Kaden et al. [28] found the plasma concentration of MMP-9 was elevated in patients with acute MI and showed a rapid normalization to baseline levels within 1 week.
In the present study, significantly positive correlations were observed between MMP-9 and TIMP-1, CRP, and LDH levels. TIMP-1 levels were positively correlated with both CRP and leukocyte count levels. Kalela et al. [27] found a positive correlation between serum MMP-9 and leukocyte count in healthy subjects. MMP-9 levels were closely correlated with the release of creatinine kinase as a marker of myocardial damage [29]. TIMP-1 was strongly correlated with CRP [35]. Tan et al. [36] observed significant and positive correlations between MMP-9 and TIMP-1. Thus, these data show that TIMP-1, in response to the activation of MMP-9, plays a protective role during the early phase of NSTEMI.
Although recent studies have reported alterations in MMP-9 and TIMP-1 levels in the early stages of NSTEMI, no study has reported alterations in IL-33 levels in the early stages of NSTEMI. Therefore, the present study is the first to demonstrate the relationship between levels of IL-33 and levels of MMP-9, TIMP-1, and CRP and leukocyte count in the early stages of NSTEMI.
The IL-33 decoy receptor sST2 was shown to be elevated in serum early after acute MI [37]. IL-33 treatment increased levels of the Th2 cytokines IL-4, IL-5, and IL-13, but decreased levels of the Th1 cytokine IFN-γ in serum [38]. In the atheromatous plaque, proinflammatory factors produced by T lymphocytes (e.g., IFN-γ) seem to play a crucial role [21,39,40]. These cytokines decrease the production of the ECM by smooth muscles, and at the same time, they increase the production of the metalloproteinases by macrophages [40]. For this reason, IL-33 has a defensive role against MMP activation and ECM destruction by inhibiting IFN-γ release.
The results of this study indicate that serum levels of IL-33 also increased in the NSTEMI group compared to the control group. We examined the time course changes of the plasma soluble IL-33 levels (on admission, and at 12, 24, and 48 hours after NSTEMI). We showed that the IL-33 levels on admission were significantly lower than those detected in the blood samples collected at 12 hours. Thus, we speculate that IL-33, following the activation of IL-33, plays a protective role during the early phase of NSTEMI. IL-33 levels were negatively correlated with MMP-9, TIMP-1, and CRP levels.
In conclusion, the results presented here suggest that the determination of serum MMP-9 and TIMP-1 could represent a sensitive marker of inflammation in patients with NSTEMI. However, the endothelial cells of patients with NSTEMI seem to have an increased response to this inflammatory stimulus, leading to higher levels of MMP-9 and a higher degree of endothelial cell injury accompanied by the release of lower levels of IL-33 when compared to both the UA and control groups. Nevertheless, we believe that combined measurement of MMP-9, TIMP-1, and IL-33 with other well-known markers of inflammation, such as CRP, might provide an index of plaque activity in NSTEMI and may prompt future investigations addressing the predictive role of MMP-9, TIMP-1, and IL-33 in the definition of the risk of acute coronary events in patients with CAD.
One limitation of this study was the small number of subjects (a total of 80 in the three groups). Another limitation was the absence of a ST MI group.
KEY MESSAGE
1. Elevated levels of matrix metalloproteinase (MMP)-9 and decreased levels of interleukin (IL)-33 and tissue inhibitor of MMP-1 play a role in the development and progression of myocardial infarction.
2. Decreased IL-33 levels may play a major role in the process that terminates in plaque rupture, thrombus formation, and an acute coronary syndrome.
Footnotes
No potential conflict of interest relevant to this article is reported.
References
- 1.Ross R. Atherosclerosis: an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 2.Libby P. Inflammation and cardiovascular disease mechanisms. Am J Clin Nutr. 2006;83:456S–460S. doi: 10.1093/ajcn/83.2.456S. [DOI] [PubMed] [Google Scholar]
- 3.Li JJ. Inflammation: an important mechanism for different clinical entities of coronary artery diseases. Chin Med J (Engl) 2005;118:1817–1826. [PubMed] [Google Scholar]
- 4.Myllarniemi M, Calderon L, Lemstrom K, Buchdunger E, Hayry P. Inhibition of platelet-derived growth factor receptor tyrosine kinase inhibits vascular smooth muscle cell migration and proliferation. FASEB J. 1997;11:1119–1126. doi: 10.1096/fasebj.11.13.9367346. [DOI] [PubMed] [Google Scholar]
- 5.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 6.Brauer PR. MMPs: role in cardiovascular development and disease. Front Biosci. 2006;11:447–478. doi: 10.2741/1810. [DOI] [PubMed] [Google Scholar]
- 7.Schonbeck U, Libby P. CD40 signaling and plaque instability. Circ Res. 2001;89:1092–1103. doi: 10.1161/hh2401.101272. [DOI] [PubMed] [Google Scholar]
- 8.Uzui H, Harpf A, Liu M, et al. Increased expression of membrane type 3-matrix metalloproteinase in human atherosclerotic plaque: role of activated macrophages and inflammatory cytokines. Circulation. 2002;106:3024–3030. doi: 10.1161/01.cir.0000041433.94868.12. [DOI] [PubMed] [Google Scholar]
- 9.Lacraz S, Nicod L, Galve-de Rochemonteix B, Baumberger C, Dayer JM, Welgus HG. Suppression of metalloproteinase biosynthesis in human alveolar macrophages by interleukin-4. J Clin Invest. 1992;90:382–388. doi: 10.1172/JCI115872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lijnen HR. Metalloproteinases in development and progression of vascular disease. Pathophysiol Haemost Thromb. 2003;33:275–281. doi: 10.1159/000083814. [DOI] [PubMed] [Google Scholar]
- 11.Hirohata S, Kusachi S, Murakami M, et al. Time dependent alterations of serum matrix metalloproteinase-1 and metalloproteinase-1 tissue inhibitor after successful reperfusion of acute myocardial infarction. Heart. 1997;78:278–284. doi: 10.1136/hrt.78.3.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kai H, Ikeda H, Yasukawa H, et al. Peripheral blood levels of matrix metalloproteases-2 and -9 are elevated in patients with acute coronary syndromes. J Am Coll Cardiol. 1998;32:368–372. doi: 10.1016/s0735-1097(98)00250-2. [DOI] [PubMed] [Google Scholar]
- 13.Etoh T, Joffs C, Deschamps AM, et al. Myocardial and interstitial matrix metalloproteinase activity after acute myocardial infarction in pigs. Am J Physiol Heart Circ Physiol. 2001;281:H987–H994. doi: 10.1152/ajpheart.2001.281.3.H987. [DOI] [PubMed] [Google Scholar]
- 14.Creemers EE, Cleutjens JP, Smits JF, Daemen MJ. Matrix metalloproteinase inhibition after myocardial infarction: a new approach to prevent heart failure? Circ Res. 2001;89:201–210. doi: 10.1161/hh1501.094396. [DOI] [PubMed] [Google Scholar]
- 15.Dinh W, Futh R, Scheffold T, et al. Increased serum levels of tissue inhibitor of metalloproteinase-1 in patients with acute myocardial infarction. Int Heart J. 2009;50:421–431. doi: 10.1536/ihj.50.421. [DOI] [PubMed] [Google Scholar]
- 16.Schmitz J, Owyang A, Oldham E, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 17.Bartunek J, Delrue L, Van Durme F, et al. Nonmyocardial production of ST2 protein in human hypertrophy and failure is related to diastolic load. J Am Coll Cardiol. 2008;52:2166–2174. doi: 10.1016/j.jacc.2008.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuchler AM, Pollheimer J, Balogh J, et al. Nuclear interleukin-33 is generally expressed in resting endothelium but rapidly lost upon angiogenic or proinflammatory activation. Am J Pathol. 2008;173:1229–1242. doi: 10.2353/ajpath.2008.080014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanada S, Hakuno D, Higgins LJ, Schreiter ER, McKenzie AN, Lee RT. IL-33 and ST2 comprise a critical biomechanically induced and cardioprotective signaling system. J Clin Invest. 2007;117:1538–1549. doi: 10.1172/JCI30634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller AM, Xu D, Asquith DL, et al. IL-33 reduces the development of atherosclerosis. J Exp Med. 2008;205:339–346. doi: 10.1084/jem.20071868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daugherty A, Rateri DL. T lymphocytes in atherosclerosis: the yin-yang of Th1 and Th2 influence on lesion formation. Circ Res. 2002;90:1039–1040. doi: 10.1161/01.res.0000021397.28936.f9. [DOI] [PubMed] [Google Scholar]
- 22.Laurat E, Poirier B, Tupin E, et al. In vivo downregulation of T helper cell 1 immune responses reduces atherogenesis in apolipoprotein E-knockout mice. Circulation. 2001;104:197–202. doi: 10.1161/01.cir.104.2.197. [DOI] [PubMed] [Google Scholar]
- 23.Pinderski LJ, Fischbein MP, Subbanagounder G, et al. Overexpression of interleukin-10 by activated T lymphocytes inhibits atherosclerosis in LDL receptor-deficient mice by altering lymphocyte and macrophage phenotypes. Circ Res. 2002;90:1064–1071. doi: 10.1161/01.res.0000018941.10726.fa. [DOI] [PubMed] [Google Scholar]
- 24.Hamm CW, Bassand JP, Agewall S, et al. ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation: the Task Force for the management of acute coronary syndromes (ACS) in patients presenting without persistent ST-segment elevation of the European Society of Cardiology (ESC) Eur Heart J. 2011;32:2999–3054. doi: 10.1093/eurheartj/ehr236. [DOI] [PubMed] [Google Scholar]
- 25.Shah PK, Falk E, Badimon JJ, et al. Human monocyte-derived macrophages induce collagen breakdown in fibrous caps of atherosclerotic plaques: potential role of matrix-degrading metalloproteinases and implications for plaque rupture. Circulation. 1995;92:1565–1569. [PubMed] [Google Scholar]
- 26.Kim SH, Kang YJ, Kim WJ, et al. TWEAK can induce pro-inflammatory cytokines and matrix metalloproteinase-9 in macrophages. Circ J. 2004;68:396–399. doi: 10.1253/circj.68.396. [DOI] [PubMed] [Google Scholar]
- 27.Kalela A, Koivu TA, Sisto T, et al. Serum matrix metalloproteinase-9 concentration in angiographically assessed coronary artery disease. Scand J Clin Lab Invest. 2002;62:337–342. doi: 10.1080/00365510260296483. [DOI] [PubMed] [Google Scholar]
- 28.Kaden JJ, Dempfle CE, Sueselbeck T, et al. Time-dependent changes in the plasma concentration of matrix metalloproteinase 9 after acute myocardial infarction. Cardiology. 2003;99:140–144. doi: 10.1159/000070670. [DOI] [PubMed] [Google Scholar]
- 29.Higo S, Uematsu M, Yamagishi M, et al. Elevation of plasma matrix metalloproteinase-9 in the culprit coronary artery in patients with acute myocardial infarction: clinical evidence from distal protection. Circ J. 2005;69:1180–1185. doi: 10.1253/circj.69.1180. [DOI] [PubMed] [Google Scholar]
- 30.Romanic AM, Burns-Kurtis CL, Gout B, Berrebi-Bertrand I, Ohlstein EH. Matrix metalloproteinase expression in cardiac myocytes following myocardial infarction in the rabbit. Life Sci. 2001;68:799–814. doi: 10.1016/s0024-3205(00)00982-6. [DOI] [PubMed] [Google Scholar]
- 31.Kameda K, Matsunaga T, Abe N, et al. Increased pericardial fluid level of matrix metalloproteinase-9 activity in patients with acute myocardial infarction: possible role in the development of cardiac rupture. Circ J. 2006;70:673–678. doi: 10.1253/circj.70.673. [DOI] [PubMed] [Google Scholar]
- 32.van den Borne SW, Cleutjens JP, Hanemaaijer R, et al. Increased matrix metalloproteinase-8 and -9 activity in patients with infarct rupture after myocardial infarction. Cardiovasc Pathol. 2009;18:37–43. doi: 10.1016/j.carpath.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 33.Inokubo Y, Hanada H, Ishizaka H, Fukushi T, Kamada T, Okumura K. Plasma levels of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 are increased in the coronary circulation in patients with acute coronary syndrome. Am Heart J. 2001;141:211–217. doi: 10.1067/mhj.2001.112238. [DOI] [PubMed] [Google Scholar]
- 34.Tziakas DN, Chalikias GK, Hatzinikolaou EI, et al. N-terminal pro-B-type natriuretic peptide and matrix metalloproteinases in early and late left ventricular remodeling after acute myocardial infarction. Am J Cardiol. 2005;96:31–34. doi: 10.1016/j.amjcard.2005.02.039. [DOI] [PubMed] [Google Scholar]
- 35.Nanni S, Melandri G, Hanemaaijer R, et al. Matrix metalloproteinases in premature coronary atherosclerosis: influence of inhibitors, inflammation, and genetic polymorphisms. Transl Res. 2007;149:137–144. doi: 10.1016/j.trsl.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 36.Tan J, Hua Q, Gao J, Fan ZX. Clinical implications of elevated serum interleukin-6, soluble CD40 ligand, metalloproteinase-9, and tissue inhibitor of metalloproteinase-1 in patients with acute ST-segment elevation myocardial infarction. Clin Cardiol. 2008;31:413–418. doi: 10.1002/clc.20254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weinberg EO, Shimpo M, De Keulenaer GW, et al. Expression and regulation of ST2, an interleukin-1 receptor family member, in cardiomyocytes and myocardial infarction. Circulation. 2002;106:2961–2966. doi: 10.1161/01.CIR.0000038705.69871.D9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McLaren JE, Michael DR, Salter RC, et al. IL-33 reduces macrophage foam cell formation. J Immunol. 2010;185:1222–1229. doi: 10.4049/jimmunol.1000520. [DOI] [PubMed] [Google Scholar]
- 39.Mehta JL, Saldeen TG, Rand K. Interactive role of infection, inflammation and traditional risk factors in atherosclerosis and coronary artery disease. J Am Coll Cardiol. 1998;31:1217–1225. doi: 10.1016/s0735-1097(98)00093-x. [DOI] [PubMed] [Google Scholar]
- 40.Shishehbor MH, Bhatt DL. Inflammation and atherosclerosis. Curr Atheroscler Rep. 2004;6:131–139. doi: 10.1007/s11883-004-0102-x. [DOI] [PubMed] [Google Scholar]