

SUMMARY
Phage G1 gp67 is a 23 kDa protein that binds to the Staphylococcus aureus (Sau) RNA polymerase (RNAP) σA subunit and blocks cell growth by inhibiting transcription. We show that gp67 has little to no effect on transcription from most promoters but is a potent inhibitor of ribosomal RNA transcription. A 2.0-Å-resolution crystal structure of the complex between gp67 and Sau σA domain 4 (σA4) explains how gp67 joins the RNAP promoter complex through σA4 without significantly affecting σA4 function. Our results indicate that gp67 forms a complex with RNAP at most, if not all, σA-dependent promoters, but selectively inhibits promoters that depend on an interaction between upstream DNA and the RNAP α-subunit C-terminal domain (αCTD). Thus, we reveal a promoter-specific transcription inhibition mechanism by which gp67 interacts with the RNAP promoter complex through one subunit (σA), and selectively affects the function of another subunit (αCTD) depending on promoter usage.
INTRODUCTION
Staphylococcus aureus (Sau) is a pathogenic Gram-positive bacterium that causes infections of the skin as well as pneumonia, meningitis, endocarditis, and sepsis (Lowy, 1998). Methicillin-resistant Sau (MRSA) infections have become increasingly common and lead to significant morbidity and mortality (Nordmann et al., 2007). MRSA strains were recently reported to also be resistant to vancomycin, the current antibiotic of last resort (Howden et al., 2010), highlighting the importance of discovering novel therapeutics that inhibit Sau growth.
Before the advent of modern antibiotics, bacteriophages (phages) were considered potential therapeutic agents because of their ability to kill specific bacterial species rapidly. As microbes become increasingly resistant to antibiotics, phages are once again being exploited for their ability to directly eliminate bacterial infections and provide novel targets for the drug discovery process (Fischetti, 2008).
Recent studies have mined phage genomes for proteins and peptides that inhibit Sau cell growth (Kwan et al., 2005; Liu et al., 2004). One such protein, phage G1 gp67 (encoded by orf67), inhibits cell growth when produced in Sau, but not in Escherichia coli (Eco), and was shown to significantly decrease RNA production in Sau (Liu et al., 2004). Subsequent work demonstrated a direct interaction between gp67 and Sau RNA polymerase (RNAP; Dehbi et al., 2009).
All transcription in prokaryotes is performed by the ~400 kDa core RNAP (subunit composition α2ββ′ω; Darst, 2001). Promoter recognition and initiation require an additional subunit, σ, which binds to the core RNAP to form the holoenzyme (Murakami and Darst, 2003). The group 1, or primary, σ factors (σ70 in Eco, σA in Sau) are responsible for the bulk of transcription during log-phase growth and are essential for viability (Gruber and Gross, 2003). In the context of the RNAP holoenzyme, the structural domains of σA directly recognize conserved core promoter elements: domain 2 of σA (σA2) recognizes the –10 element, and σA4 recognizes the –35 element (Campbell et al., 2002; Feklistov and Darst, 2011; Murakami et al., 2002a; Shultzaberger et al., 2007). Promoters may contain additional elements recognized by σ, such as the extended –10 (Keilty and Rosenberg, 1987) or discriminator element (Feklistov et al., 2006; Haugen et al., 2008b; Travers, 1984). In addition, certain promoters are dependent on an A/T-rich DNA sequence upstream of the –35 element, known as the UP element (Ross et al., 1993). The UP element is recognized by the 9 kDa C-terminal domains of the RNAP α subunits (αCTDs), which are attached to the α-N-terminal domains by ~15-residue-long, flexible unstructured linkers (Blatter et al., 1994; Gourse et al., 2000).
Previous biochemical analyses established that gp67I (1) interacts directly with Sau σA but not Eco σ70, (2) uses σA4 as its primary interaction determinant, and (3) forms a stable, ternary gp67/Sau-σA/Sau-core-RNAP complex (Dehbi et al., 2009). Using the λPL promoter with a hybrid system comprising Sau σA with Eco core RNAP, as well as Sau-σA/Sau-core-RNAP, Dehbi et al. (2009) concluded that gp67 is a Sau σA-specific anti-σ factor that blocks σA4 recognition of the –35 element and is therefore a general inhibitor of –35 element-dependent promoters.
Here, we demonstrate that gp67 is a promoter-specific transcription inhibitor. Gp67 does not inhibit transcription from the majority of Sau promoters, but is a potent inhibitor of ribosomal RNA (rRNA) transcription in vitro and in vivo. Using X-ray crystal structures of gp67 bound to Sau σA4, in vivo RNA sequencing (RNA-seq) analysis, and in vitro biochemical analyses, we show that gp67 interferes with binding of the αCTDs to DNA upstream of the promoter –35 element, and therefore inhibits promoters that depend on the αCTD/DNA interaction. Gp67 is a phage-encoded transcription factor that acts through a novel mechanism in which it joins the RNAP holoenzyme through σA4 but modulates the host transcription program by interfering with promoter DNA interactions of the αCTDs.
RESULTS
Gp67 Function Requires a Native Sau Transcription System
In contrast to Sau RNAP, Eco RNAP is well characterized biochemically, and numerous tools such as mutant and modified Eco RNAPs are available for probing mechanistic questions. Therefore, as a first step toward understanding the molecular mechanism through which gp67 inhibits RNAP, we sought to reproduce the results of Dehbi et al. (2009) using the hybrid transcription system of Sau σA with Eco core RNAP on the λPL promoter. In contrast to Dehbi et al. (2009), we did not observe any effect of gp67 on λPL transcription, even when gp67 was at a 100-fold molar excess over the hybrid holoenzyme (Figure S1A available online). Moreover, a native gel-shift analysis revealed that gp67 formed a ternary gp67/Sau-σA/Sau-core-RNAP complex (Figure S1B, lane 3) but did not interact with the hybrid Sau-σA/Eco-core-RNAP (Figure S1C, lane 1).
Because gp67 failed to inhibit or even interact with the hybrid holoenzyme, we purified Sau core RNAP (Deora and Misra, 1996) and optimized the in vitro transcription conditions (see Experimental Procedures). Few Sau promoters have been characterized in vitro, so we first investigated Sau RNAP-holoenzyme activity in vitro using Sau genomic DNA as a template. Gp67 inhibited promoter-specific Sau RNAP-holoenzyme transcription from Sau genomic DNA (Figure 1, red bars), but did not inhibit transcription by the hybrid holoenzyme Sau-σA/Eco-RNAP (Figure 1, blue bars) or by Eco RNAP holoenzyme (Figure 1, orange bars), consistent with our in vitro results on the λPL promoter (Figure S1A). To determine whether inhibition of Sau RNAP holoenzyme by gp67 was dependent on the Sau-σA/gp67 interaction, we used a bacterial two-hybrid assay (Dove and Hochs-child, 2004) to generate an otherwise functional σA mutant that could no longer interact with gp67 (Figure S1D). We found that gp67 was unable to inhibit transcription by the corresponding mutant Sau holoenzyme (Figure 1, green bars). Gp67 inhibition therefore requires a native Sau transcription system.
Figure 1. Gp67 Inhibits Transcription by the Sau RNAP Holoenzyme but Not the Eco RNAP Holoenzyme or the Hybrid Holoenzyme Sau-σA/Eco-core RNAP.
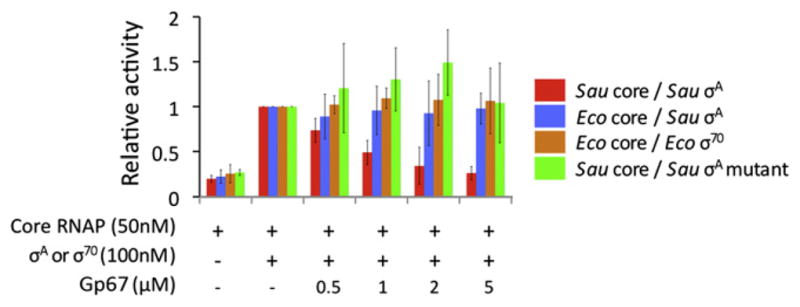
Values represent the mean of three independent experiments, and error bars denote the mean ± 1 SD.
See also Figure S1.
Gp67 Inhibits rRNA Synthesis In Vitro and In Vivo
Phage promoters have been important tools for studying RNAP function (Kadesch et al., 1982; Rosenberg et al., 1982; Stevens, 1977). The genome of the Sau-specific G1 phage has many easily identifiable promoters that closely match the –10/–35 consensuses with the optimal 17 bp spacing between them (Shultzaberger et al., 2007). One such promoter is G1-pORF05 (Figure 2A). Sau RNAP-holoenzyme has robust activity from this promoter when tested in vitro, but gp67 has no effect on this activity even at very high concentrations (Figure 2B). Because gp67 does not inhibit this –10/–35 promoter, it is unlikely that gp67 blocks recognition of the –35 element as previously proposed (Dehbi et al., 2009).
Figure 2. Gp67 Inhibits rRNA Synthesis In Vitro and In Vivo.

(A) Sequences of promoters used in subsequent experiments.
(B) Gp67 inhibits RNAP activity at Sau rrn promoters but not at a G1 phage –10/–35 promoter (pORF05). The indicated bands were quantified by phosphor-imagery; relative activity (versus holoenzyme alone) is denoted below each lane.
(C) Gp67 does not significantly decrease rRNA abundance in vivo. Cellular RNA was purified from 2 × 108 Sau RN4220 cells at different times after the induction of gp67 synthesis. The Sau σ is either wild-type (WT) or the mutant (M) unable to interact with gp67 (Figure S1D).
(D) Gp67 inhibits synthesis of rRNA in vivo. Inducer was added to Sau RN4220 cells containing pRMC2 or pRMC2-gp67, and newly synthesized RNA was labeled by the addition of [32P]-orthophosphoric acid to the growth medium. RNA was purified from 2 × 108 pRMC2 and pRMC2-gp67 cells, run on a 6% urea-PAGE gel, stained with GelRed to visualize all RNAs (left), and visualized by phosphorimagery (right).
See also Figure S2.
Gp67 inhibits bulk RNA synthesis in vivo (Liu et al., 2004) and in vitro (Figure 1). Because the major fraction of transcription in growing bacterial cells is dedicated to ribosome synthesis (Gourse et al., 1996), we examined the effect of gp67 on transcription from three Sau rRNA promoters: rrnA, rrnB-P1, and rrnB-P2 (Figure 2A). Gp67 is a potent inhibitor of in vitro transcription from these Sau rRNA promoters, and the inhibition depends on the interaction between gp67 and RNAP (Figure 2B).
To test whether gp67 inhibits rRNA synthesis in vivo, we expressed gp67 in Sau cells using an inducible expression vector (Corrigan and Foster, 2009). Upon addition of inducer, cell growth was slowed but not halted by gp67 expression (Figure S2) and rRNA levels remained unchanged (Figure 2C). Whereas messenger RNAs (mRNAs) generally have short half-lives in vivo, rRNAs are stable in prokaryotic cells (Deutscher, 2003). Therefore, to assess the effect of gp67 expression on rRNA synthesis, we used metabolic labeling to observe newly transcribed RNAs. Gp67 was expressed in Sau cells, and then newly synthesized RNAs were labeled by addition of radiolabeled phosphate to the growth medium (Wade et al., 1964). Whereas gp67 had little to no effect on the overall rRNA levels (Figures 2C and 2D, left panel), gp67 production significantly reduced new rRNA synthesis (Figure 2D, right panel). These data indicate that gp67 inhibits RNAP activity at rRNA promoters, but the rRNAs remain stable in the cells for hours after gp67 production. We conclude that gp67 is not a general inhibitor of Sau σA holoenzyme, but is a potent and direct inhibitor of rRNA transcription in vitro (Figure 2B) and in vivo (Figure 2D).
RNA-seq Reveals Promoters Inhibited by gp67 on a Genome-wide Scale
Because gp67 appears to be a promoter-specific inhibitor of Sau transcription, we wanted to determine the effect of gp67 on a wide variety of promoters in order to understand the characteristics that lead to gp67 sensitivity. For this purpose, we used RNA-seq to quantitatively compare RNA transcripts from Sau cells grown in the absence or presence of gp67. RNA was purified from Sau RN4220 cells producing gp67 as well as from control cells (Figure S2C), rRNAs were depleted, and libraries were prepared by standard procedures. The RNA was then sequenced by Illumina technology. We found that gp67 had no effect on transcription from the majority of promoters in the Sau genome (Figure 3A). Less than 4% of all transcripts were downregulated, and ~5% were upregulated by gp67 production during the growth phase.
Figure 3. RNA-seq Reveals Promoters that Are Sensitive to gp67 Inhibition In Vivo.

(A) Gp67 production in Sau RN4220 cells does not significantly inhibit RNA levels from ~91% of promoters. RNAs were sequenced directly and visualized with the Integrated Genomics Viewer. The position on the genome is shown on the horizontal axis, and the number of RNA reads per million total reads is shown on the vertical axis. Upper panel (green) represents RNA-seq data from pRMC2-gp67 cells; the lower panel (red) represents RNA-seq data from control pRMC2 cells.
(B) Gp67 production does not inhibit transcription from selected DNA replication promoters in vivo. RNA-seq data were visualized as above from the dnaA, aag, and polIII loci.
RNA-seq only reveals RNA transcript levels; it does not differentiate between direct and indirect regulation of gene expression, nor does it reveal whether differential gene expression is due to changes in promoter binding and initiation or mRNA stability. We used the Sau in vitro transcription system to test whether gp67 directly affects transcription at promoters identified by RNA-seq.
DNA replication is often a target of early phage proteins (Datta et al., 2005; Yano and Rothman-Denes, 2011). However, expression of genes involved in replication (Xu et al., 2010) was not significantly altered in the presence of gp67 in vivo (Figure 3B). Gp67 also did not inhibit the activity of RNAP at the dnaA or aag (a putative DNA repair protein) promoters in vitro, and had an effect on already weak RNAP activity at the polIII promoter only at a very high gp67 concentration (Figure 3C). In contrast, when tested on promoters that show significant levels of inhibition by gp67 in vivo (Figure 3D), gp67 was found to be a potent inhibitor of transcription in vitro (Figure 3E), indicating that, at least at these promoters, the in vivo effects are direct. Gp67 had no effect on transcript levels from Sau promoters that were previously studied using purified in vitro transcription systems (Figure S3; Rao et al., 1995; Reyes et al., 2011). Because gp67 had no effect on >90% of Sau promoters in vivo, or on the G1-pORF05, dnaA, and aag promoters in vitro, gp67 must not target general σ functions, such as –10 or –35 promoter element recognition or core RNAP interaction.
Crystal Structure of the gp67/Sau σA4 Complex
To shed light on the mechanism by which gp67 modulates Sau RNAP function, we determined X-ray crystal structures of the gp67/Sau-σA4 complex. We solved the structure from two crystal forms: form I (2.0 Å resolution) and form II (3.0 Å resolution; Tables S1 and S2; Figure S4C). Three crystallographically independent structures did not show any significant differences (root-mean-square deviation [rmsd] of α-carbon positions = ≤ 0.78 Å). Therefore, we derived all of the structural analyses using the high-resolution form I structure. Although the structure of σ4 has been well described (Campbell et al., 2002), gp67 has no sequence or structural homology to any previously described fold.
The 2.0 Å crystal structure reveals the nature of the interactions between gp67 and σA4 (Figures 4A, S4D, and S4E). The σA4 has been studied structurally in many contexts, including bound to DNA (Campbell et al., 2002), RNAP (Murakami et al., 2002b; Vassylyev et al., 2002), RNAP and DNA (Murakami et al., 2002b), and anti-σ factors (Campbell et al., 2008). Gp67 does not reorganize the conformation of σA4 (Figures 4A and S4F), as occurs in some anti-σ/σ complexes (Campbell et al., 2007; Lambert et al., 2004).
Figure 4. Cocrystal Structure of gp67/σA4.
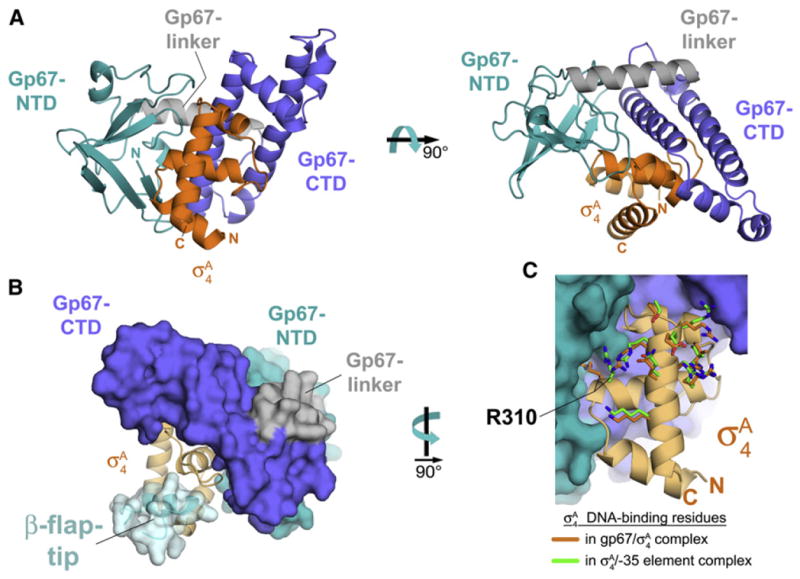
(A) Cocrystal structure of gp67 bound to σA4 (orange). Two orthogonal views are shown. Proteins are shown in ribbon format.
(B) Structural modeling indicates that the RNAP β-flap-tip/σA4 interaction is compatible with the presence of gp67. Gp67 is shown as a molecular surface, color-coded as in (A). The σA4 is shown as a light orange ribbon. The RNAP β-flap-tip, modeled by superimposing σA4 from the Thermus aquaticus RNAP holoenzyme (Murakami et al., 2002b), is shown as a light blue ribbon with a transparent molecular surface (cyan).
(C) Gp67 does not contact most residues of σA4 that interact with –35-element DNA. Shown is a close-up view of Sau σA4 (light orange ribbon) in the complex with gp67 (molecular surface, color-coded as in [A]). Side chains of residues that directly interact with –35-element DNA are shown from the gp67/Sau σA4 structure (orange) and from the superimposed Taq σA4/-35 element DNA complex (green side chains; PDB ID code 1KU7; Campbell et al., 2002). Only the side chain of Sau σA4 R310 (corresponding to Taq σA 4 R379/Eco σ70 R554) is altered through an interaction with gp67.
See also Figure S4 and Tables S1 and S2.
(C) Gp67 does not inhibit transcription from selected DNA replication promoters in vitro. The indicated bands were quantified by phosphorimagery; relative activity (versus holoenzyme alone) is denoted below each lane.
(D) Selected genes inhibited by gp67 in vivo. RNA-seq data were visualized as above from the csp1, csp2, and pstp loci.
(E) Gp67 inhibits transcription in vitro from the selected promoters that are susceptible in vivo when wild-type (WT) σA is used, but not the quadruple mutant σA that does not interact with gp67 (M). The indicated bands were quantified by phosphorimagery; relative activity (versus holoenzyme alone) is denoted below each lane.
See also Figures S2 and S3, and Table S3.
Gp67 itself comprises two domains: an N-terminal β-sheet-rich domain (gp67-NTD; Figure 4A, teal) and a C-terminal α-helical domain (gp67-CTD; Figure 4A, blue). The two domains are connected by a linker-α-helix on the surface of gp67 opposite the σA4 interaction surface (gp67-linker; Figure 4A, gray). Gp67 forms an extensive molecular interface with σA4 through both the gp67-NTD (1,032 Å 2 buried surface area) and the gp67-CTD (1,757 Å 2 buried surface area), with a total buried surface area of 2,789 Å 2. Based on the results of our two-hybrid analysis, we identified a limited number of amino acid differences between Sau σA4 and Eco σ704 that determine the specificity of gp67 binding (Figure S1D). In accord with this genetic analysis, the identified residues (Sau σA D309, E312, N313, and V335) all participate in the gp67/σA interface (Figures S4D and S4E).
Most anti-σ factors bind their cognate σ factor and block at least one of the main σ functions: core RNAP binding and/or core promoter element recognition (Campbell et al., 2008; Lambert et al., 2004; Patikoglou et al., 2007). Gp67 does not prevent core RNAP binding of Sau σA (Figure S1B), and our finding that gp67 does not inhibit transcription from the vast majority of Sau –10/–35 promoters in vivo (Figure 3A) is inconsistent with a model in which gp67 blocks recognition of the –35 element by σA4. Indeed, structural modeling, as described below, suggests that core RNAP binding and –35 element DNA binding by σA4 would be allowed in the complex with gp67 (Figures 4B and 4C).
The σA4 binds to core RNAP primarily through an interaction with the β-flap-tip-helix, a structural element of the RNAP β subunit. The σA4/β-flap-tip-helix interaction is essential to position σA4 in the proper orientation and spacing for –35 element recognition (Kuznedelov et al., 2002; Murakami et al., 2002b). One can superimpose the β-flap-tip from the RNAP holoenzyme (Murakami et al., 2002b) on the gp67/σA4 structure (by superimposing the σA4 structural core) without introducing any steric clashes with gp67 (Figure 4B).
Ten highly conserved residues of σA4 make direct contact with –35 element DNA (Campbell et al., 2002; Jain et al., 2004). All but one of these DNA-contacting residues of σA4 are unperturbed by gp67 and do not make any contacts with gp67 (Figure 4C). The one exception is Sau σA4 Arg310 (which corresponds to Taq σA Arg379/Eco σ70 Arg554). In the –35 element-bound complex, this Arg residue interacts with the DNA phosphate backbone at the –36 position just upstream of the –35 element (Campbell et al., 2002; Jain et al., 2004). In the gp67 complex, Sau σA4 Arg310 is redirected away from the DNA-binding interface and is buried in a deep pocket of gp67 (Figure 4C) where it makes extensive interactions with gp67 residues.
Using either the Taq σA4/-35 element DNA complex structure (Protein Data Bank [PDB] ID code 1KU7; Campbell et al., 2002) or an RNAP-holoenzyme open promoter complex (RPo) model to superimpose –35 element DNA on the gp67/σA4 structure reveals minor steric clashes (Figure 5). Nevertheless, based on the following points, we argue that gp67 binding to σA4 does not significantly affect recognition of the –35 promoter element by σA4, and may introduce additional protein/DNA interactions:
Figure 5. Structural Model of the gp67/RNAP Holoenzyme/Open Promoter Complex.
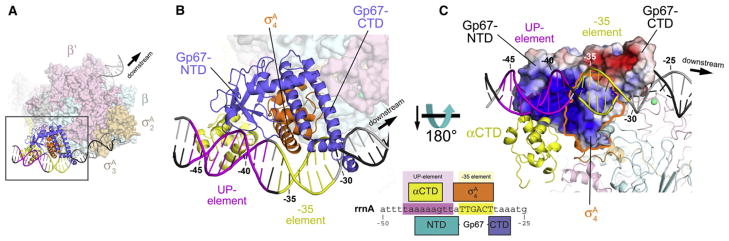
(A) Overall view. The core RNAP is shown as a molecular surface (β′, pale pink; β, pale cyan). The σA subunit is shown as a molecular surface (σA2 and σA 3, pale orange) or a ribbon (σA4, orange). Gp67 is shown as a blue ribbon. One αCTD is shown (yellow ribbon) bound to the proximal UP-element subsite, modeled as described in Jain et al. (2005). The DNA is shown as a phosphate-backbone worm, with the promoter –35 element in yellow and the proximal UP-element subsite (Estrem et al., 1998, 1999) in magenta. The boxed region is magnified in (B).
(B) Magnified view of the –35-element/proximal UP-element region. Shown as in (A). The green sphere denotes a Zn2+ ion. The DNA is numbered with respect to the Sau rrnA promoter start site (see Figure 6A). The schematic at the lower right shows the –35-element/UP-element region of the Sau rrnA promoter. The regions of close protein/DNA interaction for σA4 (orange), gp67 (blue), and αCTD (yellow) are denoted by the span of the boxes.
(C) View toward the gp67/σA4 DNA interaction surface. The RNAP is shown in ribbon format. The molecular surface of the gp67/σA4 complex is colored according to the electrostatic surface distribution (red, –5 kT; white, neutral; blue, +5 kT). The region corresponding to σA4 is outlined in orange.
See also Figure S4 and Tables S1 and S2.
Our in vitro and in vivo results indicate that gp67 is not a general inhibitor of –10/–35 promoters (Figures 2B and 3A–3C).
The minor steric clash with the DNA is at the distal end of the gp67-CTD helical tower, which has relatively high B-factors and is likely to be conformationally flexible. A small rearrangement of the end of the gp67-CTD helical tower and/or the DNA could relieve the clash and appears to facilitate an interaction in the DNA major groove.
The molecular surface of gp67 that faces the DNA in our model is very basic, particularly upstream of the –35 element (Figure 5C).
In summary, the 2-Å-resolution X-ray crystal structure of the gp67/Sau-σA4 complex, combined with structural modeling, reveals that gp67 bound to σA4 would not interfere with σA 4 binding to RNAP (Figure 4B), and would be unlikely to completely block σA4 interactions with –35-element DNA (Figures 4C and 5). These observations are consistent with our biochemical findings that gp67 forms a ternary gp67/σA/RNAP complex (Figure S1B) and does not disrupt –35-element recognition by σA4 at most –35-element-dependent promoters (Figure 3A).
Gp67 Inhibits Transcription from Promoters Containing an A/T-Rich Sequence Upstream of the –35 Element
Because gp67 is unlikely to block –35-element recognition in the context of the RNAP holoenzyme, we hypothesized that other promoter DNA elements may confer susceptibility to gp67 inhibition. We aligned all promoters tested in vitro for direct gp67 activity by their –10 and –35 elements (Figure S5A). The sensitive promoters do not share an obvious common sequence but tend to be A/T rich in the region upstream of the –35 element, where an UP element would be expected (Estrem et al., 1998, 1999), whereas this characteristic was less prominent in promoters resistant to gp67 inhibition (Figure S5A).
To test whether the region upstream of the –35 element was important for gp67 function, we constructed hybrid promoters by swapping the DNA immediately upstream of the –35 element between a gp67-sensitive promoter, rrnA, and a gp67-resistant promoter, dnaA (Figure 6A). As observed previously, gp67 inhibited transcription from rrnA (Figure 6B, lanes 1 and 2). In the absence of gp67, the hybrid rrnA(upstream dnaA) promoter showed decreased activity, similar to the gp67-inhibited rrnA (Figure 6B, lane 3), and gp67 had no additional effect on this hybrid promoter (Figure 6B, lane 4). Gp67 did not significantly affect transcription from the dnaA promoter (Figure 6B, lanes 5 and 6). However, when its upstream sequence was replaced by the rrnA A/T-rich sequence, the resulting dnaA(upstream rrnA) promoter became highly sensitive to gp67 (Figure 6B, lanes 7 and 8). The loss of activity between rrnA and rrnA(upstream dnaA) (Figure 6B, lanes 1 and 3) demonstrates that the A/T-rich sequence just upstream of the rrnA –35 element contributes significantly to rrnA activity. Moreover, these results argue that susceptibility to gp67 inhibition is mediated by a promoter feature upstream of the –35 element, and implicate an A/T-rich sequence in this susceptibility.
Figure 6. Gp67 Blocks UP-Element Utilization.

(A) Schematic of promoters used in swapping and footprinting experiments. DNA positions for the rrnA and dnaA promoters are labeled relative to the start site (+1). The –10 and –35 elements are shaded red. The putative UP-element region of the rrnA promoter is shaded green. The corresponding region of the dnaA promoter is denoted by red text.
(B) The region upstream of the –35 element is required for gp67 inhibition. In vitro transcription assays were performed from hybrid promoters constructed by swapping the region upstream of the –35 element between the rrnA (gp67-sensitive) and dnaA (gp67-resistant) promoters.
(C) DNase I footprinting. Left panel: Sau rrnA promoter with Eco RNAP and Eco RNAP-ΔαCTD. Middle panel: Sau rrnA promoter with Sau RNAP holoenzyme (±gp67). Right panel: Sau dnaA promoter with Sau RNAP holoenzyme (±gp67).
See also Figure S5.
Gp67 Alters RNAP Interactions with Promoter DNA Upstream of the –35 Element
To test whether gp67 directly modulates RNAP binding to promoters with A/T-rich elements, we used DNase I footprinting. DNase I cleaves at exposed minor grooves of the DNA double helix. Cleavage is enhanced by deformations or bends in the DNA double helix that widen the minor groove (Fox, 1997).
Because Sau rrn promoters have not been tested biochemically, and UP-element binding has not been shown in this organism, we first examined the Sau rrnA promoter by DNase I footprinting using Eco RNAP (Figure 6C, left panel). Binding and distortion of the promoter DNA by Eco RNAP are indicated by strong DNase I hypersensitive sites observed on the template strand upon Eco RNAP binding (Figure 6C; compare no RNAP, lane 1, with Eco RNAP, lane 2) between the –10 and –35 elements (at ~–29; Figure 6C, band a) and just upstream of the –35 element (at ~–40; Figure 6C, band b). DNase I hypersensitivity in these regions is commonly observed in Eco RNAP/promoter complexes (Ozoline and Tsyganov, 1995). We also determined the DNase I footprint of a mutant Eco RNAP lacking the αCTDs (Eco RNAP-ΔCTD; Figure 6C, lane 3) to assess the effects of the αCTD/UP-element interaction on the cleavage pattern. In the absence of the αCTD/UP-element interaction, DNase I hypersensitive band a was unaffected, whereas band b was relatively much less intense. In addition, new cleavage sites appeared in the region upstream of the –35 element, at approximately –46 and –52, in the middle of the expected UP-element region (–37 to –56; Estrem et al., 1998; Gourse et al., 2000).
Thus, in terms of DNase I hypersensitivity, the RNAP/promoter complex with the αCTD/UP-element interaction is characterized by very strong hypersensitivity at band b, and an absence of hypersensitive sites in the expected region of the UP element. In the absence of the αCTD/UP-element interaction, band b is much reduced and cleavage sites appear within the region of the UP element.
The DNase I footprint of Sau RNAP on the gp67-sensitive Sau rrnA promoter is very similar to the Eco RNAP footprint (Figure 6D, compare lanes 4 and 5) and displays the characteristics of the αCTD/UP-element interaction (i.e., very strong hypersensitivity at band b and absence of cleavage upstream). The DNase I footprint in the presence of gp67 (Figure 6D, lane 6) is indicative of disrupted αCTD/UP-element interactions: band a and downstream regions of the footprint show few changes, but upstream of the –35 element, the hypersensitive site at –40 (band b) is completely eliminated and hypersensitive sites appear within the UP element (at approximately –47 and –51).
The disappearance of the hypersensitive band b in the presence of gp67 could be explained by protection of the site due to the physical presence of gp67 (since our structural modeling suggests that gp67 may interact with the DNA minor groove between –37 and –44; Figure 5) or by an alteration of the conformation of the DNA induced by gp67. In view of the results comparing Eco RNAP and RNAP-ΔCTD (Figure 6C, lanes 2 and 3), disruption of the αCTD/UP-element interaction by gp67 is the best explanation for the appearance of cleavage sites within the UP element with Sau RNAP in the presence of gp67 (Figure 6D, lane 6).
The DNase I footprint of Sau RNAP (±gp67) on the gp67-insensitive dnaA promoter shows qualitatively different features compared with the footprints on the rrnA promoter. As with rrnA, RNAP binding to the dnaA promoter induces DNase I hypersensitivity at sites between the –10 and –35 elements; at the dnaA promoter, these sites are at –27, –26, and to a lesser extent –25 (Figure 6C, lanes 7–9). RNAP binding (in the absence of gp67) induces DNase I cleavage at the upstream edge of the –35 element that is not present without RNAP (Figure 6C, compare lanes 7 and 8, band b). However, band b in this case is weak (relative to band a) compared with band b of the rrnA footprints, which is very prominent (and stronger than band a). Moreover, a DNase I cleavage site at approximately –46 is not protected, suggesting that the αCTD binds weakly or not at all to this region. The weak features upstream of band a are altered in the presence of gp67; the weak band b cleavage is eliminated, and new weak cleavage sites appear, likely reflecting the disruption of weak αCTD-DNA interactions.
Thus, the DNase I footprint of Sau RNAP on the gp67-insensitive dnaA promoter does not show the distinctive features indicative of strong αCTD/UP-element interactions, suggesting that transcription from this promoter is not UP-element dependent. In the presence of gp67, alterations in the cleavage pattern just upstream of the –35 element indicate that gp67 is present in the RNAP-holoenzyme/dnaA promoter complex, even though gp67 has little to no effect on transcription output from this promoter (Figures 3C and 6B).
We also determined DNase I footprints for the rrnA(upstream dnaA) and dnaA(upstream rrnA) promoters (Figure S5B). On the gp67-insensitive rrnA(upstream dnaA) promoter, the DNase I cleavage pattern in the region upstream of the –35 element suggests weak αCTD/UP-element interactions (i.e., dramatically weakened hypersensitivity around –39, and distinct cleavage sites between –40 and –56, where one would expect protection due to αCTD binding). On the gp67-sensitive dnaA(upstream rrnA) promoter, the DNase I cleavage pattern suggests αCTD/UP-element interactions (increased hypersensitivity of band b and protection through the upstream region), which are altered in the presence of gp67.
DISCUSSION
Bacteriophages are the most abundant and diverse form of life on Earth, and exert a major influence over the biosphere. The complete genome sequences of >400 double-stranded DNA phages have been determined. Metagenomic analyses have shown that the phage population is dominated by genetic information that is not related to known sequences (Brüssow and Hendrix, 2002). Bacteriophages are dependent upon a host organism for propagation and have evolved ingenious mechanisms to subvert host cellular processes (such as transcription) for their own needs (Nechaev and Severinov, 2003). In addition to elucidating new mechanisms of bacterial RNAP regulation, the study of phage-encoded regulators can shed light on RNAP function. One can argue that most basic principles of cellular transcription regulation have been exploited by phages, and that the study of phages has revealed many of these mechanisms (Ptashne, 1992).
The use of bacteriophages, including Sau-specific phages, for the treatment or prophylaxis of bacterial infectious diseases is experiencing a resurgence, increasing the need to understand bacteriophage/host interactions. Identification and mechanistic analysis of phage proteins that bind and inhibit essential host enzymes can point to potential drug targets and mechanisms (Liu et al., 2004).
Our work on Sau phage G1 gp67 has uncovered a novel regulatory mechanism (Figure 7). Upon its expression in the cell, gp67 forms a tight complex with σA4 (Figure 4A), but in a way that does not block the two primary functions of σA4: core RNAP binding (Figure 4B) and promoter –35 element recognition (Figure 4C). Therefore, gp67 becomes a stable component of the RNAP holo-enzyme (Figure S1B) and even of RNAP-holoenzyme/promoter complexes. Nevertheless, the vast majority (–91%) of promoters in Sau are unaffected by gp67 (Figure 3A). Rather than acting as a general anti-σ factor, gp67 inhibits Sau transcription in a promoter-specific fashion by selectively targeting promoters depending on the UP-element/RNAP αCTD interaction.
Figure 7. Model for gp67 Function.
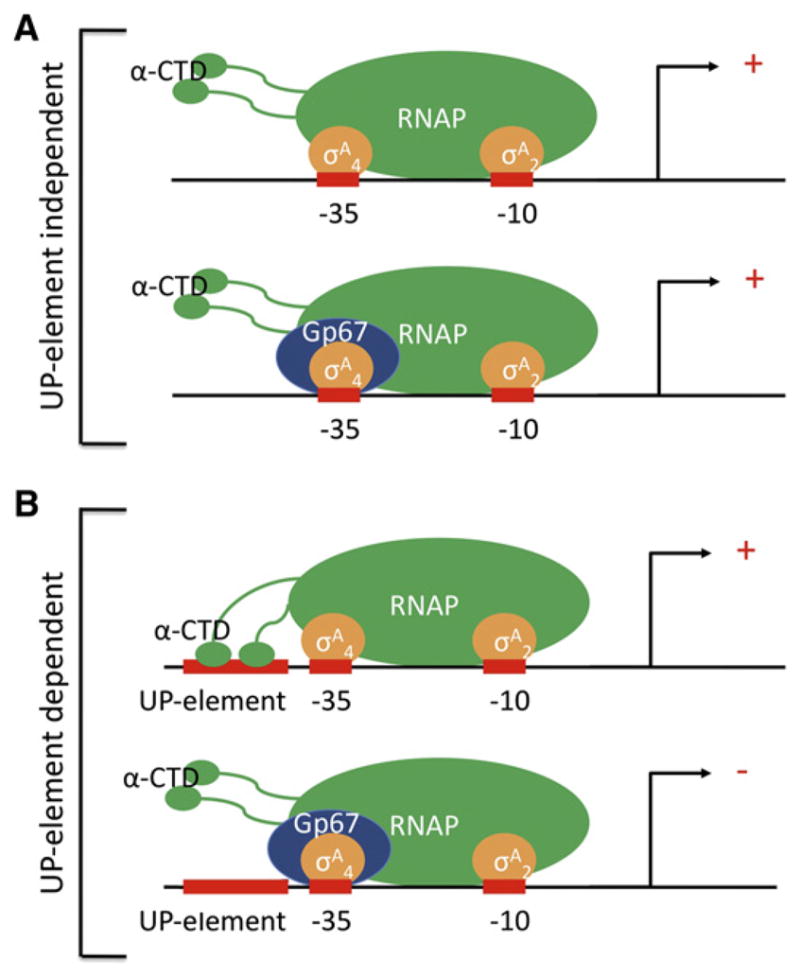
(A) At most –10/–35 promoters, which are not dependent on αCTD/UP-element interactions, gp67 joins the Sau RNAP holoenzyme through its interaction with σA4. However, gp67 does not block any of the functions of σA4 and does not inhibit RNAP.
(B) At promoters where activity depends on αCTD/UP-element interactions, gp67 joins the RNAP holoenzyme through its interaction with σA4 and inhibits transcription initiation by preventing αCTD binding to the promoter-proximal UP-element subsite, or to both proximal and distal subsites (shown).
Approximately 90 Sau promoters are downregulated by gp67, and 116 promoters are upregulated (Figures 2D and 3A). Six of these in vivo downregulated promoters (rrnA, rrnB-P1, rrnB-P2, csp1, csp2, and pstp; Figure S5A) have been tested in vitro, and all six are directly downregulated by gp67 (Figures 2B and 3E). Based on this limited sample, we postulate that gp67 directly inhibits most, if not all, of the promoters that are downregulated by gp67 expression in vivo. We examined six in vivo upregulated promoters in vitro, but did not observe any evidence for direct, gp67-mediated stimulation. Therefore, we cannot rule out the possibility that the observed in vivo upregulation was an indirect effect of inhibition of rRNA transcription, which would significantly increase the cellular concentration of available RNAP (Barker et al., 2001).
Bacterial transcription initiation is often regulated by activator proteins that typically bind DNA operators upstream of the core promoter and contact the RNAP through the αCTD or through σ4 (Browning and Busby, 2004). Thus, the mechanism of gp67-mediated inhibition of transcription initiation at UP-element-dependent promoters suggests the possibility that gp67 may interfere with factor-dependent transcription activation as well, a point that needs to be tested in future experiments.
Inhibition of Sau Cell Growth by gp67
To aid in the search for targets of small-molecule inhibitors, Xu et al., (2010) identified a set of 308 genes required for Sau growth. Among these were the rRNA genes that were shown here to be directly inhibited by gp67 (Figure 2). In addition, seven other genes inhibited by gp67 were required for cell growth in Sau. Three of these are also required for translation (Table S3).
Recent work has shown that ribosomal protein promoters and rRNA promoters are regulated by similar mechanisms (Lemke et al., 2011). Given that all but four of the required genes down-regulated by gp67 are required for a functional translational machinery, and that cell growth is known to be limited by rRNA synthesis (Gourse et al., 1996), the direct effect of gp67 on rRNA expression specifically, and on expression of the translational machinery in general, is the likely mechanism through which gp67 inhibits cell growth. Further experiments will be required to determine whether gp67 inhibition of other genes that are not involved in translation is sufficient to affect cell growth.
Sau Transcription and Transcriptional Regulation
Previous studies examined transcription and its regulation in vitro in Sau, but these studies focused on atypical, highly regulated virulence promoters (Rao et al., 1995; Reyes et al., 2011). Our analysis of gp67 function enabled us to identify σA-dependent –10/–35 promoters in Sau and to characterize Sau rRNA promoters. The molecular tools developed here will be useful for further studies of gene expression and its regulation in this important pathogen. The native Sau transcription system (Sau RNAP holoenzyme and Sau promoters) used here was critical for elucidating the mechanism of gp67 inhibition, whereas the use of a hybrid transcription system (Eco-core-RNAP/Sau-σA/Eco-RNAP promoters) in previous work led to misleading results (Dehbi et al., 2009).
Gp67 and rRNA Synthesis in Sau
The αCTD/UP-element interaction activates rRNA expression by >100-fold in Eco and by roughly 3-fold in the Gram-positive Bacillus subtilis (Krásný and Gourse, 2004). When gp67 is bound to RNAP (suppressing UP-element function), or when the UP element is removed from the Sau rRNA promoters in vitro, transcription decreases 2- to 3-fold (Figure 6B), arguing that the UP-element activation of rRNA transcription in Sau is more similar to that in B. subtilis than that in Eco. In vivo, gp67 decreases expression of rRNA promoters more dramatically (~10-fold; Figure 2D), which may be explained by the competition for RNAP binding between rRNA promoters and other promoters in the genome.
rRNA synthesis is tightly regulated in prokaryotes and is quickly inhibited upon the entry of bacteria into stationary phase (growth-rate control; Haugen et al., 2008a). In Eco, growth-rate control of rRNA synthesis involves several mechanisms, including direct modulation of the RNAP by the transcription factor DksA and the small molecule ppGpp (Paul et al., 2004; Perederina et al., 2004). Like DksA and ppGpp, gp67 is a transcription regulator that interacts with RNAP at many promoters, but affects transcriptional output from only a small subset of these promoters due to their unique features. Gp67 specifically takes advantage of the fact that full activity of the rRNA promoters depends on UP-element utilization.
Role of gp67 in the Phage Life Cycle
The genomes of many Sau phages have been sequenced (Kwan et al., 2005). The G1 phage is nearly identical to the Sau phage K (Kwan et al., 2005), and gp67 shares 100% sequence identity between these phages. Obvious gp67 homologs can be found in five firmicute-specific phages. The molecular underpinnings of these phages’ life cycles are largely unknown.
A consensus –10/–35 promoter (with an extended –10 element) that is highly active with Sau RNAP in vitro (Figure S5C) directs the synthesis of gp67, which is likely produced at high levels early after initial injection of the double-stranded phage genome into the host cell. We hypothesize that gp67 would then engage with the host RNAP (Figure S1B) and suppress production of rRNAs (Figure 2) by selectively inhibiting UP-element-dependent promoters (Figure 6) while allowing transcription from the majority of –10/–35 promoters (Figure 3), including its own promoter and other potential early phage promoters that are resistant to gp67 inhibition (e.g., porf67 [Figure S5D] and porf05 [Figures 2A and 2B]). The phage ultimately will require the use of host ribosomes to translate phage gene products. Because rRNA is stable in prokaryotic cells (Deutscher, 2003), previously formed ribosomes remain abundant in Sau cells for hours after gp67 expression (Figures 2C and 2D).
During log-phase growth, the majority of RNAP in prokaryotic cells is occupied in actively transcribing rRNA. Inhibition of rRNA transcription not only leads to arrest of cell division (Gourse et al., 1996) but also frees a large pool of host RNAP that can then be recruited to the strong phage early promoters. The T4 phage anti-σ factor, AsiA, inhibits host RNAP transcription of the host genome by blocking –35 element recognition while an additional protein, MotA, recruits the AsiA-modified RNAP complex to phage promoters (Hinton et al., 2005). The T4 phage has many additional protein factors that suppress transcription of the host genome and/or favor transcription of the phage genome in a coordinated fashion, including enzymes that ADP-ribosylate the RNAP αCTD, leading to inhibition of UP-element-dependent promoters and recruitment of RNAP to phage promoters (Tiemann et al., 2004). The phage G1 genome does not encode its own RNAP, but relies on the host RNAP for transcription during its entire life cycle, which likely requires complex coordination akin to that observed for phage T4. Therefore, although gp67 expression alone is sufficient to inhibit cell growth (Figure S3; Dehbi et al., 2009; Liu et al., 2004), the existence of additional phage proteins that control the host RNAP throughout the phage life cycle seems likely; however, this remains to be determined. Particularly interesting is the possibility that other phage proteins directly cooperate with gp67 to promote regulated transcription of the phage genome.
EXPERIMENTAL PROCEDURES
Full details of the experimental procedures used in this work are presented in the Extended Experimental Procedures.
Protein Expression and Purification
Gp67 or gp67/σA4 encoding sequences were cloned into pET-based expression vectors, transformed into Eco BL21(DE3) cells, and overexpressed, and the proteins were purified using standard methods. The purified complex was dialyzed into crystallization buffer (10 mM Tris-HCl, pH 8.0, 0.5 M NaCl) and screened for crystallization conditions. Endogenous Sau RNAP was purified from NCTC8325 cells essentially as previously described (Deora and Misra, 1996).
In Vitro Transcription Assays
In vitro transcription assays were performed using standard methods.
Genetic Analysis of the gp67/Sau-σA4 Interaction
A bacterial two-hybrid assay (Dove and Hochschild, 2004) was used to genetically dissect the interaction between gp67 and Sau σA4. Taking advantage of the fact that gp67 binds to Sau σA4 but not to Eco70 σ4, we made a series of Eco/Sau σ4 chimeras in order to define a minimal specificity-determining region (Figure S1D). Having identified four amino acid differences within this region that suffice to dictate whether or not gp67 can bind, we generated an otherwise functional Sau σA4 mutant (bearing the corresponding Eco residues at Sau positions 309, 312, 313, and 335) that did not interact with gp67. We assessed the functional integrity of this mutant by using the two-hybrid assay to test its ability to interact with the β-flap (Figure S1D), and a one-hybrid assay to test its ability to bind a –35 element (data not shown).
Gp67 Expression In Vivo
Gp67 was cloned into the Sau expression vector pRMC2 (Corrigan and Foster, 2009). pRMC2-gp67 or empty pRMC2 was then transformed into Sau strain RN4220 by electroporation (Schenk and Laddaga, 1992). Cells were then grown in trypticase soy (TS) broth containing chloramphenicol, and transgene expression was induced with 100 ng/ml anhydrotetracycline.
RNA Purification and Metabolic Labeling
RNA was purified from cells at mid-log phase growth (OD600 0.3–0.4) using the RNeasy kit from QIAGEN. In vivo labeling of nascent RNAs was carried out as previously described (Wade et al., 1964).
RNA-seq: Sample Preparation, Sequencing, and Data Analysis
Samples were amplified onto flowcells using an Illumina cBot and sequenced on an Illumina HiSeq2000 for 51 cycles according to the manufacturer’s protocols. Raw sequencing data were processed using the onboard SCS/RTA software, yielding 51 bp reads. Sequencing reads were processed using TopHat (Trapnell et al., 2009). Alignments reported from TopHat were processed by the Cufflinks software package (Trapnell et al., 2010) to determine differential expression of genes and transcripts between conditions. Expression values are reported as fragments per kilobase of gene per million mapped reads (FPKM). Data were visualized using the Integrated Genomics Viewer (Robinson et al., 2011).
Crystallization of the gp67/σA4 Complex
Crystals of the gp67/σA4 complex were grown under two different conditions at 22°C. Form I crystals grew from a crystallization solution of 0.1 M 2-ethane-sulfonic acid (MES), pH 6.5, 10% (w/v) polyethylene glycol (PEG) 5,000 monomethyl-ether, 20% 1-propanol. The crystals were soaked briefly in crystallization solution supplemented with 15% glycerol and then flash-frozen in liquid nitrogen. Form II crystals grew from a crystallization solution of 0.16 M Ca-acetate, 0.08 M Na-cacodylate, 15% (w/v) PEG 8,000, 20% glycerol. The crystals were flash-frozen in liquid nitrogen directly from the mother liquor. Crystals were formed using a 1:1 ratio of gp67/σA4 complex (10 mg/ml) and reservoir solution. Selenomethionyl-substituted protein was purified and crystallized under the same conditions.
Diffraction data were collected at the Advanced Photon Source (Argonne National Laboratory) beamline NE-CAT 24 ID and the National Synchrotron Light Source (Brookhaven National Laboratory) beamline X3A. Both structures were solved by single wavelength anomalous diffraction, and refined against the higher-resolution native data (Tables S1 and S2) to yield the final models.
DNase I Footprinting
DNase I footprinting was performed using standard methods.
Supplementary Material
Acknowledgments
We thank E.A. Campbell, R. Gourse, W. Ross, and P. Deighan for helpful discussions; K.R. Rajashankar and F. Murphy at APS NE-CAT beamline 24ID, and W. Shi at NSLS beamline X3A for support with synchrotron data collection; S. Wigneshweraraj for gifts of plasmids and helpful advice; and C. Zhao and S. Dewell of The Rockefeller University Genomics Resource Center for assistance with RNA-seq. We also thank all members of the Darst and Hochschild laboratories for helpful discussions. This work was based in part on research conducted at the APS and NSLS, supported by the Office of Basic Energy Sciences, U.S. Department of Energy. The NE-CAT beamlines at the APS are supported by award RR-15301 from the National Center for Research Resources, National Institutes of Health (NIH). This work was supported by NIH grants RO1 GM044025 to A.H. and RO1 GM053759 to S.A.D.
Footnotes
ACCESSION NUMBERS
The X-ray crystallographic coordinates and structure factor files have been deposited in the Protein Data Bank under ID Codes 4G6D (form I) and 4G94 (form II).
Supplemental Information includes Extended Experimental Procedures, five figures, and three tables and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2012.10.034.
References
- Barker MM, Gaal T, Gourse RL. Mechanism of regulation of transcription initiation by ppGpp. II Models for positive control based on properties of RNAP mutants and competition for RNAP. J Mol Biol. 2001;305:689–702. doi: 10.1006/jmbi.2000.4328. [DOI] [PubMed] [Google Scholar]
- Blatter EE, Ross W, Tang H, Gourse RL, Ebright RH. Domain organization of RNA polymerase alpha subunit: C-terminal 85 amino acids constitute a domain capable of dimerization and DNA binding. Cell. 1994;78:889–896. doi: 10.1016/s0092-8674(94)90682-3. [DOI] [PubMed] [Google Scholar]
- Browning DF, Busby SJ. The regulation of bacterial transcription initiation. Nat Rev Microbiol. 2004;2:57–65. doi: 10.1038/nrmicro787. [DOI] [PubMed] [Google Scholar]
- Brüssow H, Hendrix RW. Phage genomics: small is beautiful. Cell. 2002;108:13–16. doi: 10.1016/s0092-8674(01)00637-7. [DOI] [PubMed] [Google Scholar]
- Campbell EA, Muzzin O, Chlenov M, Sun JL, Olson CA, Weinman O, Trester-Zedlitz ML, Darst SA. Structure of the bacterial RNA polymerase promoter specificity sigma subunit. Mol Cell. 2002;9:527–539. doi: 10.1016/s1097-2765(02)00470-7. [DOI] [PubMed] [Google Scholar]
- Campbell EA, Greenwell R, Anthony JR, Wang S, Lim L, Das K, Sofia HJ, Donohue TJ, Darst SA. A conserved structural module regulates transcriptional responses to diverse stress signals in bacteria. Mol Cell. 2007;27:793–805. doi: 10.1016/j.molcel.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell EA, Westblade LF, Darst SA. Regulation of bacterial RNA polymerase sigma factor activity: a structural perspective. Curr Opin Microbiol. 2008;11:121–127. doi: 10.1016/j.mib.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrigan RM, Foster TJ. An improved tetracycline-inducible expression vector for Staphylococcus aureus. Plasmid. 2009;61:126–129. doi: 10.1016/j.plasmid.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Darst SA. Bacterial RNA polymerase. Curr Opin Struct Biol. 2001;11:155–162. doi: 10.1016/s0959-440x(00)00185-8. [DOI] [PubMed] [Google Scholar]
- Datta I, Sau S, Sil AK, Mandal NC. The bacteriophage lambda DNA replication protein P inhibits the oriC DNA- and ATP-binding functions of the DNA replication initiator protein DnaA of Escherichia coli. J Biochem Mol Biol. 2005;38:97–103. doi: 10.5483/bmbrep.2005.38.1.097. [DOI] [PubMed] [Google Scholar]
- Dehbi M, Moeck G, Arhin FF, Bauda P, Bergeron D, Kwan T, Liu J, McCarty J, Dubow M, Pelletier J. Inhibition of transcription in Staphylococcus aureus by a primary sigma factor-binding polypeptide from phage G1. J Bacteriol. 2009;191:3763–3771. doi: 10.1128/JB.00241-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deora R, Misra TK. Characterization of the primary sigma factor of Staphylococcus aureus. J Biol Chem. 1996;271:21828–21834. doi: 10.1074/jbc.271.36.21828. [DOI] [PubMed] [Google Scholar]
- Deutscher MP. Degradation of stable RNA in bacteria. J Biol Chem. 2003;278:45041–45044. doi: 10.1074/jbc.R300031200. [DOI] [PubMed] [Google Scholar]
- Dove SL, Hochschild A. A bacterial two-hybrid system based on transcription activation. Methods Mol Biol. 2004;261:231–246. doi: 10.1385/1-59259-762-9:231. [DOI] [PubMed] [Google Scholar]
- Estrem ST, Gaal T, Ross W, Gourse RL. Identification of an UP element consensus sequence for bacterial promoters. Proc Natl Acad Sci USA. 1998;95:9761–9766. doi: 10.1073/pnas.95.17.9761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrem ST, Ross W, Gaal T, Chen ZWS, Niu W, Ebright RH, Gourse RL. Bacterial promoter architecture: subsite structure of UP elements and interactions with the carboxy-terminal domain of the RNA polymerase alpha subunit. Genes Dev. 1999;13:2134–2147. doi: 10.1101/gad.13.16.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feklistov A, Darst SA. Structural basis for promoter-10 element recognition by the bacterial RNA polymerase σ subunit. Cell. 2011;147:1257–1269. doi: 10.1016/j.cell.2011.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feklistov A, Barinova N, Sevostyanova A, Heyduk E, Bass I, Vvedenskaya I, Kuznedelov K, Merkiene E, Stavrovskaya E, Klimasauskas S, et al. A basal promoter element recognized by free RNA polymerase sigma subunit determines promoter recognition by RNA polymerase holoenzyme. Mol Cell. 2006;23:97–107. doi: 10.1016/j.molcel.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Fischetti VA. Bacteriophage lysins as effective antibacterials. Curr Opin Microbiol. 2008;11:393–400. doi: 10.1016/j.mib.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox KR. DNase I footprinting. Methods Mol Biol. 1997;90:1–22. doi: 10.1385/0-89603-447-X:1. [DOI] [PubMed] [Google Scholar]
- Gourse RL, Gaal T, Bartlett MS, Appleman JA, Ross W. rRNA transcription and growth rate-dependent regulation of ribosome synthesis in Escherichia coli. Annu Rev Microbiol. 1996;50:645–677. doi: 10.1146/annurev.micro.50.1.645. [DOI] [PubMed] [Google Scholar]
- Gourse RL, Ross W, Gaal T. UPs and downs in bacterial transcription initiation: the role of the alpha subunit of RNA polymerase in promoter recognition. Mol Microbiol. 2000;37:687–695. doi: 10.1046/j.1365-2958.2000.01972.x. [DOI] [PubMed] [Google Scholar]
- Gruber TM, Gross CA. Multiple sigma subunits and the partitioning of bacterial transcription space. Annu Rev Microbiol. 2003;57:441–466. doi: 10.1146/annurev.micro.57.030502.090913. [DOI] [PubMed] [Google Scholar]
- Haugen SP, Ross W, Gourse RL. Advances in bacterial promoter recognition and its control by factors that do not bind DNA. Nat Rev Microbiol. 2008a;6:507–519. doi: 10.1038/nrmicro1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugen SP, Ross W, Manrique M, Gourse RL. Fine structure of the promoter-sigma region 1.2 interaction. Proc Natl Acad Sci USA. 2008b;105:3292–3297. doi: 10.1073/pnas.0709513105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton DM, Pande S, Wais N, Johnson XB, Vuthoori M, Makela A, Hook-Barnard I. Transcriptional takeover by sigma appropriation: remodeling of the sigma70 subunit of Escherichia coli RNA polymerase by the bacteriophage T4 activator MotA and co-activator AsiA. Microbiology. 2005;151:1729–1740. doi: 10.1099/mic.0.27972-0. [DOI] [PubMed] [Google Scholar]
- Howden BP, Davies JK, Johnson PD, Stinear TP, Grayson ML. Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: resistance mechanisms, laboratory detection, and clinical implications. Clin Microbiol Rev. 2010;23:99–139. doi: 10.1128/CMR.00042-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain D, Nickels BE, Sun L, Hochschild A, Darst SA. Structure of a ternary transcription activation complex. Mol Cell. 2004;13:45–53. doi: 10.1016/s1097-2765(03)00483-0. [DOI] [PubMed] [Google Scholar]
- Jain D, Kim Y, Maxwell KL, Beasley S, Zhang R, Gussin GN, Edwards AM, Darst SA. Crystal structure of bacteriophage λ cII and its DNA complex. Mol Cell. 2005;19:259–269. doi: 10.1016/j.molcel.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Kadesch TR, Rosenberg S, Chamberlin MJ. Binding of Escherichia coli RNA polymerase holoenzyme to bacteriophage T7 DNA. Measurements of binding at bacteriophage T7 promoter A1 using a template competition assay. J Mol Biol. 1982;155:1–29. doi: 10.1016/0022-2836(82)90489-2. [DOI] [PubMed] [Google Scholar]
- Keilty S, Rosenberg M. Constitutive function of a positively regulated promoter reveals new sequences essential for activity. J Biol Chem. 1987;262:6389–6395. [PubMed] [Google Scholar]
- Krásný L, Gourse RL. An alternative strategy for bacterial ribosome synthesis: Bacillus subtilis rRNA transcription regulation. EMBO J. 2004;23:4473–4483. doi: 10.1038/sj.emboj.7600423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznedelov K, Minakhin L, Niedziela-Majka A, Dove SL, Rogulja D, Nickels BE, Hochschild A, Heyduk T, Severinov K. A role for interaction of the RNA polymerase flap domain with the sigma subunit in promoter recognition. Science. 2002;295:855–857. doi: 10.1126/science.1066303. [DOI] [PubMed] [Google Scholar]
- Kwan T, Liu J, DuBow M, Gros P, Pelletier J. The complete genomes and proteomes of 27 Staphylococcus aureus bacteriophages. Proc Natl Acad Sci USA. 2005;102:5174–5179. doi: 10.1073/pnas.0501140102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert LJ, Wei Y, Schirf V, Demeler B, Werner MH. T4 AsiA blocks DNA recognition by remodeling sigma70 region 4. EMBO J. 2004;23:2952–2962. doi: 10.1038/sj.emboj.7600312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemke JJ, Sanchez-Vazquez P, Burgos HL, Hedberg G, Ross W, Gourse RL. Direct regulation of Escherichia coli ribosomal protein promoters by the transcription factors ppGpp and DksA. Proc Natl Acad Sci USA. 2011;108:5712–5717. doi: 10.1073/pnas.1019383108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Dehbi M, Moeck G, Arhin F, Bauda P, Bergeron D, Callejo M, Ferretti V, Ha N, Kwan T, et al. Antimicrobial drug discovery through bacteriophage genomics. Nat Biotechnol. 2004;22:185–191. doi: 10.1038/nbt932. [DOI] [PubMed] [Google Scholar]
- Lowy FD. Staphylococcus aureus infections. N Engl J Med. 1998;339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- Murakami KS, Darst SA. Bacterial RNA polymerases: the wholo story. Curr Opin Struct Biol. 2003;13:31–39. doi: 10.1016/s0959-440x(02)00005-2. [DOI] [PubMed] [Google Scholar]
- Murakami KS, Masuda S, Campbell EA, Muzzin O, Darst SA. Structural basis of transcription initiation: an RNA polymerase holoenzyme-DNA complex. Science. 2002a;296:1285–1290. doi: 10.1126/science.1069595. [DOI] [PubMed] [Google Scholar]
- Murakami KS, Masuda S, Darst SA. Structural basis of transcription initiation: RNA polymerase holoenzyme at 4 A resolution. Science. 2002b;296:1280–1284. doi: 10.1126/science.1069594. [DOI] [PubMed] [Google Scholar]
- Nechaev S, Severinov K. Bacteriophage-induced modifications of host RNA polymerase. Annu Rev Microbiol. 2003;57:301–322. doi: 10.1146/annurev.micro.57.030502.090942. [DOI] [PubMed] [Google Scholar]
- Nordmann P, Naas T, Fortineau N, Poirel L. Superbugs in the coming new decade; multidrug resistance and prospects for treatment of Staphylococcus aureus, Enterococcus spp. and Pseudomonas aeruginosa in 2010. Curr Opin Microbiol. 2007;10:436–440. doi: 10.1016/j.mib.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Ozoline ON, Tsyganov MA. Structure of open promoter complexes with Escherichia coli RNA polymerase as revealed by the DNase I footprinting technique: compilation analysis. Nucleic Acids Res. 1995;23:4533–4541. doi: 10.1093/nar/23.22.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patikoglou GA, Westblade LF, Campbell EA, Lamour V, Lane WJ, Darst SA. Crystal structure of the Escherichia coli regulator of sigma70, Rsd, in complex with sigma70 domain 4. J Mol Biol. 2007;372:649–659. doi: 10.1016/j.jmb.2007.06.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul BJ, Barker MM, Ross W, Schneider DA, Webb C, Foster JW, Gourse RL. DksA: a critical component of the transcription initiation machinery that potentiates the regulation of rRNA promoters by ppGpp and the initiating NTP. Cell. 2004;118:311–322. doi: 10.1016/j.cell.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Perederina A, Svetlov V, Vassylyeva MN, Tahirov TH, Yokoyama S, Art-simovitch I, Vassylyev DG. Regulation through the secondary channel—structural framework for ppGpp-DksA synergism during transcription. Cell. 2004;118:297–309. doi: 10.1016/j.cell.2004.06.030. [DOI] [PubMed] [Google Scholar]
- Ptashne M. A Genetic Switch, Phage Lambda and Higher Organisms. Cambridge: Cell Press; 1992. [Google Scholar]
- Rao L, Karls RK, Betley MJ. In vitro transcription of pathogenesis-related genes by purified RNA polymerase from Staphylococcus aureus. J Bacteriol. 1995;177:2609–2614. doi: 10.1128/jb.177.10.2609-2614.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes D, Andrey DO, Monod A, Kelley WL, Zhang G, Cheung AL. Coordinated regulation by AgrA, SarA, and SarR to control agr expression in Staphylococcus aureus. J Bacteriol. 2011;193:6020–6031. doi: 10.1128/JB.05436-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg S, Kadesch TR, Chamberlin MJ. Binding of Escherichia coli RNA polymerase holoenzyme to bacteriophage T7 DNA. Measurements of the rate of open complex formation at T7 promoter A. J Mol Biol. 1982;155:31–51. doi: 10.1016/0022-2836(82)90490-9. [DOI] [PubMed] [Google Scholar]
- Ross W, Gosink KK, Salomon J, Igarashi K, Zou C, Ishihama A, Severinov K, Gourse RL. A third recognition element in bacterial promoters: DNA binding by the alpha subunit of RNA polymerase. Science. 1993;262:1407–1413. doi: 10.1126/science.8248780. [DOI] [PubMed] [Google Scholar]
- Schenk S, Laddaga RA. Improved method for electroporation of Staphylococcus aureus. FEMS Microbiol Lett. 1992;73:133–138. doi: 10.1016/0378-1097(92)90596-g. [DOI] [PubMed] [Google Scholar]
- Shultzaberger RK, Chen Z, Lewis KA, Schneider TD. Anatomy of Escherichia coli sigma70 promoters. Nucleic Acids Res. 2007;35:771–788. doi: 10.1093/nar/gkl956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens A. Effect of salt on the transcription of T7 DNA by RNA polymerase from T4 phage-infected E.coli. Nucleic Acids Res. 1977;4:877–882. doi: 10.1093/nar/4.4.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiemann B, Depping R, Gineikiene E, Kaliniene L, Nivinskas R, Rüger W. ModA and ModB, two ADP-ribosyltransferases encoded by bacteriophage T4: catalytic properties and mutation analysis. J Bacteriol. 2004;186:7262–7272. doi: 10.1128/JB.186.21.7262-7272.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers AA. Conserved features of coordinately regulated E. coli promoters. Nucleic Acids Res. 1984;12:2605–2618. doi: 10.1093/nar/12.6.2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassylyev DG, Sekine S, Laptenko O, Lee J, Vassylyeva MN, Borukhov S, Yokoyama S. Crystal structure of a bacterial RNA polymerase holoenzyme at 2.6 A resolution. Nature. 2002;417:712–719. doi: 10.1038/nature752. [DOI] [PubMed] [Google Scholar]
- Wade HE, Lovett S, Robinson HK. The autodegradation of 32-P-labelled ribosomes from Escherichia coli. Biochem J. 1964;93:121–128. doi: 10.1042/bj0930121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu HH, Trawick JD, Haselbeck RJ, Forsyth RA, Yamamoto RT, Archer R, Patterson J, Allen M, Froelich JM, Taylor I, et al. Staphylococcus aureus TargetArray: comprehensive differential essential gene expression as a mechanistic tool to profile antibacterials. Antimicrob Agents Chemother. 2010;54:3659–3670. doi: 10.1128/AAC.00308-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano ST, Rothman-Denes LB. A phage-encoded inhibitor of Escherichia coli DNA replication targets the DNA polymerase clamp loader. Mol Microbiol. 2011;79:1325–1338. doi: 10.1111/j.1365-2958.2010.07526.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.