

Abstract
Numerous studies have reported that Vpr alters NF-κB signaling in various cell types, however, the findings have been largely conflicting with reports of both stimulatory and inhibitory effects of Vpr. Our aim was to investigate the role of Vpr signaling in myeloid cells using an adenovirus based expression and indicator system. Our results show that Vpr is inhibitory to NF-κB, however, this effect is dependant on the particular manner of NF-κB stimulation. Consistent with this notion, we report that Vpr has inhibitory effects that are specific to the TNF-α pathway, but not affecting the LPS pathway, suggesting that differential targets of Vpr may exist for NF-κB regulation. Further, we identify VprBP as one possible cellular component of Vpr’s regulation of IκBα in response to TNF-α stimulation. We did not identify such a role for HSP27, which instead seems to inhibit Vpr functions. Chronically HIV-1 infected U1 cells with knockdown constructs for Vpr were unexpectedly less responsive to TNF-α mediated viral replication, perhaps suggesting that other HIV-1 components may antagonize these anti-NF-κB effects in infected cells. We hypothesize that Vpr may serve an important role in the context of viral infection and immune function in vivo, through its selective inhibition of NF-κB pathways.
Introduction
Viral protein R (Vpr) is a 96 amino acid, 14 kDa protein which was originally isolated almost two decades ago (Cohen et al., 1990; Yuan et al., 1990) and is highly conserved in both human immunodeficiency virus (HIV)-1 and simian immunodeficiency virus (SIV) viruses (Emerman, 1996; Planelles et al., 1996; Tristem et al., 1992). Vpr molecular functions include nuclear import of viral pre-integration complex (PIC), induction of G2 cell cycle arrest, activation of apoptosis bystander cells, anti-apoptotic effects in infected cells, transcriptional co-activation of viral and host genes, and inhibition of nuclear factor kappa B (NF-κB), however, numerous controversies exist about these topics.
Many studies have shown that Vpr may suppress cellular immunity by modulating antigen mediated activation and cytotoxic killing of surviving T-cells, presumably, at least in part, by inhibiting NF-κB. Fore example, Vpr promotes T-helper (Th)2 cytokine interleukin (IL)-10 while suppressing the expression of Th1 cytokine IL-12 (Majumder et al., 2005; Mariani et al., 2001; Muthumani et al., 2004) by modulating NF-κB response. Vpr downregulates NF-κB inducible cytokines in a manner reversed with RU486 treatment, an antagonist for both glucocorticoid receptor (GR) and progesterone receptor (For review see: Mahajan and London, 1997), presumably suggesting that the inhibition of NF-κB via IκB induction mechanistically involves Vpr/GR interaction (Ayyavoo et al., 1997; Mirani et al., 2002; Muthumani et al., 2000). Indeed, Vpr and GR cooperate to suppress NF-κB mediated transcription by a mechanism involving the inhibition of Poly (ADP-ribose) polymerase (PARP)-1 nuclear trafficking in response tumor necrosis factor alpha (TNF-α) (Muthumani et al., 2006). It should be noted, however, that other reports have demonstrated opposite findings with regard to Vpr and NF-κB activity (Hoshino et al., 2010; Varin et al., 2005).
It is yet unknown why discrepant findings with regard to NF-κB activity are shown in different studies with Vpr. However, several binding partners of Vpr, which have been identified in previous reports, may mediate Vpr’s interactions with the NF-κB pathway. Vpr binding protein (VprBP) (Zhao et al., 1994) is an important cellular partner to Vpr and mediates binding of Vpr to ubiquitinated residues, promoting polyubiquitination of target proteins (Belzile et al., 2010), which may affect nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha (IκBα) (Schweitzer et al., 2007). Consequently, Vpr/VprBP binding mediates immune dysregulation (Richard et al., 2010; Ward et al., 2009), further suggesting that this pathway may be relevant for NF-κB suppression. Another cellular binding partner of Vpr, heat shock protein 27 (HSP27), has been shown to inhibit Vpr properties, including G2 arrest (Bukrinsky and Zhao, 2004). HSP27 is involved in NF-κB activation, presumably by binding IκBα via ubiquitin sites to promote degradation (Guo et al., 2009; Parcellier et al., 2003). In other contexts HSP27 can actually inhibit the NF-κB pathway (Carlson et al., 2007; Dodd et al., 2009). Therefore, it is possible that HSP27 may alter the effects of Vpr or allow Vpr’s stimulatory/inhibitory effects on the pathway.
In this report we evaluated the effect of Vpr derived from ectopic adenovirus mediated gene expression. The use of adenovirus allowed for a high rate of transduction while avoiding confounding variables from whole virus infection. Vpr is needed for infection of HIV-1 into macrophages, although this property may be overcome at higher concentrations of virus (For review see: Kogan and Rappaport, 2011). The presence of other viral proteins, however, also provides a challenge to investigation of Vpr’s effects on NF-κB in isolation, as these products may also impact the regulation of NF-κB in HIV-1 infected cells (Bour et al., 2001; Demarchi et al., 1996). We investigated the contribution of host function to Vpr’s effects, including cellular proteins HSP27 and VprBP by using small hairpin RNA (shRNA) constructs for these native transcripts. Finally, we evaluated the role of Vpr in a viral context as well by using shRNA for Vpr in latently infected U1 cells, however, in these experiments the role of Vpr on NF-κB and viral replication were difficult to separate completely.
Materials and Methods
Cell Culture
Peripheral blood mononuclear cells (PBMC) were isolated from heparinized whole blood or buffy coats acquired from healthy seronegative donors by density-gradient centrifugation (Histopaque-1017, Sigma-Aldrich, St. Louis, MO). PBMCs were incubated at a concentration of 4 × 106/mL in RPMI 1640 (Invitrogen, Carlsbad, CA) containing 10% Fetal bovine serum (FBS), 10% human AB serum, 2 mM L-glutamine and penicillin (50 U/ml)/streptomycin (50 ug/ml) and M-CSF (2 ng/mL) (R&D systems, Minneapolis, MN) overnight at 37°C. Non-adherent cells were removed and allowed to adhere for another 24 hours. Adherent cells were cultured in RPMI 1640 supplemented with 10% human AB serum, 2 mM L-glutamine, penicillin (50 U/ml)/streptomycin (50 ug/ml), and macrophage colony stimulating factor M-CSF (2 ng/mL) for an additional 4–6 days. Mono-Mac 1 cells are derived from peripheral monoblastic leukemia (M5a) (Sundstrom and Nilsson, 1976) and resemble mature monocytes (Collins, 1987; Tsuchiya et al., 1980). Mono-Mac 1 cells were cultivated in RPMI 1640 supplemented with 10% FBS, 2 mM L-glutamine, penicillin (50 U/ml)/streptomycin (50 ug/ml), 1x non-essential amino acids (Invitrogen) and 1 mM sodium pyruvate (Invitrogen). U937 cells are derived from pleural effusion from a patient with histiocytic lymphoma (Gendelman et al., 1988) and resemble promonocytic cells (Genois et al., 2000). U1 cells are U937 cells that were latently infected with HIV-1 due to a mutation in the Tat gene (Emiliani et al., 1998). Both cell lines were grown in the same RPMI media as the Mono-Mac 1 cells. The TMZ-BL cells were obtained from the NIH AIDS Research & Reference Reagent Program (8129). This cell line was generated from JC.53 cells by introducing separate integrated copies of the luciferase and β-galactosidase genes under control of the HIV-1 promoter. The TZM-BL cell line is highly sensitive to infection with diverse isolates of HIV-1 and can serve as an indicator for HIV-1 viral transcription activity. U87-MG cells were obtained from the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: U87MG cells from Dr. Bruce Chesebro. These are human astroglioma cells with an epithelial like morphology. U87-MG and TMZ-BL cells were grown in DMEM (Invitrogen) supplemented with 10% FBS, 2 mM L-glutamine, and penicillin (50 U/ml)/streptomycin (50 ug/ml). The compounds used for treatment of cells were as follows: recombinant human phorbol 12-myristate 13-acetate (PMA) (Sigma-Aldrich, 79346), TNF-α (R&D systems, 210-TA-010), LPS (Sigma-Aldrich, L-8274), RU486 (Sigma-Aldrich, M8046), puromycin (Sigma-Aldrich, P7255), and G418 (Sigma-Aldrich, G5013). Although 10ng/mL is a relatively high concentration of TNF-α concentrations of 5–10 ng/mL have been used by in many previous papers in this field during studies and macrophages were shown to make such concentrations in vitro (Ayyavoo et al., 1997; Muthumani et al., 2006).
Adenovirus and Lentivirus Constructs
AdNull and AdVpr constructs have been used in previously (Siddiqui et al., 2008) and have been described in detail (Deshmane et al., 2009). Ad-NF-kappa-B-Luc vector was purchased from (Vector Biolabs, Philadelphia, PA, 1740). These constructs were trasduced into U87-MG cells, Macrophages, or U937 cells using 5 moi of each virus for 42–72 hours. For co-culture experiments, we transduced 5 moi of either virus for 24 hours, and then washed off the media, trypsinized the cells, and mixed the two populations for promoter studies. In order to have one hundred percent infectivity on U37 cells, we first differentiated these cells using PMA (5 nM) for 24 hours, then for another 48 hours with new media. Lentiviruses from sigma were used to create stable U937/U1 cell lines expressing shRNA for HSP27, VprBP, or Vpr. The shNull (non-target, puromycin resistant, SHC002V), shHSP27 (1; Clone TRCN000008752, 2; TRCN000008753*), and shVprBP (1; TRCN0000129280, 2; TRCN0000129579*, 3; TRCN0000129831, 4; TRCN0000129909) were ordered from Sigma-Aldrich as pre-made lentivirus transduction particles (SHCLNV-NM_001540). To generate the shNull, shHSP27, and shVprBP cells, the cell lines were transduced with 1 moi of lentivirus and selected using puromycin (1 ug/mL) for at least one week. The shNT (non-target, neomycin resistant) and shVpr constructs were custom cloned by sigma into the pLKO.1-Neo vector. The Vpr sequences were based on oligonuclotide sequences 1, 2, and 3* (2 and 3 are specific for Vpr, 1 also cross reacts with Vif) that were used to knockdown Vpr in a previous publication (Balotta et al., 1993). To generate the shNT, or shVpr constructs cells were selected with G418 (1000 ug/mL) for one week. The double knockdown cells were generated by infecting the shNull, shHSP27, or shVprBP cells with shNT or shVpr followed by selection with G418. Knockdown was confirmed by real-time PCR. (* Used as in stable cell lines for remaining experiments)
Luciferase Assay
The luminescence was detected on an EnVision 2104 multilabel reader (Perkin Elmer) at 10 seconds post luciferin addition. The counts per second values were standardized for cell number using BCA protein values and then expressed as fold change relative to untreated cells for every individual promoter construct. We did not use renilla or other promoter constructs to standardize this data as these are driven by promoters that may potentially be influence by Vpr. Some data was combined as an average of at least three separate triplicates or experiments from three different donors. Two outliers were dropped from the luciferase assay using U937 shNull, shHSP27, and shVprBP cells (Fig. 4B) that were greatly different from the remaining eight luciferase values as these samples produced very high standard deviations (the trends were not altered).
Figure 4. Vpr mediated regulation of IκBα and NF-κB p65 signaling is altered by HSP27 and VprBP knockdown.
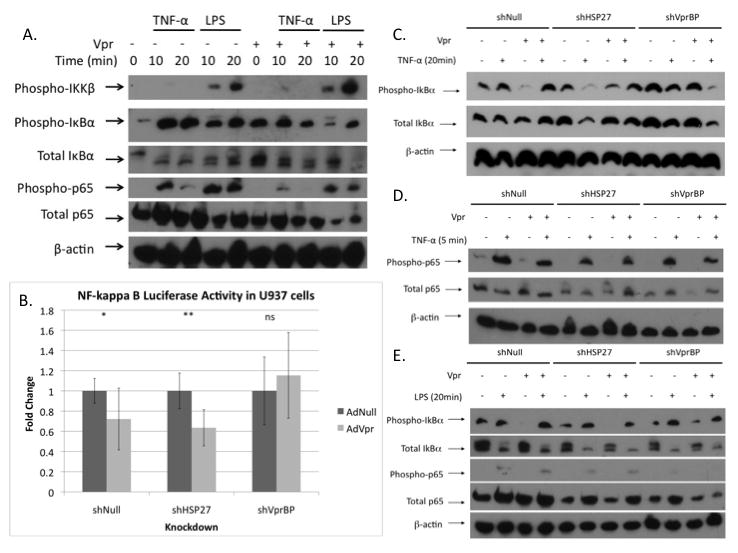
A. Effect of Vpr on NF-κB p65 phosphorylation, IκBα phosphorylation, and IκBα degradation in response to TNF-α (10 ng/mL) or LPS (50 ng/mL) treatment in PMA differentiated U937 cells. B. Induction of AdNF-κB luciferase activity in PMA differentiated shNull, shHSP27, or shVprBP cells transduced with 5 moi AdNull or AdVpr for one day and then treated with different concentrations for TNF-α (10 ng/mL) for 1 day (P= 0.039, P= 7.64×10−4, P= 0.41). Effect of Vpr on NF-κB p65 phosphorylation, IκBα phosphorylation, and IκBα degradation in response to 20 minutes of TNF-α (10 ng/mL) (C), 5 minutes of TNF-α (10 ng/mL) (D), or 20 minutes of LPS (50 ng/mL) (E) treatment in shNull, shHSP27, or shVprBP PMA differentiated U937 cells. (*P≤ .05, ** P≤ .001, ns: non-significant)
Real-Time PCR
RNA was harvested for 1×106 cells using the RT2 qPRC-Grade RNA isolation Kit (Qiagen, Duesseldorf, Germany, PA-001) and cDNA was made using the RT2 First Strand Kit (Qiagen, C-03). The assay was run using a Bio-Rad iCycler machine using 3 cycle PCR with a 50°C annealing temperature. The data was analyzed using the ΔΔCt method with primers for ribosomal L13a protein as an internal control. The primers used were as follows: VprBP F1; CGCTTTGCTTGGATGTCCTG, VprBP R1; GCTTTTAGGGTTCTGAGGCAGC, HSP27 F1; AGGAGCGGCAGGACGAGCAT, HSP27 R1; GCGACTCGAAGGTGACTGGG, Vpr F1; GACACTAGAGCTTTTAGAGG, Vpr R1; GGATAAACAGCAGTTGTTGCAG, Ribo F1; GAGGCCCCTACCACTTCC, Ribo R1; AACACCTTGAGACGGTCCAG.
Western Blot
Samples were run using Tris/Glycine buffer and transferred onto a hybond-P membrane (Amersham, GE, Fairfield, CT). Antibodies used: phospho-IκB kinase (IKK)α/β (Ser176/180) (Cell signaling, Beverly, MA, 2607, 1:1000), phospho-NF-κB p65 (Ser536) (Cell Signaling, 3033, 1:1000), NF-κB p65 (Cell signaling, C22B4, 1:1000), phospho-IκBα (Ser32) (Cell signaling 2859, 1:1000), total IκBα (Santa Cruz, Santa Cruz, CA, sc-847, 1:1000), PARP-1 ( (Santa Cruz, sc-8007, 1:1000), HSP27 (Cell Signaling, 2402, 1:1000), VprBP (abcam, ab75458, 1:1000), Lamin A (abcam, ab26300, 1:1000), β-actin (Sigma-Aldrich, A2228, 1:10,000), and α-Tubulin (Sigma-Aldrich, T6074, 1:1000). Vpr was detected using rabbit antiserum developed in our center (1:500). All antibodies were incubated overnight at 4°C in 5%BSA TTBS. The secondary antibodies mouse IgG-HPR (Santa Cruz, sc-2061, 1:10,000) or rabbit IgG-HPR (Santa Cruz, sc-2030, 1:10,000) were incubated for 1 hour at room temperature in 5% milk TTBS. The signal was detected using Super Signal West Pico Chemiluminescent Substrate (Thermo Scientific, 34080). Nuclear cytoplasmic fractions were obtained using the PE-NER nuclear and cytoplasmic extraction kit (Thermo Scientific, Waltham, MA 78833II). M-CSF ELISA was conducted performed using the Quantikine Human M-CSF Immunoassay Ka kit (R&D systems, SMC00) according to the manufactures protocols.
MTT and HIV p24 Assay
For toxicity experiments in U87-MG, Macrophage, and PMA differentiated U937 cells, cells were infected with 5 moi of adenovirus construct for 2–3 days. U1 cells were initially plated at 25 × 103 cells/96 well condition and Treated with TNF-α (.1–10 ng/mL) for 3 Days. 100uL of assayed using the NIH AIDS Research & Reference Reagent Program HIV-1 p24CA Antigen Capture Assay Kit according to the manufactures protocol. The cells were also evaluated for toxicity using an (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, a yellow tetrazole) (MTT) assay (Roche, Basel, Switzerland, 11465007001) and the p24 data was normalized to OD of the MTT assay. The optical density was read at 4 hours (Test: 550 nm, Reference: 630nm). A 2-way analysis of variance (ANOVA) was conducted for this experiment on the standardized p24/OD MTT values derived after treatment with TNF-α at 0, .1, 1, and 10 ng/ml for three days, with the data being converted to log units to assess relative differences in each condition. The three puromycin vector backbone cell lines (shNull shHSP27, and shVprBP), were defined as background, and the neomycin resistant vectors were (shNT, shVpr) were assessed as Vpr positive or negative in the analysis. Significant main effects of HSP27 background were followed up with Bonferroni corrected t-tests.
Results
In order to investigate Vpr’s effect on NF-κB signaling, we assessed the responsiveness of an adenovirus luciferase construct, which is driven by repeat NF-κB promoter sequences, to an adenovirus overexpressing Vpr in primary macrophages (Fig. 1A). In macrophages derived from five out of six donors, Vpr produced statistically significant inhibition of the NF-kappaB-Luc promoter. In donor three, however, Vpr produced a two-fold increase in NF-κB promoter activity (Data not shown). In three or more of these same donors we were also able to compare the responsiveness of NF-kappaB-Luc to TNF-α (.1–10 ng/mL) (Fig. 1B) and LPS (.5–50 ng/mL) (Fig. 1C) in the presence and absence of Vpr. While we observed significant inhibition of NF-kappB-Luc with Vpr almost every concentration of TNF-α, LPS treatment at or above 5 ng/ml showed no significant difference between the Vpr and Null conditions. As some studies have suggested that Vpr may be secreted from the cell (Levy et al., 1994), we used a co-culture model to assess the potential role of secreted Vpr on macrophages containing only NF-κB luciferase constructs by infecting the promoter and expression vectors into separate populations of cells and mixing these after stable infection (Data not shown). In the co-culture experiment, a likely primarily “extracellular” Vpr significantly promoted NF-κB activity at 0, 0.1 and 5 ng/mL concentrations of TNF-α, although some intracellular trafficking of the protein from the media into cells not directly expression the construct was also possible. Intriguingly, these experiments showed a complete reversal of the effects seen in the preceding experiments using cells co-expressing both promoter construct and expression vector for Vpr (Fig. 1A–1C), albeit the magnitudes of the effect in the co-culture experiment were mostly quite small (Data not shown).
Figure 1. Differential effects of Vpr on NF-kappa B activity in primary macrophages.
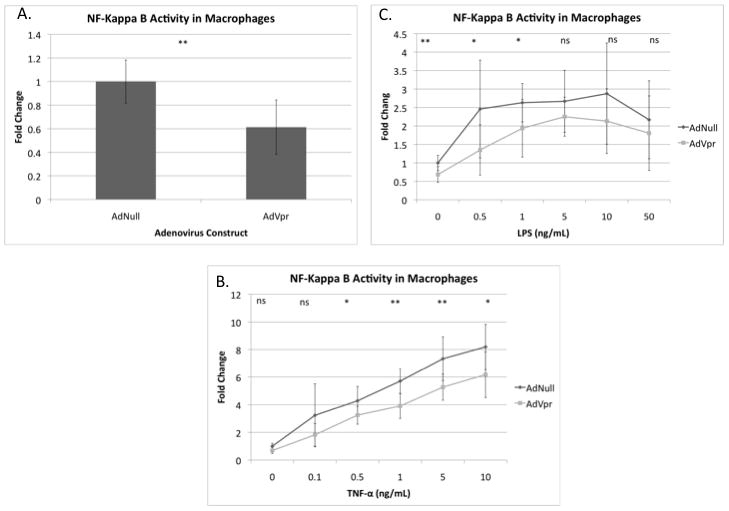
A. Primary macrophages from six separate donors were transduced with 5 moi of AdNull or AdVpr and 5 moi of AdNF-κB luciferase reporter construct for two days (Donor 3 excluded). (P= 7.9×10−8) Induction of AdNF-κB luciferase activity in primary macrophages transduced with 5 moi AdNull or AdVpr for one day and then treated with different concentrations for TNF-α for 1 day (B) or LPS for 1 day (C). Combined fold change in luciferase activity as compared to AdNull shown for donors four through six. P values were: P= 0.058, P= 0.11, P= 0.024, P= 5.9×10−4, P= 0.0052 P= 0.02, (B) and P= 1.6 × 10−5, P= 0.045, P= 0.043, P= 0.22, P= 0.19 P= 0.466 (C) respectively. Fold change in luciferase shown for donors three, five, and six. (* P≤ .05, ** P≤ .001, ns: non-significant)
As our results confirmed that Vpr exhibits inhibitory effects on NF-κB signaling, we investigated the mechanism involved in this phenomenon. Previous studies had demonstrated that Vpr binds to GR and recruits PARP-1 to cause depletion of this NF-κB co-factor from the nucleus (Muthumani et al., 2006). The report also showed that both the Vpr/GR/PARP-1 interaction and Vpr mediated NF-κB inhibition was disrupted with RU486 treatment. To test the relevance of this pathway in primary macrophages, we treated transduced macrophages with AdNull or AdVpr and four hours later treated them with media or RU486 (1 uM) for additional twenty-four hours, at which point the effect of treatment on response to THF-α was examined (Fig. 2A). Donor one was used for Figure 2A, which showed significant effects of VPR only in absence of additional TNF-α, however, we were not able to demonstrate these effect in macrophages or U87MG cells as 10ug/mL of TNF- α treatment likely saturates NF-κB making the effect of Vpr less significant at these concentrations. Vpr inhibited NF-kappaB-luc activity in the absence of TNF-α treatment and this effect was not reversed with RU486. This concentration of RU486 (1 uM), however, abrogated the effects of dexamethasone (.1 uM) on inhibition of NF-κB activity in primary macrophages in numerous other experiments in our lab looking at a variety of outcomes including promoter activity and phosphorylation of IκBα, excluding the possibility that the reagent simply did not work (Data not shown). Similarly, in TMZ-BL cells, which harbor an HIV-1 long terminal repeat (LTR) driven luciferase construct, AdVpr expression caused a reduction in luciferase activity, which was not reversed with RU486 co-treatment (Fig. 2B). Additionally, in TMZ-BL cells Vpr inhibited even TNF-α induced activation of the HIV-1 LTR, which was also not reversed with RU486 treatment in TMZ-BL cells. We next assessed the role of Vpr on IκBα response to TNF-α treatment in both macrophages (Fig. 2C) and PMA differentiated U937 cells (Fig. 2D). In both cell types, Vpr inhibited NF-κB signaling, as increased levels of IκBα, the inhibitor to NF-κB, were seen in the presence of TNF-α and Vpr, consistent with the observations in Figure 1. To assess the downstream consequences of Vpr expression on NF-κB signaling, we evaluated the nuclear/cytoplasmic trafficking of NF-κB, p65, and PARP-1, using differentiated U937 cells as a model system (Fig. 2E). Although expression of Vpr altered IκBα signaling in U937 cells (Fig. 2D), surprisingly there was no effect of Vpr on the nuclear/cytoplasmic trafficking in these same cells. Further, localization of PARP-1 was predominantly nuclear in these differentiated cells irrespective of Vpr expression; therefore, Vpr did not promote trafficking of PARP-1 to the cytoplasm. However, lower levels of nuclear PARP-1 were observed in nuclear extracts derived from cells expressing Vpr and treated with TNF-α as compared to Null/TNF-α conditions.
Figure 2. Inhibition of NF-kappa B by Vpr involves IκBα regulation but not PARP-1/GR signaling.
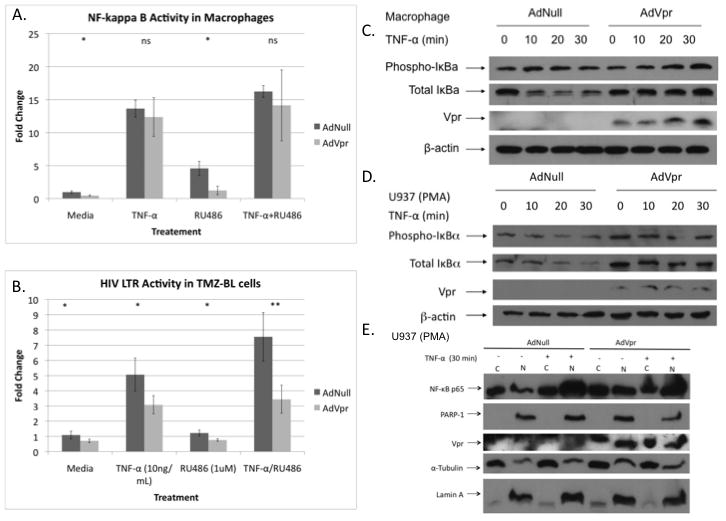
Macrophages from donor one (A) and TMZ-BL cells (B) were traduced with 5 moi of AdNull or AdVpr and 5moi of AdNF-κB luciferase reporter construct and after four hours with media or RU486 (1 uM) for one day. The cells were then treated for one additional day with TNF-α (10 ng/mL) and/or RU486 (1 uM). P values: P= 0.024, P= 0.55, P= 0.056, P= 0.57 (A) and P= 0.012, P= 0.005, P= 0.001, P= 5.86×10−4 (B) respectively. Effect of Vpr on IκBα phosphorylation and degradation in response to TNF-α (10 ng/mL) treatment in macropages (C) and PMA differentiated U937 cells. D. Effect of Vpr on PARP-1 and NF-κB translocation nuclear translocation in response to thirty minutes of TNF-α (10 ng/mL) treatment in PMA differentiated U937 cells. (* P≤ .05, ** P≤ .001, ns: non-significant)
We next compared the toxicity of Vpr expression in U87-MG cells, macrophages, and differentiated U937 cells (Fig. 3A). While Vpr was toxic in U87-MG cells, as reported previously (Siddiqui et al., 2008), there was no toxicity detected in macrophages or U937 cells, suggesting that the effects of Vpr on the NF-κB are independent of Vpr’s toxicity. We, therefore, focused our mechanistic studies on pathways that may be directly involved NF-κB signaling. As mentioned above, VprBP and HSP27 are likely to interact with both Vpr and the NF-κB intracellular signaling pathways. To investigate the role of Vpr and HSP27 on NF-κB signaling, we used shRNA lentivirus constructs to generate stable U1 and U937 cell lines knockdowns for these genes (Table 1). We screened two HSP27 and four VprBP shRNA constructs for gene knockdown in U1 cells by real-time PCR (S. Fig. 1A). We observed the greatest inhibition with HSP27 construct #2 and VprBP construct #4. However, shVprBP construct #4 consistently appeared to be toxic in U1 and U937 cells as we were not able to maintain these cells for more than one passage after selection with puromycin. As a result, we used HSP27 #2 and VprBP #2 for the remaining experiments. We then confirmed the expression level of HSP27 and VprBP in the three U937 knockdown cell lines with and without expression of Vpr (Fig. 3B–D). Expectedly, HSP27 knockdown greatly reduced the levels of the gene relative to shNull and shVprBP stable cell lines. Further, VprBP knockdown resulted reduction of the protein expression, but the reduction was less efficient than that of the shHSP27 construct. Surprisingly, there was a substantial, reproducible reduction of VprBP levels in the HSP27 knockdown conditions when Vpr was also co-expressed.
Figure 3. Toxicity of Vpr and generation of knockdown U1/U937 cells.
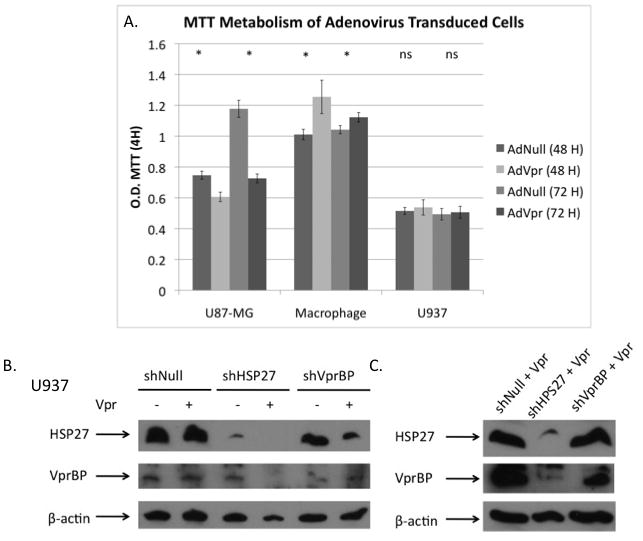
A. MTT values after 2 and 3 days of culture for U87-MG cells, macrophages, and PMA differentiated U937 cells that were transduceded with 5 moi of AdNull or AdVpr. (P= 0.004, P= 0.001, P= 0.049, P= 0.027, P= 0.16, P= 0.43). B–C. Levels of HSP27 and VprBP in AdNull and AdVpr transduced or AdVpr PMA differentiated U937 stable cell lines expressing shNull, shHSP27 #2 and shVprBP #2 shRNA.
Table 1.
Sequences used in lentivirus shRNA constructs.
| Name | Target Sequence |
|---|---|
| pLKO.1-Neo, available from Sigma-Aldrich | |
| shHSP27#1 | CCGGCCCAAGTTTCCTCCTCCCTGTCTCGAGACAGGGAGGAGGAAACTTGGGTTTTT |
| shHPS27#2 | CCGGCCGATGAGACTGCCGCCAAGTCTCGAGACTTGGCGGCAGTCTCATCGGTTTTT |
| shVprBP#1 | CCGGCGTATCGCTAATGGCATTGCACTCGAGTGCAATGCCATTAGCGATACGTTT |
| shVprBP#2 | CCGGCCTCCCATTCTTCTGCCTTTACTCGAGTAAAGGCAGAAGAATGGGAGGTTTTTTG |
| shVprBP#3 | CCGGCGAGAAACTGAGTCAAATGAACTCGAGTTCATTTGACTCAGTTTCTCGTTTTTTG |
| shVprBP#4 | CCGGGCGCCAATAAACTTTACGTCACTCGAGTGACGTAAAGTTTATTGGCGCTTTTTTG |
| pLKO.1-Neo vector, sequences from (Balotta, et al., 1993) | |
| shVpr#1 | TTCATTGTGTGGCTCCCTCTGTGGCCC |
| shVpr#2 | GCCCTAAGCCATGAAGCCATATCCTAG |
| shVpr#3 | TGGCTTCCACTCCTGCCCAAGTATCCC |
To localize the effect of Vpr on the NF-κB signaling pathway in differentiated U937 cells, we compared the effects of TNF-α (10 ng/mL) and LPS (50 ng/mL) on cells with and without Vpr (Fig. 4A). As in Figure 2D, Vpr expression resulted in increased IκBα levels in the presence of TNF-α treatment. Further, Vpr greatly inhibited the phosphorylation of NF-κB p65 in response to such treatment. However, in the response to LPS treatment, Vpr expression did not consistently increase IκBα levels while the inhibition of p65 phosphorylation was less than observed in the TNF-α treatment condition at all time points. These results suggested that Vpr has distinct intracellular effects that selectively inhibit the TNF-α but not LPS mediated induction of NF-κB in the studied presented, in apparent agreement with Figure 1B and 1C. Importantly, molecular findings independently confirm the effect of Vpr on inhibition of the NF- κB signaling in a system that does not rely on luciferase promoter activity readout. We then investigated the role of HSP27 and VprBP on Vpr’s alteration of NF-κB signaling. Expression of Vpr in U937 shHSP27 and shNull knockdown cell lines significantly decreased the NF-kappaB-Luc activity, whereas shVprBP-expressing cells had no observable effects of Vpr on luciferase activity (Fig. 4B), consistent with the idea that VprBP is an necessary component of Vpr’s inhibitory effects. Further, shVprBP/Vpr expressing cells showed lower levels of total IκBα after twenty minutes of TNF-α treatment, as would be expected if NF-κB activation proceeded in response to TNF-α, while shNull/Vpr and shHSP27/Vpr cells demonstrated higher or equal levels of IkBα after TNF-α stimulation (Fig. 4C). We assessed NF-κB p65 phosphorylation after five minutes of TNF-α treatment in these same cell lines and again demonstrated that Vpr inhibits p65 in Vpr/shNull cells (Fig. 4D). However, both shHSP27 and shVprBP cells had lower levels of NF-κB phosphorylation in response to TNF-α treatment as compared to shNull cells, making the apparent lack of effect from Vpr on this process difficult to interpret in these knockdown cells. In agreement with Figure 4A as well as the promoter experiments from macrophages in Figure 1B and 1C, twenty minutes of LPS treatment promoted IκBα degradation and NF-κB p65 phosphorylation in all three cell lines regardless of Vpr expression status, further suggesting that this process is TNF-α specific (Fig. 4E).
Considering that NF-κB is an important pathway for the activation of HIV-1 LTR (Lenardo and Baltimore, 1989; Poli et al., 1990), we next investigated the role of Vpr and NF-κB in the induction of HIV-1 expression using U1 cells, a latently infected cell line. We first generated a U1 cell line with a stable knockdown of Vpr and confirmed the reduction in RNA using real-time PCR (S. Fig. 1B). We then treated shNT and shVpr cells with TNF-α (.01–10 ng/mL) for three days (Fig. 5A). In light of our results from Figure 2B, we expected that in the absence of Vpr, less TNF-α stimulation would be required to stimulate HIV-1 production from latency, however, the opposite effect was observed. It remains unclear if the absence of Vpr in U1 cells influences NF-κB directly, viral replication directly, or both, but p24 production in response to TNF-α is certainly inhibited in the absence of Vpr in U1 cells. We compared the IκBα signaling in U937 and U1 cells in response to TNF-α treatment (Fig. 5B). Consistent with reports that HIV-1 infection increase NF-κB signaling (Chang et al., 1995), U1 cells displayed greater degradation of IκBα in response to TNF-α, preliminarily suggesting that any Vpr present in these cells may actually be less able to inhibit the pathway to a significant extent, perhaps due to the presence of other HIV-1 proteins that inhibit Vpr’s effects.
Figure 5. Effect of Vpr knockdown in U1 cells in induction of HIV-1 from latency.
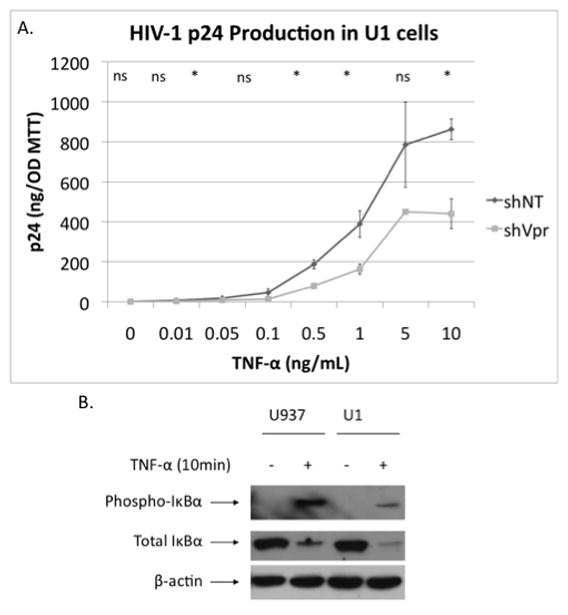
A. HIV p24 induction with TNF-α treatment of shNT and shVpr U1 cells. (P= 0.59, P= 0.17, P= 0.012, P= 0.097, P= 0.004, P= 0.017, P= 0.11, P= 0.002) B. Effect of 10 minutes of TNF-α (10 ng/mL) treatment on IκBα phosphorylation and IκBα degradation in U937 and U1 cells. (* P≤ .05, ** P≤ .001, ns: non-significant)
We also treated double knockdown U1 cell lines with TNF-α (.1–10 ng/mL) for three days. Again, the absence of Vpr (shVpr) significantly decreased virus production with TNF-α treatment at 0, 1 and 10, but not 0.1 ng/mL of TNF-α; however, there was no significant interaction of Vpr and background at any dose (Table 2). These data do not support a modification of Vpr’s effect by presence or absence of HSP27 or VprBP in an HIV-1 context, in contrast to the role of VprBP identified in NF-κB inhibition by Vpr in uninfected cells, but may support the idea that different mechanisms are at play in infected cells. The background was significant at 1 ng/ml of TNF-α treatment, with the shHSP27 background appearing to have the highest levels of virus production (Table 2). This experiment was one of three conducted, with all three experiments having shown generally similar trends (increased p24 production with shHSP27 background and decreased production with shVpr expression in all backgrounds). These effects differed in magnitude and significance at various doses of TNF-α between experiments.
Table 2.
2-way ANOVA analysis of the p24 production in U1 cells treated with TNF-α.
| TNF-α (ng/mL) | 0 | .1 | 1 | 10 |
|---|---|---|---|---|
| Background | .009 | .882 | <0.001 | <0.001 |
| Vpr | .009 | .515 | <0.001 | <0.001 |
| Background/Vpr | .731 | .200 | .413 | .700 |
Discussion
In this study we report that Vpr has inhibitory effects on NF-κB activation that are specific to the TNF-α pathway, but not the LPS pathway. This suggests that multiple targets of Vpr may exist for NF-κB regulation, which may serve different roles in the context of HIV-1 infection. Further, we identify VprBP as one possible cellular component of Vpr’s regulation of IκBα in response to TNF-α stimulation. We did not identify such a role for another of Vpr’s binding partners, HSP27, which instead seems to inhibit Vpr functions. Finally, our preliminary findings suggest NF-κB regulation by Vpr may not behave in the same manner when other HIV-1 components are present, as U1 knockdown cells for Vpr were unexpectedly less responsive to TNF-α stimulation of HIV transcription than those that had baseline Vpr expression levels. However, this last finding is mechanistically unconfirmed and may be due to effects of Vpr downstream from transcriptional regulation, for example viral replication or packaging.
Although the role of Vpr in NF-κB signaling, inflammation, viral transcription, and latency is well studied, there have been conflicting reports about these properties of Vpr. Indeed, studies using extracellular and serum derived Vpr showed that Vpr stimulates HIV-1 production (Levy et al., 1994; Levy et al., 1995). Consequently, viron derived Vpr (Roux et al., 2000) and synthetic Vpr (Varin et al., 2005) activates HIV-1 via NF-κB. Further, extracellular Vpr stimulates virus production via toll-like receptor 4 (TLR4), the receptor for lipopolysaccaride (LPS), by inducing NF-κB (Hoshino et al., 2010). Interestingly, HIV-1 infected macrophages and myeloid cells are well known to exhibit NF-κB activation (Choe et al., 2002; Roulston et al., 1995), HIV-1 infection and NF-κB activation form a positive feedback loop (Poli et al., 1990), and Tat is known to induce the HIV-1 LTR synergistically with NF-κB (Chang et al., 1995) indicating that this pathway is indeed important for viral transcription. Consistent with these reports, I kappa B alpha (IκBα) levels are known to be reduced in the presence of HIV-1 in myeloid cells (Asin et al., 1999). Our results in this study partly address these conflicting findings. We report that there are differences in the response of cells to TNF-α and LPS when Vpr is expressed: Vpr inhibits NF-κB in the presence of TNF-α but not with LPS treatment (Fig 1B, 1C). Our findings are generally consistent with the notion that Vpr inhibits NF-κB signaling within the cell (Muthumani et al., 2006). However, our experiments using a co-culture model suggest the possibility that Vpr is indeed capable of stimulating signaling extracellularly via TLR4 (LPS receptor) (Data not shown) as reported by others (Hoshino et al., 2010). The role of extracellular Vpr is a preliminary finding in our study, which is limited by numerous technical considerations including a lack of reliable methods to measure Vpr concentrations in the extracellular and intracellular samples from in vitro cultures. We hypothesize that HIV-1 is able to both suppress NF-κB to dampen immune response to virus while activating, or at very least not inhibiting, this same pathway in HIV-1 infected macrophages (discussed below).
While our study shows that Vpr clearly inhibits NF-κB, p65 nuclear/cytoplasmic trafficking was not altered in U937 cells (Fig. 2E). Further, the inhibitory effect on NF-κB signaling is quite small in U937 cells and in some macrophage donors, both in the presence and absence of TNF-α. The small changes in U937 cells may be due to altered NF-κB signaling in this cell line as it is common for this pathway to be involved in cancer transformation (Karin et al., 2002). Further, the incomplete inhibitory effects of Vpr on TNF-α mediated activation may also be explained in both macrophages and U937 cells due to the hypothetical stimulatory effects of Vpr via TLR4. Additionally, different donors may have altered susceptibility to intracellular/extracelluar effects of Vpr, if they are indeed both relevant in these cell lines. This is also one possible reason why donor 3 in Figure 1A showed increase in NF-κB activity, while other donors had inhibition of this pathway. We hypothesize that neutralization of “extracellular” Vpr with an antibody may potentiate the observed inhibition of the intracellular form and such an approach may overcome current technical limitation of Vpr quantification.
We were not able to confirm the previously described mechanism of NF-κB inhibition by Vpr due to PARP-1 exclusion to the cytoplasm (Muthumani et al., 2006). It should be noted that in contrast to the study mentioned, we failed to observe any difference in PARP-1 localization due to TNF-α treatment. It is possible that this is a mechanism that is more relevant to specific cell types, such as Hela cells there that were used in experiments by Muthumani and others. Although we did not identify this as an important aspect of Vpr regulation of NF-κB in macrophages, PARP-1 may be important in repression of the pathway in T-cells or other immune cells whose functions are disinhibited following RU486 treatment (Ayyavoo et al., 1997; Mirani et al., 2002; Muthumani et al., 2000). Further experiments are needed to discern exactly which mechanisms are at play in particular cell types.
VprBP is likely a key mediator for the inhibitory effects of Vpr on NF-κB signaling. Indeed, our results show that U937 cells that express VprBP shRNA have less inhibition of NF-κB activity (Fig. 4B) and have lower levels of IκBα with TNF-α treatment then control cells (Fig. 4C). Considering that Vpr does not affect the LPS pathway, we reason that Vpr/VprBP interaction leads to inhibition of the TNF-α specific pathway by binding to some member of the cascade upstream of IKK. However, as a consequence of Vpr/VprBP function, the degradation of another target that regulates the NF-κB pathway is also possible. Therefore, we reason that Vpr may cause inhibition of the TNF-α/NF-κB signaling directly by recruiting a member of the signaling cascade to the VprBP ubiquitination complex, or indirectly by depleting other necessary cellular genes (Fig. 6).
Figure 6. Hypothesis of Vpr function in myeloid cells.
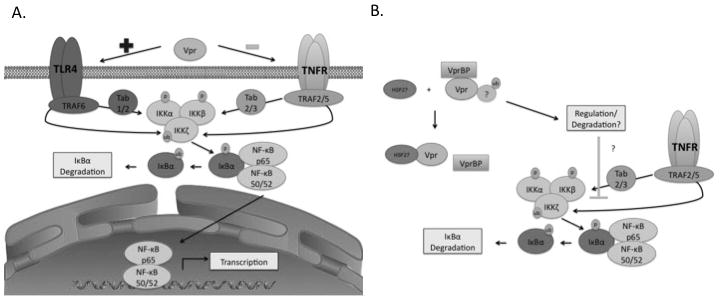
A. Vpr stimulates LPS pathways but inhibits TNF-α pathways. B. Vpr/VprBP interaction inhibits TNF-α/LPS signaling. HSP27 may disrupt the Vpr/VprBP interaction.
HSP27 knockdown did not appear to influence the role of Vpr on NF-κB (Fig. 4B-C). If anything, HSP27 knockdown seemed to potentiate the inhibitory effects of Vpr on the TNF-α/NF-κB pathway, consistent with the reports that have characterized HSP27 as an inhibitor of Vpr (Liang et al., 2007). Interestingly, VprBP seemed to be depleted in cells that lack HSP27 and express Vpr (Fig. 3B–C). This may suggest in the absence of the endogenous inhibitor for Vpr, increased Vpr/VprBP binding leads to destabilization of the complex, however, this needs to be confirmed by future studies. Finally, HSP27 knockdown seemed to produce higher levels of HIV p24 in U1 cells, however, shNull, shVprBP, and shHSP27 backgrounds were all affected by shVpr expression (Table 2). This further suggests a protective, anti-HIV-1 role for HSP27, as has been suggested in previous reports.
Our observed effects of Vpr in U1 cells on HIV-1 activation from latency were inconsistent with the findings that Vpr inhibited NF-κB in uninfected parental U937 cells (Fig. 4, Fig. 5, Table 2). As it is known that Vpr inhibition results in decreased HIV-1 replication (Balotta et al., 1993) and that Tat has been shown to induce NF-κB activity by promoting IκBα degradation (Demarchi et al., 1996), it possible that Vpr serves a different role in HIV-1 infected cells due to inhibition of on the effects on NF-κB signaling from other HIV-1 proteins such as Tat. It should be noted that U1 cells have a mutation in Tat that reduces transcription of the HIV-1 promoter (Emiliani et al., 1998), however, if Tat is indeed responsible for the de-inhibition of NF-κB, the mutation carried by the U1 cells may not abrogate this effect. Alternatively, a different HIV-1 gene may be involved. Consistent with this notion, U1 cells had greater degradation of IκBα with TNF-α treatment (Fig. 5B), however, direct effects of Vpr on HIV-1 transcription or packaging cannot be excluded and future studies are necessary to decipher these mechanisms.
Although our results do not point to one specific function of Vpr in NF-κB regulation, the roles of NF-κB as well as Vpr in viral replication have multiple, conflicting descriptions in the field. Vpr inhibits immune responses by in a mechanism that is at least partly dependent on NF-κB inactivation (Ayyavoo et al., 1997), but also activates HIV-1 from latency and is important for viral replication (Balotta et al., 1993; Chang et al., 1995; Hoshino et al., 2010; Levy et al., 1994; Levy et al., 1995; Roux et al., 2000; Varin et al., 2005). It has not been adequately addressed how Vpr is able to stimulate viral replication while simultaneously inhibiting NF-κB signaling in the host. In our study we show clear inhibitory effects of Vpr on TNF-α/NF-κB signaling of uninfected cells TMZ-BL cells (Fig. 2B), U937 cells (Fig. 4B), and macrophages (Fig 1A–1C), while sparing LPS/NF-κB activity. Consequently, if the proposed extracellular Vpr is indeed capable of stimulating the TLR4 receptor, this may produce selective activation of particular cells in vivo. A recent report has shown that TLR4 expression, while not normal for T-cells, is induced by HIV-1 on CD4+ and CD8+ T-cells and that this expression decreases as patients progress to AIDS (Miller Sanders et al., 2010). Further, HIV-1 infected macrophages have been reported to possess a Type 1 phenotype that is more responsive to LPS (Brown et al., 2008). Therefore, we believe that the differential regulation of the NF-κB pathway by Vpr may promote activation of virus replication in macrophages and perhaps infected T-cells, but inhibition of NF-κB in other bystander cells, ultimately suppression of the immune response as reported in previous work without preventing viral replication (Ayyavoo et al., 1997; Ayyavoo et al., 2002; Muthumani et al., 2002). However, the potential selectivity of the inhibitory effects for bystander cells warrants extensive further investigation.
In conclusion, this study is a case in point that Vpr/NF-κB signaling is a complex phenomenon that likely involves multiple targets. The NF-κB pathway is a particular challenge for HIV-1 virus, being both necessary for viral growth and the driver of immune response; therefore, it is intuitive that regulation of such signaling on the part of HIV-1 would require cell or context specific mechanisms. Future studies are needed to address the role of Vpr in T-cells versus macrophages, the role of intracellular or extracellular Vpr, as well as Vpr’s effects in infected versus uninfected cells. Vpr’s alteration of NF-κB signaling is very likely relevant for HIV-1 persistence and immune evasion, therefore, research in to these mechanisms may provide avenues for future therapy.
Acknowledgments
NIH Grant Support:
This work was supported by an NIH Grant:
RO1MH090910 to Jay Rappaport
“This investigation was supported by the National Institutes of Health under Ruth L. Kirschstein National Research Service Award (1T32MH079785) providing support to MK, as well as RO1 grant support to JR from NINDS and NIMH. We would also like to especially acknowledge Dr. Wenhui Hu, Dr. Tracy Fischer-Smith, Dr. Dianne Langford and Dr. Brian Wigdahl for helpful discussion on this study.
References
- Asin S, Taylor JA, Trushin S, Bren G, Paya CV. Ikappakappa mediates NF-kappaB activation in human immunodeficiency virus-infected cells. J Virol. 1999;73(5):3893–3903. doi: 10.1128/jvi.73.5.3893-3903.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyavoo V, Mahboubi A, Mahalingam S, Ramalingam R, Kudchodkar S, Williams WV, Green DR, Weiner DB. HIV-1 Vpr suppresses immune activation and apoptosis through regulation of nuclear factor kappa B. Nat Med. 1997;3(10):1117–1123. doi: 10.1038/nm1097-1117. [DOI] [PubMed] [Google Scholar]
- Ayyavoo V, Muthumani K, Kudchodkar S, Zhang D, Ramanathan P, Dayes NS, Kim JJ, Sin JI, Montaner LJ, Weiner DB. HIV-1 viral protein R compromises cellular immune function in vivo. Int Immunol. 2002;14(1):13–22. doi: 10.1093/intimm/14.1.13. [DOI] [PubMed] [Google Scholar]
- Balotta C, Lusso P, Crowley R, Gallo RC, Franchini G. Antisense phosphorothioate oligodeoxynucleotides targeted to the vpr gene inhibit human immunodeficiency virus type 1 replication in primary human macrophages. J Virol. 1993;67(7):4409–4414. doi: 10.1128/jvi.67.7.4409-4414.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belzile JP, Richard J, Rougeau N, Xiao Y, Cohen EA. HIV-1 Vpr induces the K48-linked polyubiquitination and proteasomal degradation of target cellular proteins to activate ATR and promote G2 arrest. J Virol. 2010;84(7):3320–3330. doi: 10.1128/JVI.02590-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bour S, Perrin C, Akari H, Strebel K. The human immunodeficiency virus type 1 Vpu protein inhibits NF-kappa B activation by interfering with beta TrCP-mediated degradation of Ikappa B. J Biol Chem. 2001;276(19):15920–15928. doi: 10.1074/jbc.M010533200. [DOI] [PubMed] [Google Scholar]
- Brown JN, Kohler JJ, Coberley CR, Sleasman JW, Goodenow MM. HIV-1 activates macrophages independent of Toll-like receptors. PLoS One. 2008;3(12):e3664. doi: 10.1371/journal.pone.0003664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukrinsky M, Zhao Y. Heat-shock proteins reverse the G2 arrest caused by HIV-1 viral protein R. DNA Cell Biol. 2004;23(4):223–225. doi: 10.1089/104454904773819806. [DOI] [PubMed] [Google Scholar]
- Carlson R, Vavricka S, Eloranta J, Musch M, Arvans D, Kles K, Walsh-Reitz M, Kullak-Ublick G, Chang E. fMLP induces Hsp27 expression, attenuates NF-kappaB activation, and confers intestinal epithelial cell protection. Am J Physiol Gastrointest Liver Physiol. 2007;292(4):G1070–1078. doi: 10.1152/ajpgi.00417.2006. [DOI] [PubMed] [Google Scholar]
- Chang HK, Gallo RC, Ensoli B. Regulation of Cellular Gene Expression and Function by the Human Immunodeficiency Virus Type 1 Tat Protein. J Biomed Sci. 1995;2(3):189–202. doi: 10.1007/BF02253380. [DOI] [PubMed] [Google Scholar]
- Choe W, Volsky DJ, Potash MJ. Activation of NF-kappaB by R5 and X4 human immunodeficiency virus type 1 induces macrophage inflammatory protein 1alpha and tumor necrosis factor alpha in macrophages. J Virol. 2002;76(10):5274–5277. doi: 10.1128/JVI.76.10.5274-5277.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen EA, Terwilliger EF, Jalinoos Y, Proulx J, Sodroski JG, Haseltine WA. Identification of HIV-1 vpr product and function. J Acquir Immune Defic Syndr. 1990;3(1):11–18. [PubMed] [Google Scholar]
- Collins SJ. The HL-60 promyelocytic leukemia cell line: proliferation, differentiation, and cellular oncogene expression. Blood. 1987;70(5):1233–1244. [PubMed] [Google Scholar]
- Demarchi F, d’Adda di Fagagna F, Falaschi A, Giacca M. Activation of transcription factor NF-kappaB by the Tat protein of human immunodeficiency virus type 1. J Virol. 1996;70(7):4427–4437. doi: 10.1128/jvi.70.7.4427-4437.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmane SL, Mukerjee R, Fan S, Del Valle L, Michiels C, Sweet T, Rom I, Khalili K, Rappaport J, Amini S, Sawaya BE. Activation of the oxidative stress pathway by HIV-1 Vpr leads to induction of hypoxia-inducible factor 1alpha expression. J Biol Chem. 2009;284(17):11364–11373. doi: 10.1074/jbc.M809266200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd S, Hain B, Senf S, Judge A. Hsp27 inhibits IKKbeta-induced NF-kappaB activity and skeletal muscle atrophy. FASEB J. 2009;23(10):3415–3423. doi: 10.1096/fj.08-124602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerman M. HIV-1, Vpr and the cell cycle. Curr Biol. 1996;6(9):1096–1103. doi: 10.1016/s0960-9822(02)00676-0. [DOI] [PubMed] [Google Scholar]
- Emiliani S, Fischle W, Ott M, Van Lint C, Amella CA, Verdin E. Mutations in the tat gene are responsible for human immunodeficiency virus type 1 postintegration latency in the U1 cell line. J Virol. 1998;72(2):1666–1670. doi: 10.1128/jvi.72.2.1666-1670.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendelman HE, Orenstein JM, Martin MA, Ferrua C, Mitra R, Phipps T, Wahl LA, Lane HC, Fauci AS, Burke DS, et al. Efficient isolation and propagation of human immunodeficiency virus on recombinant colony-stimulating factor 1-treated monocytes. J Exp Med. 1988;167(4):1428–1441. doi: 10.1084/jem.167.4.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genois N, Robichaud GA, Tremblay MJ. Mono Mac 1: a new in vitro model system to study HIV-1 infection in human cells of the mononuclear phagocyte series. J Leukoc Biol. 2000;68(6):854–864. [PubMed] [Google Scholar]
- Guo K, Kang N, Li Y, Sun L, Gan L, Cui F, Gao M, Liu K. Regulation of HSP27 on NF-kappaB pathway activation may be involved in metastatic hepatocellular carcinoma cells apoptosis. BMC Cancer. 2009;9:100. doi: 10.1186/1471-2407-9-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino S, Konishi M, Mori M, Shimura M, Nishitani C, Kuroki Y, Koyanagi Y, Kano S, Itabe H, Ishizaka Y. HIV-1 Vpr induces TLR4/MyD88-mediated IL-6 production and reactivates viral production from latency. J Leukoc Biol. 2010;87(6):1133–1143. doi: 10.1189/jlb.0809547. [DOI] [PubMed] [Google Scholar]
- Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2(4):301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- Kogan M, Rappaport J. HIV-1 accessory protein Vpr: relevance in the pathogenesis of HIV and potential for therapeutic intervention. Retrovirology. 2011;8:25. doi: 10.1186/1742-4690-8-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenardo MJ, Baltimore D. NF-kappa B: a pleiotropic mediator of inducible and tissue-specific gene control. Cell. 1989;58(2):227–229. doi: 10.1016/0092-8674(89)90833-7. [DOI] [PubMed] [Google Scholar]
- Levy DN, Refaeli Y, MacGregor RR, Weiner DB. Serum Vpr regulates productive infection and latency of human immunodeficiency virus type 1. Proc Natl Acad Sci U S A. 1994;91(23):10873–10877. doi: 10.1073/pnas.91.23.10873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DN, Refaeli Y, Weiner DB. Extracellular Vpr protein increases cellular permissiveness to human immunodeficiency virus replication and reactivates virus from latency. J Virol. 1995;69(2):1243–1252. doi: 10.1128/jvi.69.2.1243-1252.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang D, Benko Z, Agbottah E, Bukrinsky M, Zhao RY. Anti-vpr activities of heat shock protein 27. Mol Med. 2007;13(5–6):229–239. doi: 10.2119/2007-00004.Liang. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan DK, London SN. Mifepristone (RU486): a review. Fertil Steril. 1997;68(6):967–976. doi: 10.1016/s0015-0282(97)00189-1. [DOI] [PubMed] [Google Scholar]
- Majumder B, Janket ML, Schafer EA, Schaubert K, Huang XL, Kan-Mitchell J, Rinaldo CR, Jr, Ayyavoo V. Human immunodeficiency virus type 1 Vpr impairs dendritic cell maturation and T-cell activation: implications for viral immune escape. J Virol. 2005;79(13):7990–8003. doi: 10.1128/JVI.79.13.7990-8003.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani R, Rasala BA, Rutter G, Wiegers K, Brandt SM, Krausslich HG, Landau NR. Mouse-human heterokaryons support efficient human immunodeficiency virus type 1 assembly. J Virol. 2001;75(7):3141–3151. doi: 10.1128/JVI.75.7.3141-3151.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller Sanders C, Cruse JM, Lewis RE. Toll-like receptor and chemokine receptor expression in HIV-infected T lymphocyte subsets. Exp Mol Pathol. 2010;88(1):26–31. doi: 10.1016/j.yexmp.2009.09.006. [DOI] [PubMed] [Google Scholar]
- Mirani M, Elenkov I, Volpi S, Hiroi N, Chrousos GP, Kino T. HIV-1 protein Vpr suppresses IL-12 production from human monocytes by enhancing glucocorticoid action: potential implications of Vpr coactivator activity for the innate and cellular immunity deficits observed in HIV-1 infection. J Immunol. 2002;169(11):6361–6368. doi: 10.4049/jimmunol.169.11.6361. [DOI] [PubMed] [Google Scholar]
- Muthumani K, Bagarazzi M, Conway D, Hwang DS, Ayyavoo V, Zhang D, Manson K, Kim J, Boyer J, Weiner DB. Inclusion of Vpr accessory gene in a plasmid vaccine cocktail markedly reduces Nef vaccine effectiveness in vivo resulting in CD4 cell loss and increased viral loads in rhesus macaques. J Med Primatol. 2002;31(4–5):179–185. doi: 10.1034/j.1600-0684.2002.02004.x. [DOI] [PubMed] [Google Scholar]
- Muthumani K, Choo AY, Zong WX, Madesh M, Hwang DS, Premkumar A, Thieu KP, Emmanuel J, Kumar S, Thompson CB, Weiner DB. The HIV-1 Vpr and glucocorticoid receptor complex is a gain-of-function interaction that prevents the nuclear localization of PARP-1. Nat Cell Biol. 2006;8(2):170–179. doi: 10.1038/ncb1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthumani K, Desai BM, Hwang DS, Choo AY, Laddy DJ, Thieu KP, Rao RG, Weiner DB. HIV-1 Vpr and anti-inflammatory activity. DNA Cell Biol. 2004;23(4):239–247. doi: 10.1089/104454904773819824. [DOI] [PubMed] [Google Scholar]
- Muthumani K, Kudchodkar S, Papasavvas E, Montaner LJ, Weiner DB, Ayyavoo V. HIV-1 Vpr regulates expression of beta chemokines in human primary lymphocytes and macrophages. J Leukoc Biol. 2000;68(3):366–372. [PubMed] [Google Scholar]
- Parcellier A, Schmitt E, Gurbuxani S, Seigneurin-Berny D, Pance A, Chantôme A, Plenchette S, Khochbin S, Solary E, Garrido C. HSP27 is a ubiquitin-binding protein involved in I-kappaBalpha proteasomal degradation. Mol Cell Biol. 2003;23(16):5790–5802. doi: 10.1128/MCB.23.16.5790-5802.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planelles V, Jowett JB, Li QX, Xie Y, Hahn B, Chen IS. Vpr-induced cell cycle arrest is conserved among primate lentiviruses. J Virol. 1996;70(4):2516–2524. doi: 10.1128/jvi.70.4.2516-2524.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli G, Kinter A, Justement JS, Kehrl JH, Bressler P, Stanley S, Fauci AS. Tumor necrosis factor alpha functions in an autocrine manner in the induction of human immunodeficiency virus expression. Proc Natl Acad Sci U S A. 1990;87(2):782–785. doi: 10.1073/pnas.87.2.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J, Sindhu S, Pham TN, Belzile JP, Cohen EA. HIV-1 Vpr up-regulates expression of ligands for the activating NKG2D receptor and promotes NK cell-mediated killing. Blood. 2010;115(7):1354–1363. doi: 10.1182/blood-2009-08-237370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulston A, Lin R, Beauparlant P, Wainberg MA, Hiscott J. Regulation of human immunodeficiency virus type 1 and cytokine gene expression in myeloid cells by NF-kappa B/Rel transcription factors. Microbiol Rev. 1995;59(3):481–505. doi: 10.1128/mr.59.3.481-505.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux P, Alfieri C, Hrimech M, Cohen E, Tanner J. Activation of transcription factors NF-kappaB and NF-IL-6 by human immunodeficiency virus type 1 protein R (Vpr) induces interleukin-8 expression. J Virol. 2000;74(10):4658–4665. doi: 10.1128/jvi.74.10.4658-4665.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer K, Bozko P, Dubiel W, Naumann M. CSN controls NF-kappaB by deubiquitinylation of IkappaBalpha. EMBO J. 2007;26(6):1532–1541. doi: 10.1038/sj.emboj.7601600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui K, Del Valle L, Morellet N, Cui J, Ghafouri M, Mukerjee R, Urbanska K, Fan S, Pattillo CB, Deshmane SL, Kiani MF, Ansari R, Khalili K, Roques BP, Reiss K, Bouaziz S, Amini S, Srinivasan A, Sawaya BE. Molecular mimicry in inducing DNA damage between HIV-1 Vpr and the anticancer agent, cisplatin. Oncogene. 2008;27(1):32–43. doi: 10.1038/sj.onc.1210632. [DOI] [PubMed] [Google Scholar]
- Sundstrom C, Nilsson K. Establishment and characterization of a human histiocytic lymphoma cell line (U-937) Int J Cancer. 1976;17(5):565–577. doi: 10.1002/ijc.2910170504. [DOI] [PubMed] [Google Scholar]
- Tristem M, Marshall C, Karpas A, Hill F. Evolution of the primate lentiviruses: evidence from vpx and vpr. Embo J. 1992;11(9):3405–3412. doi: 10.1002/j.1460-2075.1992.tb05419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya S, Yamabe M, Yamaguchi Y, Kobayashi Y, Konno T, Tada K. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1) Int J Cancer. 1980;26(2):171–176. doi: 10.1002/ijc.2910260208. [DOI] [PubMed] [Google Scholar]
- Varin A, Decrion A, Sabbah E, Quivy V, Sire J, Van Lint C, Roques B, Aggarwal B, Herbein G. Synthetic Vpr protein activates activator protein-1, c-Jun N-terminal kinase, and NF-kappaB and stimulates HIV-1 transcription in promonocytic cells and primary macrophages. J Biol Chem. 2005;280(52):42557–42567. doi: 10.1074/jbc.M502211200. [DOI] [PubMed] [Google Scholar]
- Ward J, Davis Z, DeHart J, Zimmerman E, Bosque A, Brunetta E, Mavilio D, Planelles V, Barker E. HIV-1 Vpr triggers natural killer cell-mediated lysis of infected cells through activation of the ATR-mediated DNA damage response. PLoS Pathog. 2009;5(10):e1000613. doi: 10.1371/journal.ppat.1000613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, Matsuda Z, Matsuda M, Essex M, Lee TH. Human immunodeficiency virus vpr gene encodes a virion-associated protein. AIDS Res Hum Retroviruses. 1990;6(11):1265–1271. doi: 10.1089/aid.1990.6.1265. [DOI] [PubMed] [Google Scholar]
- Zhao LJ, Mukherjee S, Narayan O. Biochemical mechanism of HIV-I Vpr function. Specific interaction with a cellular protein. J Biol Chem. 1994;269(22):15577–15582. [PubMed] [Google Scholar]