

ABSTRACT
Streptococcus canis is a zoonotic pathogen capable of causing serious invasive diseases in domestic animals and humans. Surface-exposed M proteins and metabolic enzymes have been characterized as major virulence determinants in various streptococcal species. Recently, we have identified SCM, the M-like protein of S. canis, as the major receptor for miniplasminogen localized on the bacterial surface. The present study now characterizes the glycolytic enzyme enolase as an additional surface-exposed plasminogen-binding protein. According to its zoonotic properties, purified S. canis enolase binds to both human and canine plasminogen and facilitates degradation of aggregated fibrin matrices after activation with host-derived urokinase-type plasminogen activator (uPA). Unlike SCM, which binds to the C terminus of human plasminogen, the S. canis enolase interacts N terminally with the first four kringle domains of plasminogen, representing angiostatin. Radioactive binding analyses confirmed cooperative plasminogen recruitment to both surface-exposed enolase and SCM. Furthermore, despite the lack of surface protease activity via SpeB in S. canis, SCM is released and reassociated homophilically to surface-anchored SCM and heterophilically to surface-bound plasminogen. In addition to plasminogen-mediated antiphagocytic activity, reassociation of SCM to the bacterial surface significantly enhanced bacterial survival in phagocytosis analyses using human neutrophils.
IMPORTANCE
Streptococcal infections are a major issue in medical microbiology due to the increasing spread of antibiotic resistances and the limited availability of efficient vaccines. Surface-exposed glycolytic enzymes and M proteins have been characterized as major virulence factors mediating pathogen-host interaction. Since streptococcal infection mechanisms exert a subset of multicombinatorial processes, the investigation of synergistic activities mediated via different virulence factors has become a high priority. Our data clearly demonstrate that plasminogen recruitment to the Streptococcus canis surface via SCM and enolase in combination with SCM reassociation enhances bacterial survival by protecting against phagocytic killing. These data propose a new cooperative mechanism for prevention of phagocytic killing based on the synergistic activity of homophilic and heterophilic SCM binding in the presence of human plasminogen.
Introduction
Streptococcus canis is an opportunistic zoonotic pathogen that belongs to group G streptococci. S. canis colonizes mucosal surfaces and the skin as a commensal but can also cause serious invasive diseases, such as streptococcal toxic shock syndrome, necrotizing fasciitis, septicemia, and meningitis, in domestic animals and in humans (1, 2). Only scant information is available so far regarding virulence factors of S. canis. In 2010, a streptococcal protective antigen (SPASc) that shared sequence homology with SPA of other streptococcal species, such as S. equi and S. pyogenes, was described (3). A protective response of SPASc was shown in a mouse model of infection (3).
Recently, we identified the M-like protein of S. canis (SCM) as a plasminogen binding protein expressed on some clinical S. canis isolates (4). SCM is a 45-kDa fibrillar protein which is covalently anchored to the cell surface via a conserved LPxTG motif (4). The M proteins of streptococci represent major virulence factors due to their antiphagocytic activities (5–7). These antiphagocytic properties are explained by binding to a variety of different host proteins, with the most prominent one being fibrinogen (8–12). Another hypothesis relies on the M-protein-mediated formation of large bacterial aggregates, thereby inhibiting phagocytic engulfment (13). During the infection processes, several human-pathogenic streptococcal species interact with various host proteins circulating in human blood, such as plasminogen (14–17). Plasminogen is a major component of eukaryotic fibrinolysis and serves as a precursor of the broad-spectrum serine protease plasmin (18, 19). Plasminogen is composed of five highly conserved cysteine-rich triple-loop structures named kringle domains (each about 13 kDa), followed by the serine protease domain (25 kDa) at the C terminus. The first four kringle domains comprise the plasminogen derivative angiostatin, a potent antiangiogenic polypeptide, whereas kringle 5 and the serine protease domain are described as miniplasminogen.
In contrast to the M-like protein PAM (plasminogen-binding group A streptococcal M protein) of S. pyogenes, which binds to the lysine binding site of the second kringle domain (20), SCM has been shown to interact with miniplasminogen (4). This interaction promotes conversion of surface-immobilized plasminogen to proteolytically active plasmin by exploitation of the host-derived activators urokinase (uPA) and tissue-type plasminogen activator (tPA). Equipped with proteolytic plasmin activity, S. canis may degrade aggregated fibrin thrombi, thereby promoting dissemination of the bacteria in tissue (4).
For many bacteria, the presence of more than one plasminogen receptor has been reported, indicating a relevant mechanism for microbe invasion, secured by redundant plasminogen receptor expression (19). Two examples of prevalent bacterial plasminogen receptors in bacteria not expressing M or M-like proteins are enolase and glyceraldehyde-3-phosphate dehydrogenase (GAPDH, Plr/SDH), which are found on the surface of several streptococcal species, including S. pneumoniae, S. pyogenes, and oral streptococci (16, 21–24).
Here we report binding of plasminogen to SCM-negative S. canis isolates. In addition to the SCM protein, the glycolytic enzyme enolase was identified as a surface-displayed plasminogen receptor, mediating recruitment of proteolytic plasmin activity. This interaction occurs via the N-terminal part of plasminogen, comprising the kringle domains 1 to 4, whereas SCM binds to miniplasminogen. In addition to the immobilization of proteolytic activity on the streptococcal surface, we demonstrate that plasminogen binding led to resistance against phagocytosis. The reassociation of SCM to the streptococcal surface via a homophilic protein interaction further enhanced antiphagocytic activity significantly. However, the highest level of bacterial protection against phagocytosis was obtained by concerted activity of both mechanisms: SCM reassociation and synergistical plasminogen binding to enolase and SCM.
RESULTS
Non-species-specific plasminogen binding of SCM-negative S. canis isolates.
The M-like protein of S. canis (SCM) was previously identified as a plasminogen binding protein. Interestingly, in binding analysis with iodinated human plasminogen, a plasminogen binding activity of up to 10% was detected for several S. canis isolates, although no scm gene could be amplified by gene-specific PCR (Fig. 1A) (see also [4]). For the SCM-expressing strain G361, a binding activity for human plasminogen of 45.45% ± 13.24% was detected. In contrast, scm-negative strains showed reduced binding capacity, resulting in 7.11% ± 1.77% for strain G2, 6.2% ± 2.54% for strain 1022/96, 6.34% ± 2.48% for strain Sc14074-03, 6.09% ± 1.75% for strain 5520/2/10, 5.65% ± 1.75% for strain 2424/96, and 10.08% ± 8.72% for strain 5408/1/10. Similar results were detected using canine plasminogen from the natural host. Scanning electron microscopic visualization detected recruitment of human plasminogen homogenously distributed on the surface of scm-negative S. canis G2 (Fig. 1B). Photometric measurements of plasmin activity using the chromogenic substrate S-2251 demonstrated an exponential increase in plasmin activity for the SCM-positive strain G361, reaching an optical density at 405 nm (OD405) of 3.26 ± 0.01 after 180 min (Fig. 1C). In contrast, a slower but detectable conversion from plasminogen into plasmin (Fig. 1C, inset) directly contributed to the reduced plasminogen binding capacity of all tested scm-negative S. canis strains (Fig. 1A). After 180 min, the highest activity was detected for the isolate 2424/96, with an OD405 of 0.54 ± 0.02. These results demonstrate detectable plasminogen recruitment to the surface of S. canis even in the absence of SCM.
FIG 1.
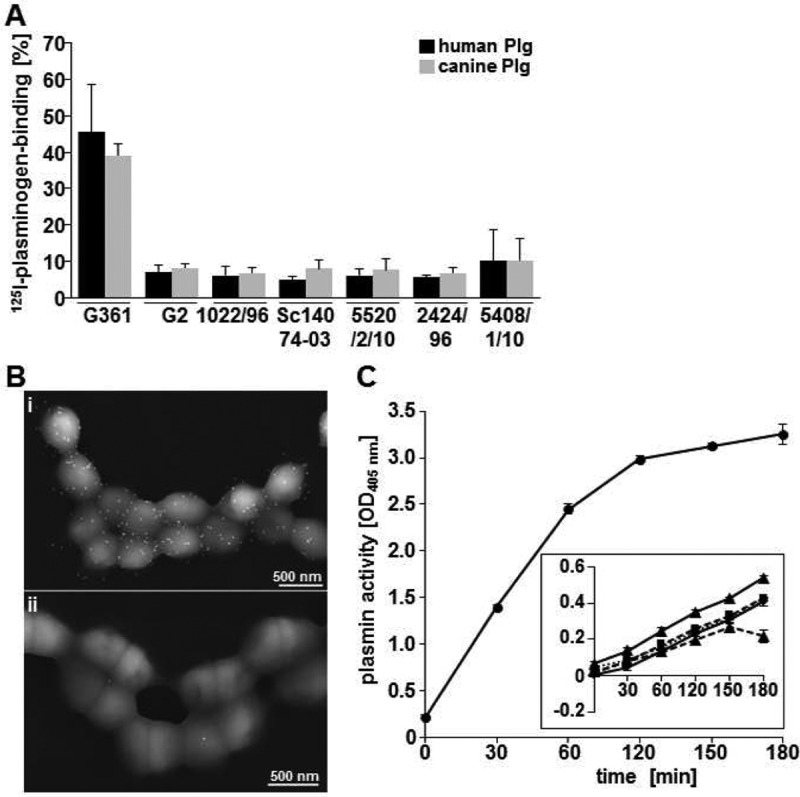
Plasminogen binding of scm-negative S. canis. (A) Binding of iodinated human plasminogen (dark bars) and canine plasminogen (grey bars) to S. canis isolates expressing SCM (G361) and not carrying the scm gene (G2,1022/96, Sc14074-03, 5520/2/10, 2424/96, or 5408/1/10) in percentages. (B) Electron microscopic visualization of plasminogen binding on the surface of scm-negative S. canis G2 using immune gold labeling. (i) Detection of plasminogen was performed with plasminogen-specific antibodies and 15-nm gold particles. (ii) The control shows minor background binding in the absence of plasminogen. (C) Photometric determination of plasmin activity of S. canis G361 using the chromogenic substrate S-2251 at time points 0 min, 30 min, 60 min, 120 min, 150 min, and 180 min. The inset illustrates only minor plasmin activity as detected for scm-negative isolates G2 (dotted line with triangles), 1022/96 (dotted line with squares), 5520/2/10 (dotted line with circles), 5468/1/10 (line with rhombus), and 2424/96 (line with triangles). Standard deviations were calculated from three independent assays using triplicates.
Enolase facilitates non-species-specific plasminogen binding of scm-negative S. canis.
Glycolytic enzymes like enolase and glyceraldehyde-3-phosphate-dehydrogenase have been identified as important plasminogen binding proteins of S. pyogenes and S. pneumoniae (16, 23, 25). Therefore, surface-exposed glycolytic enzymes were considered putative plasminogen receptors in SCM-negative S. canis isolates. Scanning electron microscopic localization of enolase using specific antibodies and protein A/G-conjugated gold detected a homogenous distribution of the glycolytic enzyme on the surface of the S. canis isolate G2 (Fig. 2A). After incubation of scm-negative S. canis G2 with plasminogen, electron microscopic detection of enolase and plasminogen using immune gold labeling with 10-nm and 15-nm gold particles visualized colocalization of both proteins on the bacterial surface (iii and iv). After expression cloning of S. canis G361 enolase, plasminogen binding to 1.0 µg and 2.0 µg purified enolase protein was analyzed by Western blot overlay. Signal detection with enhanced chemiluminescence demonstrated plasminogen binding to the 47-kDa enolase (Fig. 2B). In order to evaluate binding affinity of enolase-plasminogen interaction, surface plasmon resonance (SPR) studies were conducted. Human plasminogen was immobilized on a CM5 sensor chip, and purified S. canis enolase was injected as an analyte in different concentrations (4.0 µM, 2.0 µM, and 1.0 µM). Evaluation of the dissociation constant according to the Langmuir 1:1 binding model provided by the evaluation software determined an equilibrium dissociation constant (KD) of 8 × 10−7 M for S. canis enolase-plasminogen interaction (Fig. 2C). This dissociation constant indicated a weak but specific affinity of the glycolytic enzyme for the host protein. These results were confirmed by dot spot overlay analyses with immobilized human and canine plasminogen, respectively. Enolase protein was bound to both plasminogen isotypes in a concentration-dependent manner, demonstrating a non-species-specific binding of S. canis enolase (Fig. 2D).
FIG 2.

Enolase of S. canis as surface-displayed plasminogen binding protein (A) (i) FESEM visualization of enolase on the surface of S. canis G2 using antienolase antibodies and protein A-gold (15 nm; immune gold labeling). (ii) S. canis G2 treated with preimmune serum and protein A-gold (15 nm) exhibits no labeling. (iii and iv) Visualization of enolase using immune gold labeling (10 nm) (arrows) and visualization of plasminogen binding to enolase by plasminogen-gold (15 nm) (arrowheads). (B) Western blot analysis of plasminogen binding to purified S. canis enolase (eno) after separation of 1.0 μg and 2.0 μg protein via SDS-PAGE. Plasminogen binding was detected with plasminogen-specific antibodies and horseradish peroxidase-conjugated secondary antibodies. Visualization of binding signals was performed using enhanced chemiluminescence detection. (C) Determination of dissociation constant describing plasminogen binding to S. canis enolase via surface plasmon resonance analyses using 4.0 μM, 2.0 μM, and 1.0 μM concentrations of enolase as an analyte (KD of 8 × 10−7 M). (D) Detection of enolase binding to human (hum) and canine (can) plasminogen using the dot spot technique after immobilization of 0.07 μg, 0.13 μg, 0.25 μg, 0.5 μg, and 1.0 μg of plasminogen isotypes onto a nitrocellulose membrane. Signal detection was performed with enolase-specific antibodies followed by peroxidase-conjugated secondary antibodies and enhanced chemiluminescence detection. (E) Dot spot overlay analysis was performed after immobilization of recombinant S. canis enolase in amounts of 10.0 μg, 5.0 μg, 2.5 μg, 1.25 μg, and 0.625 μg onto a nitrocellulose membrane. Human plasminogen (Plg), miniplasminogen (mPlg), representing kringle domain 5 and the enzymatic domain, and kringle domains 1 to 4 (K1-4), representing the first four kringle domains at the plasminogen N terminus, were used for protein overlay. Binding was detected with polyclonal plasminogen-specific antibodies and peroxidase-conjugated secondary antibodies followed by enhanced chemiluminescence detection.
For identification of plasminogen domains mediating enolase binding, dot spot analyses were performed after immobilization of recombinant S. canis enolase in different amounts (10.0 µg, 5.0 µg, 2.5 µg, 1.25 µg, and 0.625 µg) on a membrane. Results of the overlay demonstrated that S. canis enolase recruited plasminogen (Plg) and angiostatin kringle domains 1 to 4 (K1-4), whereas no binding to miniplasminogen (mPlg), representing kringle domain 5 and the enzymatic domain at the carboxy terminus, could be detected (Fig. 2E). These results confirmed that S. canis enolase serves as a surface-exposed plasminogen binding protein specifically interacting with the amino-terminal kringle domains of plasminogen in a non-species-specific manner.
Enolase-bound plasmin activity mediates fibrinogen degradation and transmigration through semisynthetic fibrin thrombi.
In order to analyze enolase-mediated plasminogen activation, the recombinant purified enolase protein was immobilized on Dynabeads and applied in plasmin activity analyses (Fig. 3A). Plasminogen conversion into proteolytic active plasmin was enhanced up to an OD405 of 0.29 ± 0.02 at time point 180 min by plasminogen-coated enolase beads (Fig. 3A). Plasmin activity of plasminogen-incubated bovine serum albumin (BSA)-coupled beads reached only an OD405 of 0.12 ± 0.02 after uPA application (Fig. 3A). In addition, time-dependent degradation of the plasmin-specific substrate fibrinogen by enolase- and BSA-coated Dynabeads in the presence of plasminogen and the host-derived activator uPA was visualized by Western blot analysis. Substantial degradation of fibrinogen was observed using enolase-coated Dynabeads after 60 min of incubation (Fig. 3B). In control experiments using BSA-coupled beads, substantial degradation of fibrinogen was not detected before 240 min (Fig. 3B). In addition to plasmin-mediated cleavage of soluble purified fibrinogen, degradation of aggregated fibrin thrombi was analyzed by electron microscopy. The regular constructed fibrin network is depicted in Fig. 3C, i. No degradation activity was observed for S. canis G2 preincubated with plasminogen only (Fig. 3C, ii), whereas supplementation of uPA to plasminogen-coated bacteria resulted in dissolution of the fibrin network, forming a negative imprint at the bacterial contact points with the fibrin matrix (Fig. 3C, iii). Similar results were obtained for plasminogen-preincubated enolase-coated beads, mediating plasmin-dependent cleavage of the semisynthetic fibrin matrix after supplementation of uPA (Fig. 3C, iv to vi). Thus, the enolase-plasminogen interaction of scm-negative S. canis mediates surface-bound plasmin activity that subsequently promotes degradation of soluble fibrinogen and aggregated fibrin matrices.
FIG 3.
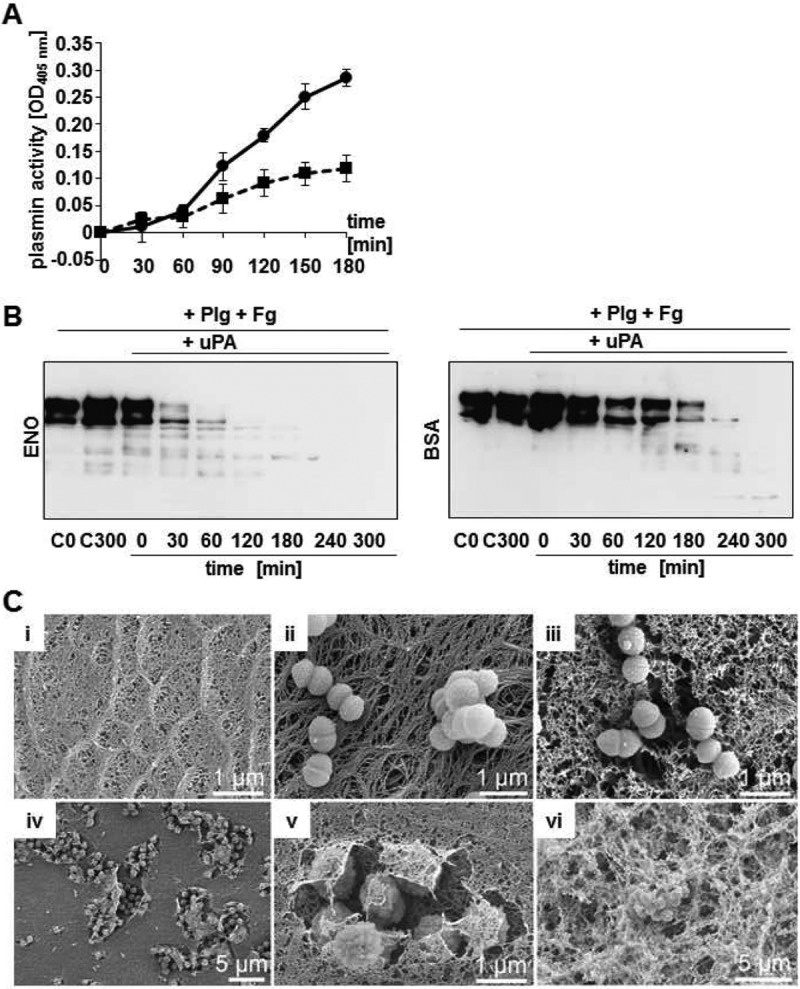
Proteolytic plasmin activity of enolase/BSA-conjugated Dynabeads and S. canis G2. (A) Determination of plasmin-mediated degradation of chromogenic substrate S-2251 after plasminogen incubation of enolase-conjugated beads (line with circles) at indicated time points. BSA-coated beads were used as a negative control and showed only weak plasmin activity after incubation with plasminogen (dotted line with squares). Graphical illustration represents mean values of a representative assay performed in triplicate after subtraction of unspecific proteolytical activity of plasminogen-incubated beads without incubation with the activator uPA. Experiments were repeated three times. (B) Time-dependent degradation of soluble fibrinogen after incubation of enolase (ENO)-conjugated Dynabeads and BSA-conjugated beads with plasminogen in the presence of urokinase (uPA) at indicated time points. Peptide signals of lower molecular weight represented degradation products. C0 and C300 represent controls of fibrinogen degradation of conjugated beads after incubation with plasminogen without uPA at time points 0 min and 300 min. (C) Electron microscopic visualization of plasmin-mediated degradation of an aggregated fibrin matrix (i) by scm-negative S. canis G2 after incubation with plasminogen in the presence of uPA (iii). In contrast, no degradation is visible without uPA (ii). Substantial fibrin degradation is detected after incubation of the matrix with plasminogen-preincubated enolase beads as visualized in different magnifications (iv to vi).
Secretion and reassociation of SCM to S. canis surface.
Release of SCM into bacterial supernatant was analyzed by Western blot analyses (Fig. 4A). SCM-specific antibodies detected the SCM protein in the supernatant of SCM-expressing G361, but no SCM signal was obtained using culture supernatant from scm-negative strain G2 (Fig. 4A). Since the cysteine protease SpeB is responsible for the release of M proteins in S. pyogenes (26), we tested SpeB-mediated proteolytic cleavage in different S. canis strains. As expected, strong proteolytic activity was detected for S. pyogenes strain A60, whereas no cysteine protease activity, and therefore no SpeB activity, was monitored on the bacterial surface of both SCM-expressing S. canis G361 and SCM-negative G2 (Fig. 4B).
FIG 4.
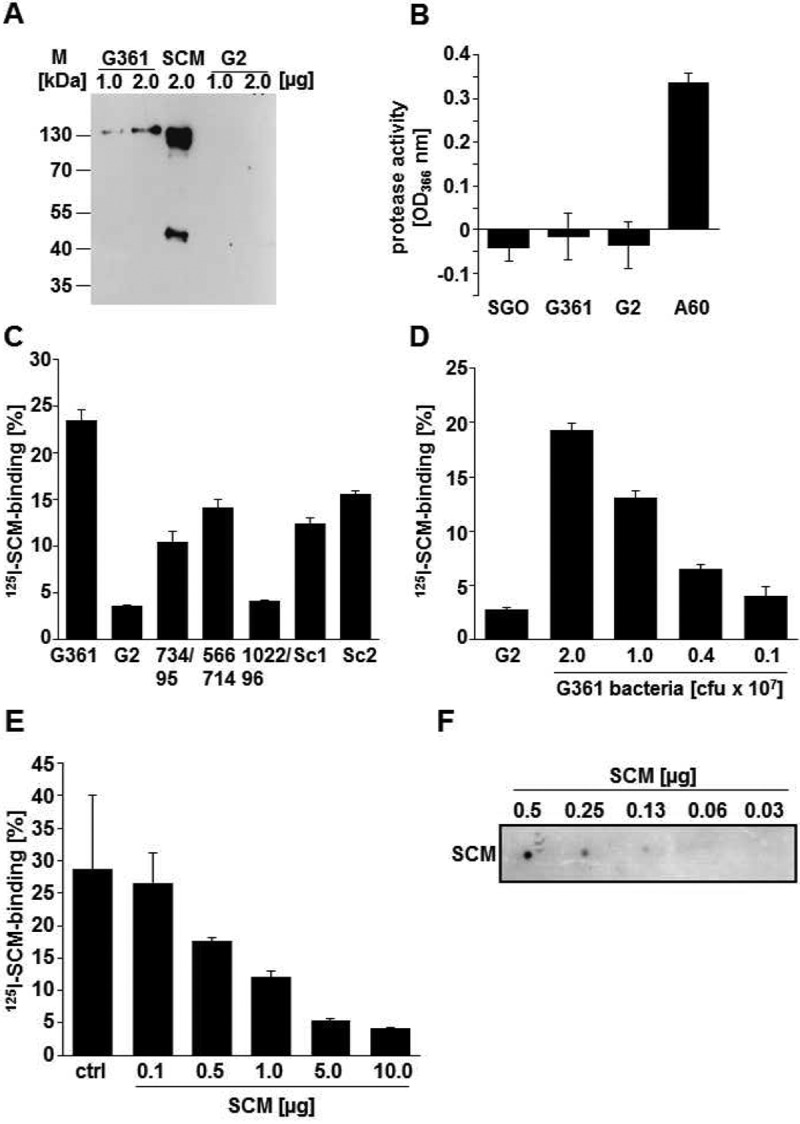
Secretion and reassociation of SCM and SCM derivatives. (A) Release of surface-expressed SCM protein into the culture supernatant was analyzed by immunoblot analyses using 1.0 μg and 2.0 μg of supernatant proteins of S. canis G361 (lanes 1 and 2) and an scm-negative G2 isolate (lanes 4 and 5). A 2.0-μg amount of the purified SCM protein was used as a positive control. Detection of SCM was performed with SCM-specific antibodies and peroxidase-conjugated secondary antibodies followed by enhanced chemiluminescence detection. (B) The cysteine protease assay was performed with S. canis isolates G361 and G2 in order to detect proteolytic activity of bacterial surface-localized proteases. Strong proteolytic activity was determined for protease-positive S. pyogenes isolate A60. The protease-negative S. gordonii isolate was applied as a negative control. (C) Binding of iodinated SCM to different S. canis isolates, including SCM-expressing S. canis G361, scm-negative G2, and isolates 734/95, 566714, 1022/96, Sc1 (4074-03), and Sc2 (6318-02). (D) Dose-dependent binding of iodinated SCM to 2.0 × 107 CFU G2 bacteria and to the G361 isolate using decreasing amounts of bacteria (2.0 × 107, 1.0 × 107, 0.4 × 107, and 0.1 × 107 CFU). (E) Inhibition of SCM binding to S. canis G361 using 0.1 μg, 0.5 μg, 1.0 μg, 5.0 μg, and 10.0 μg nonlabeled SCM protein. SCM binding to G361 bacteria without SCM protein as an inhibitor was used as a control (ctrl). (F) Dot spot overlay of 125I-SCM binding to purified nonlabeled SCM, which was immobilized in amounts of 0.5 μg, 0.25 μg, 0.125 μg, 0.06 μg, and 0.03 μg.
Reassociation of M-like proteins has been demonstrated for group A streptococci (13). A variety of different S. canis strains were tested for their ability to bind to recombinant SCM derived from strain G361 (Fig. 4C). A binding capacity of more than 10% was detected for the parental strain G361 (23.5% ± 1.5%), strain 734/95 (10.4% ± 1.3%), strain 566714 (14.1% ± 1.0%), strain Sc1 4074-03 (12.4% ± 0.8%), and strain Sc2 6318-02 (15.6% ± 0.3%). Interestingly, the scm gene was detected in the genome of all these strains as evaluated by PCR (4). In contrast, the scm-negative strains G2 and 1022/96 exhibited binding capacities of less than 5% (3.5% ± 0.1% and 4.1% ± 0.1%, respectively) and were therefore characterized as negative in SCM binding. A 2-fold dilution of the bacterial suspension from 2 × 107 bacteria down to 1 × 107 bacteria reduced the binding capacity from 19.3% ± 0.7% to 13.1% ± 0.6% (Fig. 4D). Reducing the amount of bacteria to 4 × 106 bacteria (1:5) led to a binding capacity of 6.5% ± 0.4%. Finally, a dilution of 1:10 (2 × 106) of the initial inoculum reduced the percentage of SCM binding to 4.0% ± 0.8% and was therefore similar to the result obtained by the nonbinding strain G2 (2.8% ± 0.2%). These data demonstrate a dose-dependent interaction between SCM and G361. This dose dependence of SCM binding relied on the expression of the SCM protein, since supplementation of an increasing amount of nonradiolabeled SCM reduced the binding capacity from 28.7% ± 11.3% to 4.2% ± 0.1% in the presence of excess of 10.0 µg nonlabeled SCM (Fig. 4E). Direct SCM-SCM protein interaction was tested in dot blot overlay assays after immobilization of purified SCM on a nitrocellulose membrane in amounts ranging from 0.5 µg to 0.03 µg and 125I-labeled SCM for protein overlay. As depicted in Fig. 4F, a specific SCM-SCM binding signal with decreasing intensity could be detected following decreasing amounts of immobilized SCM.
Cooperative plasminogen recruitment to S. canis surface by SCM and enolase.
In radioactive binding analyses, iodinated SCM (125I-SCM) was recruited to SCM-expressing G361 bacteria, reaching 20.48% ± 2.14% specific binding (Fig. 5A). Incubation of SCM-expressing S. canis G361 with angiostatin representing kringle domains 1 to 4 (K1-4) resulted in a slightly decreased 125I-SCM binding (16.37% ± 1.77%). A significant increase in SCM binding, to 55.02% ± 2.9% (P < 0.001), was detected after incubation of G361 bacteria with 1.0 µM plasminogen (Plg). These data provide evidence that plasminogen mediates SCM reassociation as bridging molecule via binding to the C-terminal miniplasminogen domain.
FIG 5.

Synergistic plasminogen binding by surface-exposed SCM and enolase of S. canis. (A) Binding of the iodinated SCM protein to SCM-expressing S. canis G361 after incubation with 1.0 μM human plasminogen (G361-Plg) or 1.0 μM angiostatin (G361-K1-4) was determined by gamma counting. (B) Analyses of SCM binding was performed using the SCM-expressing strains G361, H3605, and G13 and the scm-negative isolates 1022/96, G14, and G2. Preincubation of bacteria with 1.0 μM plasminogen (Plg) enhanced binding of iodinated SCM. (C) Binding of iodinated SCM (125I-SCM) and iodinated plasminogen (125I-Plg) was detected after coincubation of SCM-expressing S. canis G361 with radioactive protein and 1.0 μM nonlabeled SCM and plasminogen (Plg). Analyses were performed in triplicate in three independent assays. Percent binding was calculated related to binding of radioactive labeled SCM to fetal calf serum, which was set to 100%. P values ≤ 0.05 are marked by an asterisk. (D) (i and ii) Transmission electron microscopic visualization of SCM recruitment to the surface of scm-negative S. canis G14 was performed using SCM-specific antibodies and protein A/G-coupled gold particles. In contrast to marginal SCM detection without preincubation (i), large amounts of SCM protein signals were detected after preincubation of the bacteria with plasminogen (ii). (iii and iv) Colocalization between plasminogen and SCM was detected after incubation of G2 with plasminogen-gold (10 nm) (arrows), followed by incubation with SCM and immune labeling of SCM with protein A-gold (20 nm) (arrowheads).
The contribution of plasminogen to increased SCM reassociation was further analyzed for several SCM-positive and scm-negative S. canis isolates (Fig. 5B). A similar increase of 125I-SCM binding after incubation with plasminogen was detected for the SCM-expressing strains H3506 and G13, reaching 71.18% ± 2.25% (H3506) and 44.16% ± 2.67%, respectively (Fig. 5B, G13). Although binding of the iodinated SCM protein to scm-negative S. canis 1022/96m reaching 4.58% ± 0.22%, G14 (11.51% ± 0.68%), and G2 (4.12% ± 0.53%) were significantly less than SCM recruitment to G361 (25.69% ± 11.32%) and H3506 (43.30% ± 0.73%), plasminogen recruitment to the bacterial surface enhanced 125I-SCM binding up to 7-fold for S. canis 1022/96 and up to 3-fold for S. canis G14, reaching 31.29% ± 2.47% and 32.19% ± 1.55% binding (Fig. 5B). Further inhibition studies clearly demonstrated that nonlabeled plasminogen and nonlabeled SCM competitively inhibited binding of radioactive plasminogen, whereas nonlabeled plasminogen significantly enhanced recruitment of radioactive SCM to the bacterial surface (Fig. 5C). In contrast to the significant reduction of 125I-plasminogen binding to S. canis G361, from 94.12% ± 12.64% to 41.32% ± 5.55%, after incubation with 1.0 µM nonlabeled exogenous SCM, recruitment of iodinated SCM to S. canis G361 was only marginally inhibited by 1.0 µM nonlabeled SCM protein, reaching 15.78% ± 0.96% binding (Fig. 5C). Interestingly, incubation of S. canis G361 with nonlabeled plasminogen significantly increased binding of iodinated SCM, up to 3-fold, to 73.93% ± 19.18% (Fig. 5C). Detection of the SCM protein on the surface of scm-negative S. canis G14 by electron microscopy visualized the large amount of SCM recruitment after incubation of the bacteria with a complex consisting of SCM and plasminogen (Fig. 5D, i/ii). Furthermore, a direct interaction between Plg and SCM was observed in colocalization studies using gold-labeled Plg and gold-labeled anti-SCM antibodies (Fig. 5D, iii/iv). These data indicate that plasminogen recruitment to the S. canis surface via surface-expressed SCM or surface-exposed enolase promotes recruitment of the exogenous SCM protein.
Complex formation of plasminogen on S. canis surface promotes bacterial survival.
The contribution of SCM reassociation with plasminogen-bound S. canis to bacterial survival has been investigated by phagocytosis analyses with neutrophil granulocytes purified from human blood. Binding of plasminogen protected against phagocytic killing of both SCM-expressing S. canis G361 and scm-negative G14. Incubation of the G361 strain with 1.0 µM of purified SCM increased the bacterial survival rate from 0.33% ± 0.14% up to 34.42% ± 8.57% (Fig. 6). Although G14 is not expressing endogenous SCM, bacterial survival was significantly increased, from 0.05% ± 0.01% to 10.25% ± 2.88%, after incubation of G14 with purified SCM. Interestingly, incubation of SCM-expressing G361 with 1.0 µM plasminogen increases survival only marginally, to 4.44% ± 1.63%, whereas the same procedure with scm-negative G14 resulted in a more than 2-fold-higher survival rate of 10.67% ± 3.06%. The highest survival rates, with 59.28% ± 15.10% for G361 and 31.94% ± 12.93% for the scm-negative G14, were reached after recruitment of plasminogen followed by SCM binding to the bacterial surfaces (Fig. 6). Recruitment of SCM to the bacterial surface via plasminogen thereby mediated protection against phagocytosis and was independent of endogenous SCM expression.
FIG 6.
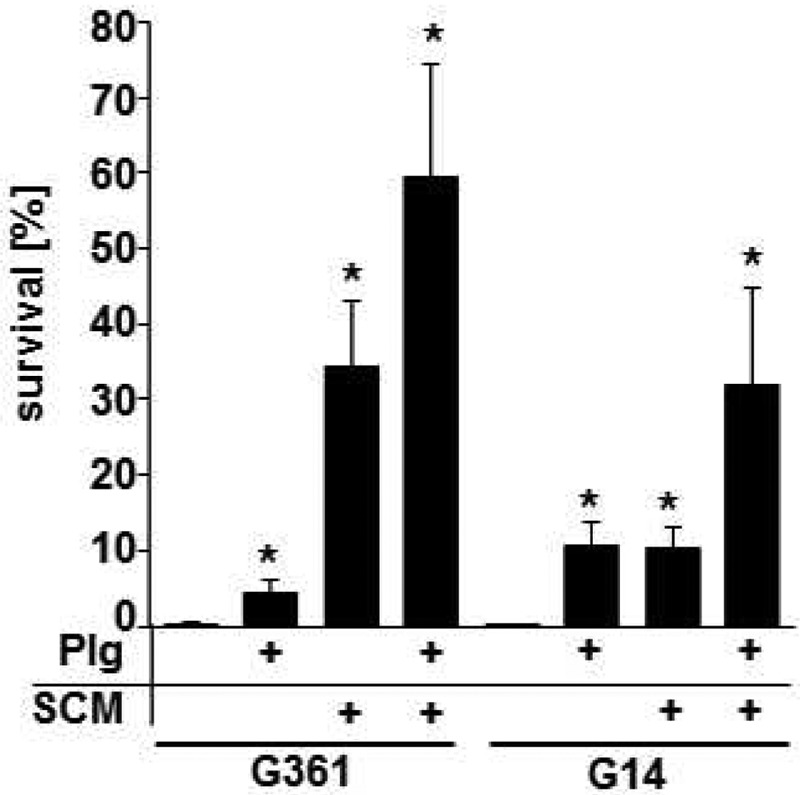
Bacterial survival in phagocytosis analysis with human blood neutrophils. Analysis of bacterial survival after infection of human blood neutrophils with SCM-expressing S. canis G361 and scm-negative G14 after incubation with 1.0 μM human plasminogen (Plg), 1.0 μM purified SCM protein, or both by determination of bacterial CFU is shown. Results represent mean values from three independent analyses performed in triplicate. P values ≤ 0.05 are marked by an asterisk.
DISCUSSION
Binding of human plasminogen and subsequent transmigration through fibrin thrombi using surface-bound plasmin activity have recently been proposed to comprise a virulence trait in zoonotic S. canis. We have previously characterized plasminogen binding to surface-displayed M-like protein (SCM) of S. canis as the responsible mechanism (4). In the present study, we characterized a moderate but substantial interaction of SCM-negative strains with both human and canine plasminogen, suggesting the presence of additional plasminogen binding proteins on the S. canis surface. From the literature, it is known that glycolytic enzymes which are displayed on the bacterial surface as moonlighting proteins have been identified as plasminogen binding proteins as well (16, 22, 27). As already demonstrated for enolase of S. pneumoniae (16), electron microscopic studies confirmed a localization of S. canis enolase on the streptococcal surface with a homogenous distribution. In addition to C-terminally localized lysine residues, an internal nonapeptide has been identified as an important plasminogen-binding motif of S. pneumoniae (28). Subsequent sequencing of the entire binding domain of S. canis enolase revealed the presence of an identical motif within S. canis enolase (data not shown), which most likely contributed to plasminogen binding. Moreover, recombinantly expressed S. canis enolase mediated a non-species-specific plasminogen binding to human and canine plasminogen. Surface plasmon resonance using recombinant S. canis enolase with immobilized human plasminogen demonstrated a specific interaction, with kinetic dissociation constants in the nanomolar range. And, finally, a direct interaction of surface-exposed enolase and externally administered plasminogen was detected in colocalization studies using immune-electron microscopy (EM). Collectively, these data strongly indicate a physiological relevance of plasminogen binding via surface-exposed enolase, especially in scm-negative S. canis.
Interestingly, conversion of plasminogen into proteolytic active plasmin was determined for both SCM-positive and -negative S. canis strains. Although plasminogen activation by SCM-negative isolates was significantly weaker, a profound plasminogen conversion was also determined using recombinant S. canis enolase. These results were confirmed by degradation analyses, using enolase-coated latex beads. Time-dependent degradation of fibrinogen indicated the recruitment of substrate-specific proteolytic activity on the bead surface. Electron microscopic visualization of reconstituted fibrin thrombi illustrated the degradation of the dense fibrin matrix by plasminogen-covered latex beads and S. canis bacteria in the presence of uPA. In addition to the results shown for recombinant enolase, these data confirmed the plasmin-mediated dissolution of fibrin thrombi by scm-negative S. canis bacteria. Degradation of fibrin thrombi and extracellular protein matrices after activation of enolase-bound plasminogen has been characterized and visualized for S. pneumoniae (29, 30). In addition to the prominent plasminogen binding proteins like PAM of S. pyogenes and SCM of S. canis, our results strongly suggest that surface-exposed glycolytic enzymes of S. canis may contribute to bacterial dissemination within the host (4, 20). Moreover, the simultaneous expression of a subset of different plasminogen binding proteins on the surface of pathogenic streptococci like S. canis emphasizes the high relevance of this interaction and suggested a possible synergistic binding activity.
Although functional consequences of plasminogen recruitment via glycolytic enzymes share similarities to M-protein-mediated plasminogen binding, the underlying molecular interaction mechanisms with respect to binding motifs elicit a high diversity. A subset of M proteins expressed by group A streptococci bind plasminogen and plasmin directly with high affinity (8) and have been well characterized in vitro (20, 31–36). For example, two internal plasminogen binding motifs (a1 and a2) mediate interaction of PAM, representing the M53 serotype of S. pyogenes, with the kringle domain 2 of plasminogen (20, 33). Meanwhile, additional plasminogen-binding motifs have been identified in M proteins of other GAS serotypes associated with both invasive and noninvasive disease. It is suggested that these M proteins interact with plasminogen kringle domains via the same mechanism and with similar affinities (37, 38). SCM of S. canis displays an exception. In a recent study, we clearly demonstrated that the interaction of SCM and plasminogen occurs via binding to miniplasminogen comprising kringle domain 5 and the serine protease domain (4).
In accordance with data demonstrating interaction of S. pneumoniae enolase with the N-terminal kringle domains of plasminogen (28), results of dot blot overlay analyses confirmed binding of S. canis enolase to angiostatin, comprising kringles 1 to 4. The distanced localization of binding sites mediating interaction with SCM and enolase gives rise to the hypothesis that cooperative binding of enolase and SCM to plasminogen may occur simultaneously, as depicted in Fig. 7. Cooperative plasminogen binding to enolase and SCM on the S. canis surface is likely to mediate efficient bacterial dissemination within the host tissue. In addition, the presence of both a homophilic self-aggregation via surface-bound SCM and a heterophilic reassociation of SCM to a plasminogen-decorated S. canis surface are indicated.
FIG 7.
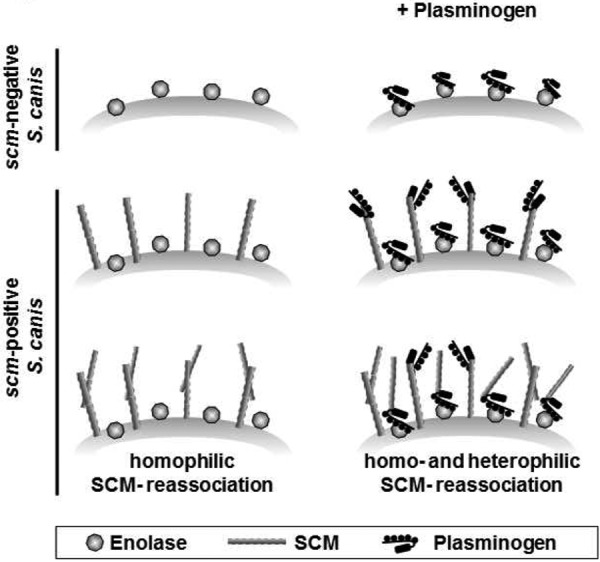
Schematic model of synergistic plasminogen binding to S. canis SCM and enolase. Plasminogen binding is mediated via surface-exposed enolase on scm-negative bacteria. Enhanced plasminogen binding is detected on scm-positive S. canis isolates expressing both enolase and M protein on their surface. Secreted SCM reassociates to the bacterial surface by homophilic interaction with surface-linked SCM in addition to heterophilic binding to plasminogen molecules recruited to surface-exposed enolase.
For decades, M proteins have been within the focus of scientific investigation and have been characterized with respect to structural and functional properties in detail. In addition to the fibrous molecular structure (39) containing repeated domains and the alpha-helical coiled-coil dimerization (40, 41), several members of the M-like protein family, such as protein H of S. pyogenes, display a homogenous molecular interaction on the bacterial surface mediating bacterial aggregation (13). In electron microscopy studies, SCM-mediated bacterial self-aggregation was visualized for S. gordonii bacteria heterologously expressing SCM on their surface but not for scm-negative S. gordonii (data not shown). The visualization of M proteins as hair-like structures protruding from the surface (42) implied the contribution of this protein in an end-to-end aggregation mechanism mediating bacterial clumping (40). Self-aggregation of streptococcal M- and M-like proteins contributed to bacterial adherence, antiphagocytic properties, and virulence in mouse models of infection (13). In accordance to the reported self-aggregation of protein H, radioactive binding assays demonstrated the significantly increased reassociation of exogenous SCM to the S. canis surface after coincubation with human plasminogen.
The antiphagocytic activity of M proteins has been characterized as one of the most important virulence mechanisms of pyogenic streptococci (43, 44). Results of the present phagocytosis analyses are in accordance with binding studies, demonstrating a significantly increased survival of bacteria decorated with SCM. Results of phagocytic analyses further demonstrated an increased bacterial survival after decoration of both SCM-expressing and scm-negative S. canis bacteria with plasminogen. These data indicate an antiphagocytic activity of bacterium-bound plasminogen. In a recently published report, antiphagocytic activity has been correlated with plasminogen binding to Bacillus anthracis spores as well (45). Plasminogen was shown to be bound to the spores via surface-exposed enolase, thereby mediating evasion from complement opsonization by C3b molecules (45). An even stronger antiphagocytic activity was determined for both SCM-expressing S. canis and scm-negative bacteria subverting plasminogen as a host-derived cofactor for heterophilic SCM reassociation. Based on similarities to key attributes of proteins from the M-like protein family, SCM was initially characterized as an M-like protein. The observed homogenous self-aggregation and functional antiphagocytic activity clearly classified SCM as an M protein.
In addition to M- and M-like-protein-mediated protection against antibacterial attack by complement (46–50), bacterial aggregation has been designated a further virulence mechanism preventing phagocytic uptake of large bacterial aggregates (13, 51). Both homophilic and heterophilic SCM self-aggregation may promote clumping of S. canis bacteria, thereby preventing phagocytic uptake. A high resistance against phagocytosis and neutrophil killing has also been reported for coaggregates between oral bacteria and other bacterial species (52). This report supports the suggestion of a cooperative aggregate formation in bacterial communities of SCM-expressing and scm-negative S. canis bacteria.
Most of the described M- and M-like proteins facilitate binding of host components like fibrinogen to the streptococcal surface (53). It is assumed that fibrinogen serves as a bridging molecule mediating bacterial aggregation (12, 44). In addition to a direct recruitment of inhibiting complement factors like C4b binding protein to the streptococcal surface (12), binding of fibrinogen to M- and M-like proteins, such as Mrp (M-related protein) of S. pyogenes, has already been shown to reduce surface deposition of complement C3b (12, 44). Streptococcal resistance against phagocytosis by neutrophils has been attributed to both mechanisms, fibrinogen-dependent bacterial aggregation (54, 55) and the hindrance of complement factor binding (9, 12, 17, 44, 56), Fibrinogen-mediated anti-phagocytic activity was not restricted only to S. pyogenes but was also demonstrated for group G streptococci (57), although no reduced binding of C3b was reported.
In conclusion, the decoration of the S. canis surface with human plasminogen via SCM as well as enolase promoted SCM reassociation and mediated antiphagocytic activity. The protection of S. canis against phagocytic killing is based on a complex surface remodeling induced by at least two synergistic mechanisms: first, binding of human plasminogen via SCM and enolase, and second, SCM self-aggregation via homophilic and heterophilic SCM reassociation. These findings extend the gain of functional benefit of host-derived proteolytic activity to bacterial survival after neutrophil uptake and shed new light on the role of plasminogen binding via SCM for S. canis virulence.
MATERIALS AND METHODS
Bacterial strains, media, and growth conditions.
S. canis strains were graciously provided by J. Verspohl, Veterinary School, Hannover, Foundation; J. F. Prescott (Department of Pathobiology, University of Guelph, Guelph, Ontario, Canada); B. W. Beall (Centers for Disease Control and Prevention, Atlanta, GA), and M. van der Linden (National Reference Centre for Streptococci, Aachen, Germany). Streptococcal strains were routinely cultivated in tryptic soy broth (TSB) at 37°C without shaking (4). Escherichia coli was grown in Luria Bertani (LB) medium at 37°C with continuous aeration (120 rpm). Where appropriate, ampicillin was added at a concentration of 100 µg ⋅ ml−1.
Proteins and sera.
Human plasminogen was purchased from Sigma. Antibodies directed against pneumococcal enolase were kindly provided by S. Hammerschmidt. Antibodies against human plasminogen were purchased from Acris. Plasminogen-depleted fibrinogen was purchased from Calbiochem. Secondary horseradish peroxidase (HRP)-conjugated anti-goat and anti-rabbit antibodies were purchased from Dako.
Expression cloning and recombinant DNA technique.
If not stated otherwise, all enzymes were obtained from New England Biolabs (NEB). Enolase was amplified from the genome of S. canis strain G361 using the primer pair eno_fwd_BamHI (ggg gga tcc ATG TCA ATT ATT ACT GAT GTT TAC GC)/eno_rev_SalI (ccc gtc gac TTA TTT TTT AAG GTT GTA GAA TGA TTT GAT GCC). The incorporated restriction sites (in bold) and random nucleotides (lowercase letters) for restriction enzyme binding allowed us to subsequently digest the PCR product and subclone it into the expression vector pGEX-6P-1. Overexpression and purification of rENO was carried out under native conditions as per the manufacturer’s instructions. Purified rENO was dialyzed against phosphate-buffered saline (PBS) (1.5 mM KH2PO4, 8.0 mM Na2HPO4, 2.7 mM KCl, and 137 mM NaCl, pH 7.4). If desired, the glutathione S-transferase (GST) tag was removed using PreScission protease (GE Healthcare). Quantification of the protein was done using the Bradford reagent (Bio-Rad), following the manufacturer’s instructions.
Radioactive binding analyses.
Labeling of plasminogen and purified SCM with 125I was performed by using Chloramin T according to the method of Hunter and Greenwood (58). Binding experiments with 125I-plasminogen and 125I-SCM were performed as described previously (4). In brief, for binding analyses, 2 × 108 bacteria were incubated with 100,000 cpm plasminogen (~15 ng) or 200,000 cpm SCM (~15 ng) for 30 min at room temperature (RT). The analyses were performed in triplicate in three independent analyses. For investigation of cooperative binding, the bacteria were incubated for 1 h at 4°C with 1.0 µM nonlabeled SCM or 1.0 µM nonlabeled plasminogen prior to incubation with 125I-labeled proteins. Binding of 125I-SCM to 20 µl heat-inactivated fetal calf serum (FCS) was used as a positive control. After 30 min of incubation, all binding reactions were precipitated with 10% trichloric acid (TCA), and reaction mixtures were centrifuged at maximum speed for 7 min. After removal of supernatant containing nonbound proteins, bacterium-bound radioactivity was determined using a 1470 Wallac Wizard gamma counter (PerkinElmer). Binding values were presented as percentages related to the determined cpm for precipitation of radioactive proteins with FCS, which was set to 100%.
Immobilization of recombinant enolase to Dynabeads.
Covalent immobilization of enolase protein and BSA to carboxylated polystyrene beads of 1 µm in diameter (Dynabeads MyOne carboxylic acid; Invitrogen) was performed essentially as described by the manufacturer using the “two-step coating procedure with NHS.” In brief, 120 µg recombinant enolase or BSA bovine serum albumin (Roth) was incubated with 300 µl beads (~3 × 109 beads) overnight at 4°C, followed by blocking of unspecific binding with BSA (Roth). Protein coupling was monitored by flow cytometry (FACSCalibur).
Plasmin activity assay.
Detection of surface-associated plasmin activity was performed essentially as described by Fulde et al. (4). In brief, triplicates of scm-positive and scm-negative S. canis strains (109 CFU ⋅ ml−1) were incubated with 40 µg ⋅ ml−1 plasminogen in 10 mM PBS for 15 min at 37°C. After removal of unbound plasminogen by washing with 10 mM PBS, uPA (500 ng) was added in order to start conversion from plasminogen to plasmin. As a control, assays without the addition of uPA determined no autocatalytic activities of plasminogen (data not shown). The chromogenic substrate d-valyl-leucyl-lysine-p-nitroanilide dihydrochloride (S-2251; Fluka) was added to a final concentration of 400 µM, and proteolytic cleavage was detected photometrically at 405 nm using an enzyme-linked immunosorbent assay (ELISA) reader (Tecan; Sunrise), following a time course of 180 min. Results were given as means for the assays with uPA after subtraction of background activity determined without uPA. For determination of plasmin activity of purified enolase protein, 10 µl of enolase-coated Dynabeads (~1 × 108 beads) were incubated for 30 min with 50 µg human plasminogen. After three washing steps with 10 mM PBS, the beads were incubated with 500 ng uPA (Haemochrom Diagnostica) and measurement was started immediately after adding 20 µl of the plasmin-specific S-2251 substrate. Plasmin activity was determined in time intervals of 30 min up to 180 min using triplicates in three independent assays. For evaluation of plasmin activity and data visualization, background plasmin activity of plasminogen-treated enolase beads without activator was determined, and mean values were subtracted from uPA-incubated probes at each time point of measurement.
Immunoblot analysis.
Canine and human plasminogen was spotted onto a nitrocellulose membrane at appropriate amounts. Membranes were blocked using 5.0% skimmed milk in 10 mM PBS as described above and incubated with 100 µg purified enolase protein overnight at 4°C. Immunoblot analysis was carried out using antienolase (1:200) antiserum and HRP-conjugated secondary antibodies (1:3,000). For detection of SCM in supernatants of S. canis isolates G361 and G2, protein amounts of culture supernatants were quantified photometrically using Bradford solution (Sigma). S. canis supernatant proteins (1.0 µg and 2.0 µg) were separated by SDS-PAGE and subjected to immunoblotting using SCM-specific antibodies. A 1.0-µg amount of purified SCM was used as a positive control. Peroxidase activity was detected by chemoluminescence using 100 mM Tris HCl, 1.25 mM 3-aminopthalhydrazide, 225 µM p-coumaric acid, and 0.01% H2O2 at pH 8.8 in water and exposed to chemoluminescence films (Hyperfilm; Amersham).
Dot blot overlay.
Purified enolase protein was immobilized on a nitrocellulose membrane using 0.625 µg, 1.25 µg, 2.5 µg, 5 µg, and 10.0 µg protein in 4.0 µl PBS. Unspecific binding sites were blocked by incubation with 10.0% skimmed milk in 10 mM PBS supplemented with 0.05% Tween 20 (PBST) overnight at 4°C. After three washing steps with PBST, the membrane was incubated with 50 µg human plasminogen, 50 µg miniplasminogen, or 50 µg angiostatin overnight at 4°C. After three additional washing steps with PBST, the membrane was incubated with polyclonal plasminogen-specific antibodies from goat (1:500; Acris) in 1.0% skimmed milk followed by incubation with horseradish peroxidase-conjugated secondary antibodies (1:3,000). The antibody detected human plasminogen, miniplasminogen, and angiostatin in similar intensities (data not shown). After extensive washing with PBST, visualization of binding signals was performed by incubation with the substrate solution containing 1 mg ⋅ ml−1 4-chloro-1-naphthol and 0.1% H2O2 in 10 mM PBS.
Surface plasmon resonance.
Binding kinetics of recombinant S. canis enolase and plasminogen (Sigma) were analyzed using the surface plasmon resonance technique as described by Fulde et al. (4). In brief, 35 µl of 1 mg ⋅ ml−1 plasminogen was immobilized on a CM5 sensor chip by a standard amine coupling procedure using 20 mM sodium acetate, pH 4.5. Binding analysis was performed using 210 µg ⋅ ml−1 purified enolase in HBS BIAcore running buffer (10 mM HEPES, 150 mM NaCl, 1.4 mM EDTA, 0.05% Tween 20, pH 7.4) at 20°C using a flow rate of 30 µl ⋅ ml−1 in all experiments. The affinity surface was regenerated between subsequent sample injections of analytes with 10 µl of 20 mM NaOH. Binding was assayed at least in duplicate using a BIAcore optical biosensor (BIAcore 2000 system) and independently prepared sensor chips. Analyses of association and dissociation of S. canis enolase and immobilized plasminogen was performed using the kinetic models included in the BIAevaluation software program, version 3.0. The experimental data were fitted globally according to the simple one-step bimolecular association reaction (1:1 Langmuir kinetic: A + B ↔ AB). All results recorded in this report were within the typical dynamic ranges of BIAevaluation 3.0 software. For each evaluation, a minimum of three data sets corresponding to plasminogen binding reactions at concentrations of 0.4 µM, 0.2 µM, and 0.1 µM enolase were analyzed.
Cysteine protease assay.
In order to determine SpeB activity on the S. canis surface, a bacterial culture was cultivated overnight at 37°C without shaking, and 2 × 109 bacteria were sedimented for 10 min at 4,200 × g at RT. The bacterial supernatant was incubated with 200 µl activation buffer containing 20 ml sodium acetate-buffer (0.6% [vol/vol] acetic acid and 8.2 g ⋅ liter−1 sodium acetate, pH 5.0), 40 µl EDTA solution (0.5 M), and 0.062 g dithiothreitol (DTT) for 30 min at 40°C, followed by supplementation with 400 µl azocasein (2% [wt/vol]) for an additional 30 min at 40°C. To stop the reaction, 120 µl trichloroacetic acid (TCA) (15% [wt/vol] in 100% acetone) was added and centrifuged for 5 min at 25,000 × g. Protease activity was determined by photometrical measurement at 366 nm (Tecan; Sunrise).
Degradation of fibrinogen.
Ten microliters of enolase-BSA-coated Dynabeads were preincubated with plasminogen and subsequently added to 4 µg of plasminogen-depleted human fibrinogen (Sigma) in a final volume of 125 µl in PBS following a time series of 30 min, 60 min, 120 min, 150 min, 180 min, 240 min, and 300 min at 37°C. Degradation was started by incubation with 500 ng uPA (Haemochrom Diagnostica), and the reaction was stopped with 50 µl of SDS-containing sample buffer. The supernatants were separated by SDS-PAGE, followed by transfer of proteins to a polyvinylidene difluoride membrane (PVDF) (Immobilon-P; Millipore) using a semidry blotting system. The membranes were blocked as described earlier prior to incubation with rabbit antiserum directed against human fibrinogen (1:2,000; Dako). HRP-conjugated anti-rabbit antibodies (1:3,000) were used, and chemiluminescence-based visualization was performed as described above.
Degradation of fibrin matrix by enolase-coated Dynabeads and scm-negative S. canis for electron microscopic visualization.
Fibrin matrix was produced on coverslips by incubating 100 µl of 50 mg ⋅ ml−1 plasminogen-depleted human fibrinogen (Millipore) in PBS with 2 µl of 1.0 KU ⋅ ml−1 thrombin (from bovine plasma; MP Biomedicals) for 10 h at 37°C. Enolase-coated Dynabeads and S. canis G2 pretreated with plasminogen as described were applied in 200 µl PBS to the fibrin matrix for 1 h. Surface-bound plasminogen was activated with uPA (500 ng). Plasminogen-coated S. canis G2 bacteria were used as a control in the absence of uPA.
Electron microscopy; preembedding labeling of surface-displayed enolase and recruited plasminogen.
S. canis was cultured in TSB broth, and 2 ml of the culture was harvested by centrifugation. Bacteria were washed with PBS, and the resulting pellet was resuspended in 200 µl of a 1:25 solution of rabbit antienolase antibodies (2.2 mg/ml IgG, generated by rabbit immunization with recombinant S. pneumoniae enolase). For detection of plasminogen binding, bacteria were incubated with human plasminogen as described for the plasmin activity assay, followed by incubation with goat anti-plasminogen IgG antibodies (80 µg ⋅ ml−1) for 1 h at 30°C. After two washes with PBS, bound antibodies were detected with protein A gold nanoparticles for enolase (15 nm in diameter) or anti-goat IgG gold nanoparticles for plasminogen (15 nm in diameter) by incubating in a 1:25 diluted stock solution. After two washes in PBS, samples were fixed with 2% glutaraldehyde for 15 min at room temperature and washed with TE buffer (20 mM Tris, 1 mM EDTA, pH 6.9) and distilled water. Then, samples were adsorbed onto carbon Butvar-coated copper grids (300 mesh) and observed in a Zeiss Merlin field emission scanning electron microscope (FESEM) at an acceleration voltage of 5 kV. Imaging was done with the high-efficiency SE2 detector. Contrast and brightness were adjusted using the software program Adobe Photoshop CS5.
For colocalization studies of (i) enolase with plasminogen and (ii) plasminogen and SCM, plasminogen gold nanoparticles were prepared by incubation of 50 µg of plasminogen with a gold nanoparticle solution, pH 6.2, with 10-nm- or 15-nm-size gold nanoparticles. After incubation for 30 min at room temperature, the gold nanoparticle complexes were centrifuged and resuspended in PBS with 0.5 mg × ml−1 polyethylene glycol (PEG) 20,000. Enolase of S. canis G2 was then localized as described above. After the last washing step, bacteria were incubated in a 1:20 dilution of the stock plasminogen gold nanoparticles (15 nm) for 30 min, washed, and treated as described above.
For colocalization with SCM, bacteria were incubated with plasminogen gold nanoparticles (10 nm) for 30 min and washed twice in PBS, followed by incubation with SCM (150 µg/ml) for 30 min and washing in PBS. Bound SCM was detected with anti-SCM IgG antibodies and protein A gold nanoparticles (20 nm). Samples were further treated as described above, with the exception that imaging was performed at an acceleration voltage of 10 kV.
Visualization of fibrin degradation.
After preparation of fibrin aggregates on coverslips and incubation with scm-negative S. canis G2 bacteria as described above, samples were fixed in 5% formaldehyde and 2% glutaraldehyde in cacodylate buffer (0.1 M cacodylate, 0.01 M CaCl2, 0.01 M MgCl2, and 0.09 M sucrose, pH 6.9) for 1 h on ice. Before dehydrating in a graded series of acetone (10, 30, 50, 70, 90, and 100%) on ice for 15 min for each step, samples were washed with TE buffer (20 mM Tris and 1 mM EDTA, pH 7.0). Samples were then subjected to critical-point drying with liquid CO2 (CPD 30; Bal-Tec). Dried samples were covered with a gold film by sputter coating (SCD 500; Bal-Tec Union) before examination in a Zeiss DSM 982 Gemini field emission scanning electron microscope (Zeiss, Germany) using the Everhart-Thornley secondary electron (SE) detector and the SE-in-lens detector in a 50:50 ratio with an acceleration voltage of 5 kV. Images were recorded on a MO-disk; contrast and brightness were adjusted by applying Adobe Photoshop CS3.
Transmission electron microscopy.
For detection of SCM recruitment on the surface of scm-negative S. canis G14, bacteria were incubated with a complex of 1.0 µM human plasminogen and 1.0 µM SCM as described for radioactive binding analyses. Bound SCM was visualized by incubation with rabbit anti-SCM IgG antibodies (80 µg/ml) and protein A gold nanoparticles (15 nm in diameter). After two washes in PBS, samples were fixed in 2% glutaraldehyde for 15 min, washed in PBS, TE buffer, and distilled water, and adsorbed onto Butvar-coated copper grids. Samples were examined in a Zeiss TEM 910 microscope at calibrated magnifications and an acceleration voltage of 80 kV. Images were recorded digitally with a Slow-Scan charge-coupled-device (CCD) camera (1,024 × 1,024; ProScan, Scheuring, Germany) using the ITEM software program (Olympus Soft Imaging Solutions, Münster, Germany). Contrast and brightness were adjusted with Adobe Photoshop CS5.
Bacterial survival after phagocytosis by neutrophil granulocytes.
Neutrophil granulocytes (polymorphonuclear leukocytes [PMN]) were purified from human blood using a Polymorphprep (Axis-Shield) two-phase density column. In brief, 10 ml human blood was diluted 1:2 with PBS and loaded onto a column. The column was centrifuged for 35 min at 450 × g. The column layer containing PMN was recovered and sedimented (250 × g) for 10 min. Erythrocyte contamination was removed by osmotic lysis with 9 ml ice-cold water for 30 s followed by addition of 1 ml 100 mM PBS and 5 ml Hanks buffered salt solution (HBSS) (Invitrogen). After centrifugation at 4°C for 10 min (250 × g), the PMN were resuspended in 1 ml HBSS supplemented with 0.15 mM CaCl2 and 1.0 mM MgCl2. Granulocytes (2 × 105) were incubated with 5 × 106 S. canis bacteria for 30 min at 37°C with 5.0% CO2. For opsonophagocytosis analyses, bacteria were preincubated with 1.0 µM plasminogen, 1.0 µM SCM, and 1.0 µM of an SCM-plasminogen complex for 30 min at 37°C with 300-rpm shaking. The bacteria were washed with PBS prior to incubation with granulocytes. Bacterial survival was determined by cultivation after plating of serial dilution on blood agar plates. Experiments were performed in triplicate in three independent analyses using granulocytes from three different donors.
ACKNOWLEDGMENTS
We are grateful to Ina Schleicher, Franziska Voigt, Andreas Raschka, and Sarah Podlech for technical assistance. Christine M. Gillen is gratefully acknowledged for help in purification of recombinant enolase.
The study was supported in part by a fund from the European Community’s Seventh Framework Programme under grant agreement no. HEALTH-F3-2009-223111 and by a grant from the BMBF, Clinical Research Unit Pneumonia (to K.T.P.).
Footnotes
Citation Fulde M, Rohde M, Polok A, Preissner KT, Chhatwal GS, Bergmann S. 2013. Cooperative plasminogen recruitment to the surface of Streptococcus canis via M protein and enolase enhances bacterial survival. mBio 4(2):e00629-12. doi:10.1128/mBio.00629-12.
REFERENCES
- 1. DeWinter LM, Low DE, Prescott JF. 1999. Virulence of Streptococcus canis from canine streptococcal toxic shock syndrome and necrotizing fasciitis. Vet. Microbiol. 70:95–110 [DOI] [PubMed] [Google Scholar]
- 2. Bert F, Lambert-Zechovsky N. 1997. Septicemia caused by Streptococcus canis in a human. J. Clin. Microbiol. 35:777–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yang J, Liu Y, Xu J, Li B. 2010. Characterization of a new protective antigen of Streptococcus canis. Vet. Res. Commun. 34:413–421 [DOI] [PubMed] [Google Scholar]
- 4. Fulde M, Rohde M, Hitzmann A, Preissner KT, Nitsche-Schmitz DP, Nerlich A, Chhatwal GS, Bergmann S. 2011. SCM, a novel M-like protein from Streptococcus canis, binds (mini)-plasminogen with high affinity and facilitates bacterial transmigration. Biochem. J. 434:523–535 [DOI] [PubMed] [Google Scholar]
- 5. Bisno AL, Craven DE, McCabe WR. 1987. M proteins of group G streptococci isolated from bacteremic human infections. Infect. Immun. 55:753–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sheoran AS, Sponseller BT, Holmes MA, Timoney JF. 1997. Serum and mucosal antibody isotype responses to M-like protein (SeM) of Streptococcus equi in convalescent and vaccinated horses. Vet. Immunol. Immunopathol. 59:239–251 [DOI] [PubMed] [Google Scholar]
- 7. Carlsson F, Berggård K, Stålhammar-Carlemalm M, Lindahl G. 2003. Evasion of phagocytosis through cooperation between two ligand-binding regions in Streptococcus pyogenes M protein. J. Exp. Med. 198:1057–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Oehmcke S, Shannon O, Mörgelin M, Herwald H. 2010. Streptococcal M proteins and their role as virulence determinants. Clin. Chim. Acta 411:1172–1180 [DOI] [PubMed] [Google Scholar]
- 9. Whitnack E, Beachey EH. 1982. Antiopsonic activity of fibrinogen bound to M protein on the surface of group A streptococci. J. Clin. Invest. 69:1042–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Whitnack E, Beachey EH. 1985. Inhibition of complement-mediated opsonization and phagocytosis of Streptococcus pyogenes by D fragments of fibrinogen and fibrin bound to cell surface M protein. J. Exp. Med. 162:1983–1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Horstmann RD, Sievertsen HJ, Leippe M, Fischetti VA. 1992. Role of fibrinogen in complement inhibition by streptococcal M protein. Infect. Immun. 60:5036–5041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carlsson F, Sandin C, Lindahl G. 2005. Human fibrinogen bound to Streptococcus pyogenes M protein inhibits complement deposition via the classical pathway. Mol. Microbiol. 56:28–39 [DOI] [PubMed] [Google Scholar]
- 13. Frick IM, Mörgelin M, Björck L. 2000. Virulent aggregates of Streptococcus pyogenes are generated by homophilic protein-protein interactions. Mol. Microbiol. 37:1232–1247 [DOI] [PubMed] [Google Scholar]
- 14. Sandin C, Carlsson F, Lindahl G. 2006. Binding of human plasma proteins to Streptococcus pyogenes M protein determines the location of opsonic and non-opsonic epitopes. Mol. Microbiol. 59:20–30 [DOI] [PubMed] [Google Scholar]
- 15. Kinnby B, Booth NA, Svensäter G. 2008. Plasminogen binding by oral streptococci from dental plaque and inflammatory lesions. Microbiology 154:924–931 [DOI] [PubMed] [Google Scholar]
- 16. Bergmann S, Rohde M, Chhatwal GS, Hammerschmidt S. 2001. Alpha-enolase of Streptococcus pneumoniae is a plasmin(ogen)-binding protein displayed on the bacterial cell surface. Mol. Microbiol. 40:1273–1287 [DOI] [PubMed] [Google Scholar]
- 17. Ringdahl U, Sjöbring U. 2000. Analysis of plasminogen-binding M proteins of Streptococcus pyogenes. Methods 21:143–150 [DOI] [PubMed] [Google Scholar]
- 18. Pöllänen J, Stephens RW, Vaheri A. 1991. Directed plasminogen activation at the surface of normal and malignant cells. Adv. Cancer Res. 57:273–328 [DOI] [PubMed] [Google Scholar]
- 19. Lähteenmäki K, Kuusela P, Korhonen TK. 2001. Bacterial plasminogen activators and receptors. FEMS Microbiol. Rev. 25:531–552 [DOI] [PubMed] [Google Scholar]
- 20. Berge A, Sjöbring U. 1993. Pam, a novel plasminogen-binding protein from Streptococcus pyogenes. J. Biol. Chem. 268:25417–25424 [PubMed] [Google Scholar]
- 21. Jones MN, Holt RG. 2007. Cloning and characterization of an alpha-enolase of the oral pathogen Streptococcus mutans that binds human plasminogen. Biochem. Biophys. Res. Commun. 364:924–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bergmann S, Rohde M, Hammerschmidt S. 2004. Glyceraldehyde-3-phosphate dehydrogenase of Streptococcus pneumoniae is a surface-displayed plasminogen-binding protein. Infect. Immun. 72:2416–2419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pancholi V, Fischetti VA. 1998. Alpha-enolase, a novel strong plasmin(ogen) binding protein on the surface of pathogenic streptococci. J. Biol. Chem. 273:14503–14515 [DOI] [PubMed] [Google Scholar]
- 24. Winram SB, Lottenberg R. 1996. The plasmin-binding protein Plr of group A streptococci is identified as glyceraldehyde-3-phosphate dehydrogenase. Microbiology 142:2311–2320 [DOI] [PubMed] [Google Scholar]
- 25. Lottenberg R, Broder CC, Boyle MD, Kain SJ, Schroeder BL, Curtiss R., III 1992. Cloning, sequence analysis, and expression in Escherichia coli of a streptococcal plasmin receptor. J. Bacteriol. 174:5204–5210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Eriksson A, Norgren M. 2003. Cleavage of antigen-bound immunoglobulin G by SpeB contributes to streptococcal persistence in opsonizing blood. Infect. Immun. 71:211–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pancholi V, Fischetti VA. 1992. A major surface protein on group A streptococci is a glyceraldehyde-3-phosphate-dehydrogenase with multiple binding activity. J. Exp. Med. 176:415–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bergmann S, Wild D, Diekmann O, Frank R, Bracht D, Chhatwal GS, Hammerschmidt S. 2003. Identification of a novel plasmin(ogen)-binding motif in surface displayed alpha-enolase of Streptococcus pneumoniae. Mol. Microbiol. 49:411–423 [DOI] [PubMed] [Google Scholar]
- 29. Bergmann S, Rohde M, Preissner KT, Hammerschmidt S. 2005. The nine residue plasminogen-binding motif of the pneumococcal enolase is the major cofactor of plasmin-mediated degradation of extracellular matrix, dissolution of fibrin and transmigration. Thromb. Haemost. 94:304–311 [DOI] [PubMed] [Google Scholar]
- 30. Bergmann S, Hammerschmidt S. 2007. Fibrinolysis and host response in bacterial infections. Thromb. Haemost. 98:512–520 [PubMed] [Google Scholar]
- 31. Sjöbring U, Ringdahl U, Ruggeri ZM. 2002. Induction of platelet thrombi by bacteria and antibodies. Blood 100:4470–4477 [DOI] [PubMed] [Google Scholar]
- 32. Wistedt AC, Ringdahl U, Müller-Esterl W, Sjøbring U. 1995. Identification of a plasminogen-binding motif in PAM, a bacterial surface protein. Mol. Microbiol. 18:569–578 [DOI] [PubMed] [Google Scholar]
- 33. Wistedt AC, Kotarsky H, Marti D, Ringdahl U, Castellino FJ, Schaller J, Sjöbring U. 1998. Kringle 2 mediates high affinity binding of plasminogen to an internal sequence in streptococcal surface protein pam. J. Biol. Chem. 273:24420–24424 [DOI] [PubMed] [Google Scholar]
- 34. Sanderson-Smith ML, Walker MJ, Ranson M. 2006. The maintenance of high affinity plasminogen binding by group A streptococcal plasminogen-binding M-like protein is mediated by arginine and histidine residues within the a1 and a2 repeat domains. J. Biol. Chem. 281:25965–25971 [DOI] [PubMed] [Google Scholar]
- 35. Sanderson-Smith ML, Dowton M, Ranson M, Walker MJ. 2007. The plasminogen-binding group A streptococcal M protein-related protein Prp binds plasminogen via arginine and histidine residues. J. Bacteriol. 189:1435–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ringdahl U, Svensson M, Wistedt AC, Renné T, Kellner R, Müller-Esterl W, Sjöbring U. 1998. Molecular co-operation between protein PAM and streptokinase for plasmin acquisition by Streptococcus pyogenes. J. Biol. Chem. 273:6424–6430 [DOI] [PubMed] [Google Scholar]
- 37. McKay FC, McArthur JD, Sanderson-Smith ML, Gardam S, Currie BJ, Sriprakash KS, Fagan PK, Towers RJ, Batzloff MR, Chhatwal GS, Ranson M, Walker MJ. 2004. Plasminogen binding by group A streptococcal isolates from a region of hyperendemicity for streptococcal skin infection and a high incidence of invasive infection. Infect. Immun. 72:364–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sanderson-Smith ML, Dinkla K, Cole JN, Cork AJ, Maamary PG, McArthur JD, Chhatwal GS, Walker MJ. 2008. M protein-mediated plasminogen binding is essential for the virulence of an invasive Streptococcus pyogenes isolate. FASEB J. 22:2715–2722 [DOI] [PubMed] [Google Scholar]
- 39. Fischetti VA. 1989. Streptococcal M protein: molecular design and biological behavior. Clin. Microbiol. Rev. 2:285–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Phillips GN, Jr, Flicker PF, Cohen C, Manjula BN, Fischetti VA. 1981. Streptococcal M protein: alpha-helical coiled-coil structure and arrangement on the cell surface. Proc. Natl. Acad. Sci. U. S. A. 78:4689–4693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nilson BH, Frick IM, Akesson P, Forsén S, Björck L, Akerström B, Wikström M. 1995. Structure and stability of protein H and the M1 protein from Streptococcus pyogenes. Implications for other surface proteins of gram-positive bacteria. Biochemistry 34:13688–13698 [DOI] [PubMed] [Google Scholar]
- 42. Swanson J, Hsu KC, Gotschlich EC. 1969. Electron microscopic studies on streptococci. I. M antigen. J. Exp. Med 130:1063–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lancefield RC. 1962. Current knowledge of type-specific M antigens of group A streptococci. J. Immunol. 89:307–313 [PubMed] [Google Scholar]
- 44. Courtney HS, Hasty DL, Dale JB. 2006. Anti-phagocytic mechanisms of Streptococcus pyogenes: binding of fibrinogen to M-related protein. Mol. Microbiol. 59:936–947 [DOI] [PubMed] [Google Scholar]
- 45. Chung MC, Tonry JH, Narayanan A, Manes NP, Mackie RS, Gutting B, Mukherjee DV, Popova TG, Kashanchi F, Bailey CL, Popov SG. 2011. Bacillus anthracis interacts with plasmin(ogen) to evade C3b-dependent innate immunity. PLoS ONE 6:e18119 http://dx.doi.org/10.1371/journal.pone.0018119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bisno AL. 1979. Alternate complement pathway activation by group A streptococci: role of M-protein. Infect. Immun. 26:1172–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Peterson PK, Schmeling D, Cleary PP, Wilkinson BJ, Kim Y, Quie PG. 1979. Inhibition of alternative complement pathway opsonization by group A streptococcal M protein. J. Infect. Dis. 139:575–585 [DOI] [PubMed] [Google Scholar]
- 48. Horstmann RD, Sievertsen HJ, Knobloch J, Fischetti VA. 1988. Antiphagocytic activity of streptococcal M protein: selective binding of complement control protein factor H. Proc. Natl. Acad. Sci. U. S. A. 85:1657–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Thern A, Stenberg L, Dahlbäck B, Lindahl G. 1995. Ig-binding surface proteins of Streptococcus pyogenes also bind human C4b-binding protein (C4BP), a regulatory component of the complement system. J. Immunol. 154:375–386 [PubMed] [Google Scholar]
- 50. Berge A, Kihlberg BM, Sjöholm AG, Björck L. 1997. Streptococcal protein H forms soluble complement-activating complexes with IgG, but inhibits complement activation by IgG-coated targets. J. Biol. Chem. 272:20774–20781 [DOI] [PubMed] [Google Scholar]
- 51. Galdiero F, Romano Carratelli C, Nuzzo I, Bentivoglio C, Galdiero M. 1988. Phagocytosis of bacterial aggregates by granulocytes. Eur. J. Epidemiol. 4:456–460 [DOI] [PubMed] [Google Scholar]
- 52. Ochiai K, Kurita-Ochiai T, Kamino Y, Ikeda T. 1993. Effect of co-aggregation on the pathogenicity of oral bacteria. J. Med. Microbiol. 39:183–190 [DOI] [PubMed] [Google Scholar]
- 53. Smeesters PR, McMillan DJ, Sriprakash KS. 2010. The streptococcal M protein: a highly versatile molecule. Trends Microbiol. 18:275–282 [DOI] [PubMed] [Google Scholar]
- 54. Meehan M, Lynagh Y, Woods C, Owen P. 2001. The fibrinogen-binding protein (FgBP) of Streptococcus equi subsp. equi additionally binds IgG and contributes to virulence in a mouse model. Microbiology 147:3311–3322 [DOI] [PubMed] [Google Scholar]
- 55. Baiano JC, Tumbol RA, Umapathy A, Barnes AC. 2008. Identification and molecular characterisation of a fibrinogen binding protein from Streptococcus iniae. BMC Microbiol. 8:67 http://dx.doi.org/10.1186/1471-2180-8-67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Thern A, Wastfelt M, Lindahl G. 1998. Expression of two different antiphagocytic M proteins by Streptococcus pyogenes of the OF+ lineage. J. Immunol. 160:860–869 [PubMed] [Google Scholar]
- 57. Campo RE, Schultz DR, Bisno AL. 1995. M proteins of group G streptococci: mechanisms of resistance to phagocytosis. J. Infect. Dis. 171:601–606 [DOI] [PubMed] [Google Scholar]
- 58. Hunter WM, Greenwood FC. 1962. Preparation of iodine-131 labelled human growth hormone of high specific activity. Nature 194:495–496 [DOI] [PubMed] [Google Scholar]