

Abstract
Water-soluble compounds of the type [AuCl(PR3)] with alkyl-bis-(m-sulfonated-phenyl)-(mC6H4SO3Na)2 and dialkyl-(m-sulfonated-phenyl)-(mC6H4SO3Na) (R = nBu, Cp) phosphanes have been prepared. Dialkyl-phosphane compounds generate water-soluble nanoparticles of 10-15 nm radius when dissolved in water. These air-stable complexes have been evaluated as catalysts in the synthesis of propargylamines via a three-component coupling reaction of aldehydes, amines and alkynes in water. The antimicrobial activity of the new complexes against Gram-positive and Gram-negative bacteria and yeast has been evaluated. The new compounds display moderate to high antibacterial activity. The more lipophilic compounds are also potent against fungi.
Their cytotoxic properties have been analyzed in vitro utilizing human Jurkat T-cell acute lymphoblastic leukemia cells. Compounds with dialkyl-(m-sulfonated-phenyl)-(mC6H4SO3Na) phosphanes displayed moderate to high cytotoxicity on this cell line. Death cell mechanism occurs mainly by early apoptosis. The catalytic/biological activity of the previously described compound with commercial m-trisulfonated-triphenylphosphine [AuCl(TPPTS)] (6) has been also evaluated to compare the effects of the higher basicity and lipophilicity of the alkyl- and di-alkyl-(m-sulfonated-phenyl) phosphanes on these new compounds.
Keywords: water-soluble gold, nanoparticles, recyclable catalysts, antimicrobial, cytotoxicity, apoptosis
Introduction
Gold(I) compounds with phosphanes have been known for many years.[1] Most of gold compounds in the oxidation state +1 are two coordinate, linear, and 14 electron species. Other complexes with different coordination numbers have also been characterized although they are not that common. They are usually three-coordinate trigonal-planar complexes and four-coordinate gold(I) complexes (including those with diphosphane chelating ligands).[2] Several gold(I) compounds with phosphanes display interesting structural properties, dynamic behaviour in solution or luminescent properties.[2] Many of these compounds have allowed for the observation of aurophilicity or Au…Au interactions (due to relativistic effects).[3] An isolobal relationship of [Au(PPh3)]+ cations and H+ and Li+ has been proposed based on the behaviour of [Au(PPh3)]+ fragments in many homo and hetero-metallic clusters;[4] some of them having a very unusual coordination number for the central atom (e.g. C and O).[5] The isolobality can be exploited in the application of organometallic compounds of gold(I) [AuR(PPh3)] as a less toxic, air stable and room temperature alternative to organolithium, organomagnesium and organomercury compounds. Thus these complexes can be used as transmetallating agents to other metallic centers.[6] Gold(I) complexes with phosphanes have displayed toxicity against different tumour cell lines while targeting mitochondria.[7] They have also displayed antimalarial properties,[8] moderate antimicrobial activities[9] and have found applications as antireumathic pharmaceuticals (auranofin).[10] More recently an increasing number of papers based on the high catalytic activity of mainly [Au(PR3)]+ cations and other gold(I)-phosphane derivatives in different chemical processes have been reported.[11]
As green chemistry principles are being incorporated in academic and industrial settings, the development of water-based reactions and water-soluble catalysts has become extremely important. Since water is the biologically most relevant solvent, the synthesis of metal-based drugs with enough solubility and stability in water or physiological media is still a matter of increasing interest.
Gold(I) compounds with water-soluble phosphanes (di, tri or even four-coordinated) have been described.[12] Some of the phosphanes used are depicted in figure 1. Sulfonated aryl phosphanes (abbreviated TPPMS, TPPDS, TPPTS),[13] mono o di-phosphanes containing pyridyl groups such as DnPYPE, DnPYPP[14] or maleic acid (DPMAA),[15] phosphanes like 1,3,5-triaza-7-phosphaneadamantane (TPA) or 3,7-diacetyl-1,3,7-triaza-5-phosphabicyclo[3.3.1] nonane (DAPTA)[16] have been employed in the preparation of gold compounds with simple halide ligands (Cl, Br) or other coordination/organometallic complexes with thiolato, thionato, or alkynyl ligands. More recently, phosphanes containing hydroxyl groups[17] and water-soluble cyclodiphosphazanes[18] have been employed in the preparation of gold(I) compounds. Some of the compounds have been structurally characterized (affording linear coordinated or polymeric structures depending on the phosphine) in the solid state. Some of the complexes with these phosphanes display interesting photoluminescence[13,15] and cytotoxic properties against different tumour cell lines. [14,15,16j,17,18]
Figure 1.
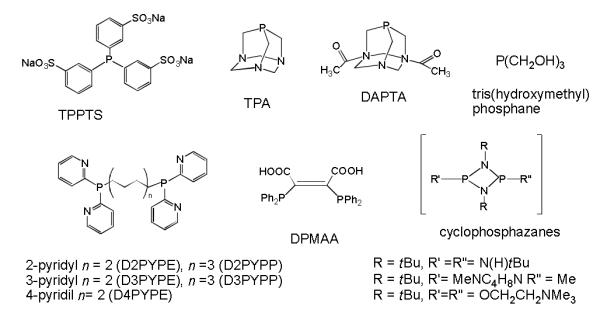
Water-soluble phosphanes used in the preparation of gold(I) compounds.
[AuCl(TPPTS)] is a good co-catalyst in the bi-phasic palladium-catalysed alkynylation reaction[19a] while [AuR(TPPTS)] compounds (R = alkynyl) are efficient and recyclable catalysts in the hydration of alkynes. [20] Surprisingly studies on the microbial activity of gold compounds with water soluble phosphanes have been neglected.
The increasing interest on homogeneous catalysis in water as alternative reaction media with the recovery of the catalyst as well as the development of gold catalysis (a current scientific hotspot) [21] warrants the study of new water-soluble gold compounds. The study of the biological properties of these compounds becomes relevant in the search of new metallo-pharmaceuticals.
We describe here the synthesis of new water-soluble gold(I) compounds that contain the recently described alkyl-bis(m-sulfonated-phenyl)- and dialkyl-(m-sulfonated-phenyl)-phosphanes. [22] The incorporation of other organic groups such alkyls (cyclopentanyl and buthyl) in these sulfonated phosphanes can modulate not only the electronic and steric parameters of these phosphanes (increasing their basicity) but their hydrophilicity/lipophilicity properties. This makes the new compounds good candidates to study their application in homogeneous catalysis in water and, to explore some biological properties oriented to their use as pharmaceuticals. We have chosen a three-component coupling reaction of aldehydes, amines and alkynes in water to synthesize propargylamines which are extremely versatile building blocks for organic synthesis.[23,24] We have also evaluated the antimicrobial properties of these compounds against bacteria (gram negative and gram positive) and yeasts. The cytotoxic and apoptotic properties of these complexes have been evaluated in vitro utilizing human Jurkat T-cell acute lymphoblastic leukemia cells. We have used the previously described [AuCl(TPPTS)][19a] compound 6 with commercially available phosphane [P(mC6H4SO3Na)3] (TPPTS) 1 in order to compare the behaviour of alkyl-susbtituted and unsubstituted sulfonated phosphanes in the catalytic/biological properties of the water-soluble gold compounds.
Results and Discussion
Synthesis of Gold(I) Compounds with Alkyl-bis(m-sulfonated- phenyl)- and Dialkyl-(m-sulfonated-phenyl)-Phosphanes
The preparation of the new complexes and compound [AuCl(TPPTS)] (6)[19a] is achieved by displacing the labile ligand tht (tetrahydrothiophene) from the starting gold (I) material [AuCl(tht)] by the water-soluble phosphanes (Scheme 1).[19] The complexes are obtained as white solids that are stable at air. They are all soluble in water (with solubilities in the range of 125 to 75 g/L, see experimental) but they have different stabilities in solution. The most stable compounds (in water solution and at RT) are derivatives with three aryl-sulfonated groups in the phosphane 6, or one alkyl group and two aryl-sulfonated groups (7, 8). Complexes with two alkyl groups and one sulfonated aryl group (9, 10) display a different behaviour. The new compounds have been characterized by different spectroscopic (31P{1H}, 13C{1H}, 1H, NMR), spectrometric (ESI-MS) and analytical techniques (C, H, S microanalysis). 31P{1H} NMR spectroscopy is quite useful in the characterization and study of the stability of these complexes over time. In all cases the chemical shifts correspond to usual Au(I)-phosphane chemical shifts for linear compounds. The 31P{1H} NMR data for the free phosphane ligands, for 6 and new gold(I) compounds 7-10 are collected in table 1 (see also supporting information).
Scheme 1.
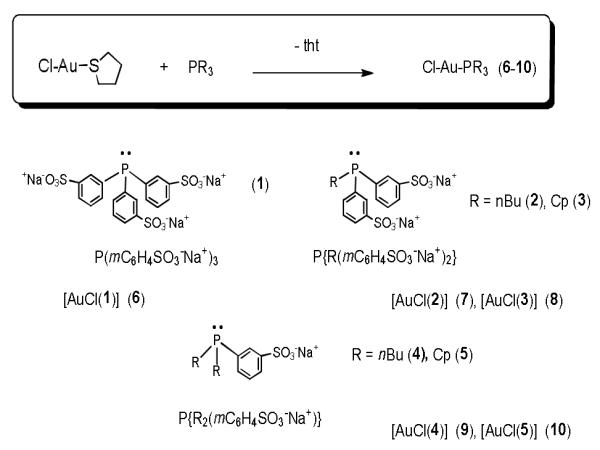
Preparation of the new water-soluble compounds and complex 6.[19]
Table 1.
31P{1H} NMR chemical shifts (ppm) in D2O (25 °C) for the water-soluble phosphane ligands 1, 2-5[22] and for the new gold(I)-phosphane complexes 6-10.
| Free Ligands |
1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| δ31P{1H} NMR |
− 5.88 (s) |
− 15.22 (s) |
−3.11 (s) |
−23.56 (s) |
3.00 (s) |
| [AuClL] | 6 | 7 | 8 | 9 | 10 |
| δ31P{1H} NMR |
33.40 (s) |
30.40 (s) |
46.06 (s) |
29.68 (s) |
56.77 (s) |
Compounds 1-3 are stable during 48 hours in aqueous solution at RT as shown by 31P{1H} NMR spectroscopy. After this time a little decomposition to metallic gold can be observed but the signals in the 31P{1H} NMR spectra remain the same for weeks. Compounds with the more basic dialkyl-(m-sulfonated-phenyl)-(mC6H4SO3Na) phosphanes 9 and 10 have a different behaviour. 9 and 10 can be obtained as analytical pure white compounds (see experimental) but when dissolved in water after a few minutes they give a solution of intense purple colour indicative of water-soluble colloids. The 31P{1H} NMR spectra does not change much over time and only small signals (singlets) accompanying the main singlet at 51.94 (9) and 65.61 (10) ppm can be seen after 24 h at RT. In the case of 10 the extra signal at 65.61 ppm become a major signal after 48 hours (see supporting information). While we do not have information about the nature of these decomposition products, complexes 9 and 10 are stable enough at RT during 24 h to perform the biological tests described here.
Unfortunately attempts to get crystals of enough quality for X-ray diffraction studies (6-10) were unsuccessful.
We characterized these purple solutions to test for the presence of gold colloids. Indeed, TEM analysis confirmed the formation of homogeneous nanoparticles with a 10-15 nm radius (Figure 2, see experimental section for details). Smaller dimensions (2-3 nm) have been previously observed for fluorous-soluble gold nanoparticles that were generated from gold(I)-fluorous phosphane derivatives.[25]
Figure 2.

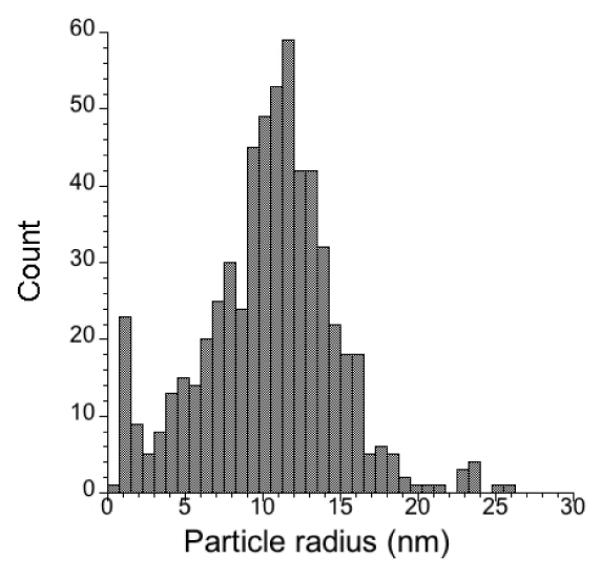
a) TEM field-view image at 10,000x of nano-9. The insert shows a detail at 50,000 x.
(b) Histogram of particle count as a function of particle radius (nm) in the image
Catalytic Activity in the Synthesis of Propargylamines via a Three-component Coupling Reaction of Aldehydes, Amines and Alkynes
We used the water-soluble compounds in a coupling reaction with formation of C-C and C-N bonds. The reaction is a one-pot reaction with a three-component of aldehyde, alkyne, and amines via C-H activation. Gold salts like AuCl or AuBr3 had resulted very good catalysts for this reaction (in amounts of 5 or 1 mol%).[24a] However these salts are hygroscopic, highly acidic and can not be handled at air. The reactions were generally run at 100 °C and water was obtained as the only byproduct of the reaction (Scheme 2).
Scheme 2.
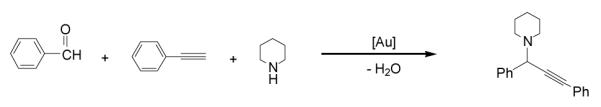
Three-component coupling of benzaldehyde, piperidine and phenylacetylene catalyzed by gold.[23,24]
More recently other gold catalysts have been employed in this reaction.[23,24b-g] Gold(III)-salen complexes afforded excellent yields at 40 °C and when chiral prolinol derivatives were used as the amine component, excellent diastereoselectivities (up to 99:1) were obtained.[24c] A gold(III) C,O metallocycle (with a phosphinamide ligand) has also resulted highly efficient for this three-component coupling.[24g] The synthesis of quinoline derivatives can be achieved by a sequential catalytic process by AuCl3/CuBr (5 mol%/30 mol%).[24e] The recyclability of the system was achieved for 4 runs with gold nanoparticles (10 mol %) at 75-80 °C.[24d] Supported gold catalysts (layered double-supported gold[24b] and gold nanoparticles supported on nanocrystaline ZrO2 and CeO2, 2.8 mol%[24f]) have resulted highly active, selective (high diastereoselectivities for chiral amines) and recyclable in this A3 coupling in water at 100 °C.[24f] However, apart from the report on AuCl by Liu, there were no reports on the catalytic activity of gold(I) compounds in these reactions.
We tested the most stable of our compounds (6-8) under different reaction conditions in a biphasic system consisting of 1 mL of H2O (where we dissolved the gold compounds) and the organic reagents. The separation of the gold catalyst after the reaction had taken place was by simple decantation of the two layers. The results of these studies are collected in table 2. We found good yields when reactions were run at 100 °C and with 5 mol% of catalysts (entries 2,7 and 9). Yields improve by addition of NaPF6 (which should facilitate the removal of chloride from the gold compounds). Moreover with compound 6 we found good conversions at room temperature if reactions were run during 42 hours (entry 5). When reactions were run at RT or 40 °C we did not observe decomposition to metallic gold (we observed this decomposition at 100 °C) and we decided to recycle the water layers from these runs in order to study the recyclability of the system.
Table 2.
Three-component coupling of benzaldehyde, piperidine and phenylacetylene catalyzed by water-soluble gold compounds (6-8).
| Run [a] |
Catalyst | Co- catalyst |
Temp. (°C) |
Time (h) |
Yield (%) |
Conv. (%)[b] |
|---|---|---|---|---|---|---|
| 1 | 6 (1 mol%) | --- | 100 | 12 | 17 | 54 |
| 2 | 6 (5 mol%) | --- | 100 | 17 | 62 | 44 |
| 3 | 6 (5 mol%) | --- | 40 | 42 | 71 | 100 |
| 4 | 6 (5 mol%) | NaPF6 (5 mol%) |
100 | 12 | 78 | 56 |
| 5 | 6 (5 mol%) | --- | RT | 42 | 81 | 98 |
| 6 | 6 (2.5 mol%) | NaPF6 (5 mol%) TPPTS (5 mol%) |
100 | 12 | 78 | 56 |
| 7 | 7 (5 mol%) | --- | 100 | 17 | 99 | 85 |
| 8 | 7 (7 mol%) | --- | RT | 42 | 12 | 70 |
| 9 | 8 (5 mol%) | NaPF6 (5 mol%) |
100 | 12 | 94 | 93 |
| 10 | 8 (5 mol%) | NaPF6 (5 mol%) |
50 | 24 | 48 | 91 |
| 11 | 8 (5 mol%) | NaPF6 (5 mol%) |
40 | 21 | 25 | 88 |
| 12 | 8 (7 mol%) | --- | RT | 42 | 39 | 97 |
| 13 | 6 (7 mol%) | NaPF6 (7 mol%) |
RT | 42 | 75 | 96 |
| 14 | RPa (run 13) | --- | RT | 42 | 65 | 83 |
| 15 | RPa (run 14) | --- | RT | 42 | 53 | 77 |
| 16 | RPa (run 15) | --- | RT | 42 | 38 | 85 |
| 17 | 6 (7 mol%) | --- | RT | 93 | 91 | 87 |
| 18 | RPa (run 17) | --- | RT | 42 | 88 | 85 |
| 19 | RPa (run 18) | --- | RT | 42 | 83 | 80 |
| 20 | RPa (run 19) | --- | RT | 42 | 77 | 74 |
RP: recycled water phase containing the gold catalyst.
Determined by 1H NMR analysis of crude reaction mixture based on benzaldehyde conversion.
The study of recycling catalyst 6 is collected in entries 13-16. With 7 mol% of 1 and 7 mol% of NaPF6 the catalyst can be separated and re-used 4 times. However, the yields of product decrease steadily from run to run from 75 to 38 (4th run). Addition of NaPF6 was however not necessary and we could separate catalyst 6 and recycle it with higher conversion and less decrease in the yield/conversion from run to run (entries 17-19) of product. It seems that at RT the addition of NaPF6 is not as beneficial as at higher temperatures.
While preparing this manuscript a report on the recyclability of an organogold(III) complex was reported.[23] The compound of the type [Au(C-N)Cl2] (N-CH = 2-phenylpyridine) was an efficient and selective catalyst (1 mol%) for this three-component coupling reaction in water at 40 °C. Notably this complex could be repeatedly used for 10 cycles, leading to an overall turnover number of 812.[23] While our system is not so efficient, it has allowed for the synthesis of propargylamines at RT. These compounds are stable in water in mild conditions (up to 40 °C) for several hours and this may allow for their use as catalysts in some other processes in the future in which a higher basicity of the phosphanes coordinated to the gold centres are preferred.
Biological Activity: Antimicrobial Activity and Cytotoxic Properties In Vitro
Antimicrobial Activity
The antimicrobial activity of the new compounds as well as that of compound 6 were evaluated against yeast (Saccharomyces cerevisiae) Gram-negative (Escherichia coli and Salmonella tiphymurium) and Gram-positive (Bacillus cereus and Staphyloccocus aureus) bacteria. The results of the experiments are collected in table 3. They all have moderate to high antibacterial activity. The compounds with the highest lipophilicity (9 and 10) are also potent against fungi.
Table 3.
Toxicity assesment of compounds 6-10 against microbial organisms.[a]
| Compound |
S.
Cerevisiae |
E.
Coli |
S.
Tiphymurium |
B. Cereus |
S.
Aureus |
|---|---|---|---|---|---|
| 6 | --- | 10 | 100 | 10 | 100 |
| 7 | --- | 100 | 100 | 100 | 100 |
| 8 | --- | 100 | 10 | 100 | 100 |
| 9 | 100 | 100 | 100 | 1 | 1 |
| 10 | 100 | 100 | 10 | 10 | 10 |
Minimun number of μ required to create a zone of clearing of 0.5 cm diameter on a lawn of fungal or bacterial cells: 100, 10, 1 or --- no toxicity.
Saccharomyces cerevisiae appears resistant to this class of compounds. The plasma membrane appears to be an effective barrier to the more hydrophylic compounds (6-8), whereas the more lipophilic compounds with dialkyl-(m-sulfonated-phenyl)-(mC6H4SO3Na) phosphanes (9, 10) are able to penetrate the plasma membrane to suppress growth. Compounds 9 and 10 suppress yeast growth in a zone of about 2.5 cm diameter. Further testing with other fungal species is required to see if this pattern of resistance is uniform across the fungi.
Among the bacteria we found that Gram-positives are 100-fold more sensitive to [AuCl{PR2(mC6H4SO3Na)}] than Gram-negatives, whether R is cyclopentanyl (9) or n-butyl (10). Gram negatives are moderately resistant, showing growth suppression at only at 100 ug/disk, however individual species may selective sensitivity to a particular molecule. Thus Escherichia coli was more sensitive to [AuCl{PmC6H4SO3Na}3] (6) whereas the cyclopentanyl group in [AuCl{PCp(mC6H4SO3Na)}] (8) and [AuCl{PCp2(mC6H4SO3Na)}] (10) increases toxicity to Salmonella typhimurium. For both Gram-negative organisms clearance zones were 1.0-1.4 cm dia. regardless of quantity of each compound.
The behaviour for Bacillus cereus and Staphyloccocus aureus are nearly identical and compounds 9 and 10 (the more lipophilic) resulted the more toxic to these organisms. For both compounds the zones of growth suppression exceeded 2.0 cm diameter at 100 μg per disk. The relative difference in toxicity between 9 and 10 could be due to the fact that the phosphine in compound 9 contains a bulkier Cp group (since both phosphanes should have similar lipophilicity).
A similar general trend (more toxic for Gram-positive bacteria than Gram-negative or fungi) has been noted previously with auranofin[9a] and some other gold(I) phosphane derivatives. [9b-d,9f,g] However caution has to be applied when generalizing the results since many factors are involved in the antimicrobial activities. Thus the nature of other ligands coordinated to the gold centers (besides the phosphanes) plays a decisive role. For instance a more complicated pattern is found for gold-thiol-phosphane derivatives and the antimicrobial potency can be higher for Gram-negatives or fungi depending on the combination thiol-phosphane.[9a]
An explanation for the behavior displayed by the new complexes 7-10 could be as follows: if these compounds interfere with respiration (a mitochondrial function) then the fungi should be most resistant because electron transport is shielded inside internal organelles, whereas in bacteria electron transport occurs at the plasma membrane. Gram-negatives have a protective outer second membrane; therefore the compounds do not have ready access to the electron transport chain. Finally, the most lipophilic (hydrophobic) molecules are the most effective for Gram-positives. This is explained by the ability of 9 and 10 to partition into or through the membrane, whereas 6, the most highly charged compound, is less likely to pass through the plasma membrane.
Cytotoxic Properties and Apoptosis Studies
The cytotoxic properties of gold(I) 6-10 compounds were analyzed in vitro according to the procedures described by Montoya et al[26] and our laboratories[27] utilizing Jurkat T-cell acute lymphoblastic leukemia cells. Before use, all tested compounds were dissolved in physiological saline solution (NaCl 0.9%), and dilutions of each compound were then added to the cells in normal growth medium. All the tested complexes have proven, by 31P NMR studies, to be stable in D2O over 24 hours or more. 9 and 10 give stable water-soluble nanoparticles when dissolved in water.
Cis-platin was used as a positive control as previously reported by our laboratories with organogold(III) complexes containing iminophosphorane ligands.[27]
Compounds with dialkyl-(m-sulfonated-phenyl)-(mC6H4SO3Na) phosphanes that produced nanoparticles when dissolved in water displayed moderate to high cytotoxicity on this cell line. It appears that an increase in the lipophilic properties of the phosphanes increases the cytotoxicity of this type of compounds. The cytotoxicity of 9 was not much higher than that displayed by cis- platin. Compound 10 was the most cytotoxic compound of all tested, even more than cis-platin for this cell line. The IC50 values for 9 and 10 are similar to those found for some organogold(III) complexes with iminophosphorane C,N skeletons and chloride and/or water-soluble phosphanes recently described by us[27] but not as high as those with the combination of iminophosphorane and dithiocarbamate ligands (1-2 μM). [27] Very recently Bindoli and co-workers have reported that well-known gold(I) phosphane complexes such S-triethylphosphanegold(I)-2,3,4,6-tetra-O-acetyl-1-thio-B-D-glucopyranoside (auranofin) and triethylphosphane gold(I) chloride induced apoptosis in Jurkat T cells.[28] Although IC50 values were not provided different experiments were run with 1 μM of gold compounds for which % cell mortality was between 20 and 27. This apoptosis appeared to be triggered by inhibition exerted by the gold(I) compounds on the cytosolic and mitochondrial isoforms of thioredoxin reductase, which resulted in an increase in H2O2. However, no significant lipid peroxidation or nitric oxide formation were observed after incubation with gold(I) complexes, indicating that the cells had not been subjected to extensive oxidative stress. Polymeric gold(I) compound aurothiomalate was poorly effective, both in inhibiting thioredoxine reductase and in inducing apoptosis.[28]
In order to gain some insight of the type of cell death that complexes 6-10 induce in this cancer cell line, we performed some apoptosis assays with Jurkat cells (see experimental for details). As cells may undergo programmed cell death (apoptosis) or necrosis, the mode of death mediated by our compounds was investigated. In early stages of apoptosis, one of the significant biochemical features is loss of plasma membrane phospholipid asymmetry, due to translocation of phosphatidylserine (PS) from cytoplasmic to extracellular side. This characteristic allows detection of externalized PS by the specific binding of Annexin V (FITC-conjugatated). Initiation of cell death will eventually result in the permeabilization of the cell membrane, allowing PI to stain DNA within the nucleus. The results of these studies are summarized in figure 3.
Figure 3.
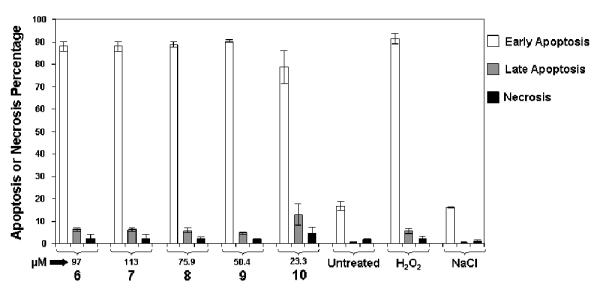
Preferential induction of early apoptosis in Jurkat cells after treatment with the gold (I) complexes. The IC50 concentration shown in table 4 was used for each gold compound.
All five compounds tested demonstrated that the inflicted cell damage was preferentially due to apoptosis instead of necrosis. The preferential apoptosis induced is markedly much higher than that for the gold(I) phosphane compounds described by Bandoli and coworkers.[28] Apparently, compound 10 may be slightly more toxic than the other four compounds tested, since it induces higher levels of late apoptosis during 16 h of incubation time. As described in the experimental section, three controls were included which were untreated cells, and H2O2 and physiological saline treated samples. There was no significant difference between untreated cells, as compared with cells treated with sodium chloride. There is no significant difference between untreated cells, as compared with cell treated with sodium chloride. Auranofin is known to also induce apoptosis in HL-60 cells,[29] ovarian cancer cells[30] and cardiac tissue.[31] Other gold(I) phosphane or phosphite complexes such as water-soluble compounds with the D2PYPP diphosphane ligand,[32] cyclodiphosphazanes[17] or compounds with bis(phosphite) ligands containing mesocyclic thioether moieties[33] have induced apoptosis in HeLa, breast cancer and/or HCT-116 cell lines by activation of p53 protein.
We also performed a study of the cytotoxicity of compound 10 after having being dissolved in the media and kept in the absence of light and at - 20°C for 30 days. In figure 4, the cytotoxicity of a freshly prepared solution of 10 is sample 1. The cytotoxicity of this complex after 30 days at − 20°C is shown in sample 2. We also measured the citotoxicity of solutions that were kept in the dark and at − 20°C for 28 days and then kept at RT for two extra days either in the dark (sample 4) or in the presence of artificial light (sample 3). Basically the cytotoxicity of these solutions decreases to ca. half of the value of that for a freshly prepared solution of compound 10.
Figure 4.
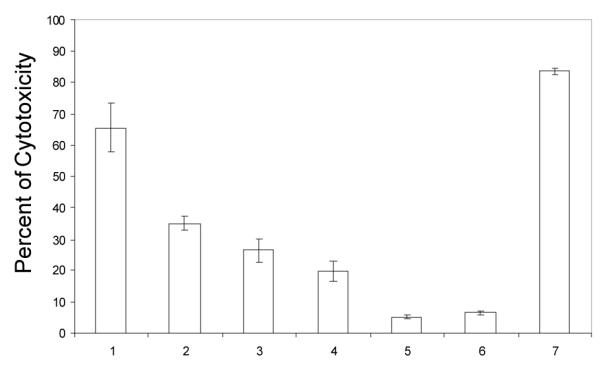
Testing cytotoxic activity against Jurkat cells of compound 10 after exposure to artificial light at room temperature and time of being dissolved. 1) freshly prepared sample of 10 (25 μM in saline solution); 2) solution of 10 kept at − 20°C during 30 days in the dark; 3) solution of 10 kept at − 20°C during 28 days in the dark plus 2 additional days at RT in the presence of light; 4) solution of 10 kept at −20°C during 28 days in the dark plus 2 additional days at RT in the dark; 5) untreated cells; 6) cells treated with 1 μL of NaCl (0.9%); 7) cells treated with 500 μM of H2O2 (positive control). Incubation time 22 h.
Compounds 6-10 give [AuPR3]+ species in solution which may exert an effect to pass more easily through the lipid bilayer of membranes and, possibly, mitochondria like it happens for some delocalized lipophilic cations (DLCs) of gold(I).[33] The degree of lipophilicity of the ligands has been recently connected to the in vitro cytotoxicity. A higher lipophilicity of the gold(I) drugs with phosphines results in a higher in vitro antitumor and hepatotoxicity potency.[34] We believe that further modification of the gold(I) compounds described here by exchange of chloride by other organic or inorganic ligands may render promising stable water-soluble cytotoxic compounds with an optimized lipophilicity value. Interestingly, gold nanoparticles have been found to enhance the anti-leukemia action of a 6-mercaptopurine chemotherapeutic agent.[34] The most cytotoxic compounds described here (9 and 10) generate water-soluble nanoparticles when dissolved in water. Further biochemical assays will be performed in order to determine the mechanism that triggers apoptosis for these complexes as well as their toxicity against normal human cells.
Conclusions
We have prepared gold(I) compounds with water-soluble phosphanes with higher basicity and lipophilicity than previously reported [AuCl{P(mC6H4SO3Na)3}] (6)[19a] by introduction of one or two alkyl groups (R = nBu or Cp) in the sulfonated aryl phosphane ([PR(mC6H4SO3Na)2] or [PR2(mC6H4SO3Na)). The complexes with two alkyl groups in the phosphanes (9 and 10) produce nanoparticles when dissolved in water as opposed to 6 or compounds with just one alkyl group (7 and 8). While these compounds have shown to be not as efficient catalysts as 6 (the most hydrophilic complex) in a biphasic three-component coupling reaction in the synthesis of propargylamines, they have displayed interesting biological activities (antimicrobial and anticancer). They all have moderate to high antimicrobial activity. The compounds with the highest lipophilicity (9 and 10) are the most potent for Gram-positive bacteria and fungi. Also the higher lipophilicity of, especially compounds with dialkyl-(m-sulfonated-phenyl)-(mC6H4SO3Na) phosphanes (9 and 10) makes them the most cytotoxic of the group (6-10) in vitro against human Jurkat T-cell acute lymphoblastic leukemia cells. 10 is even more cytotoxic than cis-platin for this cell line. Death cell mechanism occurs mainly by early apoptosis for all the compounds studied (6-10) like for some other gold(I)-phosphane complexes (although compounds 6-10 are more apoptotic than those described previously).
The higher basicity of the new gold complexes can be further exploited for some other homogeneous biphasic catalytic processes. Subsequent modification of these complexes by exchange of chloride for other ligands may afford complexes with an optimized lipophilicity value with potential applications as more efficient antimicrobial and anticancer agents.
Experimental Section
1. Sythesis and Characterization of the Gold(I) Water-soluble Complexes
Solvents were purified by use of a PureSolv purification unit from Innovative Technology, Inc.; all other chemicals were used as received. Elemental analyses were carried out by Atlantic Microlab, Inc. (US). The 1H, 13C{1H} and 31P{1H} NMR spectra were recorded in D2O solutions at 25 °C on a Bruker 400 spectrometer (δ, ppm; J, Hz); 1H and 13C{1H} were referenced using the solvent signal as internal standard while 31P{1H} was externally referenced to H3PO4 (85%). The mass spectra (ESI) were recorded from acetone or water solutions by the mass spectrometry facility of the University of California Riverside (US). The preparation[19] of compound 6 is slightly different to that previously reported.[19a] Phosphanes 2-5 [PR(mC6H4SO3Na}2] and [PR2(mC6H4SO3Na}] (R = nBu, Cp) were prepared as recently described.[22] [AuCl(tht)] was prepared following the procedure of Uson et al.[36] The rest of chemicals and solvents were purchased from Sigma-Aldrich.
[AuCl{P(mC6H4SO3Na}3][19a] (6)
To a solution of [AuCl(tht)] (0.192 g, 0.6 mmol) in 15 mL of dichloromethane a solution of [P(mC6H4SO3Na)3] (0.341g, 0.6mmol) in 5 mL of acetone and 9 drops of water was added drop-wise. The reaction mixture was stirred at RT during 30 min and all the solvents removed under vacuum. By addition of 5 mL of acetone/10 mL of diethyl ether compound 6 was obtained as a white solid that was filtered-off and dried under vacuum. Yield: 0.480 g, 99%. Solubility in H2O: 125 g/L. 31P{1H} NMR (D2O) δ = 33.4 (s).
[AuCl{PnBu(mC6H4SO3Na)2}] (7)
To a solution of [AuCl(tht)] (0.0962 g/ 0.3 mmol) in 15 mL of dichloromethane a solution of [PnBu(mC6H4SO3Na)2] (0.134 g/0.3 mmol) in 5 mL of acetone and 9 drops of water was added drop-wise. The reaction mixture was stirred at RT during 30 min and all the solvents removed under vacuum. By addition of 5 mL of acetone/10 mL of diethyl ether compound 7 was obtained as a white solid. Yield: 0.195 g, 96%. Solubility in H2O: 125 g/L. Anal. Calc. for 7 C12H15AuClNa2PO6S2 (678.81): C, 28.31; H, 2.52; S, 9.44; found: C, 27.75; H, 3.15; S, 9.33; MS(ESI-) [M/Z]: 654.94 [M - Na]−. 31P{1H} NMR (D2O) δ = 30.40 (s); 1H NMR (D2O) δ = 0.71 (br, 3H, CH3 Bu), 1.33 (m, br, 4H, -CH2-CH2-CH2-P), 2.53 (br, 2H, -CH2-P), 7.53 (br, 2H, H5, mC6H4SO3Na,), 7.67 (br, 2H, H6, mC6H4SO3Na), 7.87 (br, 2H, H4, mC6H4SO3Na), 7.97 (d, 2H, H2, mC6H4SO3Na, 3JP-H = 12). 13C{1H} NMR (D2O) δ = 12.99 (s, CH3, Bu), 23.21 (d, 3JP-C = 16.4 Hz, -CH2-CH2-P, Bu), 27.05 (s, CH3-CH2-), 40.10 (s, -CH2-P), 129.26 (br, C2, mC6H4SO3Na), 129.72 (d, 1JP-C = 12.68 Hz, C1, mC6H4SO3Na), 130.16 (br, C4, mC6H4SO3Na), 130.29 (br, C5, mC6H4SO3Na), 136.14 (d, 2JP-C = 12.98 Hz, C6, mC6H4SO3Na), 143.83 (br, C3, m-C6H4-SO3Na).
[AuCl{PCp(mC6H4SO3Na)2}] (8)
To a solution of [AuCl(tht)] (0.0962 g/ 0.3 mmol) in 15 mL of dichloromethane a solution of [PCp(mC6H4SO3Na)2] (0.136 g/0.3 mmol) in 5 mL of acetone and 9 drops of water was added drop-wise. The reaction mixture was stirred at RT during 30 min and all the solvents removed under vacuum. By addition of 5 mL of acetone/10 mL of diethyl ether compound 8 was obtained as a white solid. Yield: 0.188 g, 92%. Solubility in H2O: 125 g/L. Anal. Calc. for 8 C17H16AuClNa2PO6S2 (700.33): C, 29.15; H, 2.30; S, 9.15; Found: C, 29.11; H, 3.22; S, 10.1; MS(ESI-) [M/Z]: 666.94 [M - Na]−. 31P{1H} NMR (D2O) δ = 45.06 (s); 1H NMR (D2O) δ = 1.4-1.6 (br m, 9H, Cp), 7.51 (t, 2H, H5, m-C6H4-SO3Na, 3JH-H = 8), 7.76 (q, 2H, H6, m-C6H4-SO3Na), 7.82 (d, 2H, H4, m-C6H4-SO3Na, 3JH-H = 16), 8.11 (d, 2H, H2, m-C6H4-SO3Na, 3JP-H = 12). 13C{1H} NMR (D2O) δ = 26.5 (d, 2JP-C = 9.33 Hz, Cb, Cp), 30.31 (d, 3JP-C = 5.82 Hz, Cc, Cp), 34.51 (d, 1JP-C = 38.47 Hz, Ca, Cp), 129.24 (br, C2, mC6H4SO3Na), 129.87 (d, 1JP-C = 75.68 Hz, C1, mC6H4SO3Na), 129.93 (br, C4, mC6H4SO3Na), 130.10 (br, C5, mC6H4SO3Na), 136.44 (d, 2JP-C = 12.98 Hz, C6, mC6H4SO3Na), 143.83 (d, 3JP-C = 10.72 Hz, C3, mC6H4SO3Na).
[AuCl{PnBu2(m-C6H4SO3Na)}] (9)
To a solution of [AuCl(tht)] (0.178 g, 0.556 mmol) in 6 mL of dry acetonitrile a solution of [PnBu2(mC6H4SO3Na)] (0.181g, 0.556 mmol) in 1.5 mL of deoxygenated of water was added drop-wise. The reaction mixture was stirred under argon at RT during 30 min and all the solvents removed under vacuum. By addition of 5 mL of acetone/10 mL of diethyl ether compound 9 was obtained as a white solid. Yield: 0.22 g, 71%. Solubility in H2O: 75 g/L. Anal. Calc. for 9 C14H22AuClNaPO3S (556.77): C, 30.20; H, 3.98; S, 5.76; Found: C, 30.11; H, 4.20; S, 6.01. MS(ESI-) [M/Z]: 533.03.94 [M - Na]−. 31P{1H} NMR (D2O) δ = 29.68 (s); 1H NMR (D2O) δ = 0.61 (br, 6H, CH3 Bu), 1.27 (m, br, 8H, -CH2-CH2-CH2-P), 2.07 (br, 4H, - CH2-P), 7.43 (br, H, H5, mC6H4SO3Na,), 7.5-8.05 (vbr, 3H, H6, H4, H2, mC6H4SO3Na). 13C{1H} NMR (D2O) δ = 12.99 (s, CH3, Bu), 23.21 (d, 3JP-C = 16.4 Hz, -CH2-CH2-P, Bu), 27.05 (s, CH3-CH2-), 40.10 (s, -CH2-P), 129.26 (br, C2, mC6H4SO3Na), 129.72 (d, 1JP-C = 12.68 Hz, C1, mC6H4SO3Na), 130.16 (br, C4, mC6H4SO3Na), 130.29 (br, C5, mC6H4SO3Na), 136.14 (d, 2JP-C = 12.98 Hz, C6, mC6H4SO3Na), 143.83 (br, C3, mC6H4SO3Na).
[AuCl{PCp2(mC6H4SO3Na)}] (10)
To a solution of [AuCl(tht)] (0.0961 g/ 0.3mmol) in 6 mL of dry acetonitrile a solution of [PCp2(mC6H4SO3Na)] (0.1021g/ 0.3mmol) in 1.5 mL of desoxygenated of water was added drop-wise. The reaction mixture was stirred under argon at RT during 30 min and all the solvents removed under vacuum. By addition of 5 mL of acetone/10 mL of diethyl ether compound 10 was obtained as a white solid. Yield: 0.0468g, 92%. Solubility in H2O: 75 g/L. Anal. Calc. for 10 C16H20AuClNaPO3S (578.78): C, 33.20; H, 3.48; S, 5.54; Found: C, 33.19; H, 4.43; S, 5.60; MS(ESI-) [M/Z]: 557.03 [M - Na]−. 31P{1H} NMR (D2O) δ = 56.77 (s); 1H NMR (D2O) δ = 0.85-1.99 (v br m, 10H, Cp), 7.46 (m, 1H, H5, mC6H4SO3Na), 7.84 (m, 2H, H6, H4, mC6H4SO3Na), 7.82 (d, 2H, H4, mC6H4SO3Na, 3JH-H = 16), 8.04 (d, 1H, H2, mC6H4SO3Na, 3JP-H = 10.8). 13C{1H} NMR (D2O) δ = 25.63 (d, 2JP-C = 7.02 Hz, Cb, Cp), 26.54 (d, 2JP-C = 6.172 Hz, Cb, Cp’), 30.51 (br, Cc, Cp), 30.90 (br, Cc, Cp’), 36.05 (br, Ca, Cp), 36.42 (br, Ca, Cp’), 129.29 (br, C2, mC6H4SO3Na), 129.60 (br, C1, C4, mC6H4SO3Na), 130.14 (br, C5, mC6H4SO3Na), 130.10 (br, C5, mC6H4SO3Na), 136.78 (br, C6, mC6H4SO3Na), 144.58 (d, 3JP-C = 8.78 Hz, C3, mC6H4SO3Na).
2. General Procedure for the Gold(I) Catalyzed A3 Coupling of Benzaldehyde, Piperidine, and Phenylacetylene
In a Shlenk or Kontex (for reactions at temperature higher than room temperature) tube, under N2 (g), the appropriate amount of the catalyst was dissolved in 1 mL of deoxygenated water. Then phenylacetylene (150 μL, 1.5 mmol), benzaldehyde (100 μL, 1.0 mmol), and piperidine (105 μL, 1.1 mmol) were added. The reaction mixtures were allowed to stir for the time and temperature specified in table 1. Once the reaction had taken place, 2-3 mL of Et2O was added and the two layers (water/organic) were separated. Another 20 mL of Et2O were added to the organic layer and it was washed with 10 mL of a brine solution and 2×10 mL of H2O. After drying with anhydrous MgSO4 the organic solvents were completely removed and the residue was weighted (yield) and analyzed by NMR (conversion). When recycling the catalyst we used the aqueous layer and added extra 0.5 mL of deoxygenated water.
3. Antimicrobial Assays
The gold compounds were tested for microbial toxicity in a Kirby-Bauer disk diffusion assay. Compounds 6-10 were solvated in methanol to a concentration of 10 mg/mL and serially diluted in methanol by factors of 10 to create solutions ranging down to 0.1 mg/mL. 10 μL of a solution was aliquoted onto a paper filter disk (5 mm diameter × 1 mm thickness) that were then vacuum dried and stored at − 20°C prior to the experiment. For the assay, 100 μL of a cell suspension containing 3*107 cells/ml were spread uniformly on a Mueller-Hinton (MH for bacteria) or Yeast Extract (YPD for fungi) agar plate (100 mm × 15 mm). After spreading 4 paper disks impregnated with either 100, 10, 1, or 0.1 μg of a compound were placed on the agar surface with a solvent control in the center of the plate. The plates were incubated at either 30°C (fungi) or 37°C (bacteria) for 48 h and resulting zones of growth suppression were measured.
4. Cytotoxicity Assay
Jurkat cells, non-adherent human T leukemia lymphoblast-like cell line (American Type Culture Collection), were seeded at 100,000 cells/well using RPMI media (HyClone, Logan, UT) supplemented with antibiotics and 10% heat-inactivated newborn calf serum (also referred as complete media). After overnight incubation, cells were exposed for 22 h to several concentrations of chemical compounds. Cells from each individual well, were collected in a ice-cold tube and placed on ice, and washed with cold complete media, and then, with cold PBS. After centrifugation at 1,400 rpm by 5 min at 4°C, the supernatant was removed and the cell pellets resuspended in 500 μL of staining solution, containing 2 μg/mL propidium iodide dissolved in FACS buffer (PBS, 0.5 mM EDTA, 2% heat inactivated fetal bovine serum, and 0.1% sodium azide). The cells were then incubated in the dark at room temperature for 15 min and analyzed by flow cytometry, using Cytomic FC 500 (Beckman-Coulter, Miami, FL). The data were acquired and analyzed using CXP software (Beckman-Coulter).
To calculate the IC50, the average of PI-positive cells (dead cells) obtained from triplicates of untreated cells, was subtracted from each experimental point to obtain normalized values, and also, to eliminate culture inherent effects. In these studies, only untreated cells that were at 90% viability or higher were utilized. The average of cytotoxicity (annotated as a percentage) of three independent experiments was plotted against chemical compound concentration in an xy (scatter) chart function (Microsoft Excel). The IC50 values were then calculated from the triplicate values using the linear regression plot with its respective R-squared value between data points, to calculate the concentration of compounds disrupting 50% plasma membrane of the test cells. For each experimental set, three controls were prepared in triplicate and these were the following; (1) untreated cells, (2) cells treated with hydrogen peroxide, and (3) cells treated with physiolocal saline. Hydrogen peroxide, which is a well known as inducer of apoptosis and necrosis,[37] was added to a final concentration of 500 μM as a positive control of cell death, and utilized to ensure that the flow cytometer was calibrated properly. Physiological saline solution (NaCl 0.9%), the diluent of chemical compounds, was tested at the same concentration as contained in the experimental samples, as a control for non-specific diluent effects. The average of triplicate values from untreated cells was used as 100% of viability. No significant differences were observed in cell treated with physiological saline solution when comparing with untreated cells.
5. Apoptosis Assay
Jurkat cells were seeded and cultured as described above and exposed for 16 h to IC50 concentrations of the chemical compounds as determined by the cytotoxicity assays. Cells from each individual well were collected and washed as described above. After centrifugation at 1,400 rpm for 5 min at 4°C, the media and PBS was then removed. The staining procedure was performed by resuspending the cell pellets in 100 μL binding buffer (0.1 M HEPES, pH 7.4; 140 mM NaCl; 2.55 mM CaCl2) containing 1 μL of 25 μg/mL Annexin V-FITC (Beckman Coulter, Miami, FL) and 5 μL of 250 μg/mL PI. After incubation for 15 min on ice in the dark, 400 μl of ice-cold binding buffer were added to cell suspensions, gently homogenized and immediately analyzed by flow cytometry. For each sample, 10,000 individual events were acquired and analyzed using CXP software (Beckman Coulter, Miami, FL). After exposure of cells to the chemical compounds, all subsequent procedures were carried out on ice or at 4°C to arrest or slow down progression of cell damage. Prior to data acquisition, the flow cytometer was setup and calibrated utilizing unstained, single- (PI or Annexin V-FITC) and double- (Annexin V-FITC plus PI) stained cells. The same 3 control groups (as described in the previous section) were used that included untreated cells, treated with 250 μM H2O2, and treated with 4 μL of physiological saline solution.
6. Electron Microscopy and Image Analysis
A 2 mL aliquot of compound 9 in aqueous solution was applied onto a glow discharged carbon coated copper grid and blotted. The specimen was imaged with a JEOL-2100 transmission electron microscope and images were recorded on a Tietz 2kx2k CCD camera at a nominal magnification of 50,000x. At this magnification the effective pixel size at the specimen was 3.06Å. Particle selection and measurement were performed with the program ImageJ.[38]
Supplementary Material
Table 4.
IC50 values of compounds 6-10 compared to cis-platin.
IC50 is defined as the concentration of drug required to disrupt the plasma membrane of 50% of cell population, compared to untreated cells, after 22 hours of incubation. Cells with compromised plasma membrane were monitored using Propidium iodide (PI) and flow cytometry. Cis-platin was used as reference compound.
Acknowledgments
We thank Brooklyn College (start-up budget M.C.), the Professional Staff Congress-City University of New York (PSC-CUNY) research award PSCOOC-38-78 (M.C.), National Institutes of Health (NIH)-National Institute of General Medical Sciences (NIGMS)- Support of Competitive Research (SCORE)-Minority Biomedical Research Support (MBRS) Grant number 3S06GM076168-02S1 (R.O.), National Science Foundation (NSF) Grant number MCB-0546087 (I.U-B.) for financial support and the Arnold and Ruth T. Kaufman Chemistry Fund at Brooklyn College for a summer research undergraduate award (C.L.). We thank the staff of the Cell Culture and High Throughput Screening (HTS) Core Facility for services and facilities provided. This core facility is supported by grant 5G12RR008124 to the Border Biomedical Research Center (BBRC), granted to the University of Texas at El Paso from the National Center for Research Resources (NCRR) of the NIH. We also thank the New York Structural Biology Center for access to their electron microscope. We are indebted to Prof. István T. Horváth (Eötvös University, Budapest, Hungary) for the generous donation of the water-soluble phosphanes used in this work. We also thank undergraduate student Tasmin Chowdhury and high school student Syeda Tabassum (recipient of the American Chemical Society-Summer Educational Experience for the Disadvantaged ACS-SEED program 2007-08) for their assistance with some of the experiments.
Dedicated to Prof. Emeritus Martin A. Bennett in recognition of his outstanding contributions to organometallic and inorganic chemistry
Footnotes
Supporting Information: Selected 31P{1H} NMR spectra of compounds 1-10.
Supporting information for this article is available on the WWW under http://www.eurjic.org/ or from the author.
References
- [1].Kowala C, Sawn JM. Australian J. Chem. 1966;19:547–554. [Google Scholar]
- [2].Gimeno MC, Laguna A. Chem Rev. 1997;97:511–522. doi: 10.1021/cr960361q. [DOI] [PubMed] [Google Scholar]
- [3].Schmidbaur H, Schier A. Chem. Soc. Rev. 2008;37:1931–1951. doi: 10.1039/b708845k. [DOI] [PubMed] [Google Scholar]
- [4].A recent example: Schulz-Dobrik M, Jansen M. Angew. Chem. Int. Ed. 2008;47:2256–2259. doi: 10.1002/anie.200705373.
- [5] a).Roesch N, Goerling A, Ellis DE, Schmidbaur H. Angew. Chem. 1989;101:1410–1412. [Google Scholar]; Angew. Chem. Int. Ed. Engl. 1989;28:1357–1359. [Google Scholar]; b) Schmidbaur H, Hofreiter S, Paul M. Nature. 1995;377:503–504. [Google Scholar]
- [6] a).Contel M, Stol M, Casado MA, van Klink GPM, Ellis DD, Spek AL, van Koten G. Organometallics. 2002;21:4556–4559. [Google Scholar]; b) Casado R, Contel M, Laguna M, Romero P, Sanz S. J. Am. Chem. Soc. 2003;125:11925–11935. doi: 10.1021/ja036049x. [DOI] [PubMed] [Google Scholar]; c) Aguilar D, Contel M, Navarro R, Urriolabeitia EP. Organometallics. 2007;26:4604–4611. [Google Scholar]
- [7] a).Two recent examples: Caruso F, Pettinari C, Paduano F, Villa R, Marchetti F, Monti E, Rossi M. J. Med. Chem. 2008;51:1584–1591. doi: 10.1021/jm700978a. refs. therein.. Barreiro E, Casas JC, Couce MD, Sánchez A, Sánchez-González A, Sordo J, Varela JM, Vázquez López EM. J. Inorg. Biochem. 2008;102:184–192. doi: 10.1016/j.jinorgbio.2007.07.034.
- [8] a).Navarro M, Pérez H, Sánchez-Delgado RA. J. Med. Chem. 1997;40:1937–1939. doi: 10.1021/jm9607358. [DOI] [PubMed] [Google Scholar]; b) Navarro M, Vásquez F, Roberto A, Pérez H, Sinou V, Schrevel J. J. Med. Chem. 2004;47:5204–5209. doi: 10.1021/jm049792o. [DOI] [PubMed] [Google Scholar]
- [9] a).Novelli F, Recine M, Sparatore F, Juliano C. Il Farmaco. 1999:232–236. doi: 10.1016/s0014-827x(99)00019-1. [DOI] [PubMed] [Google Scholar]; b) Nomiya K, Noguchi R, Ohsawa K, Tsuda K, Oda M. J. Inorg. Biochem. 2000;78:363–370. doi: 10.1016/s0162-0134(00)00065-9. [DOI] [PubMed] [Google Scholar]; c) Nomiya K, Noguchi R, Shigeta T, Kondoh Y, Tsuda K, Ohsawa K, Kasuga NC, Oda M. Bull. Chem. Soc. Jpn. 2000;73:1143–1152. [Google Scholar]; d) Nomiya K, Noguchi R, Oda M. Inorg. Chim. Acta. 2000;298:24–32. [Google Scholar]; e) Nomiya K, Yamamoto S, Noguchi R, Yokoyama H, Kasuga NC, Ohyama K, Kato C. J. Inorg. Biochem. 2003;95:208–220. doi: 10.1016/s0162-0134(03)00125-9. [DOI] [PubMed] [Google Scholar]; f) Henderson W, Nicholson BK, Tienkink ERT. Inorg. Chim. Acta. 2006;359:204–214. [Google Scholar]; g) Marques LL, de Oliveira GM, Lang ES, de Campos MMA, Gris LRS. Inorg. Chem. Commun. 2007;10:1083–1087. [Google Scholar]
- [10].Tiekink ERT. Bioinorg. Chem. Appl. 2003;1:53–67. doi: 10.1155/S1565363303000050. refs. therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11] a).For instance: Hashmi ASK. Chem. Rev. 2007;107:3180. doi: 10.1021/cr000436x. refs. therein.. Gorin DJ, Sherry BD, Toste D. Chem. Rev. 2008;108:3351–3378. doi: 10.1021/cr068430g. refs. therein.
- [12].Mohr F, Sanz S, Vergara E, Cerrada E, Laguna M. Gold Bull. 2006;39:212–215. [Google Scholar]
- [13] a.Fackler JP, Jr., Grant TA, Hanson BE, Staples RJ. Gold Bull. 1999;32:20–23. [Google Scholar]; b) Assefa Z, Forward JM, Grant TA, Staples RJ, Hanson BE, Mohamed AA, Fackler JP., Jr. Inorg. Chim. Acta. 2003;352:31–45. [Google Scholar]
- [14] a).Berners-Price SJ, Bowen RJ, Galettis P, Healy PC, McKeage MJ. Coord. Chem. Rev. 1999;185-186:823–836. refs. therein. [Google Scholar]; b) Humphreys AS, Filipovska A, Berners-Price SJ, Koutsantonis GA, Skelton BW, White AH. Dalton Trans. 2007:4943–4950. doi: 10.1039/b705008a. [DOI] [PubMed] [Google Scholar]
- [15].Berners-Price SJ, Bowen RJ, Fernandez MA, Layh M, Lesueur WJ, Mahepal S, Mtotywa MM, Sue RE, van Rensburgh CEJ. Inorg. Chim. Acta. 2005;358:4237–4246. [Google Scholar]
- [16] a).Fackler JP, Jr., Staples RJ, Assefa RJZ. J. Chem. Soc. Chem. Commun. 1994:431–432. [Google Scholar]; b) Assefa Z, McBurnett BG, Staples RJ, Fackler JP, Jr., Assmann B, Angermaier K, Schidbaur H. Inorg. Chem. 1995;34:75–83. [Google Scholar]; c) Forward JM, Fackler JP, Jr., Staples RJ. Organometallics. 1995;14:4194–4198. [Google Scholar]; d) Assefa Z, McBurnett BG, Staples RJ, Fackler JP., Jr. Inorg. Chem. 1995;34:4965–4972. [Google Scholar]; d) Forward JM, Assefa Z, Staples RJ, Fackler JP., Jr. Inorg. Chem. 1995;34:6330–6336. doi: 10.1021/ic950560c. [DOI] [PubMed] [Google Scholar]; e) Forward JM, Assefa Z, Staples RJ, Fackler JP., Jr. Inorg. Chem. 1996;35:16–22. doi: 10.1021/ic950560c. [DOI] [PubMed] [Google Scholar]; f) Assefa Z, Staples RJ, Fackler JP., Jr. Acta Cryst., Sect. C: Cryst. Struct. Commun. 1996;C52:305–307. [Google Scholar]; g) Assefa Z, Omary AM, McBurnett BG, Mohamed AA, Patterson HH, Staples RJ, Fackler JP., Jr. Inorg. Chem. 2002;41:6274–6280. doi: 10.1021/ic025784r. [DOI] [PubMed] [Google Scholar]; h) Mohamed AA, Grant T, Staples RJ, Fackler JP., Jr. Inorg. Chim. Acta. 2004;357:1761–1766. [Google Scholar]; i) Vergara E, Miranda S, Mohr F, Cerrada E, Tiekink ERT, Romero P, Mendía A, Laguna M. Eur. J. Inorg. Chem. 2007:2926–2933. [Google Scholar]; j) Miranda S, Vergara E, Mohr F, de Vos D, Cerrada E, Mendía A, Laguna M. Inorg. Chem. 2008;47:5641–5648. doi: 10.1021/ic7021903. [DOI] [PubMed] [Google Scholar]
- [17].Suresh D, Balakrishna MS, Rathinasamy K, Panda D, Mobin SM. Dalton Trans. 2008:2812–2814. doi: 10.1039/b804026p. [DOI] [PubMed] [Google Scholar]
- [18].Pillarsetty N, Katti KK, Hoffman TJ, Volkert WA, Katti KV, Kamei H, Koide T. J. Med. Chem. 2003;46:1130–1132. doi: 10.1021/jm025615g. [DOI] [PubMed] [Google Scholar]
- [19] a).Jones LA, Sanz S, Laguna M. Catal. Today. 2007;122:403–406. [Google Scholar]; b) Hashmi ASK, Loos A, Littmann A, Braun I, Knight J, Doherty S, Rominger F. Adv. Synth. Catal. 2009;351:576–582. [Google Scholar]
- [20].Sanz S, Jones LA, Mohr F, Laguna M. Organometallics. 2007;26:952–957. [Google Scholar]
- [21] a).Nolan SP. Nature. 2007;445:496–497. doi: 10.1038/445496a. [DOI] [PubMed] [Google Scholar]; b) Hashmi ASK. Nature. 2007;449:292–293. doi: 10.1038/449292a. [DOI] [PubMed] [Google Scholar]; c) Hashmi ASK. J. Organomet. Chem. 2009;694:481. [Google Scholar]
- [22] a).P(nBu)(mC6H4SO3Na)2, P(nBu)2(mC6H4SO3Na): Mika LT, Orha L, Farkas N, Horváth IT. Organometallics. 2009;28:1593–1596.. P(Cp)(mC6H4SO3Na)2, P(Cp)2(mC6H4SO3Na): similar procedure Horváth IT, et al. to be published.
- [23].Lo VK-Y, Kung KK-Y, Wong M-K, Che C-M. J. Organomet. Chem. 2009;694:583–591. refs. therein. [Google Scholar]
- [24] a).Wei C, Li C-J. J. Am. Chem. Soc. 2003;125:9584–9585. doi: 10.1021/ja0359299. [DOI] [PubMed] [Google Scholar]; b) Kantam ML, Prakash BV, Reddy CRV, Sreedhar B. Synlett. 2005:2329–2332. [Google Scholar]; c) Lo VK-Y, Liu Y, Wong M-K, Che C-M. Org. Lett. 2006;8:1529–1532. doi: 10.1021/ol0528641. [DOI] [PubMed] [Google Scholar]; d) Kidwai M, Bansal V, Kumar A, Mozumdar S. Green Chem. 2007;9:742–745. [Google Scholar]; e) Xiao F, Chen Y, Liu Y, Wang J. Tetrahedron. 2008;64:2755–2761. [Google Scholar]; f) Zhang X, Corma A. Angew. Chem. Int. Ed. 2008;47:4358–4361. doi: 10.1002/anie.200800098. [DOI] [PubMed] [Google Scholar]; g) Oña-Burgos P, Fernández I, Roces L, Torre Fernández L, García-Granda S, López Ortiz F. Organometallics. 2009;28:1739–1747. [Google Scholar]
- [25].Lantos D, Contel M, Larrea A, Szabó D, Horváth IT. QSAR Comb. Sci. 2006;25:719–722. [Google Scholar]
- [26].Montoya J, Varela-Ramírez A, Estrada A, Martínez LE, Garza K, Aguilera RJ. J. Biochem. Biophys. Res. Commun. 2004;325:1517–1523. doi: 10.1016/j.bbrc.2004.10.196. [DOI] [PubMed] [Google Scholar]
- [27].Shaik N, Martinez A, Augustin I, Giovinazzo H, Varela-Ramírez A, Sanaú M, Aguilera RJ, Contel M. Inorg. Chem. 2009;48:1577–1587. doi: 10.1021/ic801925k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Rigobello MP, Folda A, Dani B, Menabo R, Scutari G, Bindoli A. Eur. J. Pharmacol. 2008;582:26–34. doi: 10.1016/j.ejphar.2007.12.026. [DOI] [PubMed] [Google Scholar]
- [29].Suresh D, Balakrishna MS, Rathinasamy K, Panda D, Mague JT. Dalton Trans. 2008:2285–2292. doi: 10.1039/b719904j. [DOI] [PubMed] [Google Scholar]
- [30].Park SJ, Kim I-S. Br. J. Pharmacol. 2005;146:506–513. doi: 10.1038/sj.bjp.0706360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Marzano C, Gandin V, Folda A, Scutari G, Bindoli A, Rigobello MP. Free Radic. Biol. Med. 2007;42:872–881. doi: 10.1016/j.freeradbiomed.2006.12.021. [DOI] [PubMed] [Google Scholar]
- [32].Venardos K, Harrison G, Headrick J, Perkins A. Clin. Exp. Pharmacol. Physiol. 2004;31:289–294. doi: 10.1111/j.1440-1681.2004.03993.x. [DOI] [PubMed] [Google Scholar]
- [33].Rackham O, Nichols SJ, Leedman PJ, Berners-Price SJ, Filipovska A. Biochem. Pharmacol. 2007;74:992–1002. doi: 10.1016/j.bcp.2007.07.022. [DOI] [PubMed] [Google Scholar]
- [34].Liu JJ, Galettis P, Farr A, Mahraj L, Samarasinha H, McGechan AC, Baguley BC, Bowen RJ, Berners-Price SJ, McKeage MJ. J. Inorg. Biochem. 2008;102:303–310. doi: 10.1016/j.jinorgbio.2007.09.003. [DOI] [PubMed] [Google Scholar]
- [35].Podsiadlo P, Sinani VA, Bahng JH, Kam NWS, Lee J, Kotov NA. Langmuir. 2008;24:568–574. doi: 10.1021/la702782k. [DOI] [PubMed] [Google Scholar]
- [36].Uson R, Laguna A, Laguna M. Inorg. Synth. 1989;26:85–91. [Google Scholar]
- [37].Miyoshi N, Oubrahim H, Chock PB, Stadtman ER. PNAS. 2006;103:1727–1731. doi: 10.1073/pnas.0510346103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Abramoff MD, Magelhaes PJ, Ram SJ. Biophotonics Internatational. 2004;11:36–42. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.