

Abstract
Drug resistance is problematic in microbial disease, viral disease and cancer. Understanding at the outset that resistance will impact the effectiveness of any new drug that is developed for these disease categories is imperative. In this Feature, we detail approaches that have been taken with selected drug targets to reduce the susceptibility of new drugs to resistance mechanisms. We will also define the concepts of robust drugs and resilient targets, and discuss how the design of robust drugs and the selection of resilient targets can lead to successful strategies for combating resistance.
Introduction
Drug resistance is defined, in a clinical setting, as the point at which administration of the drug can no longer safely treat the disease state owing to an induced change in the drug target or an inability of the drug to reach the target. With an antimicrobial agent, clinical resistance occurs when the minimum inhibitory concentration (MIC) of the drug, for a given microbial strain, exceeds the concentration of drug that can safely be administered. Resistance to a drug can arise by: (i) mutation of the gene (or gene cluster); (ii) acquisition of extrachromosomal DNA, or a transposable plasmid, that carries the resistance gene or genes; (iii) upregulation of the target or; (iv) upregulation of an efflux mechanism.
We have recently suggested [1] an approach for combating drug resistance which involves the selection of resilient drug targets [2] that are evolutionally constrained and the development of robust drugs [3] that are less susceptible to the development of resistance. A drug target is likely to develop resistance if it cannot easily tolerate change and maintain function. Many drug targets are enzymes. Because these enzymes perform highly constrained and crucial chemical reactions, they can represent resilient targets that are less susceptible to drug resistance. However, disrupting the therapeutic target activity is necessary but not sufficient for developing a drug that avoids resistance.
A robust inhibitor is one that successfully inhibits a resilient target, and one that does not lose effectiveness quickly owing to resistance. Such an inhibitor could only bind to crucial regions within the target that would be essential for function and, thus, intolerant to change. The use of high-resolution structures and evolutionary constraints aids the design of robust inhibitors. By choosing resilient targets and designing robust inhibitors the Institute for Drug Resistance (IDR) [1] proposes to focus on drug resistance in drug design strategies, and develop a new generation of more-effective therapeutics.
This Feature will focus on selected, potentially resilient drug targets and describe efforts to produce drugs with various degrees of robustness for these targets in the disease areas of cancer and bacterial and viral infections. The highlighted drug targets include: BCR-ABL kinase; epithelial growth factor receptor (EGFR) kinase and platelet-derived growth factor (PDGFR) kinase in cancer; dihydrofolate reductase (DHFR) and DNA gyrase/topoisomerase IV in bacteria; and human immunodeficiency virus (HIV) protease and hepatitis C (NS3/4A) protease in viruses. Because enzymes are evolutionally constrained to catalyze a chemical reaction, they have the potential of being resilient targets; however, properly identifying robust inhibitors remains a challenge for the field.
Drug resistance in enzyme targets of cancer
Drug resistance is now a widespread problem in cancer and is particularly problematic with kinase inhibitors, proteasome inhibitors [4,5] and monoclonal antibodies [6]. Of these, kinase inhibitors comprise the largest class of anticancer agents where drug resistance has significantly limited treatment. In this article the focus will be on the challenges relating to developing robust drugs that overcome resistance to inhibitors of the kinase domains of BCR-ABL, EGFR and PDGF.
BCR-ABL inhibitors
Gleevec® (imatinib) shows remarkable efficacy by achieving clinical remission in chronic myeloid leukemia (CML). Imatinib inhibits the constitutively active kinase domain, c-Abl, of the fusion protein BCR-ABL [7] existing in CML patients. Evidence from biochemical and crystallographic studies [8] shows that imatinib selectively binds a unique conformation of the activation loop in which a conserved phenylalanine, a member of the Asp-Phe-Gly (DFG) trio of amino acids, undergoes an extensive 10 Å conformational change, into what is called the DFG-out conformation [9,10]. Imatinib binding maintains the kinase in the inactivated state. Interestingly, many kinases cannot adopt an inactive conformation [11]; therefore, targeting this state yields inhibitors with selectivity over other kinases. Crystal structures of other kinases in the DGF-out conformation include: KIT (mast/stem cell growth factor receptor); Aurora A; EGFR (covered in this article); p38 or mitogen-activated protein kinase (MAPK)14; kinase insert domain receptor (KDR); and BRAF [9].
However, the success of imatinib was limited because of the selection of resistant mutants. Specifically, the BCR-ABL variant with a T315I gatekeeper mutation in the ATP-binding site was observed in 10–20% of CML patients after failure of imatinib [12,13]. Consequently, there are ~200 follow-on inhibitors of BCR-ABL kinase [14–16]. Unfortunately, it has been difficult to develop robust inhibitors against the mutant variant of BCR-ABL. Two recently approved agents, dasatinib and nilotinib (Figure 1a), have shown success against imatinib-resistant CML in clinical trials. However, the BCR-ABL1 T315I variant also confers resistance to nilotinib and dasatanib [17–21]. Thus, the problem of resistance to this class of inhibitors is still unsolved. Approaches that are being taken to develop robust drugs that are effective against resistant BCR-ABL include: (i) the design of compounds to inhibit the mutated enzyme; (ii) allosteric inhibitors that bind at a site different from the ATP-binding site; (iii) inhibitors that target other tyrosine kinases and simultaneously inhibit the T315I variant of BCR-ABL; and (iv) development of other dual or multi-kinase inhibitors that simultaneously inhibit the ABL and SFK (SRC family of kinases) families [15]. The SFKs are thought to be involved in the proliferation of BCR-ABL1-expressing cell lines [15,16,22]. The outcomes of these approaches will be crucial to the successful management of CML.
Figure 1.


(a) Structures of first- and second-generation BCR-ABL kinase inhibitors. (b) Structures of EGFR tyrosine kinase inhibitors. (c) Structures of PDGFR inhibitors. (d) Inhibitors that target the GyrA/ParA subunit of gyrase and/or TopoIV. (e) Benzimidazole GyrB/ParE inhibitors.
EGFR inhibitors
Because EGFR, a receptor tyrosine kinase, plays a crucial part in cellular signaling leading to growth, proliferation and metastasis [23–30], some investigators believed that inhibitors of its function would serve as leads for antineoplastic agents. Gefitinib (Figure 1b) was approved in 2003 for the treatment of non-small-cell lung cancer (NSCLC) after failure of platinum-based and docetaxel therapies, but did not significantly improve survival [31]. In 2004 erlotinib (Figure 1b) was also approved for NSCLC (after failure of one other agent) and approved in 2005 for pancreatic cancer in combination with gemcitabine. Both compounds are anilinoquinazolines that compete with ATP to bind the active site of the EGFR tyrosine kinase; a co-crystal structure of erlotinib bound to the EGFR tyrosine kinase [32] reveals the details of that interaction (Figure 2a).
Figure 2. Crystal structures of EGFR (green) with bound inhibitors (purple or magenta).

(a) Erlotinib competes with ATP in the active site of EGFR; (b) HKI272 forms a covalent link with Cys 797 to eliminate the equilibrium between inhibitor and ATP.
Genetic evidence shows that resistance occurs in response to gefinitib and erlotinib in EGFR through mutation of the ‘gatekeeper’ residue T790M [33,34], similar to mechanisms of resistance that occur with mutants T315I (BCR-ABL/imatinib [35]), T6741 (PDGFR/imatinib [36]) and T670I (c-KIT/imatinib [37]). Interestingly, there is evidence that the T790M mutation exists before the initiation of therapy [34] and that cells with the mutation are then selected during therapy. Originally, it was believed that the T790M mutation introduced steric bulk that interfered with the binding of the inhibitors [38]. However, crystal structures and biochemical experiments with the EGFR T790M and T790M/L858R variants bound to inhibitors reveal that the T790M mutation increases ATP affinity, especially in the context of the L858R mutation that activates the kinase [39]. By increasing affinity for ATP, the T790M mutation is considered to be a generic mutation that will reduce the potency of any ATP-competitive inhibitor.
The development of irreversible inhibitors such as HKI272 (Figures 1b and 2b), which is now in Phase II trials [33,40,41], can overcome the effects of the T790M mutation. The irreversible inhibitors have a Michael acceptor that forms a covalent bond with a cysteine thiol in the active site pocket, thus eliminating the equilibrium condition with ATP. HKI272 has been shown to inhibit EGFR function even in the presence of the T790M mutation, but could be subject to resistance via a C797 mutation [40].
PDGFR inhibitors
PDGFR tyrosine kinase inhibitors have been investigated as antineoplastic agents that inhibit angiogenesis. PDGFR inhibitors are also crucial in treating hypereosinophilic syndrome (HES), a disease in which the patient has a prolonged state of eosinophilia that can lead to organ dysfunction and death [36]. Some patients with this disease respond favorably to treatment with imatinib, because they harbor a fused Fip1-like 1 (FIP1L1)-PDGFRα gene that generates an activated kinase. After initial treatment with imatinib, resistance is once again selected with the mutation of the gatekeeper residue T674I in the kinase domain of PDGFR. Interestingly, in an analogous fashion as EGFR, the gatekeeper mutation in many kinases, including PDGFR, does not exert a steric effect but has a role in favoring an activated kinase [42]. Several compounds in different stages of development are successful at overcoming the T674I variant (Figure 1c). Ki11502 [43] is a potent multi-kinase inhibitor of PDGFRα/β and inhibits T674I as well as KIT, KDR and FLT3. Nilotinib [44] and EXEL0862 [45] also inhibit the growth of cells transfected with FIP1L1-PDGFRα harboring the T674I mutation. PKC412 (midostaurin) [36], a natural product isolated from Streptomyces, is reported to be an effective treatment for FIP1L1-PDGFRα-induced disease and of imatinib-induced resistance. Although it is still ATP-competitive, PKC412 could have a different binding mode than imatinib, making it less susceptible to imatinib-specific resistance. Sorafenib is an inhibitor of the RAF-1, B-RAF, vascular endothelial growth factor receptor (VEGFR) and PDGFR tyrosine kinases and is also active against cells harboring the T674I mutation [46]. The inhibitor HG78501 is reported to bind kinases with the T674I mutation [47]. A co-crystal structure of HG78501 with Src kinase shows that the compound forms a H-bond with a neighboring methionine (Met 341 in Src), possibly explaining how it overcomes the effects of the mutation.
Drug resistance in enzyme targets of antibiotics
Antibacterial agents, like antineoplastic agents, target fast-growing proliferative cells. Although the targets differ, the occurrence of resistance is common to both classes of drugs and the development of robust drugs to overcome resistant variants is an active area of research. In this article, we highlight efforts to develop robust drugs against dihydrofolate reductase (DHFR) and gyrase/topoisomerase IV.
Dihydrofolate reductase and trimethoprim resistance
Trimethoprim (TMP) is a potent inhibitor of bacterial species with the essential enzyme dihydrofolate reductase (DHFR), which plays a crucial part in the folate biosynthetic pathway. TMP, co-administered with sulfamethoxazole (SMZ), is an effective therapeutic agent used to treat community-acquired methicillin-resistant Staphylococcus aureus (CAMRSA). However, epidemiological surveys show that ~28% of MRSA strains are TMP-resistant [48–50]. A common resistance mechanism involves the acquisition of one of two sets of chromosomal mutations in the DHFR gene, either H30N/F98Y or F98Y/H149R. Structures of wild-type and mutant (F98Y and H30N/F98Y) Staphylococcus aureus DHFR with several novel propargyl-linked antifolates show that the structural effects of the resistance mutations are subtle [51,52].
Development of compounds that have more interactions within the DHFR active site has been the predominant strategy to overcome TMP resistance. Iclaprim, which underwent Phase III trials but did not continue thereafter in development, achieved nanomolar affinity for the wild-type and mutant enzymes and showed a lower propensity for resistance [53]. Crystal structures of iclaprim bound to the wild-type and mutant enzymes show additional hydrophobic interactions in a highly conserved area of the substrate binding site [54]. Reported propargyl-based inhibitors also interact more substantially with the active site [51,52] than TMP and are less affected by the mutations that cause TMP resistance; further experiments to assess their propensity for resistance are ongoing.
DNA gyrase and topoisomerase IV inhibitors
In Gram-negative and Gram-positive bacteria gyrase and topoisomerase IV, respectively, are important drug targets [55] that serve parallel functions and are often targeted with the same compounds. To perform the processes of transcription, replication, repair and recombination, topoisomerases catalyze either a transient single-strand break (Type I) or a double-strand break (Type II) in supercoiled DNA. In bacteria, gyrase introduces negative supercoils for the entire chromosome, creating condensed packages of genetic material that can be divided correctly during cell division [56,57].
Fluoroquinolones are an established class of drugs (Figure 1d) that stabilize the covalent DNA-gyrase complex and the homologous DNA-topoIV complex. The prototypical quinolone antibiotic nalidixic acid was discovered in the early 1960s and has since been replaced by compounds with higher potency and improved dosing modalities such as moxifloxicin and ciprofloxacin.
Resistance has emerged to the quinolone antibiotics in Gram-positive and Gram-negative strains. A summary of hospital surveys from 1997 to 2001 showed an expanding rate of resistance across infectious species [58]. Resistance mechanisms include increasing drug efflux or mutating the genes that code for DNA gyrase and topoisomerase IV. Structural studies of a DNA-topoIV cleavage complex with bound moxifloxicin (Figure 3) reveal the effects of amino acid mutations in the quinolone-resistance-determining region (QRDR), located in the GyrA subunit [59]. Other drug-binding models attempt to explain the effects of other resistance-conferring mutations [60,61]. Compounds that inhibit DNA cleavage via a new binding mechanism that evades the fluoroquinolone resistance mutations and compounds that simultaneously inhibit GyrB/ParE ATPase function represent promising new approaches to overcome resistance.
Figure 3.
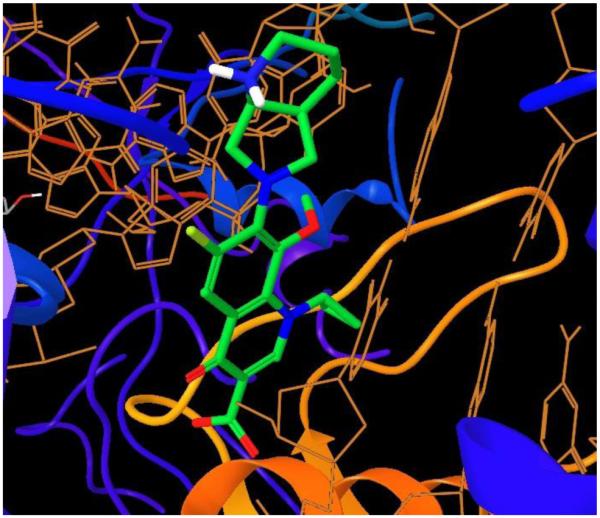
Crystal structure of moxifloxicin bound to GyrA [59]
The topoIV/GyrA subunit inhibitor GSK299423 (Figure 4) inhibits the turnover of the DNA cleaved complex by stabilizing the enzyme–DNA complex prior to cleavage and inhibiting separation of the DNA strands. In this way, GSK299423 employs a mechanism that is significantly different from that of the fluoroquinolones [62]. The binding site for GSK299423 is highly conserved in bacteria and does not overlap with the known binding sites for fluoroquinolones, creating a potent inhibitor that shows antibacterial activity against a range of resistant strains with mutations in DNA gyrase and topoisomerase IV.
Figure 4.
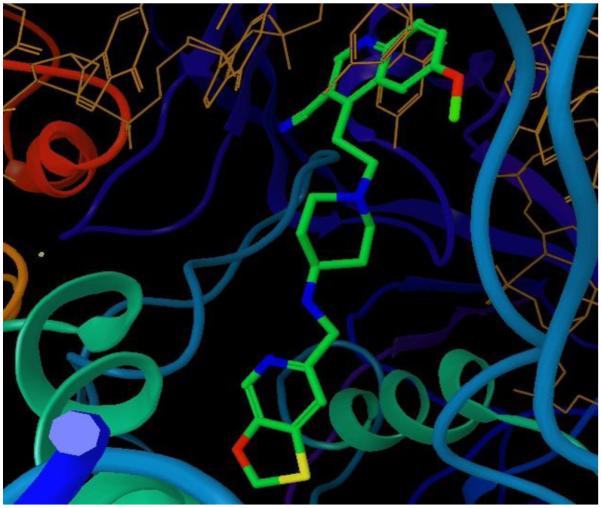
Crystal structure of GSK299423 bound to ParB [62]
Using the topology of the binding sites of DNA gyrase (GyrB) and topoisomerase IV (ParE), (reviewed in Ref. [63]), novel classes of inhibitors of the ATPase activity have been designed to circumvent the resistance observed with quinolones. Interestingly, these compounds also overcome the resistance associated with the coumarins and cyclothialidines [64,65] that bind in a region that overlaps the ATP-binding site [66,67]. These ATPase inhibitors inhibit gyrase and topoisomerase IV simultaneously, thus yielding an advantage in that the emergence of resistance would require the unlikely occurrence of simultaneous mutations. Combinations of virtual screening and HTS led to compound A (Figure 1e), which was of modest potency. Compound A was optimized using structure-based design to arrive at B, a compound that displayed nanomolar Ki values against S. aureus and Escherichia coli gyrase and topoisomerase IV, and impressive MIC90 values of 0.03–0.12 μg/ml against a range of bacteria. This compound has since spawned a wide variety of related chemotypes from a number of other companies [65,68–70], such as compound C, recently reported by Pfizer [69].
Drug resistance in enzyme targets induced by antiviral agents
Most antiviral therapy is rightly focused on vaccine development. However, for a growing number of viruses vaccines have remained elusive, or the virus evolves so quickly that their scope is limited. Although some successful antiviral agents target viral entry, several target the viral enzymes that include: proteases, transcriptases, integrases and neuraminidases. As with all quickly evolving diseases there are challenges in developing inhibitors that are robust; one strategy is to target enzymes that are evolutionarily constrained. The viral proteases, which cleave a number of diverse substrates, represent an example of such a class and, to retain activity, the protease must cleave all the substrates.
HIV protease inhibitors
HIV protease, which cleaves Gag and Gag-Pro-Pol polyproteins at ten varied sites necessary for the maturation of virus [71], is a major therapeutic target for antiviral drugs. During the past 20 years, structure-based drug discovery efforts have led to the development of nine approved competitive active site protease inhibitors (PIs). These inhibitors are the most potent anti-HIV drugs and essential components of highly active antiretroviral therapy (HAART) [72,73].
The development of drug resistance is a major reason for the failure of protease inhibitor therapy. The virus accumulates many mutations within the protease that prevent PIs from binding to the protease. More than half the residues within the protease mutate in different combinations and lead to drug resistance [74,75]. Drug resistance is a subtle change in the balance of recognition events: the protease is still able to recognize and process the natural substrate sites in the Gag and Gag-Pro-Pol polyprotein, at the same time no longer being effectively inhibited by the competitive inhibitor. When resistance emerges the interactions of the protease with an inhibitor are significantly altered, whereas the interactions with a natural substrate are maintained.
Crystallographic studies of the wild-type protease bound to inhibitor molecules have shown that most of the PIs occupy a similar volume and contact similar residues within the active site of the protease. Drug resistance occurs where inhibitor atoms protrude beyond the substrate envelope and contact protease residues [76]. Thus, mutations at these sites would specifically impact inhibitor binding while maintaining substrate cleavage. The observation that many of the drug-resistant mutations in the active site do not contact the substrates has led to the development of the substrate envelope hypothesis: inhibitors that fit well within the substrate envelope would be less susceptible to drug resistance because a mutation that affects inhibitor binding would simultaneously impact the recognition and processing of the majority of the substrates [76]. Of the currently prescribed inhibitors the most potent is darunivar (DRV) and, although not designed with the substrate envelope constraint, DRV fits well within this volume [2,77]. DRV in combination with ritinovir has a high genetic barrier to resistance [78].
Developing robust HIV-1 PIs that avoid drug resistance has proven a challenging task, and the substrate envelope hypothesis provides an approach to solving this problem. A survey of five approved drugs using quantitative measures of the bound inhibitor outside the substrate envelope indicated that the exterior volume of the inhibitors correlated with the loss of affinity to mutant proteases [79]. The ability of the substrate envelope to correlate with resistance mutations prompted the prospective design of new inhibitors with a lower likelihood of developing resistance [3,80–83]. These studies assist in validating the use of the substrate envelope hypothesis [84] in structure-based drug design of novel HIV PIs and have yielded several leads for potential new drugs.
Hepatitis C NS/3A protease inhibitors
Drug resistance is also a major obstacle in the treatment of hepatitis C virus (HCV). An estimated 180 million people worldwide are infected with HCV [85]. The essential HCV NS3/4A protease is an attractive therapeutic target responsible for cleaving at least four sites along the viral polyprotein. Many PIs are currently in clinical trials; however, multi-drug resistance is widespread and arises very quickly. A recent study [86] compares the co-crystal structures of substrate structures with co-crystal structures of inhibitor complexes and shows that, as in the case of HIV-1 protease [77,79,81–83,87,88], primary drug resistance occurs in HCV NS3/4A where the inhibitors protrude away from the substrate envelope.
These findings suggest a general model for using the substrate envelope approach to predict patterns of drug resistance in other quickly evolving diseases. For drug resistance to occur, mutations must selectively weaken enzyme affinity for an inhibitor without significantly altering its activity. Mutations occur outside the substrate envelope to achieve this effect, because these molecular changes can selectively alter inhibitor binding without compromising enzyme activity. Whenever the interaction of a drug target with its biological substrates can be structurally characterized, we predict that drugs designed to fit within the substrate envelope will be less susceptible to resistance. Structure-based design strategies can incorporate this as an added constraint to develop inhibitors that fit within the substrate envelope. As a general paradigm, design efforts incorporating the substrate envelope approach can lead to the development of more-robust inhibitors that are less susceptible to resistance.
Concluding remarks
Drug resistance negatively impacts the lives of millions of patients and costs our society billions of dollars by limiting the longevity of many of our most potent drugs. Drug resistance develops so rapidly that new drugs quickly become less effective, as witnessed by the evolution of resistance to existing anticancer and antimicrobial agents (antibiotics, antivirals, antifungals and antiprotozoals). A recent survey of European intensive care doctors indicated that “50% have treated at least one patient with a gram-negative pathogen that was totally or almost totally resistant to all antibiotics during the previous six months” [89].
Drug resistance research has been very much a disease-specific endeavor, which has limited intellectual progress and breakthroughs. However, as shown here, we have highlighted many parallels among quickly evolving diseases, such as cancer and infectious disease. It is clear that no single approach to develop drugs that are less prone to resistance will work for every drug target, and that a number of approaches will need to be implemented to develop successfully new drugs that will work on targets where resistance is an issue. One such approach that we have highlighted in this Feature is to define targets that are resilient and to then develop robust inhibitors for those targets.
Acknowledgements
The authors thank John D. Williams, PhD, and Bing Li, PhD, of Microbiotix, for technical assistance with this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Peet NP. Drug resistance: a growing problem. Drug Discov Today. 2010;15:583–586. doi: 10.1016/j.drudis.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 2.Lefebvre E, Schiffer CA. Resilience to resistance of HIV-1 protease inhibitors: profile of darunavir. AIDS Rev. 2008;10:131–142. [PMC free article] [PubMed] [Google Scholar]
- 3.Nalam MN, Schiffer CA. New approaches to HIV protease inhibitor drug design II: testing the substrate envelope hypothesis to avoid drug resistance and discover robust inhibitors. Curr Opin HIV AIDS. 2008;3:642–646. doi: 10.1097/COH.0b013e3283136cee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oerlemans R, et al. Molecular basis of bortezomib resistance: proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood. 2008;112:2489–2499. doi: 10.1182/blood-2007-08-104950. [DOI] [PubMed] [Google Scholar]
- 5.Lu S, et al. Overexpression of the PSMB5 gene contributes to bortezomib resistance in T-lymphoblastic lymphoma/leukemia cells derived from Jurkat line. Exp Hematol. 2008;36:1278–1284. doi: 10.1016/j.exphem.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 6.Kute T, et al. Development of Herceptin resistance in breast cancer cells. Cytometry A. 2004;57:86–93. doi: 10.1002/cyto.a.10095. [DOI] [PubMed] [Google Scholar]
- 7.Shah NP, et al. Transient potent BCR-ABL inhibition is sufficient to commit chronic myeloid leukemia cells irreversibly to apoptosis. Cancer Cell. 2008;14:485–493. doi: 10.1016/j.ccr.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 8.Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109:275–282. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- 9.Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol. 2006;2:358–364. doi: 10.1038/nchembio799. [DOI] [PubMed] [Google Scholar]
- 10.Hubbard SR, et al. Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature. 1994;372:746–754. doi: 10.1038/372746a0. [DOI] [PubMed] [Google Scholar]
- 11.Nigg EA. Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol. 2001;2:21–32. doi: 10.1038/35048096. [DOI] [PubMed] [Google Scholar]
- 12.Druker BJ, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344:1038–1042. doi: 10.1056/NEJM200104053441402. [DOI] [PubMed] [Google Scholar]
- 13.Gorre ME, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 14.Sawyers CL. Even better kinase inhibitors for chronic myeloid leukemia. N Engl J Med. 2010;362:2314–2315. doi: 10.1056/NEJMe1004430. [DOI] [PubMed] [Google Scholar]
- 15.Quintas-Cardama A, et al. Flying under the radar: the new wave of BCR-ABL inhibitors. Nat Rev Drug Discov. 2007;6:834–848. doi: 10.1038/nrd2324. [DOI] [PubMed] [Google Scholar]
- 16.Patel D, et al. BCR ABL kinase inhibitors for cancer therapy. Int J Pharm Sci Drug Res. 2010;2:80–90. [Google Scholar]
- 17.Kantarjian H, et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med. 2006;354:2542–2551. doi: 10.1056/NEJMoa055104. [DOI] [PubMed] [Google Scholar]
- 18.Talpaz M, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354:2531–2541. doi: 10.1056/NEJMoa055229. [DOI] [PubMed] [Google Scholar]
- 19.Lombardo LJ, et al. Discovery of N-(2-chloro-6-methyl-phenyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004;47:6658–6661. doi: 10.1021/jm049486a. [DOI] [PubMed] [Google Scholar]
- 20.Weisberg E, et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7:129–141. doi: 10.1016/j.ccr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 21.Shah NP, et al. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305:399–401. doi: 10.1126/science.1099480. [DOI] [PubMed] [Google Scholar]
- 22.Stanglmaier M, et al. The interaction of the Bcr-Abl tyrosine kinase with the Src kinase Hck is mediated by multiple binding domains. Leukemia. 2003;17:283–289. doi: 10.1038/sj.leu.2402778. [DOI] [PubMed] [Google Scholar]
- 23.Coussens L, et al. Tyrosine kinase receptor with extensive homology to EGF receptor shares chromosomal location with neu oncogene. Science. 1985;230:1132–1139. doi: 10.1126/science.2999974. [DOI] [PubMed] [Google Scholar]
- 24.Hynes N, Lane H. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 25.King C, et al. Amplification of a novel v-erbB-related gene in a human mammary carcinoma. Science. 1985;229:974–976. doi: 10.1126/science.2992089. [DOI] [PubMed] [Google Scholar]
- 26.Lin C, et al. Expression cloning of human EGF receptor complementary DNA: gene amplification and three related messenger RNA products in A431 cells. Science. 1984;224:843–848. doi: 10.1126/science.6326261. [DOI] [PubMed] [Google Scholar]
- 27.Olayioye M, et al. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000;19:3159–3167. doi: 10.1093/emboj/19.13.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schechter A, et al. The neu gene: an erbB-homologous gene distinct from and unlinked to the gene encoding the EGF receptor. Science. 1985;229:976–978. doi: 10.1126/science.2992090. [DOI] [PubMed] [Google Scholar]
- 29.Schlessinger J. Common and distinct elements in cellular signaling via EGF and FGF receptors. Science. 2004;306:1506–1507. doi: 10.1126/science.1105396. [DOI] [PubMed] [Google Scholar]
- 30.Yarden Y, Sliwkowski M. Untangling the ErbB signaling network. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 31.Thatcher N, et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer) Lancet. 2005;366:1527–1537. doi: 10.1016/S0140-6736(05)67625-8. [DOI] [PubMed] [Google Scholar]
- 32.Stamos J, et al. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem. 2002;277:46265–46272. doi: 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- 33.Kwak E, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci. 2005;102:7665–7670. doi: 10.1073/pnas.0502860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pao W, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Medicine. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gorre M, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 36.Cools J, et al. A tyrosine kinsae created by fusion of the PDGFRα and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348:1201–1214. doi: 10.1056/NEJMoa025217. [DOI] [PubMed] [Google Scholar]
- 37.Tamborini E, et al. A new mutation in the KIT ATP pocket causes acquired resistance to imatinib in a gastrointestinal stromal tumor patient. Gastroenterology. 2004;127:294–299. doi: 10.1053/j.gastro.2004.02.021. [DOI] [PubMed] [Google Scholar]
- 38.Kobayashi S, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 39.Yun C, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci. 2008;105:2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Godin-Heymann N, et al. The T790M “gatekeeper” mutation in EGFR mediates resistance to low concentrations of an irreversible EGFR inhibitor. Mol Cancer Ther. 2008;7:874–879. doi: 10.1158/1535-7163.MCT-07-2387. [DOI] [PubMed] [Google Scholar]
- 41.Singh J, et al. Targeted covalent drugs of the kinase family. Curr Op Chem Biol. 2010;14:475–480. doi: 10.1016/j.cbpa.2010.06.168. [DOI] [PubMed] [Google Scholar]
- 42.Azam M, et al. Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat Struct Mol Biol. 2008;15:1109–1118. doi: 10.1038/nsmb.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nishioka C, et al. Ki11502, a novel multitargeted receptor tyrosine kinase inhibitor, induces growth arrest and apoptosis of human leukemia cells in vitro and in vivo. Blood. 2008;111:5086–5092. doi: 10.1182/blood-2007-06-098079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Verstovsek S, et al. Activity of AMN107, a novel aminopyrimidine tyrosine kinase inhibitor, against human FIP1L1-PDGFRα-expressing cells. Leuk Res. 2006;30:1499–1505. doi: 10.1016/j.leukres.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 45.Pan J, et al. The novel tyrosine kinase inhibitor EXEL-0862 induces apoptosis in human FIP1L1-PDGRFα-expressing cells through caspase-3-mediated cleavage of Mcl-1. Leukemia. 2007;21:1395–1404. doi: 10.1038/sj.leu.2404714. [DOI] [PubMed] [Google Scholar]
- 46.Lierman E, et al. FIP1L1-PDGFRα D842V, a novel panresistant mutant, emerging after treatment of FIP1L1-PDGFRα T674I eosinophilic leukemia with single agent sorafenib. Leukemia. 2009;23:845–851. doi: 10.1038/leu.2009.2. [DOI] [PubMed] [Google Scholar]
- 47.Weisberg E, et al. Discovery of a small-molecule type II inhibitor of wild-type and gatekeeper mutants of BCR-ABL, PDGFRa, Kit, and Src kinases: novel type II inhibitors of gatekeeper mutants. Blood. 2010;115:4206–4216. doi: 10.1182/blood-2009-11-251751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dale G, et al. A single amino acid substitution in Staphylococcus aureus dihydrofolate reductase determines trimethoprim resistance. J Mol Biol. 1997;266:23–30. doi: 10.1006/jmbi.1996.0770. [DOI] [PubMed] [Google Scholar]
- 49.Drew RH. Emerging options for treatment of invasive, multidrug-resistant Staphylococcus aureus infections. Pharmacotherapy. 2007;27:227–249. doi: 10.1592/phco.27.2.227. [DOI] [PubMed] [Google Scholar]
- 50.Proctor RA. Role of folate antagonists in the treatment of methicillin-resistant Staphylococcus aureus infection. Clin Infect Dis. 2008;46:584–593. doi: 10.1086/525536. [DOI] [PubMed] [Google Scholar]
- 51.Frey K, et al. Crystal structures of wild-type and mutant methicillin-resistant Staphylococcus aureus dihydrofolate reductase reveal an alternative conformation of NADPH that may be linked to trimethoprim resistance. J Mol Biol. 2009;387:1298–1308. doi: 10.1016/j.jmb.2009.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frey K, et al. Towards the understanding of resistance mechanisms in clinically isolated trimethoprim-resistant, methicillin-resistant Staphylococcus aureus dihydrofolate reductase. J Struc Biol. 2010;170:93–97. doi: 10.1016/j.jsb.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schneider P, et al. Iclaprim, a novel diaminopyrimidine with potent activity on trimethoprim sensitive and resistant bacteria. Bioorg Med Chem Lett. 2003;13:4217–4221. doi: 10.1016/j.bmcl.2003.07.023. [DOI] [PubMed] [Google Scholar]
- 54.Oefner C, et al. Increased hydrophobic interactions of iclaprim with Staphylococcus aureus dihydrofolate reductase are responsible for the increase in affinity and antibacterial activity. J Antimicrob Chemother. 2009;63:687–698. doi: 10.1093/jac/dkp024. [DOI] [PubMed] [Google Scholar]
- 55.Blanche F, et al. Differential behaviors of Staphylococcus aureus and Escherichia coli type II DNA topoisomerases. Antimicrob Agents Chemother. 1996;40:2714–2720. doi: 10.1128/aac.40.12.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Holmes VF, Cozzarelli NR. Closing the ring: links between SMC proteins and chromosome partitioning, condensation, and supercoiling. Proc Natl Acad Sci U S A. 2000;97:1322–1324. doi: 10.1073/pnas.040576797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sawitzke JA, Austin S. Suppression of chromosome segregation defects of Escherichia coli muk mutants by mutations in topoisomerase I. Proc Natl Acad Sci U S A. 2000;97:1671–1676. doi: 10.1073/pnas.030528397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jacoby GA. Mechanisms of resistance to quinolones. Clin Infect Dis. 2005;41(Suppl. 2):120–126. doi: 10.1086/428052. [DOI] [PubMed] [Google Scholar]
- 59.Laponogov I, et al. Structural insight into the quinolone–DNA cleavage complex of type IIA topoisomerases. Nature Structural & Molecular Biology. 2009;16:667–669. doi: 10.1038/nsmb.1604. [DOI] [PubMed] [Google Scholar]
- 60.Drlica K, et al. Quinolones: action and resistance updated. Current Topics in Medicinal Chemistry. 2009;9:981–998. doi: 10.2174/156802609789630947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vila J, et al. Association between double mutation in gyrA gene of ciprofloxacin-resistant clinical isolates of Escherichia coli and MICs. Antimicrob. Agents Chemother. 1994;38:2477–2479. doi: 10.1128/aac.38.10.2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bax BD, et al. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature. 2010;466:935–940. doi: 10.1038/nature09197. [DOI] [PubMed] [Google Scholar]
- 63.Maxwell A, Lawson DM. The ATP-binding site of type II topoisomerases as a target for antibacterial drugs. Curr Top Med Chem. 2003;3:283–303. doi: 10.2174/1568026033452500. [DOI] [PubMed] [Google Scholar]
- 64.Ali JA, et al. The 43-kilodalton N-terminal fragment of the DNA gyrase B protein hydrolyzes ATP and binds coumarin drugs. Biochemistry. 1993;32:2717–2724. doi: 10.1021/bi00061a033. [DOI] [PubMed] [Google Scholar]
- 65.East SP, et al. DNA gyrase (GyrB)/topoisomerase IV (ParE) inhibitors: synthesis and antibacterial activity. Bioorg Med Chem Lett. 2009;19:894–899. doi: 10.1016/j.bmcl.2008.11.102. [DOI] [PubMed] [Google Scholar]
- 66.Annedi SC, Kotra LP. RU-79115 (Aventis Pharma) Curr Opin Invest Drugs. 2001;2:752–754. [PubMed] [Google Scholar]
- 67.Angehrn P, et al. New antibacterial agents derived from the DNA gyrase inhibitor cyclothialidine. J Med Chem. 2004;47:1487–1513. doi: 10.1021/jm0310232. [DOI] [PubMed] [Google Scholar]
- 68.Oblak M, et al. Discovery and development of ATPase inhibitors of DNA gyrase as antibacterial agents. Curr Med Chem. 2007;14:2033–2047. doi: 10.2174/092986707781368414. [DOI] [PubMed] [Google Scholar]
- 69.Starr JT, et al. 5-(2-Pyrimidinyl)-imidazo[1,2-a]pyridines are antibacterial agents targeting the ATPase domains of DNA gyrase and topoisomerase IV. Bioorg Med Chem Lett. 2009;19:5302–5306. doi: 10.1016/j.bmcl.2009.07.141. [DOI] [PubMed] [Google Scholar]
- 70.Tanitame A, et al. Synthesis and antibacterial activity of a novel series of DNA gyrase inhibitors: 5-[(E)-2-arylvinyl]pyrazoles. Bioorg Med Chem Lett. 2005;15:4299–4303. doi: 10.1016/j.bmcl.2005.06.103. [DOI] [PubMed] [Google Scholar]
- 71.Kohl NE, et al. Active human immunodeficiency virus protease is required for viral infectivity. Proc Natl Acad Sci U.S.A. 1988;85:4686–4690. doi: 10.1073/pnas.85.13.4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gulick RM, et al. 3-Year suppression of HIV viremia with indinavir, zidovudine, and lamivudine. Ann Intern Med. 2000;133:35–39. doi: 10.7326/0003-4819-133-1-200007040-00007. [DOI] [PubMed] [Google Scholar]
- 73.Bartlett JA, et al. Overview of the effectiveness of triple combination therapy in antiretroviral-naive HIV-1 infected adults. AIDS. 2001;15:1369–1377. doi: 10.1097/00002030-200107270-00006. [DOI] [PubMed] [Google Scholar]
- 74.Stanford HIV Drug Resistance Database. Available at http://hivdb.Stanford.edu.
- 75.Wu TD, et al. Mutation patterns and structural correlates in human immunodeficiency virus type 1 protease following different protease inhibitor treatments. J Virol. 2003;77:4836–4847. doi: 10.1128/JVI.77.8.4836-4847.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.King NM, et al. Structural and thermodynamic basis for the binding of TMC114, a next-generation human immunodeficiency virus type 1 protease inhibitor. J Virol. 2004;78:12012–12021. doi: 10.1128/JVI.78.21.12012-12021.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.King NM, et al. Combating susceptibility to drug resistance: lessons from HIV-1 protease. Chem Biol. 2004;11:1333–1338. doi: 10.1016/j.chembiol.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 78.Winters B, et al. Development of Virco®TYPE resistance analysis, including clinical cut-offs, for TMC114. Antivir Ther. 2006;11:S180. [Google Scholar]
- 79.Chellappan S, et al. Design of mutation-resistant HIV protease inhibitors with the substrate envelope hypothesis. Chem Biol Drug Des. 2007;69:298–313. doi: 10.1111/j.1747-0285.2007.00514.x. [DOI] [PubMed] [Google Scholar]
- 80.Nalam MN, et al. Evaluating the substrate-envelope hypothesis: structural analysis of novel HIV-1 protease inhibitors designed to be robust against drug resistance. J Virol. 2008;84:5368–5378. doi: 10.1128/JVI.02531-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ali A, et al. Discovery of HIV-1 protease inhibitors with picomolar affinities incorporating N-aryl-oxazolidinone-5-carboxamides as novel P2 ligands. J Med Chem. 2006;49:7342–7356. doi: 10.1021/jm060666p. [DOI] [PubMed] [Google Scholar]
- 82.Chellappan S, et al. Evaluation of the substrate envelope hypothesis for inhibitors of HIV-1 protease. Proteins. 2007;68:561–567. doi: 10.1002/prot.21431. [DOI] [PubMed] [Google Scholar]
- 83.Altman MD, et al. HIV-1 protease inhibitors from inverse design in the substrate envelope exhibit subnanomolar binding to drug-resistant variants. J Am Chem Soc. 2008;130:6099–6113. doi: 10.1021/ja076558p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nalam MNL, et al. Evaluating the substrate-envelope hypothesis: Structural analysis of novel HIV-1 protease inhibitors designed to be robust against drug resistance. J Virol. 2010;84:5368–5378. doi: 10.1128/JVI.02531-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.World Health Organization Initiative for Vaccine Research. Hepatitis C. Available at http://www.who.int/vaccine_research/diseases/viral_cancers/en/index2.html.
- 86.Romano KP, et al. Drug resistance against HCV NS3/4A inhibitors is defined by the balance of substrate recognition versus inhibitor binding. Proc Natl Acad Sci U S A. 2010 doi: 10.1073/pnas.1006370107. doi:10.1073/pnas.1006370107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Altman MD, et al. Computational design and experimental study of tighter binding peptides to an inactivated mutant of HIV-1 protease. Proteins. 2008;70:678–694. doi: 10.1002/prot.21514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Prabu-Jeyabalan M, et al. Substrate envelope and drug resistance: crystal structure of RO1 in complex with wild-type human immunodeficiency virus type 1 protease. Antimicrob Agents Chemother. 2006;50:1518–1521. doi: 10.1128/AAC.50.4.1518-1521.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Benowitz AB, et al. Antibacterial drug discovery in the age of resistance. Microbe. 2010;5:390–396. [Google Scholar]