

Abstract
Objectives
The angiogenic drive in skeletal muscle ischemia remains poorly understood. Innate inflammatory pathways are activated during tissue injury and repair, suggesting that this highly conserved pathway may be involved in ischemia-induced angiogenesis. We hypothesize that one of the endogenous ligands for innate immune signaling, high mobility group box 1 (HMGB1), in combination with autophagic responses to hypoxia or nutrient deprivation plays an important role in angiogenesis.
Methods
Human dermal microvascular endothelial cells (EC) were cultured in normoxia or hypoxia (1% oxygen). Immunocytochemical analysis of HMGB1 subcellular localization, evaluation of tube formation, and Western blot analysis of myotubule light-chain 3 (LC3I) conversion to LC3II, as a marker of autophagy, were conducted. 3-methyladenine (3MA), chloroquine (CQ), or rapamycin were administered to inhibit or promote autophagy, respectively. In vivo, a murine hind-limb ischemia model was performed. Muscle samples were collected at 4 hours to evaluate for nuclear HMGB1 and at 14 days to examine endothelial density. Perfusion recovery in the hind-limbs was calculated by laser Doppler perfusion imaging (LDPI).
Results
Hypoxic EC exhibited reduced nuclear HMGB1 staining compared with normoxic cells (mean fluorescence intensity 186.9 ± 17.1 vs. 236.0 ± 1.6, respectively, P = 0.01) with a concomitant increase in cytosolic staining. HMGB1 treatment of ECs enhanced tube formation, an angiogenic phenotype of ECs. Neutralization of endogenous HMGB1 markedly impaired tube formation and inhibited LC3II formation. Inhibition of autophagy with 3MA or CQ abrogated tube formation while its induction with rapamycin enhanced tubing and promoted HMGB1 translocation. In vivo, ischemic skeletal muscle showed reduced the numbers of HMGB1 positive myocyte nuclei compared with nonischemic muscle (34.9% ± 1.9 vs. 51.7% ± 2.0, respectively, P<0.001). Injection of HMGB1 into ischemic hind-limbs increased perfusion recovery by 21% and increased EC density (49.2 ± 4.1vs. 34.2 ± 3.4 EC/HPF, respectively; p=0.02) at 14 days compared to control treated hind-limbs.
Conclusion
Nuclear release of HMGB1 and autophagy occur in ECs in response to hypoxia or serum depletion. HMGB1 and autophagy are necessary and likely play an interdependent role in promoting the angiogenic behavior of ECs. In vivo, HMGB1 promotes perfusion recovery and increased EC density after ischemic injury. These findings are the first to suggest a possible mechanistic link between autophagy and HMGB1 in EC angiogenic behavior and support the importance of innate immune pathways in angiogenesis.
Introduction
Peripheral arterial disease (PAD) affects approximately 5 million adults over the age of 40 in the United States1. The magnitude of the arterial occlusive disease as well as the degree of collateralization determines the severity of symptoms and limb viability. Patients lacking adequate collateral formation can develop critical limb ischemia, risking limb loss if surgical revascularization is not performed.2
Therapeutic angiogenesis has been studied in both peripheral and myocardial perfusion. Agents such as vascular endothelial growth factor (VEGF) have been administered intramuscularly with modest success, 3–5 but leaky vascular networks form that produce significant edema.5, 6 Also, upon withdrawal of the growth factors, the collateral vasculature regresses.7 Thus, further study is required before we can effectively manipulate this process for therapeutic purposes. A great deal of information about the molecular signals for angiogenesis has been derived from studies in tumor biology. 6, 8, 9 Less is known about these signals in the setting of skeletal muscle ischemia. Inflammation is one important mediator of angiogenesis10 and recent evidence suggests that angiogenic proteins such as angiopoeitin-2 may be involved in regulating inflammatory responses in endothelial cells (EC).11
Innate immunity is a highly conserved inflammatory pathway representing the first line of defense against pathogens.10 Its involvement in angiogenesis is suggested by the finding that the anti-angiogenic actions of angiostatin may be mediated through the regulation of innate immune responses.12 Innate immunity utilizes pattern recognition receptors (PRR) that recognize both microbial and endogenous antigens, alerting the organism of infection or injury.13 High mobility group box-1 (HMGB1), an abundant nuclear protein, has been identified as an important endogenous signaling molecule that is actively secreted by macrophages or passively released by injured or necrotic cells.13–16 HMGB1 interacts with PRRs like the Toll-like receptors (TLR) 2,4, and 9 as well as the receptor for advanced glycation end-products (RAGE).13 It is released during hypoxia17 and it mediates lethality in murine sepsis18 and remote organ damage after traumatic tissue injury.19, 20 HMGB1 has also been shown to induce EC migration in culture and EC sprouting in chick chorioallantoic membrane.21 Recent evidence suggests that HMGB1 regulates autophagy, a process of cellular content recycling essential for survival during nutrient deprivation.22 In the setting of lower extremity ischemia, we hypothesize that skeletal muscle may be a local source of HMGB1 release and that HMGB1 plays an essential role in angiogenesis. In this study, we investigated the role of HMGB1 in promoting EC angiogenic behavior in vitro and following muscle ischemia in vivo.
Materials and Methods
Reagents
Recombinant HMGB1 (rHMGB1) was isolated from yeast as described23 and used at 1μg/ml unless otherwise specified. HMGB1 formulation buffer (25mM Tris chloride pH 8, 150mM KCl, 2mM dithiothreitol, 10% glycerol) was used as a control for HMGB1 administration. Monoclonal (2g7) and polyclonal HMGB1 neutralizing antibodies (generous gifts from Dr. Kevin Tracey, Feinstein Institute for Medical Research, Manhasset NY) were developed in rabbit and prepared as described.18 Doses used in these experiments have been shown to attenuate murine sepsis.24 Rabbit polyclonal IgG served as the control (Sigma, St. Louis, MO). Growth-factor reduced (GFR) Matrigel (BD Biosciences, San Jose, CA) was stored at 4oC and allowed to solidify for 30 min at 37 oC prior to use. Antibodies were obtained from: Abcam, Cambridge, MA (HMGB1); Santa Cruz Biotechnology, Inc., Santa Cruz, CA (CD31); Novus Biologicals, Littleton, CO (LC3). Other reagents include: AEC chromogen substrate (ScyTek, Logan, UT), Pierce BCA protein assay (ThermoScientific, Rockford, IL), and 3-methyladenine (3MA), chloroquine (CQ) and rapamycin (Sigma).
Cell culture
Human dermal microvascular endothelial cells (HDMVECs; VEC Technologies, Rensselaer, NY) were cultured in a 1:1 mix of MCDB131 complete media (VEC Technologies) and DMEM with 5% fetal bovine serum (FBS), penicillin, streptomycin (P/S) and L-glutamine. Cells were used between passages 3–12. Serum depletion was performed in 0.5–1% FBS/DMEM. The cells were cultured under either normoxia or hypoxia (1% oxygen in a 95% nitrogen and 5% CO2 gas mixture in a hypoxia chamber; COY Laboratory Products, Inc., Grass Lake, MI).
Endothelial tube formation and proliferation
HDMVECs were seeded on Matrigel at 35,000 cells/well with rHMGB1 or control buffer in depleted media in either normoxia or hypoxia. Formation of tube-like structures, designated as EC “tubes”, was digitally captured with a Nikon inverted microscope and an Olympus DP25 camera. Network formation was evaluated after 18 hours in 4 non-overlapping fields at 20x magnification. The number of tubes per HPF and the number of complete boxes formed by the tubes (termed “network complexity”) were calculated. This measurement accounts for the number of branch points and tube integrity. HMGB1 was neutralized with polyclonal (300 μg/ml) or 2g7 (20 μg/ml) antibody. 3MA (0.2–4mM), CQ (5–200μM), or rapamycin (0–200nM) were added 30 minutes after cells were seeded to inhibit or promote autophagy, respectively. Images were obtained using a 4 or 10x objective after 4 (rapamycin) or 18 hours (3MA, CQ, anti-HMGB1). Proliferation was measured using 3H-thymidine incorporation after 24 hrs under serum depleted conditions.
Time lapse imaging of tube formation
HDMVECs plated on Matrigel on angioslides (ibidi, Munich, German) in serum depleted or replete (5%) DMEM. 2g7 or nonspecific IgG were additionally added. The slides were mounted in a temperature and CO2 controlled microincubator (Zeiss, Thornwood, NY) on an inverted Olympus IX81 microscope with motorized stage and CCD camera (Q-Imaging, Retiga EXi; Surrey, BC). Differential interference contrast (DIC) images were collected from the same location in each well every 5 min for 6 hrs using a 10x objective. Images were compiled into movies using MetaMorph (Molecular Devices, Downington, PA). The perpendicular distance at the midpoint of each tube within the image obtained at 6 hrs was measured as a representation of tube thickness.
In vitro immunocytochemistry
HDMVECs were plated on gelatin-coated coverslips and incubated in normoxia or hypoxia in serum depleted or repleted conditions. Cells were fixed (2% paraformaldehyde), permeabilized (0.1% Triton X), blocked with 2% BSA, and incubated with anti-HMGB1 antibody, phalloidin, and DAPI followed by Cy3-conjugated secondary antibody. Four images per coverslip were obtained (60x) using an Olympus Provis II microscope and Magnafire image acquisition software. Intensity of HMGB1 nuclear staining was determined after color separation by dividing total nuclear Cy3 mean fluorescent intensity (MFI; Metamorph) by nuclei count.
Western blot analysis
Cells were lysed in phenylmethanesulfonylfluoride (PMSF), and protein quantified with BCA assay. Nuclear and cytoplasmic fractions were separated as described17 for HMGB1 blotting. Endothelial cell LC3 was detected in whole cellular lysates 6 hrs after treatment. Densitometry was performed using Vision Works (UVP, Upland, CA).
Murine hind-limb ischemia model
All procedures conformed to the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health, and was in accordance with the policies of the Institutional Animal Use and Care Committee of the University of Pittsburgh (Protocol #0911093). Male C3H/HeOuJ mice (Stock # 000635, Jackson Laboratories, Bar Harbor, ME) were used at 12 wks of age (20–40 gm).
Mice were anesthetized with pentobarbitol (70mg/kg) and inhaled isofluorane <1%. Bilateral groins were shaved and prepped with iodine prep solution. Through a transverse incision, the right external iliac and femoral veins and arteries and all visible branches were ligated as described.3 Left femoral vessels were exposed but not ligated. Post-operative analgesia was maintained with buprenorphine 0.1mg/kg every 12 hrs. Mice were sacrificed at 4 hrs or 14 days and hind-limb muscles were collected. In separate experiments, mice were injected with HMGB1 (20μg) or control buffer in the right calf muscles immediately following arterial ligation. Left hind-limbs received control buffer only. Laser Doppler perfusion imaging (LDPI, Perimed, Stockholm Sweden) was performed 1 and 14 days after ischemia.
Quantification of myocyte nuclear HMGB1 and vascular density
Serial sections (8μm) were obtained from each muscle, de-paraffinized, blocked with avidin and biotin, and incubated with primary antibody overnight. Muscle harvested at 4 hrs was stained for HMGB1. Muscle obtained at 14 days was stained for CD31 to quantify EC/vascular density. After washing, sections were incubated with biotinylated goat-anti-rat secondary antibody. Antigen detection was performed by adding AEC chromogen substrate with hematoxylin counterstain. Non-overlapping images from 3 sections, 60μm apart were obtained with a 40x objective using a Nikon Eclipse 800 microscope attached to an Olympus camera. Percent of HMGB1 positive staining myocyte nuclei at 4 hrs of ischemia was determined using Metamorph.
Statistical analysis
Analysis of variance (ANOVA) was used to compare multiple groups, while t-test was used to compare means between two treatments (SigmaStat, San Jose, CA). Data was expressed as mean ± standard error of the mean (SEM). P<.05 determined statistical significance.
Results
Nuclear HMGB1 is released from stressed ECs
A variety of cells respond to stress by translocating HMGB1 out of the nucleus.22 To determine if ECs respond to angiogenic conditions such as hypoxia or serum deprivation by releasing nuclear HMGB1, we localized HMGB1 in HDMVECs using immunocytochemistry. After 6 hrs of hypoxia in normal serum conditions, nuclear HMGB1 levels were reduced compared with normoxic cells (186.9 ± 17.1 vs. 236.0 ± 1.6, respectively; mean ± SEM, N = 4; P = 0.01) (Figure 1A). There was a concomitant increase in cytosolic HMGB1 in cells exposed to hypoxia. In normoxia, cells in normal serum exhibited greater nuclear HMGB1 staining than cells cultured in serum depleted conditions (226.0 ± 5.5 vs. 236.0 ± 4.6, respectively; N = 4; P = 0.04). Similarly, cytosolic HMGB1 staining was increased in serum starved cells. These findings were confirmed by WB whre cytoplasmic HMGB1 levels were low under resting conditions but increased following serum deprivation or hypoxia (Figure 1B; representative of 3 experiments), although nuclear fractions did not differ at this time point (not shown). Nuclear fractions of HDMVECs cultured in 1% serum demonstrated a modest but significant loss of HMGB1 after 3 hours of hypoxia and after 24 hours in both normoxic and hypoxic conditions (Figure 1C).
Figure 1.

Exogenous HMGB1 enhance endothelial tube formation
Hypoxic (Table 1, Figure 2A), and serum deprived (Figure 2C) HDMVECs exhibited increased tube formation compared with normoxic, and serum repleted cells. Because both conditions induce nuclear HMGB1 release, the ability of HMGB1 to promote EC angiogenic behavior was examined. Supplemental rHMGB1 increased EC tubing behavior under hypoxic conditions with greater tube numbers and complexity (Figure 2A). In normoxia, the effect of rHMGB1 was limited. To assess whether the effects of HMGB1 on tube formation was due to a proliferative response, HDMVEC proliferation over 24 hours was evaluated in the presence of HMGB1 in normoxia and hypoxia, all under serum depeleted conditions. Hypoxia increased EC proliferation compared to normoxia (1.83 ± 0.27 fold over normoxia; P<0.01). rHMGB1 significantly increased cell proliferation in hypoxia but not in normoxia (2.37± 0.46 vs 1.04 ± 0.06 fold over normoxia controls; N = 3 experiments; P<0.01).
Table 1.
| Normoxia | Hypoxia | |||
|---|---|---|---|---|
| Structures/HPF | rHMGB1 | Control | rHMGB1 | Control |
| Tubes | 34.9 ± 2.3a | 25.2 ± 2.0 | 52.2 ± 2.9b | 45.9 ± 2.2c |
| Boxes | 18.3 ± 1.2b | 13.9 ± 1.1 | 35.2 ± 2.1b | 26.4 ± 1.2c |
N=4 experiments
ŠP=.04 compared to control
ŠP<0.001 compared to control
ŠP<0.001 compared to normoxia
Figure 2.
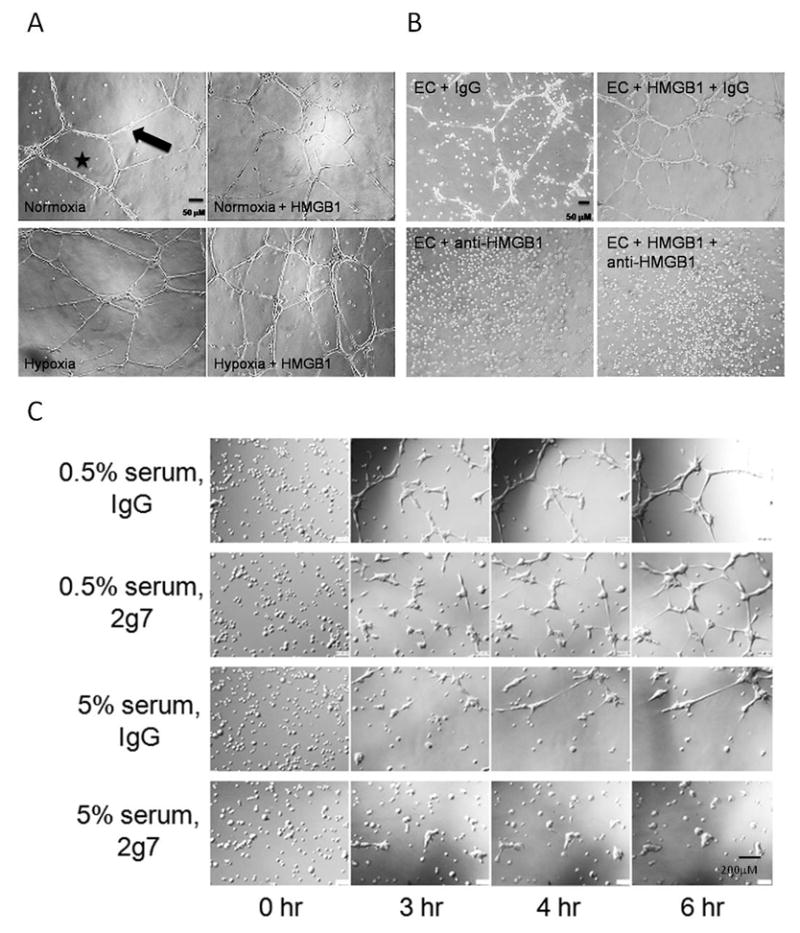
Inhibition of HMGB1 impaired endothelial tube formation
HDMVECs treated with anti-HMGB1 blocking antibodies under hypoxic conditions exhibited markedly impaired tube formation in the presence or absence of rHMGB1 (Figure 2B). Nonspecific IgG did not affect tubing. Similar results were seen in normoxia (not shown). Tube formation was imaged in real time over 6hr with 2g7 antibody or control IgG in 5% or 0.5% serum. Images from the same area within the well obtained at 0, 3, 4, and 6 hours are compiled in Figure 2C. In low serum, ECs aggregated into distinct foci from which tubes extended and formed networks. Anti-HMGB1 antibody impaired tube formation in 0.5% serum with delayed tubing and shorter tubes. Tube thickness also differed significantly between IgG and 2g7 treated cells with more robust tubes in the IgG treated group (12.2 ± 1.6 vs. 5.6 ± 0.8 μm, respectively; P<0.01; N=6 wells/group). In 5% serum, tube formation was markedly impaired at all time points and was completely inhibited in the presence of 2g7.
HMGB1 neutralizing antibody blocks LC3II formation
Our data demonstrated that EC tube formation requires endogenous HMGB1. The angiogenic process was also more robust in nutrient or oxygen deprived conditions, suggesting that energy conservation mechanisms such as autophagy may be involved in angiogenic processes. To assess EC autophagy, we examined the conversion of LC3I to LC3II, a critical step in autophagosome formation and marker of autophagy, under conditions of hypoxia or serum deprivation as well as with HMGB1 modulation (Figure 3).22 HDMVECs cultured in 10% serum exhibited low levels of LC3II while serum-depletion increased LC3II formation (Figure 3A). Inhibition of autophagy with 3MA reduced LC3II levels in low serum conditions. Similarly, 2g7 reduced LC3II levels under serum deprivation (Figure 3A). While exogenous rHMGB1 did not appear to alter LC3II formation, 2g7 significantly reduced LC3II in the presence of 0.1 and 1 μg/ml rHMGB1 (Figure 3B). These findings suggest that ECs exposed to serum deprivation undergo autophagy, and this process is, in part, regulated by endogenous HMGB1.
Figure 3.
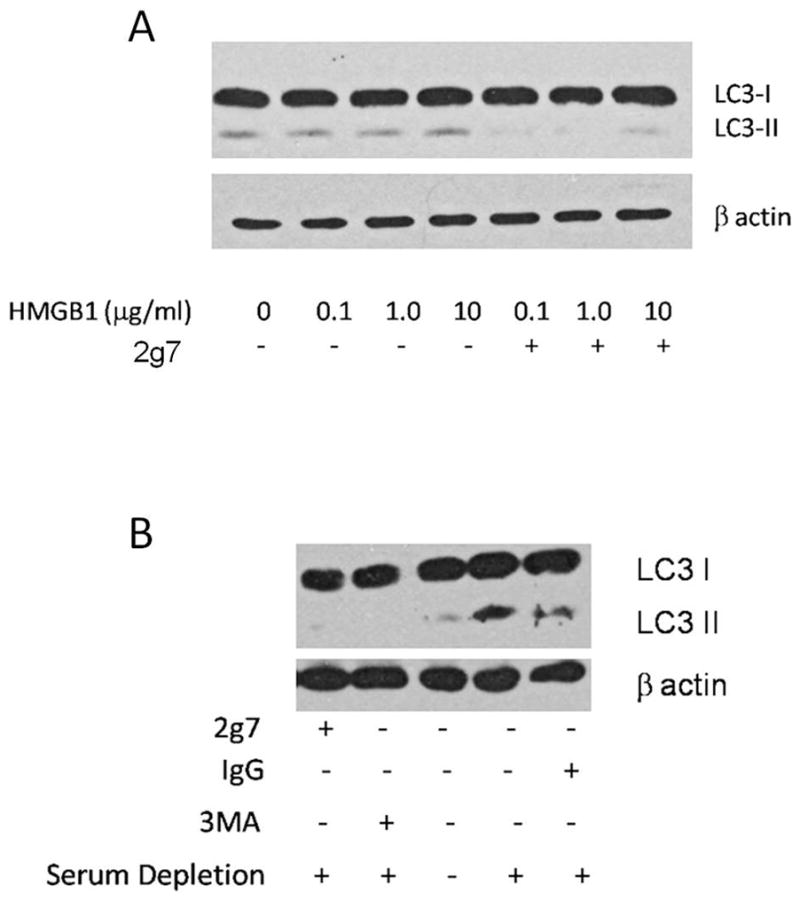
Modulation of autophagy alters endothelial tube formation
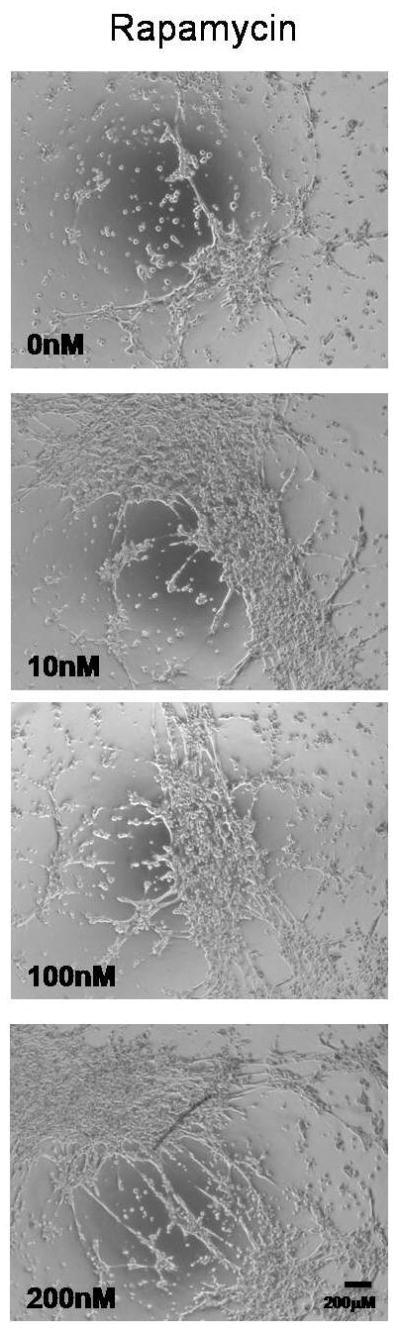
To investigate a role for autophagy in EC tube formation, HDMVECs were treated with 3MA or CQ, both inhibitors of autophagy, under conditions of serum deprivation. Both 3MA and CQ impaired EC tube formation (Figure 4A and B) in a dose dependent fashion, demonstrating a significant loss of network complexity (Figure 4B). Alternatively, induction of autophagy with rapamycin25 stimulated EC tube formation in a dose dependent manner (Figure 5). In combination, these findings suggest that autophagy promotes and is required for EC tube formation.
Figure 4.

Figure 5.
EC HMGB1 translocation from the nucleus is regulated by autophagy
Our data show that both HMGB1 and autophagy are involved in EC angiogenic behavior in vitro. To determine the relationship between HMGB1 and autophagy, we examined the ability of 3MA and rapamycin to modulate nuclear HMGB1 levels. Rapamycin decreased levels of nuclear HMGB1 in ECs cultured in normoxia and serum replete conditions compared to cells not treated with rapamycin (Figure 6A; 26.1 ± 15.9 vs. 127.7± 6.5; P<.001; data represents 4 experiments). Hypoxic, serum deprived cells treated with 3MA (4 mM) had significantly higher levels of nuclear HMGB1 staining than similarly cultured cells without 3MA (MFI 121.3± 3.9 vs. 81.9± 16.7; P=.04). These findings were confirmed by WB where cytosolic HMGB1 was increased by rapamycin, hypoxia, or 1% serum (Figure 6B). However, while 3MA promoted nuclear retention of HMGB1 by immunocytochemistry (Figure 6A), it did not reduce cytoplasmic levels of HMGB1 by WB (Figure 6C). These findings suggest that autophagy initiated by hypoxia, serum depletion, and rapamycin is a stimulus for nuclear HMGB1 release in ECs. but inhibition of autophagosome formation by 3MA does not significant block HMGB1 release.
Figure 6.

HMGB1 is released by ischemic skeletal muscle in vivo
Skeletal muscle ischemia is a strong stimulus for peripheral angiogenesis in vivo. To determine if ischemia can induce HMGB1 release from muscle cells, samples of muscle from ischemic hind-limbs after femoral artery ligation and muscle from non-ischemic controls were stained for HMGB1(Figure 7A). Four hours after arterial ligation, nuclear HMGB1 staining was reduced in the myocytes from the ischemic hind-limbs compared to the control hind-limbs (34.9% ± 1.9 vs. 51.7% ± 2.0, respectively, P<0.001).
Figure 7.

Intramuscular rHMGB1 administration improves hind-limb perfusion after ischemia
To determine if HMGB1 has pro-angiogenic effects in the setting of muscle ischemia, HeOuJ mice were injected with either rHMGB1 (20μg) or a comparable volume of control buffer into the right calf muscle before arterial ligation. To control for the injection, the same volume of control buffer was also injected into the left, nonischemic calf muscle. By LDPI at 2 weeks (Figure 7B,C), perfusion was significantly improved in rHMGB1 treated mice. EC density was determined by CD31 staining in samples of muscle collected 2 weeks after injury, (Figure 7D). and was greater in rHMGB1 treated ischemic hind-limbs than in controls (49.2 ± 4.1/HPF (N=7) vs. 34.2 ± 3.4/HPF (N=5), respectively; p=0.02).
Discussion
This study adds to the literature supporting a role for HMGB1 in ischemia mediated angiogenesis.21, 26–30 De Mori et al reported that intramuscular administration of HMGB1 enhanced muscle regeneration and improved perfusion in a murine hind-limb ischemia model.26 HMGB1 is upregulated in tumor but not normal endothelium.27 Recently, Bescetti et al demonstrated that exogenous HMGB1 promotes neovascularization and improved blood flow by LDPI in diabetic mice.30 HMGB1 has been shown in other systems to be released from cell nuclei by stimuli such as hypoxia through active secretion as well as passive release. 17 Our data confirm similar responses in hypoxic ECs and skeletal muscle and builds on the existing literature by showing that hypoxia mediates endogenous HMGB1 translocation from these cells. HMGB1 release from stressed ECs may represent an active secretion but precise mechanistic studies were not performed. In the ischemic skeletal muscle, we predict that passive release by damaged or dying cells predominates.. Additionally, rHMGB1 promotes perfusion recovery and increases endothelial density in ischemic hind-limbs.
HMGB1 may have qualitative effects on blood vessel formation, which is supported by our in vitro data. Recombinant HMGB1 increased the complexity of EC tube networks under both normoxia and hypoxia in a manner that cannot be explained by increased proliferation alone. rHMGB1 in normoxia was not pro-proliferative. HMGB1 neutralization inhibited EC tube formation under both normoxia and hypoxia, supporting a role for this endogenous danger signal in the angiogenic behavior of EC. A critical role for HMGB1 in angiogenesis has been recently suggested by Wake et al who reported that histidine-rich glycoproteins block HMGB1-heparin complex-induced vessel sprouting in matrigel plugs.31 Our time-lapse imaging demonstrated that HMGB1 neutralization impaired tube extension and thickness without interfering with EC migration into distinct foci. We used measures of branching, tube length, and tube thickness to quantify the different elements that contribute to healthy EC tube formation. This was done because there is a lack of standardized methodology to quantify in vitro angiogenesis. Although the mechanisms governing tube formation are still poorly understood, processes regulating cell adhesion, migration, and cytoskeletal rearrangement are likely to be involved. HMGB1 is critical for axonal elongation in the central nervous system,32 a process that bears marked similarity to angiogenesis.
Our experiments confirm that the signals that promote release of nuclear HMGB1 in HDMVECs (hypoxia, serum depletion) induce EC tube formation as well as autophagy. Autophagy involves lysosomal degradation of long-lived proteins under stress conditions such as hypoxia, injury, or starvation and is important for repair and energy conservation.33 In mouse embryonic fibroblasts (MEF), autophagy has recently been shown to be required for HMGB1 translocation to the cytoplasm. In turn, cytoplasmic HMGB1 sustains autophagy in these same cells.22 We showed that neutralization of HMGB1 inhibits LC3 II formation, suggesting that HMGB1 may be required to sustain autophagy in EC, if not to initiate it. We also demonstrate that treatment of EC with 3MA and CQ, specific inhibitors of PI3KC3 activity in autophagy,34 attenuates EC tube formation in serum depleted conditions. In hepatoma cells, 3MA specifically blocks protein degradation induced by amino acid depletion, a critical process to provide energy in a stressed state.35 it is possible that autophagy provides the energy for normal EC tube formation under starved or hypoxic conditions. Inhibiting autophagy through HMGB1 neutralization could compromise tube formationby reducing the availability of energy required for this demanding process. Higher serum concentrations that do not provide the autophagic drive also impair tube formation.
HMGB1 translocation within other cell types has been linked to autophagy. 36 Our experiments demonstrate that, while HMGB1 may be important for autophagosome formation in HDMVECs, autophagy may further promote HMGB1 release from the nucleus. Rapamycin stimulated HMGB1 nuclear loss from nonstressed ECs. 3MA increased nuclear HMGB1 levels in hypoxic ECs but there was still detectible HMGB1 in the cytoplasm. These findings suggest that stimuli such as starvation and hypoxia initiate HMGB1 translocation that can be further enhanced during autophagy. However, we did observe that both HMGB1 translocation and autophagy are required for EC tube formation. Inhibiting HMGB1 or autophagy significantly impaired tube formation. In vivo, skeletal muscle ischemia was a strong stimulus for HMGB1 secretion from myocyte nuclei. Exogenous HMGB1 stimulated better perfusion recovery and suggested greater vascularity with more endothelial structures observed, suggesting a similar role for HMGB1 in promoting angiogenesis in vivo. Further studies to identify functional vascular structures are needed to confirm these findings. While autophagy is known to occur in fasting muscles37 and has recently become a topic of rigorous investigation,38 the co-dependence of autophagy and HMGB1 release in skeletal muscle was not evaluated in our studies. The impact of HMGB1 and autophagy on neovessel integrity and tissue repair after ischemic injury will be the focus of future studies.
In summary, we demonstrate the novel finding that both HMGB1 and autophagy are necessary and co-dependent factors in EC tube formation. In addition, administration of HMGB1 to ischemic muscle improved limb perfusion and increased endothelial density after ischemia. These findings show how essential innate tissue responses to stress and injury are in angiogenesis. These studies provide further insight into this important adaptive process and may elucidate therapeutic options for patients with critical limb ischemia. While the focus of this study was on the in vitro regulation of angiogenesis by autophagy and HMGB1, future studies will establish the roles for autophagy and HMGB1 in angiogenesis in vivo. The role of innate immune receptors, such as the toll like receptors, in mediating these responses will also be investigated.
Acknowledgments
Funding:
This work was supported by AHA Established Investigator Award [Grant number 0640112N to ET]; VA Merit Award (ET); Foundation for Accelerated Vascular Research Wylie Scholar Award (US); Competitive Medical Research Fund from University of Pittsburgh (US, CJB); and the Clinical and Translational Science Institute, University of Pittsburgh Medical Center, [Grant Number 1 ULI RR024153 to US] from the National Center for Research Resources (NCRR), a component of the National Institutes of Health.
We thank Richard Shapiro for providing recombinant HMGB1.
Footnotes
Conflict of Interest:
None Declared
References
- 1.Selvin E, Erlinger TP. Prevalence of and risk factors for peripheral arterial disease in the United States: results from the National Health and Nutrition Examination Survey, 1999–2000. Circulation. 2004;110(6):738–743. doi: 10.1161/01.CIR.0000137913.26087.F0. [DOI] [PubMed] [Google Scholar]
- 2.Marston WA, Davies SW, Armstrong B, Farber MA, Mendes RC, Fulton JJ, et al. Natural history of limbs with arterial insufficiency and chronic ulceration treated without revascularization. J Vasc Surg. 2006;44(1):108–114. doi: 10.1016/j.jvs.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 3.Messina LM, Brevetti LS, Chang DS, Paek R, Sarkar R. Therapeutic angiogenesis for critical limb ischemia: invited commentary. J Control Release. 2002;78(1–3):285–294. doi: 10.1016/s0168-3659(01)00501-6. [DOI] [PubMed] [Google Scholar]
- 4.Powell RJ, Dormandy J, Simons M, Morishita R, Annex BH. Therapeutic angiogenesis for critical limb ischemia: design of the hepatocyte growth factor therapeutic angiogenesis clinical trial. Vasc Med. 2004;9(3):193–198. doi: 10.1191/1358863x04vm557oa. [DOI] [PubMed] [Google Scholar]
- 5.Masaki I, Yonemitsu Y, Yamashita A, Sata S, Tanii M, Komori K, et al. Angiogenic gene therapy for experimental critical limb ischemia: acceleration of limb loss by overexpression of vascular endothelial growth factor 165 but not of fibroblast growth factor-2. Circ Res. 2002;90(9):966–973. doi: 10.1161/01.res.0000019540.41697.60. [DOI] [PubMed] [Google Scholar]
- 6.Jain RK. Molecular regulation of vessel maturation. Nat Med. 2003;9(6):685–693. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- 7.Baluk P, Lee CG, Link H, Ator E, Haskell A, Elias JA, et al. Regulated angiogenesis and vascular regression in mice overexpressing vascular endothelial growth factor in airways. Am J Pathol. 2004;165(4):1071–1085. doi: 10.1016/S0002-9440(10)63369-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438(7070):932–936. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 9.Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6(4):389–395. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- 10.Frantz S, Vincent KA, Feron O, Kelly RA. Innate immunity and angiogenesis. Circ Res. 2005;96(1):15–26. doi: 10.1161/01.RES.0000153188.68898.ac. [DOI] [PubMed] [Google Scholar]
- 11.Fiedler U, Reiss Y, Scharpfenecker M, Grunow V, Koidl S, Thurston G, et al. Angiopoietin-2 sensitizes endothelial cells to TNF-alpha and has a crucial role in the induction of inflammation. Nat Med. 2006;12(2):235–239. doi: 10.1038/nm1351. [DOI] [PubMed] [Google Scholar]
- 12.Albini A, Brigati C, Ventura A, Lorusso G, Pinter M, Morini M, et al. Angiostatin anti-angiogenesis requires IL-12: the innate immune system as a key target. J Transl Med. 2009;7:5. doi: 10.1186/1479-5876-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5(4):331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 14.Yang H, Wang H, Czura CJ, Tracey KJ. The cytokine activity of HMGB1. J Leukoc Biol. 2005;78(1):1–8. doi: 10.1189/jlb.1104648. [DOI] [PubMed] [Google Scholar]
- 15.Dumitriu IE, Baruah P, Manfredi AA, Bianchi ME, Rovere-Querini P. HMGB1: guiding immunity from within. Trends Immunol. 2005;26(7):381–387. doi: 10.1016/j.it.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 16.Bianchi ME. HMGB1 loves company. J Leukoc Biol. 2009 doi: 10.1189/jlb.1008585. [DOI] [PubMed] [Google Scholar]
- 17.Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med. 2005;201(7):1135–1143. doi: 10.1084/jem.20042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285(5425):248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 19.Mollen KP, Levy RM, Prince JM, Hoffman RA, Scott MJ, Kaczorowski DJ, et al. Systemic inflammation and end organ damage following trauma involves functional TLR4 signaling in both bone marrow-derived cells and parenchymal cells. J Leukoc Biol. 2008;83(1):80–88. doi: 10.1189/jlb.0407201. [DOI] [PubMed] [Google Scholar]
- 20.Levy RM, Mollen KP, Prince JM, Kaczorowski DJ, Vallabhaneni R, Liu S, et al. Systemic inflammation and remote organ injury following trauma require HMGB1. Am J Physiol Regul Integr Comp Physiol. 2007;293(4):R1538–1544. doi: 10.1152/ajpregu.00272.2007. [DOI] [PubMed] [Google Scholar]
- 21.Mitola S, Belleri M, Urbinati C, Coltrini D, Sparatore B, Pedrazzi M, et al. Cutting edge: extracellular high mobility group box-1 protein is a proangiogenic cytokine. J Immunol. 2006;176(1):12–15. doi: 10.4049/jimmunol.176.1.12. [DOI] [PubMed] [Google Scholar]
- 22.Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. 190(5):881–892. doi: 10.1083/jcb.200911078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ngamkitidechakul C, Twining SS. Buffered non-fermenter system for lab-scale production of secreted recombinant His-tagged proteins in Saccharomyces cerevisiae. Biotechniques. 2002;33(6):1296–1300. doi: 10.2144/02336pt02. [DOI] [PubMed] [Google Scholar]
- 24.Yang H, Ochani M, Li J, Qiang X, Tanovic M, Harris HE, et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci U S A. 2004;101(1):296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bae D, Lu S, Taglienti CA, Mercurio AM. Metabolic stress induces the lysosomal degradation of neuropilin-1 but not neuropilin-2. J Biol Chem. 2008;283(42):28074–28080. doi: 10.1074/jbc.M804203200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Mori R, Straino S, Di Carlo A, Mangoni A, Pompilio G, Palumbo R, et al. Multiple effects of high mobility group box protein 1 in skeletal muscle regeneration. Arterioscler Thromb Vasc Biol. 2007;27(11):2377–2383. doi: 10.1161/ATVBAHA.107.153429. [DOI] [PubMed] [Google Scholar]
- 27.van Beijnum JR, Dings RP, van der Linden E, Zwaans BM, Ramaekers FC, Mayo KH, et al. Gene expression of tumor angiogenesis dissected: specific targeting of colon cancer angiogenic vasculature. Blood. 2006;108(7):2339–2348. doi: 10.1182/blood-2006-02-004291. [DOI] [PubMed] [Google Scholar]
- 28.Schlueter C, Weber H, Meyer B, Rogalla P, Roser K, Hauke S, et al. Angiogenetic signaling through hypoxia: HMGB1: an angiogenetic switch molecule. Am J Pathol. 2005;166(4):1259–1263. doi: 10.1016/S0002-9440(10)62344-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang CL, Shu MG, Qi HW, Li LW. Inhibition of tumor angiogenesis by HMGB1 A box peptide. Med Hypotheses. 2008;70(2):343–345. doi: 10.1016/j.mehy.2007.05.024. [DOI] [PubMed] [Google Scholar]
- 30.Biscetti F, Straface G, De Cristofaro R, Lancellotti S, Rizzo P, Arena V, et al. High-Mobility Group Box 1 Protein Promotes Angiogenesis after Peripheral Ischemia in Diabetic Mice through a VEGF-dependent Mechanism. Diabetes. doi: 10.2337/db09-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wake H, Mori S, Liu K, Takahashi HK, Nishibori M. Histidine-rich glycoprotein inhibited high mobility group box 1 in complex with heparin-induced angiogenesis in matrigel plug assay. Eur J Pharmacol. 2009;623(1–3):89–95. doi: 10.1016/j.ejphar.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 32.Rong LL, Yan SF, Wendt T, Hans D, Pachydaki S, Bucciarelli LG, et al. RAGE modulates peripheral nerve regeneration via recruitment of both inflammatory and axonal outgrowth pathways. FASEB J. 2004;18(15):1818–1825. doi: 10.1096/fj.04-1900com. [DOI] [PubMed] [Google Scholar]
- 33.Tang D, Kang R, Xiao W, Zhang H, Lotze MT, Wang H, et al. Quercetin prevents LPS-induced high-mobility group box 1 release and proinflammatory function. Am J Respir Cell Mol Biol. 2009;41(6):651–660. doi: 10.1165/rcmb.2008-0119OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seglen PO, Gordon PB. 3-Methyladenine: Specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proceedings of the National Academy of Sciences. 1982;79(6):1889–1892. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shigemitsu K, Tsujishita Y, Hara K, Nanahoshi M, Avruch J, Yonezawa K. Regulation of Translational Effectors by Amino Acid and Mammalian Target of Rapamycin Signaling Pathways. Journal of Biological Chemistry. 1999;274(2):1058–1065. doi: 10.1074/jbc.274.2.1058. [DOI] [PubMed] [Google Scholar]
- 36.Thorburn J, Frankel AE, Thorburn A. Regulation of HMGB1 release by autophagy. Autophagy. 2009;5(2):247–249. doi: 10.4161/auto.5.2.7552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ogata T, Oishi Y, Higuchi M, Muraoka I. Fasting-related autophagic response in slow-and fast-twitch skeletal muscle. Biochem Biophys Res Commun. 394(1):136–140. doi: 10.1016/j.bbrc.2010.02.130. [DOI] [PubMed] [Google Scholar]
- 38.Sandri M. Autophagy in skeletal muscle. FEBS Lett. 584(7):1411–1416. doi: 10.1016/j.febslet.2010.01.056. [DOI] [PubMed] [Google Scholar]