

Abstract
Follicular helper T (TFH) cells are essential for B-cell maturation and immunoglobulin production after immunization with thymus-dependent antigens. Nevertheless, the development and function of TFH cells have been less clearly defined than classic CD4+ effector T-cell subsets, including T-helper-1 (TH1), TH2 and TH17 cells. As such, our understanding of the genesis of TFH cells in humans and their role in the development of autoimmunity remains incomplete. However, evidence from animal models of systemic lupus erythematosus (SLE) and patients with systemic autoimmune diseases suggests that these cells are necessary for pathogenic autoantibody production, in a manner analogous to their role in promotion of B-cell maturation during normal immune responses. In this Review, I discuss the findings that have increased our knowledge of TFH-cell development and function in normal and aberrant immune responses. Such information might improve our understanding of autoimmune diseases, such as SLE, and highlights the potential of TFH cells as therapeutic targets in these diseases.
Introduction
CD4+ T cells have a crucial role in helping B cells produce antibodies in response to challenge with foreign antigens. The interaction between these cell types typically occurs in germinal centres (GCs) located within the B-cell follicles of secondary lymphoid organs—sites of immunoglobulin affinity maturation and isotype switching. GCs are formed during T-cell-dependent (thymus-dependent) immune responses, which involve a specialized CD4+ T-cell subset, follicular helper T (TFH) cells. The TFH cells localize to B-cell follicles and provide B cells with important survival and differentiation signals, via proteins including CD40 ligand (CD40L, also known as CD154), programmed death-1 (PD-1), and IL-21. TFH cells also produce factors essential for B-cell selection and maturation into memory B cells or long-lived antibody-secreting plasma cells. During T-cell-dependent immune responses, extrafollicular foci of plasmablasts form in the red pulp of the spleen and the medullary cords in lymph nodes; this process also requires CD4+ T cells with features characteristic of TFH cells. Similarly, pathogenic autoantibodies seem to be produced via both GC and extrafollicular pathways in systemic autoimmune diseases. These activities of TFH cells—and cells with similar properties that promote extrafollicular responses—differ from those of conventional CD4+ effector T cells. When aberrantly regulated, cells of the ‘classical’ effector T-helper-1 (TH1), TH2 and TH17 subsets can migrate to the periphery, where they augment inflammation, as occurs in the kidney in systemic lupus erythematosus (SLE) or in the brain in multiple sclerosis, or in allergic responses—in the asthmatic lung, for example. The influence of TFH cells on B-cell responses plays an equally important part in the development and perpetuation of systemic autoimmunity. In this Review, I describe the development and characteristics of TFH cells, and discuss the functions of these cells during normal immune responses and in autoimmune disease in mice and humans.
Effector CD4+ T helper cells
T helper cells are central to the regulation of immune responses. In primary immune responses, CD4+ T cells promote immunoglobulin affinity maturation and class switching in B cells, and inflammatory and allergic events in parenchymal tissues. CD4+ T-cell subsets that possess either B helper or inflammatory (or allergic) activity differentiate from a common naive CD4+ T-cell precursor after antigen stimulation in secondary lymphoid tissues.1 In order to provide B-cell help, T cells must migrate to B-cell follicles and ultimately GCs (or extrafollicular foci) in secondary lymphoid organs, whereas inflammatory T-cell subsets localize to peripheral tissues in response to inflammation or to allergic stimuli. Development of the specialized functions of each of the CD4+ T-cell subsets is determined by specific cell–cell interactions and cytokines, which regulate differentiation by driving expression of particular transcription factors. The transcription factor produced subsequently controls expression of the repertoire of surface-bound and soluble factors that dictate cell function, as well as chemokine receptors and adhesion molecules that regulate localization to specific tissues. Thus, separable effector T-cell subsets can be defined by lineage-specific transcription factor expression, cytokine production, and subsequent immune function (Figure 1).2
Figure 1.

The CD4+ T cell development paradigm. Differentiation of naive CD4+ T cells into different T-helper-cell subsets is dependent on factors present in the local environment, most prominently cytokines. The specific stimulatory conditions influence transcription factor expression, which determines the differentiation program that the T cell will follow and thus the cytokines that it will subsequently produce. The pattern of cytokine expression characterizes the individual T-helper-cell subsets and dictates their function in host defenses. Whereas the cytokine signals that promote the development of TH1, TH2, TH17, and TREG cells are well-defined, less is known about those that drive TFH-cell formation, but seem to include IL-6 and IL-21.76 Abbreviations: BCL6, B-cell lymphoma 6; FOXP3, forkhead box protein 3; GATA3, GATA-binding factor 3; RORα, retinoid-related orphan receptor-α; RORγt, retinoid-related orphan receptor-γt; TBX21, T-box transcription factor TBX21; TFH, follicular helper T; TGF-β, transforming growth factor-β; TH1, T-helper-1; TH2, T-helper-2; TH17, T-helper-17; TREG, regulatory T.
Classical CD4+ T helper cells
TH1 cells
TH1 cells express the lineage-specific transcription factor T-box transcription factor TBX21 (also known as TBET), which is required for IFN-γ synthesis (Figure 1).3 IFN-γ is necessary for protection from intracellular bacterial infections and initiation of physiological and pathological delayed-type hypersensitivity.2
TH2 cells
TH2 cells are characterized by the expression of GATA-binding factor 3 (GATA3), a transcription factor that drives production of the cytokines IL-4, IL-5, and IL-13 (Figure 1).4 These cytokines are essential for clearance of helminths (worms) and other extracellular pathogens and, in particular genetic and environmental contexts, are drivers of pathological allergic responses.
TH17 cells
TH17 cell differentiation occurs in response to IL-6 and transforming growth factor-β. The TH17 cell phenotype is dictated by retinoid-related orphan receptor (ROR)-γt (RORγt)—induced by hypoxia-inducible factor5—and RORα, which control transcription of IL-17 (Figure 1).6,7 IL-17 promotes neutrophil recruitment and activation, and is key to the elimination of extracellular bacteria, such as streptococci, and fungi. TH17 cells can also produce IL-22,8 a cytokine also secreted by other CD4+ T-cell subsets, which is required for regulation of host defences by cell populations at epithelial barriers.9 Like their TH1 and TH2 cell counterparts, TH17 cells promote autoimmunity when abnormally activated, contributing to tissue inflammation in classical T-cell-mediated diseases such as multiple sclerosis,10 inflammatory arthritis,11 and, as more recently demonstrated, in end-organ damage in SLE.12 Furthermore, IL-22-producing TH17 cells have a causal role in the development of pathogenic skin lesions in inflammatory skin disorders.9
Together with cytotoxic CD8+ T cells that are necessary for viral immunity, the classical CD4+ effector T-cell subsets coordinate responses to the range of pathogens —viruses, worms, fungi and intracellular and extracellular bacteria—encountered by mammals. When aberrantly regulated, these cells promote tissue injury in a variety of autoimmune, inflammatory or allergic diseases.
CD4+ TFH cells
Antibody production is crucial for pathogen clearance and prevention of re-infection, with B-cell responses to most protein antigens strictly T-cell-dependent; therefore, CD4+ T-helper-cell-dependent B-cell help is essential. 13 Collaboration between T cells and B cells during a T-cell-dependent immune response results in the formation of extrafollicular foci of antibody secreting cells14,15 and GCs within B-cell follicles.14,16,17
After T cells and B cells have been primed by antigen encounter, interactions that initiate development of extrafollicular foci or GCs occur at the border of the T-cell zone and B-cell follicle (the T-cell–B-cell border) in the spleen,14,16,18 or in the interfollicular region of lymph nodes.19 In order to appreciate how TFH cells are identified, one must first understand the migration of activated T cells and B cells to these sites of interaction: patterns of expression of receptors that enable such movement, as well as the expression of other surface proteins associated with migratory processes, are used to characterize TFH cells in healthy individuals and patients with disease using flow cytometry. In addition, analysis of transcriptional regulation and protein expression is important to understand TFH-cell function. In the following sections, I will elaborate these concepts in more detail, mainly on the basis of data obtained in mice.
Migration and identification of TFH cells
Cells destined to develop a TFH-cell phenotype undergo antigen priming by dendritic cells (DCs) in T-cell zones of secondary lymph nodes (Figure 2). Antigen priming is accompanied by co-stimulatory signals, such as those propagated through B7 interactions with CD28 and inducible T-cell co-stimulator (ICOS)—which is upregulated on T cells upon their activation—and its ligand, ICOSL, expressed on DCs.20 These stimuli lead to changes in expression of chemokine receptors on activated T cells; for example, CC-chemokine receptor 7 (CCR7) is downregulated. Initial entry of naive T cells into secondary lymphoid organs from the circulation requires engagement of CCR7 by its chemokine ligands CC-chemokine ligand (CCL) 19 and CCL21 expressed on high endothelial venules (in lymph nodes) and on T-cell zone reticular cells (in the spleen and lymph nodes).21–24 Coincident with CCR7 downregulation, expression of the cell adhesion molecule P-selectin glycoprotein ligand-1 (PSGL1) is reduced.25 PSGL1 is also expressed on naive T cells and engages CCL19 and CCL21 to facilitate T-cell zone entry.26 These changes in chemokine receptor, and presumably in PSGL1 expression, release activated T cells from the chemotactic influence of CCL19 and CCL21,21–24 enabling T-cell zone egress.
Figure 2.

TFH-cell development and TFH-cell–B-cell collaboration in extrafollicular and GC responses. Naive T cells migrate to the T-cell zones of secondary lymphoid organs (here, the spleen) following a chemokine gradient of CCL19 and CCL21 that engages CCR7 on their surface (1). B cells localize to the B-cell follicle, directed by CXCL13 that engages CXCR5 on their plasma membrane (1). Upon antigen activation and co-stimulation by dendritic cells, nascent TFH cells upregulate CXCR5, downregulate CCR7 and migrate to the T-cell–B-cell border (or interfollicular regions of lymph nodes), where they contact antigen-activated B cells that move towards the T-cell zone after upregulating CCR7 (2). The outcome of this interaction is development of extrafollicular foci (3), where short-lived plasmablasts produce antibodies, or GCs in the B-cell follicle (3). In both structures, TFH cells promote B-cell maturation, inducing class switching and affinity selection, via cytokines —particularly IL-21 and IL-4—and cell-bound molecules—including CD40 ligand (4). TFH-cell and GC B-cell development and function are dependent on the transcriptional regulator BCL6. The GC response leads to memory B cell and plasma cell formation (5). Abbreviations: BCL6, B-cell lymphoma 6; CCL19, CC-chemokine ligand 19; CCL21, CC-chemokine ligand 21; CCR7, CC-chemokine receptor 7; CXCL13, CXC-chemokine ligand 13; CXCR5, CXC-chemokine receptor 5; GC, germinal centre; TFH, follicular helper T.
In addition to downregulation of CCR7 and PSGL1, concomitant and sustained CXC-chemokine receptor 5 (CXCR5) upregulation27,28 enables newly activated T cells to enter the B-cell follicle, by following a gradient of its ligand CXC-chemokine ligand (CXCL) 13 (also known as B-lymphocyte chemoattractant; BLC) expressed therein (Figure 2).29,30 Thus, TFH cells have a characteristic surface phenotype, characterized by low CCR7 and PSGL1, and high CXCR5 expression, which is dictated by their need to emigrate from their initial site of development in the T-cell zone to their site of function in the B-cell follicle.
TFH cells continue to express ICOS after activation by DCs to enable subsequent B-cell driven maintenance and expansion.20,24,31–34 Indeed, ICOS is necessary for both the GC35 and extrafollicular responses;36 in the absence of ICOS signalling, TFH-cell development is markedly impaired.31,32,37,38 TFH cells also upregulate PD-1 as they develop in the T-cell zone, a molecule essential for their eventual function in GCs.39 Collectively, these cell surface markers enable TFH cells to be identified as a CCR7lo PSGL1lo CXCR5hi ICOShi PD-1hi cell population. This characteristic phenotype—in addition to providing a migratory history within secondary lymphoid organs and helping to determine function—is important for flow cytometric and microscopic identification of TFH cells in studies of immunity and autoimmunity.
In an analogous manner to TFH cells, antigen activation of B cells causes them to upregulate CCR7 and subsequently migrate toward the T-cell zone,40 where they can interact with T cells. The Epstein–Barr virus-induced G-protein-coupled receptor 2 (EBI2) also has an important role in the proper positioning of B cells within the follicle, enabling migration of recently activated B cells to the outer follicular regions and T-cell–B-cell border via an oxysterol gradient.41,42 After interacting with T cells in the T-cell–B-cell border or interfollicular regions, antigen-primed B cells downregulate EBI243,44 and maintain expression of CXCR5. Thus, by following a CXCL13 gradient, B cells traffic back to the B-cell follicle and eventually to GCs, where they meet their CXCR5-expressing TFH cell counterparts (Figure 2).
At the sites of initial T-cell–B-cell contact in the interfollicular region of lymph nodes or along the T-cell zone/B-cell follicle border in the spleen, SAP (SLAM-associated protein)—an adaptor molecule for the signalling of SLAM (signalling lymphocytic activation molecule) family receptors—expressed by TFH cells is required for their stable interaction with primed B cells; lack of SAP results in failed GC formation and impaired humoral immunity.45–47 At these initial contact points, T cells also provide their cognate B cells with CD40L and cytokine signals, such as those induced by IL-4 (Figure 3),48 promoting a maturation process—including initiation of class switch recombination—that is completed after they localize to GCs. In return, B cells aid TFH-cell expansion and development via ICOSL-induced and antigen-induced signalling (Figure 3); however, provision of antigen by B cells, in contrast to presentation by DCs, does not seem to be a limiting factor for TFH-cell maturation.49
Figure 3.
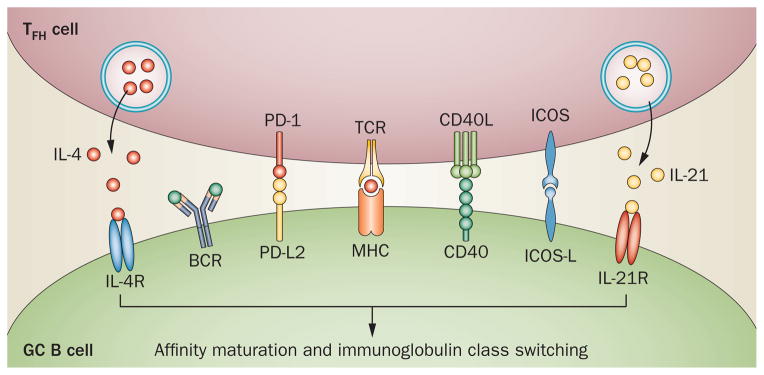
The interaction between TFH cells and GC B cells. The former express molecules critical for survival and for maturation of the latter to memory B cells and plasma cells that produce class-switched immunoglobulin, including CD40L signaling via CD40 and the cytokines IL-4 and IL-21 signaling via their receptors. PD-1 on TFH cells, via binding to its ligands on GC B cells, PD-L1 and especially PD-L2, promotes survival and selection of the latter cells. Abbreviations: BCR, B-cell receptor; CD40L, CD40 ligand; GC, germinal centre; ICOS, inducible T-cell co-stimulator; ICOS-L, inducible T-cell co-stimulator ligand; Ig, immunoglobulins; IL-4R, IL-4 receptor; IL-21R, IL-21 receptor; MHC, major histocompatibility complex; PD-1, programmed death-1; PD-L1, programmed death ligand 1; PD-L2, programmed death ligand 2; TCR, T-cell receptor; TFH, follicular helper T.
TFH cells remain in secondary lymphoid organs after having been activated in the T-cell zone and migrating to sites of T-cell–B-cell contact. By contrast, effector T helper cells eventually downregulate the expression of CCR7,50 and acquire the ability to express appropriate selectin ligands, integrins and chemokine receptors for lymphoid organ exit and trafficking into inflamed tissues;51–53 expression of the latter molecules is sufficient to dictate peripheral migration of CCR7hi effector cells.54
TFH-cell development and B-cell lymphoma 6
B-cell lymphoma 6 (BCL6) is a highly conserved, zinc-finger domain-containing transcriptional repressor that was originally identified in GC B cells and is necessary for GC formation.55,56 BCL6 is also selectively expressed in TFH cells compared with other CD4+ T-cell subsets.32,57 In a manner analogous to the roles of the transcription factors TBX21, GATA3, and RORγt and RORα in the differentiation of TH1, TH2, and TH17 cells, respectively,2,3–6 T-cell-intrinsic activity of BCL6 is required for TFH-cell development and T-cell dependent GC responses (Figure 1);33,58,59 overexpression of this protein is sufficient to dictate the TFH-cell phenotype. 33 Under the influence of appropriate cytokine signals, including the diminished negative regulatory effect of IL-2-induced signal transducer and activator of transcription 5 activity,60 BCL6 is initially upregulated in CD4+ T cells after antigen and ICOS stimulation by DCs in T-cell zones.19,20,34,61 In the nascent TFH cells, BCL6 then represses a program of gene expression that enables differentiation of other CD4+ effector T-cell subsets, effectively blocking their development.33,58,59 Concurrently, BCL6 promotes expression of molecules that are required for the TFH-cell phenotype, including those necessary for their migration and function such as CXCR5, PD-1 and CXCR4, at least to some extent by repressing expression of inhibitory microRNAs (miRNAs).59 BCL6 also downregulates expression of molecules required for T-cell zone retention, promoting egress of TFH cells toward the B-cell follicle.25 The detection of BCL6 expression in TFH cells, and dissection of its function therein, provided evidence that these cells represent a CD4+ T-helper-cell subset independent of the TH1, TH2 and TH17 lineages.
TFH-cell function in normal immune responses
TFH cells have an essential role in provision of B-cell help during the response to T-cell-dependent antigens, which relies on their localization to the B-cell follicle62 and ultimately to the GC.63–65 The latter is the primary site for B-cell somatic mutation with affinity maturation17,66 and immunoglobulin class switching,67 and thus subsequent emergence of B-cell memory and long-lived plasma cells that produce high-affinity antibodies. B-cell help in the GC is mediated partly by a cytokine produced by TFH cells, IL-21,57 which binds IL-21R on GC B cells and cooperates with B-cell receptor and CD40 engagement to promote B-cell proliferation and maturation (Figure 3).68–73 IL-21 is essential for GC development and maintenance, and subsequent development of antibody-forming cells.74–76 Furthermore, in conjunction with IL-4, IL-21 drives immunoglobulin class switching.77
As the IL-4 example indicates, TFH cells also secrete cytokines—in addition to the their canonical cytokine, IL-21—that are associated with TH1, TH2 and, under certain circumstances, TH17 cells.78–80 This flexibility —or plasticity—in TFH-cell differentiation, presumably dictated at a transcriptional level,81 enables the secretion of cytokines that regulate production of immunoglobulin isotypes appropriate for clearance of the invading pathogen; for example, IFN-γ secretion crucial for B-cell class switching to inflammatory immunoglobulin isotypes, or IL-4-dependent IgE synthesis that aids clearance of helminths.
Other molecules produced by TFH cells are also necessary for their function. The inhibitory receptor PD-1 is highly expressed on this T-cell subset, and signalling through its ligands PD-L1 (B7-H1) and PD-L2 (B7-DC) on GC B cells ensures the ultimate quality of the TFH-cell-driven response.39 PD-1, in concert with IL-21 and IL-4, is required for downstream B-cell function, survival in GCs and, ultimately, robust production of plasma cells.39 By contrast, abrogation of PD-1 signalling, especially early in the GC response, enhanced TFH-cell numbers and initial immunoglobulin responses.82 These findings demonstrate the complexity of signalling via this pathway, and highlight the need to consider both beneficial and adverse events that might be associated with potential therapeutics targeting PD-1.
As noted above, CD40L expressed on TFH cells also has a role in GC development and function;35 deletion of the gene encoding this protein—for example in human hyper-IgM syndrome83 or experimentally in mice84 —results in an absence of GC formation. Similarly to IL-4 and ICOS signalling, the function of CD40L in B-cell maturation after immunization with T-cell-dependent antigens is first manifest at initial sites of T-cell–B-cell contact, before GC entry of nascent TFH cells and maturing B cells;48 therefore, the signals that are typically associated with the mature GC response could initially be important at sites of early T-cell–B-cell collaboration, with these interactions continuing as GCs develop and mature.
Extrafollicular T helper cells
Extrafollicular foci in secondary lymphoid organs are sites where short-lived plasma cells emerge after immunization with T-cell-dependent (or with thymus-independent) antigens in conventional immune responses. 14,15 In comparison with our understanding of TFH cells, knowledge of the T helper cells that drive the extrafollicular response is relatively limited. Within 2 days after primary immunization, CD4+ T cells proliferate in the T-cell zone,85 together with the B cells destined to join the extrafollicular response as plasmablasts. 15 Whereas initial interactions between DCs and T cells required for the extrafollicular response are CD40L–CD40-independent, subsequent B-cell proliferation and plasmablast formation are dependent on these proteins,48 and on ICOS.36,86
The CD4+ T cells that promote the extrafollicular response have been defined in mice; like TFH cells, they upregulate BCL6, are dependent on ICOS for their development and express molecules necessary for B-cell maturation, including PD-1, CD40L and IL-21 (Figure 4).36,87 Such cells are found in conjunction with B cells at the T-cell–B-cell border after immunization.87
Figure 4.

Subsets of T cells that promote or regulate B-cell help. TFH cells, critical for GC development and maturation, are reliant upon the transcription factor BCL6 for differentiation and function, as are extrafollicular T-helper cells that promote formation of the extrafollicular plasmablast response. Follicular helper iNKT cells, also reliant upon BCL6 for development, provide cognate help for B-cell maturation in extrafollicular foci and GCs—similar to TFH and extrafollicular T-helper cells, respectively—resulting in the production of class-switched and affinity-matured antibodies to lipids. By contrast, TFR cells regulate the TFH and GC B-cell responses, providing a brake on development of autoimmune reactions. As for their B-cell helper counterparts, TFR cells require BCL6 for development, but upregulate the BCL6 repressor, BLIMP-1, as well as expressing molecules associated with TREG-cell development and function, including FOXP3, CTLA4, GITR and IL-10. Abbreviations: BCL6, B-cell lymphoma 6; BLIMP-1, B-lymphocyte-induced maturation protein 1; CD40L, CD40 ligand; CTLA4, cytotoxic T-lymphocyte-associated antigen 4; CXCR4, CXC-chemokine receptor 4; CXCR5, CXC-chemokine receptor 5; FOXP3, forkhead box protein 3; GC, germinal center; GITR, glucocorticoid-induced TNF-receptor-related protein; ICOS, inducible T-cell co-stimulator; iNKT, invariant natural killer T; PD-1, programmed death-1; TCR, T-cell receptor; TFH, follicular helper T; TFR, follicular regulatory T; TREG, regulatory T.
Follicular helper iNKT cells
In addition to development of antibodies targeting proteins after immunization with thymus-dependent antigens, B cells also produce class-switched antibodies to lipids, which represent a crucial component of immune responses to pathogens. Lipid-driven B-cell help is provided by invariant natural killer T (iNKT) cells, which recognize lipids presented in the context of the non-classical antigen-presenting molecule CD1d expressed on B cells (among other cell types).88,89 The discovery of GC-resident iNKT cells in 2011 has offered further mechanistic insights into the anti-lipid response.90,91 Similar to classical TFH cells, these follicular helper iNKT cells are dependent on BCL6 and B-cell interactions for development (Figure 4).90 Furthermore, they provide cognate help for B-cell maturation in extrafollicular foci and GCs via IL-21, resulting in the production of class-switched and affinity-matured antibodies to lipids.90,91 Although follicular helper iNKT cells did not promote long-lived plasma cells or long-term humoral immunity, in contrast to classical TFH cells, they were needed for rapid extrafollicular-foci-dependent and GC-dependent immunoglobulin responses to lipid antigens.90,91
Follicular regulatory T cells
Logically, regulatory pathways need to be engaged to prevent loss of self-tolerance in GCs and subsequent autoantibody production,92 given the rapid emergence of somatically mutating B-cell clones at these site following T-cell-dependent antigen immunization.17,66 The identification of follicular regulatory T (TFR) cells highlights a novel mechanism that controls the normal GC response and presumably prevents emergence of autoreactive B-cell clones.93–96
TFR cells are a thymically-derived population comprising some 5–25% of GC T cells in mice and humans.93,95 This CD4+ T-cell subset demonstrates features of the natural regulatory T (TREG)-cell phenotype, including forkhead box protein 3 expression and immunosuppressive capacity, while expressing markers of classical TFH cells, including CXCR5 and PD-1,93–95 but not IL-21 (Figure 4).93 Similar to TFH cells, TFR-cell development also depends on BCL6 expression93,95 and B-cell interactions, partially mediated by SAP.93
In contrast to the helper capacity of TFH cells, TFR cells necessarily function to limit potentially pathogenic GC responses, including the expansion of non-antigen specific and potentially autoreactive B cells,93 via inhibition of TFH cells and GC B-cell maturation; evidence also suggests that TFR cells regulate plasma cell development.94–97 In addition to expression of molecules associated with TREG cells—such as cytotoxic T-lymphocyte-associated antigen 4 and glucocorticoid-induced TNF receptor-related protein—that possibly contribute to their suppressive capacity, TFR cells also abundantly express IL-10,93,95 which might be important for the regulatory role of these cells in the GC.96
T cells and B-cell help in humans
Phenotype of germinal centre TFH cells
Our current understanding of TFH cell biology and function, as discussed above, is mainly derived from investigations performed in mice; however, data on similar T-cell populations that provide help to B cells in humans do exist. Immunohistochemical studies performed nearly three decades ago identified a population of CD4+ GC T cells that stained for the NK cell marker CD57.98–100 CD57hi GC T cells are CXCR5hi and CCR7lo (indicating that they have exited the T-cell zone), and efficiently help B cells produce antibodies in vitro.100 Nonetheless, CD57lo T cells that contain stores of preformed CD40L are also found in the GC,101 and CD57lo CXCR5hi ICOShi cells are as capable of providing B-cell help in vitro as their CD57hi counterparts;102 thus, CD57 is no longer commonly used as a marker of human TFH cells. The CXCR5hi ICOShi T-helper-cell subset also expresses CXCL13 (as do mouse TFH cells), as well as molecules that distinguish them from other CD4+ effector T-cell subsets, including transcription factors that are components of the Notch signalling pathway.102 CD4+ T cells lacking expression of the α-subunit of the IL-7 receptor (IL-7Rα; CD127), which is important for development and homeostatic proliferation of lymphocytes and for defining terminally differentiated effector cells,103 are also exclusively located in GCs and are efficient in helping B cells to produce antibodies in vitro in a CD40L-dependent manner.100 These findings suggest a scenario in which GC T helper cells in humans express CXCR5, PD-1, ICOS, CD40L and CXCL13 (Figure 4), and have low (if any) expression of CCR7 and PSGL1, analogous to mouse TFH cells, with downregulation of IL-7Rα.
Circulating TFH cells
Peripheral blood is the most convenient tissue for analyses of control individuals and patients with autoimmune diseases; therefore, whether TFH cells reside in the circulation and, if so, how their phenotype reflects what is seen in secondary lymphoid tissues are important considerations. In this regard, studies have focused upon characterization of CD4+ T cells that have the capacity to recirculate to secondary lymphoid organs, where they can interact with B cells. Such recirculating central memory cells are defined by cell surface markers, specifically CCR7 and L-selectin—both of which are necessary for re-entry into lymph nodes—and CXCR5, indicative of the ability to enter B-cell follicles and provide B-cell help.63–65,104 Although conveying a memory phenotype, the circulating CXCR5hi cells might be relatively short-lived compared with the CXCR5lo memory population.63 Upon exposure to CCL19 and CCL21 in vitro, CXCR5hi cells downregulate CCR7 —similarly to TFH cells that necessarily need to emigrate to the B-cell follicle from the T-cell zone—suggesting that they have the capacity to become TFH cells after re-entry into secondary lymphoid organs.63 Circulating CXCR5hi T helper cells can be further characterized as TH1, TH2 or TH17 memory cells, depending on expression of their signature cytokines—IFN-γ, IL-4 and IL-17, respectively—and chemokine receptors—CXCR3, CCR4 and CCR6, respectively.105 Both the TH2 and TH17 cells can help B cells produce antibodies in vitro, in an IL-21-dependent manner, whereas those with a TH1 phenotype cannot.105 Furthermore, the TH2 and TH17 subsets are expanded in patients with juvenile dermatomyositis, 105 suggesting that an altered TFH-cell regulation might play a part in the pathogenesis of systemic autoimmune diseases. Interestingly, a CXCR5hi CD4+ central memory T-cell population has also been identified in mice, and these cells have the capacity to recirculate to lymphoid tissue and promote naive B-cell activation.106 Although expression of CXCR5 and the resultant capacity to enter B-cell follicles correlates with B-cell helper function, one should note that a population of tonsillar CXCR5lo ICOSlo cells, which reside outside B-cell follicles, can also provide help to naive and memory B cells through CD40L, IL-21 and IL-10 secretion in vitro.104,107 Similarly, CXCR5lo cells from the peripheral blood can also provide B-cell help, suggesting that at least the portion of the cells with such helper capacity are derived from a CXCR5hi cell subset.65
These findings are intriguing; however, the relationship between circulating CXCR5hi memory cells and TFH cells localized to B-cell follicles is not entirely clear. This dilemma stems partly from the natural history of the GC response, and presumably the TFH-cell response; as the purpose of GCs is development of persistent memory B cells and long-lived plasma cells, these structures naturally disappear as these functions are accomplished during the days and weeks following antigen challenge. By contrast, whether TFH cells persist after the GC response remains unknown, although CD4+ T cells are found co-localized with memory B cells in GC-like structures in B-cell follicles months after immunization in mice.108 Although detailed characterization of these persistent GC CD4+ T cells has not been performed, memory T helper cells that promote B-cell responses have been identified in the peripheral blood of humans105 (albeit relatively short-lived compared with the CXCR5lo memory pool63) and in secondary lymphoid organs of mice,106 as noted above. These data suggest that such cells could represent TFH-cell precursors that can gain TFH-cell functionality after antigen rechallenge,106,109 whereas TFH cells per se do not preferentially develop into long-lived memory cells.110 The relationship of such circulating CXCR5hi memory T cells to TFH cells—and thus their potential capacity to generate immunological memory —is highly relevant to the chronicity of human autoimmunity, given that such CXCR5hi cells are expanded in the peripheral blood of patients with systemic autoimmune diseases;105,111 this topic is discussed in more detail in the following sections.
TFH cells in systemic autoimmunity
TFH cells in mouse models of lupus
Mouse models have been instructive in highlighting the role of TFH cells in promotion of systemic autoimmunity. The sanroque mouse exemplifies this paradigm:112 these animals demonstrate spontaneous GC formation (as is common in mouse models of lupus113) and enhanced TFH-cell development in the setting of increased ICOS and IL-21 production. Dysregulation of ICOS in this model results from a chemically-induced point mutation in the gene encoding the ubiquitin ligase roquin, a negative regulator of ICOS expression, which suppresses ICOS expression.112 The sanroque mutation is associated with systemic autoimmunity characterized by autoantibody production and immune-complex-mediated glomerulonephritis112—features of human SLE. ICOS-induced TFH-cell dysfunction and subsequent abnormal B-cell selection in GCs have been shown to be responsible for the SLE-like phenotype in sanroque mice.114 Nevertheless, other work has indicated that the sanroque mutation contributes to autoimmunity in an as yet undetermined manner; a deletion in the gene encoding roquin that prevented its expression did not result in the development of autoimmunity, despite evidence of increased ICOS expression and immune dysregulation in this model.115 Indeed, while IL-21 does not contribute to the lupus phenotype in sanroque mice,114 other models do support a role for TFH cells and IL-21 in disease pathogenesis: for example, the BXSB. Yaa model, in which SLE-like autoimmunity is promoted by excessive signalling induced by self-RNA autoantigens as a result of duplication of the gene encoding TLR7.116 Like sanroque animals, BXSB. Yaa mice have spontaneous GC formation with marked TFH-cell expansion and IL-21 production.71 TFH cells also expand beyond GCs to populate extrafollicular sites as BXSB. Yaa animals age.117 The excessive autoantibody production and immune-complex-mediated glomerulonephritis that characterize this strain are abrogated with genetic disruption of IL-21 signaling,117 presumably secondary to requirement of this cytokine for B-cell maturation.74,75 Although TFH cells undoubtedly contribute to the lupus phenotype in BXSB. Yaa mice, CD4+ T cells with a TFH-cell-like phenotype that reside in extrafollicular areas might also promote disease.117 The role of IL-21 in potentiation of SLE-like disease is substantiated by the prophylactic effectiveness of blockade of this cytokine in other lupus-prone mice with polygenic disease;118 whether therapeutic blockade after disease onset would be beneficial remains unclear.
Other TFH-cell cytokines could be pivotal in development of the lupus phenotype. For example, production of IFN-γ-dependent inflammatory IgG isotypes is the hallmark of lupus in mice; these antibodies deposit in peripheral tissues, including the kidney, and initiate pathways that lead to tissue injury.119 Similarly, IFN-γ-dependent responses might be an important factor in the pathogenesis of SLE in humans.120
CD40L signalling, like that of IL-21, is necessary for TFH-cell-dependent B-cell maturation in GCs, and blockade of this pathway is therapeutically appealing. Indeed, genetic disruption of CD40L or administration of anti-CD40L antibodies is effective in the reduction of lupus in a number of mouse models.121–123 Moreover, anti-CD40L therapy has shown promise in a study that enrolled a small number of patients with SLE, with diminished pathogenic autoantibody production and a subsequent reduction in disease activity measures and immune-complex-mediated glomerulonephritis being reported after treatment.124 The therapeutic effects of CD40L blockade in these individuals were presumably secondary to the abrogation of pathogenic TFH-cell–B-cell interactions that occur in SLE, as evidenced by an observed inhibition of GC B-cell maturation.124 TFH-cell-independent effects might contribute to the benefit observed with anti-CD40L therapy: activated platelets also upregulate this molecule in SLE, enabling them to interact with CD40 expressed on monocytes, leading to their differentiation in to DCs with the potential to activate autoreactive T cells.125 However, anti-CD40L therapy was complicated by the development of thromboembolic disease, apparently triggered by binding of the antibody to platelet Fc receptors together with engagement of the surface-associated CD40L and resultant platelet aggregation.126 Nevertheless, these data suggest that development of different therapeutic approaches to interference with CD40 signalling might be beneficial in SLE.
A role for TFH-cell expansion and subsequent aberrant GC responses in SLE is also supported by the results of ICOS blockade. Inhibition of ICOS signalling, via administration of anti-ICOSL antibodies or genetic ablation of ICOS, interrupts TFH-cell development and GC formation, reduces autoantibody formation and glomerulonephritis, and abrogates the disease phenotype in mouse polygenic lupus models.36,127 Whether therapeutic blockade of ICOS signalling after disease onset in humans would be beneficial is not clear at present; nonetheless, the therapeutic potential demonstrated in mouse models of lupus127 has led to initiation of early-stage clinical trials of anti-ICOSL agents in patients with SLE.
Extrafollicular T helper cells in mouse lupus
Autoantibody production in lupus-prone mouse strains also arises at extrafollicular foci within the spleen and lymph nodes, which represent important sites of T-cell migration and maturation and somatic immunoglobulin mutation of autoreactive B cells in these animals and other models of inflammatory disease.36,117,128,129 In lupus-prone MRL-Faslpr mice, extrafollicular T helper cells express ICOS, CD40L and IL-21, drive class switch recombination by upregulating activation-induced cytidine deaminase in plasmablasts, and promote antibody production by autoreactive B cells (dependant on IL-21 and CD40L).36,130 These findings are similar to those relating to extrafollicular T helper cells identified in BXSB. Yaa mice.117 Likewise, (NZB × NZW)F1 animals also spontaneously produce extrafollicular T cells with a helper phenotype.36 Features of these cells—expression of IL-21, CD40L and ICOS, and their requirement for B-cell maturation—are shared with TFH cells; however, unlike TFH cells, extrafollicular CD4+ T cells in MRL-Faslpr mice lack expression of CXCR5.36 In the absence of CXCR5, expression of CXCR4 by these cells presumably enables their movement to the extrafollicular locations that they colonize, via attraction to its ligand CXCL12 (also known as stromal-cell-derived factor-1) expressed in the splenic red pulp and the medullary cords of lymph nodes.131 Although B-cell maturation and autoantibody production as a result of extrafollicular foci formation is important in mouse models of lupus,128 the role of this pathway in human SLE remains unknown. Given the contribution of the extrafollicular response in promotion of systemic autoimmunity, consideration of this population of B-cell helpers in the development of new therapeutic approaches will be important. As the characteristics of extrafollicular T helper cells overlap with those of TFH cells, therapies that affect the function of the latter will probably disrupt extrafollicular responses as well.
TFH cells in human autoimmunity
TFH cells are clearly crucial to the pathogenesis of lupus in mice, whereas knowledge of the involvement of these cells in human SLE remains relatively limited. Their role in promotion of this disease has largely been extrapolated from the observation that the GC B-cell maturation pathway is aberrantly regulated,124,132 and from analysis of T cells isolated from the blood of patients with SLE (which might be confounded by the incomplete understanding of circulating TFH cells, as discussed above). Abnormal GC reactions in patients with SLE can be detected by analysis of circulating B cells, with a class-switched CD38hi antibody-secreting population being expanded in patients with active disease.124 As noted above, intervention with anti-CD40L antibodies caused the disappearance of this plasma cell subset and a corresponding decrease in anti-double-stranded DNA antibody levels, indicating reliance upon T-cell help for development of these cells.124
ICOShi cells are found in the blood of patients with SLE;111,133 however, this co-stimulator is upregulated after CD4+ T-cell activation, thus the ICOShi phenotype is not a specific identifying feature of TFH cells. Circulating CXCR5hi PD-1hi ICOShi cells, referred to as circulating TFH (cTFH) cells, have been detected in the blood of up to one-third of patients with SLE or Sjögren’s syndrome.111 The presence of cTFH cells in patients with SLE correlated with a more severe disease phenotype, although not with disease activity per se.111 Of interest, cTFH cells phenotypically resembled classical lymphoid TFH cells, but did not express BCL6 and IL-21, and their ability to deliver B-cell help was not discussed.111 However, BCL6 is downregulated in mouse TFH cells as the GC response progresses,61 but is rapidly re-expressed after T-cell receptor stimulation of circulating CXCR5hi cells.104 These findings suggest that cTFH cells could represent blood-borne TFH-cell precursors with the potential for rapid CXCR5-mediated follicular access and B-cell helper functions.104,105 Moreover, the high levels of ICOS and PD-1 in cTFH cells indicate that these cells might be targeted by therapies that abrogate T-cell–B-cell collaboration, as indicated by the effectiveness of blockade of ICOS signalling in mouse models of lupus.127
T-cell–B-cell aggregates and ectopic GCs have been found in the kidneys of patients with lupus nephritis;134 T-cell-associated centroblasts—GC B cells—and plasmablasts within these GCs and aggregates, respectively, seemed to be capable of clonal expansion and somatic hypermutation.134 Ectopic lymphoid structures can also develop in other chronic inflammatory conditions, and have been observed within the synovium in rheumatoid arthritis and spondyloarthritis.135,136 Indeed, development of tertiary lymphoid structures containing T cells phenotypically resembling TFH cells is not uncommon in chronic autoimmune and inflammatory diseases, and might be a direct result of the ongoing inflammatory process underlying these conditions.136 Presumably, T-helper cells in nonlymphoid tissues can promote B-cell maturation and synthesis of potentially pathogenic autoantibody production, thus potentiating tissue injury, as the lupus nephritis example suggests. However, local antibody production in synovial tissue was not found to be pathogenic in a number of patients with rheumatoid arthritis.136 Regardless of these data, the responsiveness of TFH cells in tertiary lymphoid organs to therapeutic agents designed to disrupt their more classical roles in GC maturation in the spleen and lymph nodes should be considered. This awareness seems especially important given the role of TFH cells resident within secondary lymphoid organs in systemic autoimmunity112,114 and inflammatory arthritis.137
Conclusions
Experiments from several laboratories indicate that autoreactive T cells are necessary for full penetrance of autoantibody production and disease in SLE and mouse lupus. Nevertheless, the cellular mechanisms that promote autoreactive collaboration between CD4+ T cells and B cells, with resultant effector function, remain incompletely defined. Understanding of the events that initiate or sustain aberrant T-cell–B-cell communication and the subsequent pathogenic outcomes of such interaction, including autoantibody production and inflammation, is essential, as interference in these processes is likely to be therapeutically important. Issues of therapeutic toxicity or efficacy could be encountered in the development of agents targeting these pathways; therefore, definition of the mechanistic basis of these collaborative interactions will be crucial. Dissection of the potentially separable roles of effector CD4+ T cells and TFH cells in diseases in which autoantibodies are pathogenic, such as SLE and other systemic autoimmune syndromes, offers a means to better understand disease biology; this knowledge is essential to maintain the pipeline of therapeutic targets, and might ultimately lead to the development of novel treatments.
Review criteria.
The PubMed database was searched for full-text, English-language original and review articles published between 1980 and January 2012. The search terms used, either alone or in combination, were: “arthritis”; “B-cell follicles”; “B cells”; “BCL6”; “CD4 T cells”; “CXCR5”; “follicular helper”; “germinal centre”; “lupus”; “PD-1”; “regulatory T cells”; and “interleukin-21”. The reference lists of identified articles were searched for further papers.
Key points.
Follicular helper T (TFH) cells—a subset of CD4+ T cells—are located within the B-cell follicles of secondary lymphoid organs
TFH cells are important regulators of B-cell maturation within germinal centres during normal immune responses
Characterization of the developmental program of TFH cells could aid their identification and provide insight into the function of these cells in normal and autoimmune responses
The transcription factor B-cell lymphoma 6 is both necessary and sufficient for development of TFH cells, controlling expression of molecules essential for TFH-cell trafficking and function
TFH cells promote pathogenic autoantibody production in systemic autoimmunity, and potentially represent novel therapeutic targets in autoimmune diseases
Acknowledgments
This work was partially supported by NIH grants R01 AR40072, R01 AR44076 and P30 AR053495, and by Rheuminations, Inc. and the Alliance for Lupus Research. The author also acknowledges the many helpful discussions at lab meetings and other forums with his past and present trainees.
Footnotes
Competing interests
The author declares no competing interests.
References
- 1.Campbell DJ, Kim CH, Butcher EC. Separable effector T cell populations specialized for B cell help or tissue inflammation. Nat Immunol. 2001;2:876–881. doi: 10.1038/ni0901-876. [DOI] [PubMed] [Google Scholar]
- 2.Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol. 2002;2:933–944. doi: 10.1038/nri954. [DOI] [PubMed] [Google Scholar]
- 3.Szabo SJ, et al. A novel transcription factor, T-bet, directs TH1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 4.Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for TH2 cytokine gene expression in CD4 T cells. Cell. 1997;89:587–596. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- 5.Dang EV, et al. Control of T(H)17/T(REG) balance by hypoxia-inducible factor 1. Cell. 2011;146:772–784. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ivanov II, et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T Helper Cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 7.Yang XO, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors RORα and RORγ. Immunity. 2008;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liang SC, et al. Interleukin (IL)-22 and IL-17 are coexpressed by TH17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006;203:2271–2279. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eyerich S, et al. TH22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J Clin Invest. 2009;119:3573–3585. doi: 10.1172/JCI40202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Langrish CL, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koenders MI, et al. Blocking of interleukin-17 during reactivation of experimental arthritis prevents joint inflammation and bone erosion by decreasing RANKL and interleukin-1. Am J Pathol. 2005;167:141–149. doi: 10.1016/S0002-9440(10)62961-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crispín JC, Tsokos GC. Interleukin-17-producing T cells in lupus. Curr Opin Rheum. 2010;22:499–503. doi: 10.1097/BOR.0b013e32833c62b0. [DOI] [PubMed] [Google Scholar]
- 13.Parker DC. T cell-dependent B cell activation. Annu Rev Immunol. 1993;11:331–360. doi: 10.1146/annurev.iy.11.040193.001555. [DOI] [PubMed] [Google Scholar]
- 14.Liu YJ, Zhang J, Lane PJ, Chan EY, MacLennan IC. Sites of specific B cell activation in primary and secondary responses to T cell-dependent and T cell-independent antigens. Eur J Immunol. 1991;21:2951–2962. doi: 10.1002/eji.1830211209. [DOI] [PubMed] [Google Scholar]
- 15.MacLennan IC, et al. Extrafollicular antibody responses. Immunol Rev. 2003;194:8–18. doi: 10.1034/j.1600-065x.2003.00058.x. [DOI] [PubMed] [Google Scholar]
- 16.Jacob J, Kassir R, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. I. The architecture and dynamics of responding cell populations. J Exp Med. 1991;173:1165–1175. doi: 10.1084/jem.173.5.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berek C, Berger A, Apel M. Maturation of the immune response in germinal centers. Cell. 1991;67:1121–1129. doi: 10.1016/0092-8674(91)90289-b. [DOI] [PubMed] [Google Scholar]
- 18.Coffey F, Alabyev B, Manser T. Initial clonal expansion of germinal center B cells takes place at the perimeter of follicles. Immunity. 2009;30:599–609. doi: 10.1016/j.immuni.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kerfoot SM, et al. Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity. 2011;34:947–960. doi: 10.1016/j.immuni.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi YS, et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity. 2011;34:932–946. doi: 10.1016/j.immuni.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Willimann K, et al. The chemokine SLC is expressed in T cell areas of lymph nodes and mucosal lymphoid tissues and attracts activated T cells via CCR7. Eur J Immunol. 1998;28:2025–2034. doi: 10.1002/(SICI)1521-4141(199806)28:06<2025::AID-IMMU2025>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 22.Nagira M, et al. A lymphocyte-specific CC chemokine, secondary lymphoid tissue chemokine (SLC), is a highly efficient chemoattractant for B cells and activated T cells. Eur J Immunol. 1998;28:1516–1523. doi: 10.1002/(SICI)1521-4141(199805)28:05<1516::AID-IMMU1516>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 23.Luther SA, et al. Differing activities of homeostatic chemokines CCL19, CCL21, and CXCL12 in lymphocyte and dendritic cell recruitment and lymphoid neogenesis. J Immunol. 2002;169:424–433. doi: 10.4049/jimmunol.169.1.424. [DOI] [PubMed] [Google Scholar]
- 24.Haynes NM, et al. Role of CXCR5 and CCR7 in follicular TH cell positioning and appearance of a programmed cell death gene-1High germinal center-associated subpopulation. J Immunol. 2007;179:5099–5108. doi: 10.4049/jimmunol.179.8.5099. [DOI] [PubMed] [Google Scholar]
- 25.Poholek AC, et al. In vivo regulation of Bcl6 and T follicular helper cell development. J Immunol. 2010;185:313–326. doi: 10.4049/jimmunol.0904023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Veerman KM, et al. Interaction of the selectin ligand PSGL-1 with chemokines CCL21 and CCL19 facilitates efficient homing of T cells to secondary lymphoid organs. Nat Immunol. 2007;8:532–539. doi: 10.1038/ni1456. [DOI] [PubMed] [Google Scholar]
- 27.Ansel KM, McHeyzer-Williams LJ, Ngo VN, McHeyzer-Williams MG, Cyster JG. In vivo-activated CD4 T cells upregulate CXC chemokine receptor 5 and reprogram their response to lymphoid chemokines. J Exp Med. 1999;190:1123–1134. doi: 10.1084/jem.190.8.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walker LS, et al. Compromised OX40 function in CD28-deficient mice is linked with failure to develop CXC chemokine receptor 5-positive CD4 cells and germinal centers. J Exp Med. 1999;190:1115–1122. doi: 10.1084/jem.190.8.1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gunn MD, et al. A B-cell-homing chemokine made in lymphoid follicles activates Burkitt’s lymphoma receptor-1. Nature. 1998;391:799–803. doi: 10.1038/35876. [DOI] [PubMed] [Google Scholar]
- 30.Hardtke S, Ohl L, Forster R. Balanced expression of CXCR5 and CCR7 on follicular T helper cells determines their transient positioning to lymph node follicles and is essential for efficient B-cell help. Blood. 2005;106:1924–1931. doi: 10.1182/blood-2004-11-4494. [DOI] [PubMed] [Google Scholar]
- 31.Akiba H, et al. The role of ICOS in the CXCR5+ follicular B helper T cell maintenance in vivo. J Immunol. 2005;175:2340–2348. doi: 10.4049/jimmunol.175.4.2340. [DOI] [PubMed] [Google Scholar]
- 32.Nurieva RI, et al. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity. 2008;29:138–149. doi: 10.1016/j.immuni.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnston RJ, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science. 2009;325:1006–1010. doi: 10.1126/science.1175870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baumjohann D, Okada T, Ansel KM. Cutting edge: distinct waves of BCL6 expression during T follicular helper cell development. J Immunol. 2011;187:2089–2092. doi: 10.4049/jimmunol.1101393. [DOI] [PubMed] [Google Scholar]
- 35.Dong C, Temann UA, Flavell RA. Cutting edge: critical role of inducible costimulator in germinal center reactions. J Immunol. 2001;166:3659–3662. doi: 10.4049/jimmunol.166.6.3659. [DOI] [PubMed] [Google Scholar]
- 36.Odegard JM, et al. ICOS-dependent extrafollicular helper T cells elicit IgG production via IL-21 in systemic autoimmunity. J Exp Med. 2008;205:2873–2886. doi: 10.1084/jem.20080840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bossaller L, et al. ICOS deficiency is associated with a severe reduction of CXCR5+ CD4 germinal center TH cells. J Immunol. 2006;177:4927–4932. doi: 10.4049/jimmunol.177.7.4927. [DOI] [PubMed] [Google Scholar]
- 38.Burmeister Y, et al. ICOS controls the pool size of effector-memory and regulatory T cells. J Immunol. 2008;180:774–782. doi: 10.4049/jimmunol.180.2.774. [DOI] [PubMed] [Google Scholar]
- 39.Good-Jacobson KL, et al. PD-1 regulates germinal center B cell survival and the formation and affinity of long-lived plasma cells. Nat Immunol. 2010;11:535–542. doi: 10.1038/ni.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reif K, et al. Balanced responsiveness to chemoattractants from adjacent zones determines B-cell position. Nature. 2002;416:94–99. doi: 10.1038/416094a. [DOI] [PubMed] [Google Scholar]
- 41.Hannedouche S, et al. Oxysterols direct immune cell migration via EBI2. Nature. 2011;475:524–527. doi: 10.1038/nature10280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu C, et al. Oxysterols direct B-cell migration through EBI2. Nature. 2011;475:519–523. doi: 10.1038/nature10226. [DOI] [PubMed] [Google Scholar]
- 43.Pereira JP, Kelly LM, Xu Y, Cyster JG. EBI2 mediates B cell segregation between the outer and centre follicle. Nature. 2009;460:1122–1126. doi: 10.1038/nature08226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gatto D, Paus D, Basten A, Mackay CR, Brink R. Guidance of B Cells by the orphan G protein-coupled receptor EBI2 shapes humoral immune responses. Immunity. 2009;31:259–269. doi: 10.1016/j.immuni.2009.06.016. [DOI] [PubMed] [Google Scholar]
- 45.Crotty S, Kersh EN, Cannons J, Schwartzberg PL, Ahmed R. SAP is required for generating long-term humoral immunity. Nature. 2003;421:282–287. doi: 10.1038/nature01318. [DOI] [PubMed] [Google Scholar]
- 46.Cannons JL, et al. SAP regulates T cell-mediated help for humoral immunity by a mechanism distinct from cytokine regulation. J Exp Med. 2006;203:1551–1565. doi: 10.1084/jem.20052097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qi H, Cannons JL, Klauschen F, Schwartzberg PL, Germain RN. SAP-controlled T-B cell interactions underlie germinal centre formation. Nature. 2008;455:764–769. doi: 10.1038/nature07345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cunningham AF, Serre K, Mohr E, Khan M, Toellner KM. Loss of CD154 impairs the TH2 extrafollicular plasma cell response but not early T cell proliferation and interleukin-4 induction. Immunology. 2004;113:187–193. doi: 10.1111/j.1365-2567.2004.01951.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deenick EK, et al. Follicular helper T cell differentiation requires continuous antigen presentation that is independent of unique B cell signaling. Immunity. 2010;33:241–253. doi: 10.1016/j.immuni.2010.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schaerli P, Loetscher P, Moser B. Cutting edge: induction of follicular homing precedes effector TH cell development. J Immunol. 2001;167:6082–6086. doi: 10.4049/jimmunol.167.11.6082. [DOI] [PubMed] [Google Scholar]
- 51.Springer TA. Traffic signals on endothelium for lymphocyte recirculation and leukocyte emigration. Annu Rev Physiol. 1995;57:827–872. doi: 10.1146/annurev.ph.57.030195.004143. [DOI] [PubMed] [Google Scholar]
- 52.Austrup F, et al. P- and E-selectin mediate recruitment of T-helper-1 but not T-helper-2 cells into inflammed tissues. Nature. 1997;385:81–83. doi: 10.1038/385081a0. [DOI] [PubMed] [Google Scholar]
- 53.Ley K, Kansas GS. Selectins in T-cell recruitment to non-lymphoid tissues and sites of inflammation. Nat Rev Immunol. 2004;4:325–335. doi: 10.1038/nri1351. [DOI] [PubMed] [Google Scholar]
- 54.Kim CH, et al. Rules of chemokine receptor association with T cell polarization in vivo. J Clin Invest. 2001;108:1331–1339. doi: 10.1172/JCI13543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dent AL, Shaffer AL, Yu X, Allman D, Staudt LM. Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science. 1997;276:589–592. doi: 10.1126/science.276.5312.589. [DOI] [PubMed] [Google Scholar]
- 56.Ye BH, et al. The BCL-6 proto-oncogene controls germinal-centre formation and TH2-type inflammation. Nat Genet. 1997;16:161–170. doi: 10.1038/ng0697-161. [DOI] [PubMed] [Google Scholar]
- 57.Chtanova T, et al. T follicular helper cells express a distinctive transcriptional profile, reflecting their role as non-TH1/TH2 effector cells that provide help for B cells. J Immunol. 2004;173:68–78. doi: 10.4049/jimmunol.173.1.68. [DOI] [PubMed] [Google Scholar]
- 58.Nurieva RI, et al. Bcl6 mediates the development of T follicular helper cells. Science. 2009;325:1001–1005. doi: 10.1126/science.1176676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yu D, et al. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity. 2009;31:457–468. doi: 10.1016/j.immuni.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 60.Johnston RJ, Choi YS, Diamond JA, Yang JA, Crotty S. STAT5 is a potent negative regulator of TFH cell differentiation. J Exp Med. doi: 10.1084/jem.20111174. http://dx.doi.org/10.1084/jem.20111174. [DOI] [PMC free article] [PubMed]
- 61.Kitano M, et al. Bcl6 protein expression shapes pre-germinal center B cell dynamics and follicular helper T cell heterogeneity. Immunity. 2011;34:961–972. doi: 10.1016/j.immuni.2011.03.025. [DOI] [PubMed] [Google Scholar]
- 62.Garside P, et al. Visualization of specific B and T lymphocyte interactions in the lymph node. Science. 1998;281:96–99. doi: 10.1126/science.281.5373.96. [DOI] [PubMed] [Google Scholar]
- 63.Breitfeld D, et al. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med. 2000;192:1545–1552. doi: 10.1084/jem.192.11.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schaerli P, et al. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J Exp Med. 2000;192:1553–1562. doi: 10.1084/jem.192.11.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim CH, et al. Subspecialization of CXCR5+ T cells: B helper activity is focused in a germinal center-localized subset of CXCR5+ T cells. J Exp Med. 2001;193:1373–1381. doi: 10.1084/jem.193.12.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jacob J, Kelsoe G, Rajewsky K, Weiss U. Intraclonal generation of antibody mutants in germinal centres. Nature. 1991;354:389–392. doi: 10.1038/354389a0. [DOI] [PubMed] [Google Scholar]
- 67.Liu YJ, et al. Within germinal centers, isotype switching of immunoglobulin genes occurs after the onset of somatic mutation. Immunity. 1996;4:241–250. doi: 10.1016/s1074-7613(00)80432-x. [DOI] [PubMed] [Google Scholar]
- 68.Parrish-Novak J, et al. Interleukin 21 and its receptor are involved in NK cell expansion and regulation of lymphocyte function. Nature. 2000;408:57–63. doi: 10.1038/35040504. [DOI] [PubMed] [Google Scholar]
- 69.Mehta DS, et al. IL-21 induces the apoptosis of resting and activated primary B cells. J Immunol. 2003;170:4111–4118. doi: 10.4049/jimmunol.170.8.4111. [DOI] [PubMed] [Google Scholar]
- 70.Jin H, Carrio R, Yu A, Malek TR. Distinct activation signals determine whether IL-21 induces B cell costimulation, growth arrest, or Bim-dependent apoptosis. J Immunol. 2004;173:657–665. doi: 10.4049/jimmunol.173.1.657. [DOI] [PubMed] [Google Scholar]
- 71.Ozaki K, et al. Regulation of B cell differentiation and plasma cell generation by IL-21, a novel inducer of Blimp-1 and Bcl-6. J Immunol. 2004;173:5361–5371. doi: 10.4049/jimmunol.173.9.5361. [DOI] [PubMed] [Google Scholar]
- 72.Ettinger R, et al. IL-21 induces differentiation of human naive and memory B cells into antibody-secreting plasma cells. J Immunol. 2005;175:7867–7879. doi: 10.4049/jimmunol.175.12.7867. [DOI] [PubMed] [Google Scholar]
- 73.Kuchen S, et al. Essential role of IL-21 in B cell activation, expansion, and plasma cell generation during CD4+ T cell-B cell collaboration. J Immunol. 2007;179:5886–5896. doi: 10.4049/jimmunol.179.9.5886. [DOI] [PubMed] [Google Scholar]
- 74.Linterman MA, et al. IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J Exp Med. 2010;207:353–363. doi: 10.1084/jem.20091738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zotos D, et al. IL-21 regulates germinal center B cell differentiation and proliferation through a B cell-intrinsic mechanism. J Exp Med. 2010;207:365–378. doi: 10.1084/jem.20091777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Eto D, et al. IL-21 and IL-6 are critical for different aspects of B cell immunity and redundantly induce optimal follicular helper CD4 T cell (TFH) differentiation. PLoS One. 2011;6:e17739. doi: 10.1371/journal.pone.0017739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ozaki K, et al. A critical role for IL-21 in regulating immunoglobulin production. Science. 2002;298:1630–1634. doi: 10.1126/science.1077002. [DOI] [PubMed] [Google Scholar]
- 78.Hsu HC, et al. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol. 2008;9:166–175. doi: 10.1038/ni1552. [DOI] [PubMed] [Google Scholar]
- 79.Reinhardt RL, Liang HE, Locksley RM. Cytokine-secreting follicular T cells shape the antibody repertoire. Nat Immunol. 2009;10:385–393. doi: 10.1038/ni.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.King IL, Mohrs M. IL-4-producing CD4+ T cells in reactive lymph nodes during helminth infection are T follicular helper cells. J Exp Med. 2009;206:1001–1007. doi: 10.1084/jem.20090313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bauquet AT, et al. The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and TH-17 cells. Nat Immunol. 2009;10:167–175. doi: 10.1038/ni.1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hams E, et al. Blockade of B7-H1 (programmed death ligand 1) enhances humoral immunity by positively regulating the generation of T follicular helper cells. J Immunol. 2011;186:5648–5655. doi: 10.4049/jimmunol.1003161. [DOI] [PubMed] [Google Scholar]
- 83.Mempel TR, Henrickson SE, Von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. 2004;427:154–159. doi: 10.1038/nature02238. [DOI] [PubMed] [Google Scholar]
- 84.Callard RE, Armitage RJ, Fanslow WC, Spriggs MK. CD40 ligand and its role in X-linked hyper-IgM syndrome. Immunol Today. 1993;14:559–564. doi: 10.1016/0167-5699(93)90188-Q. [DOI] [PubMed] [Google Scholar]
- 85.Xu J, et al. Mice deficient for the CD40 ligand. Immunity. 1994;1:423–431. doi: 10.1016/1074-7613(94)90073-6. [DOI] [PubMed] [Google Scholar]
- 86.McAdam AJ, et al. ICOS is critical for CD40-mediated antibody class switching. Nature. 2001;409:102–105. doi: 10.1038/35051107. [DOI] [PubMed] [Google Scholar]
- 87.Lee SK, et al. B cell priming for extrafollicular antibody responses requires Bcl-6 expression by T cells. J Exp Med. 2011;208:1377–1388. doi: 10.1084/jem.20102065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Barral P, et al. B cell receptor-mediated uptake of CD1d-restricted antigen augments antibody responses by recruiting invariant NKT cell help in vivo. Proc Natl Acad Sci USA. 2008;105:8345–8350. doi: 10.1073/pnas.0802968105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Leadbetter EA, et al. NK T cells provide lipid antigen-specific cognate help for B cells. Proc Natl Acad Sci USA. 2008;105:8339–8344. doi: 10.1073/pnas.0801375105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.King IL, et al. Invariant natural killer T cells direct B cell responses to cognate lipid antigen in an IL-21-dependent manner. Nat Immunol. 2011;13:44–50. doi: 10.1038/ni.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chang PP, et al. Identification of Bcl-6-dependent follicular helper NKT cells that provide cognate help for B cell responses. Nat Immunol. 2011;13:35–43. doi: 10.1038/ni.2166. [DOI] [PubMed] [Google Scholar]
- 92.Vinuesa CG, Sanz I, Cook MC. Dysregulation of germinal centres in autoimmune disease. Nat Rev Immunol. 2009;9:845–857. doi: 10.1038/nri2637. [DOI] [PubMed] [Google Scholar]
- 93.Linterman MA, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med. 2011;17:975–982. doi: 10.1038/nm.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wollenberg I, et al. Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J Immunol. 2011;187:4553–4560. doi: 10.4049/jimmunol.1101328. [DOI] [PubMed] [Google Scholar]
- 95.Chung Y, et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med. 2011;17:983–988. doi: 10.1038/nm.2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Alexander CM, et al. T regulatory cells participate in the control of germinal centre reactions. Immunology. 2011;133:452–468. doi: 10.1111/j.1365-2567.2011.03456.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jang E, et al. Foxp3+ regulatory T cells control humoral autoimmunity by suppressing the development of long-lived plasma cells. J Immunol. 2011;186:1546–1553. doi: 10.4049/jimmunol.1002942. [DOI] [PubMed] [Google Scholar]
- 98.Ritchie AW, James K, Micklem HS. The distribution and possible significance of cells identified in human lymphoid tissue by the monoclonal antibody HNK-1. Clin Exp Immunol. 1983;51:439–447. [PMC free article] [PubMed] [Google Scholar]
- 99.Banerjee D, Thibert RF. Natural killer-like cells found in B-cell compartments of human lymphoid tissues. Nature. 1983;304:270–272. doi: 10.1038/304270a0. [DOI] [PubMed] [Google Scholar]
- 100.Kim JR, Lim HW, Kang SG, Hillsamer P, Kim CH. Human CD57+ germinal center-T cells are the major helpers for GC-B cells and induce class switch recombination. BMC Immunol. 2005;6:3. doi: 10.1186/1471-2172-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Casamayor-Palleja M, Khan M, MacLennan IC. A subset of CD4+ memory T cells contains preformed CD40 ligand that is rapidly but transiently expressed on their surface after activation through the T cell receptor complex. J Exp Med. 1995;181:1293–1301. doi: 10.1084/jem.181.4.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rasheed AU, Rahn HP, Sallusto F, Lipp M, Müller G. Follicular B helper T cell activity is confined to CXCR5hiICOShi CD4 T cells and is independent of CD57 expression. Eur J Immunol. 2006;36:1892–1903. doi: 10.1002/eji.200636136. [DOI] [PubMed] [Google Scholar]
- 103.Lim HW, Kim CH. Loss of IL-7 receptor α on CD4+ T cells defines terminally differentiated B cell-helping effector T cells in a B cell-rich lymphoid tissue. J Immunol. 2007;179:7448–7456. doi: 10.4049/jimmunol.179.11.7448. [DOI] [PubMed] [Google Scholar]
- 104.Chevalier N, et al. CXCR5 expressing human central memory CD4 T cells and their relevance for humoral immune responses. J Immunol. 2011;186:5556–5568. doi: 10.4049/jimmunol.1002828. [DOI] [PubMed] [Google Scholar]
- 105.Morita R, et al. Human blood CXCR5+CD4+ T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity. 2011;34:108–121. doi: 10.1016/j.immuni.2010.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.MacLeod MK, et al. Memory CD4 T cells that express CXCR5 provide accelerated help to B cells. J Immunol. 2011;186:2889–2896. doi: 10.4049/jimmunol.1002955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bentebibel SE, Schmitt N, Banchereau J, Ueno H. Human tonsil B-cell lymphoma 6 (BCL6)-expressing CD4+ T-cell subset specialized for B-cell help outside germinal centers. Proc Natl Acad Sci USA. 2011;108:E488–E497. doi: 10.1073/pnas.1100898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dogan I, et al. Multiple layers of B cell memory with different effector functions. Nat Immunol. 2009;10:1292–1299. doi: 10.1038/ni.1814. [DOI] [PubMed] [Google Scholar]
- 109.Pepper M, Pagán AJ, Igyártó BZ, Taylor JJ, Jenkins MK. Opposing signals from the Bcl6 transcription factor and the interleukin-2 receptor generate T helper 1 central and effector memory cells. Immunity. 2011;35:583–595. doi: 10.1016/j.immuni.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Marshall HD, et al. Differential expression of Ly6C and T-bet distinguish effector and memory TH1 CD4+ cell properties during viral infection. Immunity. 2011;35:633–646. doi: 10.1016/j.immuni.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Simpson N, et al. Expansion of circulating T cells resembling follicular helper T cells is a fixed phenotype that identifies a subset of severe systemic lupus erythematosus. Arthritis Rheum. 2010;62:234–244. doi: 10.1002/art.25032. [DOI] [PubMed] [Google Scholar]
- 112.Vinuesa CG, et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature. 2005;435:452–458. doi: 10.1038/nature03555. [DOI] [PubMed] [Google Scholar]
- 113.Luzina IG, et al. Spontaneous formation of germinal centers in autoimmune mice. J Leukoc Biol. 2001;70:578–584. [PubMed] [Google Scholar]
- 114.Linterman MA, et al. Follicular helper T cells are required for systemic autoimmunity. J Exp Med. 2009;206:561–576. doi: 10.1084/jem.20081886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bertossi A, et al. Loss of Roquin induces early death and immune deregulation but not autoimmunity. J Exp Med. 2011;208:1749–1756. doi: 10.1084/jem.20110578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pisitkun P, et al. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006;312:1669–1672. doi: 10.1126/science.1124978. [DOI] [PubMed] [Google Scholar]
- 117.Bubier JA, et al. A critical role for IL-21 receptor signaling in the pathogenesis of systemic lupus erythematosus in BXSB-Yaa mice. Proc Natl Acad Sci USA. 2009;106:1518–1523. doi: 10.1073/pnas.0807309106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Herber D, et al. IL-21 has a pathogenic role in a lupus-prone mouse model and its blockade with IL-21R.Fc reduces disease progression. J Immunol. 2007;178:3822–3830. doi: 10.4049/jimmunol.178.6.3822. [DOI] [PubMed] [Google Scholar]
- 119.Peng SL, Moslehi J, Craft J. Roles of interferon-γ and interleukin-4 in murine lupus. J Clin Invest. 1997;99:1936–1946. doi: 10.1172/JCI119361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Masutani K, et al. Predominance of TH1 immune response in diffuse proliferative lupus nephritis. Arthritis Rheum. 2001;44:2097–2106. doi: 10.1002/1529-0131(200109)44:9<2097::AID-ART360>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 121.Mohan C, Shi Y, Laman JD, Datta SK. Interaction between CD40 and its ligand gp39 in the development of murine lupus nephritis. J Immunol. 1995;154:1470–1480. [PubMed] [Google Scholar]
- 122.Ma J, et al. Autoimmune lpr/lpr mice deficient in CD40 ligand: spontaneous Ig class switching with dichotomy of autoantibody responses. J Immunol. 1996;157:417–426. [PubMed] [Google Scholar]
- 123.Daikh DI, Finck BK, Linsley PS, Hollenbaugh D, Wofsy D. Long-term inhibition of murine lupus by brief simultaneous blockade of the B7/CD28 and CD40/gp39 costimulation pathways. J Immunol. 1997;159:3104–3108. [PubMed] [Google Scholar]
- 124.Grammer AC, et al. Abnormal germinal center reactions in systemic lupus erythematosus demonstrated by blockade of CD154–CD40 interactions. J Clin Invest. 2003;112:1506–1520. doi: 10.1172/JCI19301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Duffau P, et al. Platelet CD154 potentiates interferon-α secretion by plasmacytoid dendritic cells in systemic lupus erythematosus. Sci Transl Med. 2010;2:47ra63. doi: 10.1126/scitranslmed.3001001. [DOI] [PubMed] [Google Scholar]
- 126.Peters AL, Stunz LL, Bishop GA. CD40 and autoimmunity: the dark side of a great activator. Semin Immunol. 2009;21:293–300. doi: 10.1016/j.smim.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Iwai H, et al. Involvement of inducible costimulator-B7 homologous protein costimulatory pathway in murine lupus nephritis. J Immunol. 2003;171:2848–2854. doi: 10.4049/jimmunol.171.6.2848. [DOI] [PubMed] [Google Scholar]
- 128.William J, Euler C, Christensen S, Shlomchik MJ. Evolution of autoantibody responses via somatic hypermutation outside of germinal centers. Science. 2002;297:2066–2070. doi: 10.1126/science.1073924. [DOI] [PubMed] [Google Scholar]
- 129.Hoyer BF, et al. Short-lived plasmablasts and long-lived plasma cells contribute to chronic humoral autoimmunity in NZB/W mice. J Exp Med. 2004;199:1577–1584. doi: 10.1084/jem.20040168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rankin AL, et al. IL-21 receptor is required for the systemic accumulation of activated B and T lymphocytes in MRL/MpJ-Faslpr/lpr/J mice. J Immunol. doi: 10.4049/jimmunol.1003871. http://dx.doi.org/10.4049/jimmunol.1003871. [DOI] [PMC free article] [PubMed]
- 131.Hargreaves DC, et al. A coordinated change in chemokine responsiveness guides plasma cell movements. J Exp Med. 2001;194:45–56. doi: 10.1084/jem.194.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cappione A, 3rd, et al. Germinal center exclusion of autoreactive B cells is defective in human systemic lupus erythematosus. J Clin Invest. 2005;115:3205–3216. doi: 10.1172/JCI24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yang JH, et al. Expression and function of inducible costimulator on peripheral blood T cells in patients with systemic lupus erythematosus. Rheumatology (Oxford) 2005;44:1245–1254. doi: 10.1093/rheumatology/keh724. [DOI] [PubMed] [Google Scholar]
- 134.Chang A, et al. In situ B cell-mediated immune responses and tubulointerstitial inflammation in human lupus nephritis. J Immunol. 2011;186:1849–1860. doi: 10.4049/jimmunol.1001983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Weyand CM, Kang YM, Kurtin PJ, Goronzy JJ. The power of the third dimension: tissue architecture and autoimmunity in rheumatoid arthritis. Curr Opin Rheumatol. 2003;15:259–266. doi: 10.1097/00002281-200305000-00013. [DOI] [PubMed] [Google Scholar]
- 136.Cantaert T, et al. B lymphocyte autoimmunity in rheumatoid synovitis is independent of ectopic lymphoid neogenesis. J Immunol. 2008;181:785–794. doi: 10.4049/jimmunol.181.1.785. [DOI] [PubMed] [Google Scholar]
- 137.Victoratos P, Kollias G. Induction of autoantibody-mediated spontaneous arthritis critically depends on follicular dendritic cells. Immunity. 2009;30:130–142. doi: 10.1016/j.immuni.2008.10.019. [DOI] [PubMed] [Google Scholar]