

The crystal structure of M. tuberculosis l,d-transpeptidase (LdtMt2; Rv2518c) has been determined in both ligand-free and meropenem-bound forms. The detailed view of the interactions between meropenem and LdtMt2 will be useful in structure-guided discovery of new antituberculosis drugs.
Keywords: Rv2518c; Mt2594; LdtMt2; l,d-transpeptidases; Mycobacterium tuberculosis; meropenem; carbapenem; peptidoglycans; antituberculosis drug discovery
Abstract
Difficulty in the treatment of tuberculosis and growing drug resistance in Mycobacterium tuberculosis (Mtb) are a global health issue. Carbapenems inactivate l,d-transpeptidases; meropenem, when administered with clavulanate, showed in vivo activity against extensively drug-resistant Mtb strains. LdtMt2 (Rv2518c), one of two functional l,d-transpeptidases in Mtb, is predominantly expressed over LdtMt1 (Rv0116c). Here, the crystal structure of N-terminally truncated LdtMt2 (residues Leu131–Ala408) is reported in both ligand-free and meropenem-bound forms. The structure of meropenem-inhibited LdtMt2 provides a detailed structural view of the interactions between a carbapenem drug and Mtb l,d-transpeptidase. The structures revealed that the catalytic l,d-transpeptidase domain of LdtMt2 is preceded by a bacterial immunogloblin-like Big_5 domain and is followed by an extended C-terminal tail that interacts with both domains. Furthermore, it is shown using mass analyses that meropenem acts as a suicide inhibitor of LdtMt2. Upon acylation of the catalytic Cys354 by meropenem, the ‘active-site lid’ undergoes a large conformational change to partially cover the active site so that the bound meropenem is accessible to the bulk solvent via three narrow paths. This work will facilitate structure-guided discovery of l,d-transpeptidase inhibitors as novel antituberculosis drugs against drug-resistant Mtb.
1. Introduction
Mycobacterium tuberculosis (Mtb), which is a highly successful intracellular pathogen, infects nearly one-third of the world’s population. It causes tuberculosis (TB), which claims the lives of millions of people every year (Dye & Williams, 2010 ▶). The treatment of TB is difficult and requires many months of taking a combination of several anti-TB drugs. The growing incidence of multidrug-resistant and extensively drug-resistant (XDR) strains of Mtb poses a global health problem (Chiang et al., 2010 ▶). The enormous success of Mtb is based on three capabilities: (i) its reprogramming of macrophages after primary infection/phagocytosis to prevent its own destruction, (ii) its initiation of the formation of well organized granulomas comprising different immune cells to create a confined environment for the host–pathogen standoff and (iii) its transition into a stage of dormancy (nonreplicative state) by shutting down its own central metabolism and by terminating replication, thereby rendering itself extremely resistant to host defence and drug treatment (Gengenbacher & Kaufmann, 2012 ▶). An important issue to consider in the development of new anti-TB therapeutics is the phenotypic drug resistance of Mtb organisms in the nonreplicative state, which are genetically indistinguishable but distinct from actively multiplying Mtb organisms. An example of their major differences is the nature of peptidoglycan cross-linking.
The unusual mycolic acid-containing cell wall of Mtb accounts for up to 40% of the dry mass of the cell and the degree of peptidoglycan cross-linking is unusually high (∼70–80%; Goffin & Ghuysen, 2002 ▶; Almeida Da Silva & Palomino, 2011 ▶). The peptidoglycan structure of Mtb from a stationary-phase culture revealed a high content (80%) of nonclassical 3→3 cross-links generated by l,d-transpeptidation (Lavollay et al., 2008 ▶), whereas the classical 4→3 cross-links are predominantly formed by the d,d-transpeptidase activity of penicillin-binding proteins during the exponential phase of growth (Goffin & Ghuysen, 2002 ▶; Wietzerbin et al., 1974 ▶). l,d-Transpeptidases and d,d-transpeptidases are unrelated to each other in amino-acid sequence. Among five paralogues of l,d-transpeptidase in Mtb, two functional l,d-transpeptidases, LdtMt1 (Rv0116c; 251 amino acids) and LdtMt2 (Rv2518c; MT2594; 408 amino acids), have been shown to generate 3→3 cross-links connecting two meso-diaminopimelic acid (meso-DAP) residues at the third position of the stem peptides in vitro (Lavollay et al., 2008 ▶; Gupta et al., 2010 ▶). Of the two Mtb genes that encode functional l,d-transpeptidases, ldtMt2 is predominantly expressed at an at least tenfold higher level than ldtMt1 in all phases of growth (Gupta et al., 2010 ▶). Furthermore, the loss of LdtMt2 leads to altered colony morphology, loss of virulence and increased susceptibility to amoxicillin–clavulanate during the chronic phase of infection, supporting the idea that 3→3 cross-linking by LdtMt2 is vital to the physiology of the peptidoglycan and is essential for the virulence of Mtb in the cheronic phase (Gupta et al., 2010 ▶).
β-Lactam antibiotics are analogues of d-alanyl-d-alanine, which forms the terminal amino-acid residues on the precursor NAM/NAG-peptide subunits of the nascent peptidoglycan layer. The structural similarity between β-lactam antibiotics and d-alanyl-d-alanine facilitates their binding to the active site of penicillin-binding proteins and their inhibition of the d,d-tranpeptidase activity. β-Lactams were previously thought to be ineffective against Mtb primarily as a consequence of their rapid hydrolysis by the endogenous mycobacterial β-lactamase (BlaC), which shows no similarity in sequence or structure to l,d-transpeptidases. However, the discovery that clavulanic acid acts as an irreversible inhibitor of β-lactamases has made functional l,d-transpeptidases of Mtb an attractive target for the development of drugs against Mtb in the dormant state (Labia et al., 1985 ▶). Carbapenems, a specific class of β-lactam antibiotics (Supplementary Fig. S11), have been shown to inactivate l,d-transpeptidases from Enterococcus faecium (Mainardi et al., 2007 ▶; Dubée, Arthur et al., 2012 ▶) and Mtb (LdtMt1; Dubée, Triboulet et al., 2012 ▶). LdtMt2, the major l,d-transpeptidase in Mtb, is also likely to be a target of carbapenems and is considered to be physiologically more important than LdtMt1. A combination of meropenem and clavulanate showed in vivo activity against XDR strains of Mtb (Hugonnet et al., 2009 ▶) as well as against H37Rv strains (Hugonnet et al., 2009 ▶; Veziris et al., 2011 ▶; England et al., 2012 ▶), further suggesting that meropenem, one of the carbapenems, may inhibit Mtb LdtMt2. Despite the potential importance of Mtb l,d-transpeptidase as a novel anti-TB drug target, no structural information on the detailed interactions of carbapenems with any Mtb l,d-transpeptidase is presently available. After submitting our manuscript, the crystal structure of Mtb LdtMt2 containing a bound peptidoglycan fragment was published (Erdemli et al., 2012 ▶).
Here, we report the crystal structure of an N-terminally truncated Mtb LdtMt2 (LdtMt2Δ130) that encompasses residues Leu131–Ala408. In this structure, the catalytic l,d-transpeptidase domain (residues Asp251–Val378) is preceded by a bacterial immunoglobulin-like (Ig-like) Big_5 domain (residues His150–Gly250) and followed by an extended C-terminal tail (residues Asn379–Ala408) that interacts with both domains. We have determined the structure of LdtMt2Δ130 in both ligand-free and drug-bound forms: (i) the apo form, (ii) a mercury-derivatized ligand-free form and (iii) a meropenem-complexed form. Cys354, His336 and Ser337 form the catalytic triad in the active site of the l,d-transpeptidase domain. In the meropenem-complexed structure meropenem is covalently bound to Cys354, mimicking the acyl-enzyme intermediate, and the carbonyl O atom of the opened β-lactam ring is stabilized by the oxyanion hole. In the ligand-free mercury-derivatized model a winding loop containing a two-stranded β-sheet which encompasses residues His300–Asp323 (‘the active-site lid’) is in the open conformation. In the open conformation the catalytic cysteine (Cys354) is exposed to the bulk solvent, while His336 and Ser337 are buried. Upon the acylation of LdtMt2 by meropenem, the active-site lid undergoes a large conformational change and partially covers the catalytic Cys354 so that the bound meropenem is accessible to the bulk solvent via three narrow paths. This study provides new structural insights into the irreversible inhibition of Mtb LdtMt2 by meropenem. It will facilitate the structure-based discovery of Mtb l,d-transpeptidase inhibitors as a novel anti-TB drugs against drug-resistant Mtb.
2. Materials and methods
2.1. Protein expression and purification
Several constructs [residues 1–408 (full length), 55–408, 131–408 and 153–408] of the LdtMt2 gene from Mtb strain H37Rv were PCR-amplified and cloned into the expression vector pET-21a(+) (Novagen) using NdeI and XhoI restriction enzymes. The recombinant proteins fused with a hexahistidine-containing tag (LEHHHHHH) at the C-terminus were overexpressed in Escherichia coli Rosetta2 (DE3) pLysS cells using Luria broth culture medium. Protein expression was induced using 0.5 mM isopropyl β-d-1-thiogalactopyranoside and the cells were incubated for an additional 20 h at 303 K following growth to mid-log phase at 310 K. All constructs except for 153–408 were expressed in E. coli. Only the 131–408 construct (LdtMt2Δ130) gave well diffracting crystals. The cells were lysed by sonication in buffer A (50 mM Tris–HCl pH 7.9, 500 mM NaCl, 50 mM imidazole) containing 5%(v/v) glycerol and 1 mM phenylmethylsulfonyl fluoride. The crude lysate was centrifuged at 36 000g for 1 h. The supernatant was applied onto a HiTrap Chelating HP affinity chromatography column (GE Healthcare) previously equilibrated with buffer A. Upon elution with a gradient of imidazole in the same buffer, recombinant LdtMt2Δ130 protein eluted at an imidazole concentration of 120–180 mM. The eluted protein was applied onto a HiLoad XK-16 Superdex 200 prep-grade column (GE Healthcare) previously equilibrated with 20 mM Tris–HCl pH 7.9, 200 mM NaCl.
2.2. Crystallization
Fractions containing recombinant LdtMt2Δ130 were pooled and concentrated to 15.7 mg ml−1 (0.50 mM) using a YM10 ultrafiltration membrane (Amicon) for crystallization. Crystals were grown by the sitting-drop vapour-diffusion method at 296 K by mixing 1 µl protein solution and 1 µl reservoir solution. Crystals of LdtMt2Δ130 in the apo form were obtained using a reservoir solution consisting of 50 mM calcium chloride, 100 mM bis-tris pH 6.5, 30%(v/v) polyethylene glycol monomethyl ether 550. They grew to approximate dimensions of 0.3 × 0.3 × 0.2 mm within a few days. Crystals of LdtMt2Δ130 pre-incubated with meropenem (50.2 mM) for 30 min were obtained using a reservoir solution consisting of 200 mM lithium chloride, 20%(w/v) polyethylene glycol 3350. They grew to approximate dimensions of 0.2 × 0.2 × 0.1 mm within a few days.
2.3. X-ray data collection and phasing
X-ray diffraction data were collected from a crystal of LdtMt2Δ130 in the apo form using a Quantum 270 CCD detector system (Area Detector Systems Corporation, Poway, California, USA) on beamline NE3A of Photon Factory (PF), Japan. The crystals of apo LdtMt2Δ130 belonged to space group C2, with unit-cell parameters a = 135.6, b = 58.6, c = 40.9 Å, β = 94.4°. One monomer is present in the asymmetric unit, giving a Matthews parameter and solvent fraction of 2.59 Å3 Da−1 and 52.6%, respectively. To collect anomalous diffraction data, a crystal of apo LdtMt2Δ130 was dipped for 20 min into 5 µl of a heavy-atom-containing cryoprotectant solution that consisted of 25%(v/v) glycerol and 20 mM ethylmercury thiosalicylate (EMTS) added to the reservoir solution. Single-wavelength anomalous diffraction (SAD) data were collected from the mercury-derivative crystal of LdtMt2Δ130 at 100 K using a Saturn A200 CCD detector system (Rigaku, Japan) on beamline 26B1 of SPring-8, Japan. The raw data were processed and scaled using the HKL-2000 program suite (Otwinowski & Minor, 1997 ▶). The mercury-derivative crystals of LdtMt2Δ130 belonged to space group C2, with unit-cell parameters a = 135.7, b = 58.4, c = 41.0 Å, β = 94.3°. One monomer is present in the asymmetric unit, giving a Matthews parameter and solvent fraction of 2.59 Å3 Da−1 and 52.6%, respectively. One mercury site was located per LdtMt2Δ130 monomer and the SAD phases were calculated using the AutoSol program from PHENIX (Adams et al., 2010 ▶). X-ray diffraction data were collected from a crystal of meropenem-complexed LdtMt2Δ130 using an ADSC Quantum 270 CCD detector system on beamline BL-1A of the Photon Factory (PF), Japan. The crystals of meropenem-complexed LdtMt2Δ130 belonged to space group P212121, with unit-cell parameters a = 68.9, b = 73.4, c = 104.1 Å. Two monomers are present in the asymmetric unit, giving a Matthews parameter and solvent fraction of 2.08 Å3 Da−1 and 40.9%, respectively. Data-collection and phasing statistics are summarized in Table 1 ▶.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Data set | Apo form | Hg derivative (peak) | Meropenem complex |
|---|---|---|---|
| Data collection | |||
| Space group | C2 | C2 | P212121 |
| Unit-cell parameters | |||
| a (Å) | 135.6 | 135.7 | 68.9 |
| b (Å) | 58.6 | 58.4 | 73.4 |
| c (Å) | 40.9 | 41.0 | 104.1 |
| α = γ (°) | 90.0 | 90.0 | 90.0 |
| β (°) | 94.4 | 94.3 | 90.0 |
| X-ray wavelength (Å) | 1.00000 | 1.00600 | 1.00000 |
| Resolution range (Å) | 20.0–1.80 (1.83–1.80) | 50.0–1.79 (1.82–1.79) | 50.0–2.00 (2.03–2.00) |
| Total No. of reflections | 114133 (3344) | 218163 (7927)† | 254969 (11833) |
| No. of unique reflections | 29426 (1045) | 58915 (2557)† | 34693 (1621) |
| Completeness (%) | 97.9 (70.4) | 99.3 (86.8)† | 95.5 (91.2) |
| 〈I/σ(I)〉 | 46.5 (9.1) | 43.0 (7.4)† | 31.0 (4.3) |
| R merge ‡ (%) | 4.6 (14.4) | 4.6 (14.9)† | 12.8 (89.1) |
| SAD phasing | |||
| Figure of merit (before/after density modification) | 0.41/0.60 | ||
| Model refinement | |||
| PDB code | 4gsq | 4gsr | 4gsu |
| Resolution range (Å) | 20.0–1.80 | 20.0–1.79 | 20.0–2.00 |
| R work/R free § (%) | 19.7/23.3 | 19.6/22.6 | 18.9/23.2 |
| No. of non-H atoms/average B factor (Å2) | |||
| Protein | 1889/23.5 | 2014/23.1 | 4112/26.4 |
| Water oxygen | 206/32.9 | 230/36.3 | 228/33.0 |
| Meropenem | — | — | 52/65.5 |
| EMTS | — | 11/38.0 | — |
| Glycerol | 6/53.3 | 12/44.1 | — |
| Calcium ion | 1/65.2 | — | — |
| Wilson B factor (Å2) | 21.9 | 20.4 | 23.5 |
| R.m.s. deviations from ideal geometry | |||
| Bond lengths (Å) | 0.009 | 0.007 | 0.010 |
| Bond angles (°) | 1.29 | 1.15 | 1.40 |
| R.m.s. Z-scores¶ | |||
| Bond lengths | 0.450 | 0.335 | 0.491 |
| Bond angles | 0.590 | 0.512 | 0.622 |
| Ramachandran plot†† (%) | |||
| Favoured/outliers | 98.8/0.0 | 98.5/0.0 | 97.9/0.2 |
| Poor rotamers†† (%) | 1.00 | 0.47 | 0.91 |
Friedel pairs were treated as separate observations.
R
merge = 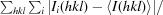
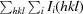 , where I(hkl) is the intensity of reflection hkl,
, where I(hkl) is the intensity of reflection hkl, 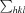 is the sum over all reflections and
is the sum over all reflections and 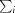 is the sum over i measurements of reflection hkl.
is the sum over i measurements of reflection hkl.
R
work = 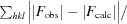
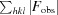 , where R
free is calculated for a randomly chosen 5% of reflections which were not used for structure refinement and R
work is calculated for the remaining reflections.
, where R
free is calculated for a randomly chosen 5% of reflections which were not used for structure refinement and R
work is calculated for the remaining reflections.
Values obtained using REFMAC.
Values obtained using MolProbity.
2.4. Model building and refinement
The SAD-phased electron-density map of LdtMt2Δ130 was interpreted using the automatic model-building program RESOLVE (Terwilliger, 2003 ▶) to build an initial model. Subsequent model building was performed manually using the program Coot (Emsley et al., 2010 ▶). The model of the mercury-derivatized LdtMt2Δ130 was refined with the program REFMAC (Murshudov et al., 2011 ▶), including bulk-solvent correction. 5% of the data were randomly set aside as test data for the calculation of R free (Brünger, 1992 ▶). The model of the mercury-derivatized LdtMt2Δ130 was used to determine the structures of the apo form and the meropenem complex of LdtMt2Δ130 by molecular replacement. A cross-rotational search followed by a translational search was performed utilizing the program MOLREP (Vagin & Teplyakov, 2010 ▶). The stereochemistry of the refined models was evaluated using MolProbity (Chen et al., 2010 ▶). Refinement statistics are summarized in Table 1 ▶.
2.5. Mass spectrometry
All mass spectra were acquired using a linear ion-trap mass spectrometer (Thermo Scientific, USA). The recombinant LdtMt2Δ130 protein (51 µM) was incubated with meropenem (5.1 mM) dissolved in 10 mM Tris–HCl pH 7.9. All samples were injected at 2 µl min−1 in 50% acetonitrile containing 0.1% formic acid. The average molecular mass of each protein sample was determined for multiply charged ions of charge states from +18 to +27. The experimental molecular mass of the apoprotein was calculated to be 31 263.8 g mol−1, showing a 0.05% error compared with the theoretical mass of 31 246.8 g mol−1. All experimental molecular masses showed a similar shift (16–18 Da) compared with the theoretical masses (Supplementary Table S1).
2.6. Equilibrium sedimentation
Equilibrium-sedimentation studies were performed in 20 mM Tris–HCl buffer pH 7.9 containing 200 mM NaCl at 293 K using a Beckman ProteomeLab XL-A analytical ultracentrifuge. LdtMt2Δ130 samples were measured at 230, 235 and 280 nm at two different speeds (24 000 and 28 000 rev min−1) using three different protein concentrations (0.80, 1.60 and 2.40 µM). All measured data can be fitted well to a monomer model and representative results for apo and meropenem-complexed LdtMt2Δ130 are presented. The LdtMt2Δ130 protein concentrations were calculated using an ∊280 nm value of 62 910 M −1 cm−1.
2.7. Accession codes
The coordinates and structure factors have been deposited in the Protein Data Bank under accession codes 4gsq, 4gsr and 4gsu for apo, mercury-derivatized and meropenem-complexed LdtMt2Δ130, respectively.
3. Results and discussion
3.1. Overall structure of LdtMt2
Of the constructs tested, we could only obtain well diffracting crystals of the 131–408 construct (LdtMt2Δ130). The crystal structure of LdtMt2Δ130 was solved by SAD phasing (Table 1 ▶). We refined three models of LdtMt2Δ130: (i) the apo form, (ii) a mercury-derivatized ligand-free form and (iii) a meropenem-complexed form (Table 1 ▶). The apo and mercury-derivatized crystals contained one monomer of LdtMt2Δ130 per asymmetric unit, whereas the crystals of the meropenem complex contained two monomers (chains A and B) in the asymmetric unit. In these models, the N-terminal residues (Leu131–Ala149 in the apo form, Leu131–Thr145 in the mercury derivative and Leu131–Gln140 in the meropenem complex for both chains A and B) and C-terminal affinity tag (LEHHHHHH) are disordered. In the apo model, the internal region of the polypeptide chain (Ser305–Tyr318) is also disordered. The mercury compound is bound around Phe215 in the EMTS-derivatized crystal. Each chain of the meropenem complex contains one meropenem linked to Cys354 by a thioester bond.
LdtMt2Δ130 is roughly C-shaped, with approximate dimensions of 65 × 45 × 30 Å (Fig. 1 ▶). LdtMt2Δ130 can be divided into an N-terminal domain (NTD; residues Leu131–Gly250), a catalytic l,d-transpeptidase (Ldt) domain (residues Asp251–Val378) and a C-terminal tail (residues Asn379–Ala408) (Figs. 1 ▶ a–1c). The bulk of NTD is folded into a bacterial Ig-like Big_5 domain (His150–Gly250); it comprises a helix (αA) and two antiparallel β-sheets (Fig. 1 ▶). The Ldt domain contains two curved β-sheets (β16↑–β15↑–β14↓–β11↑ and β11↑–β17↓–β8↓–β9↑–β10↓), with the β11 strand being shared, as well as one α-helix (αB) and two additional antiparallel β-strands (β12↑–β13↓) (Fig. 1 ▶ c). In the meropenem-complexed structure (in both chains A and B), residues His300–Asp323 containing two antiparallel β-strands (β12↑–β13↓) cover the active site of the Ldt domain (‘the active-site lid’), but they are in the open conformation in the mercury-derivatized ligand-free model (Fig. 2 ▶ a). The C-terminal tail, which is composed of extended loops and a short α-helix (αC), runs across both the Big_5 and Ldt domains (coloured blue in Figs. 1 ▶ a–1c).
Figure 1.

Overall structure of LdtMt2Δ130. (a) Ribbon diagram of meropenem-complexed LdtMt2Δ130. The NTD and the Ldt domain are shown in green and yellow, respectively. The active-site lid (His300–Asp323) and the C-terminal tail (Asn379–Ala408) are coloured red and blue, respectively. Meropenem bound to the Ldt domain is shown as a stick model. (b) Domains of Mtb LdtMt2 coloured as in (a). TM, transmembrane helix. (c) Topology diagram of LdtMt2Δ130 coloured as in (a). (d) Electrostatic surface diagram of meropenem-complexed LdtMt2Δ130. Blue and red indicate positive and negative electrostatic potentials at neutral pH, respectively.
Figure 2.

Structural comparisons and domain interactions in LdtMt2Δ130. (a) A superposition of the four chains in the three LdtMt2Δ130 models and a plot of the Cα r.m.s. deviations between any pair of chains averaged over the six pairwise comparisons. The apo, mercury-derivatized and meropenem-complexed LdtMt2 (chains A and B) are coloured orange, magenta, yellow and blue, respectively. (b) Interactions between the C-terminal tail and two domains coloured as in Fig. 1 ▶(a). The enlarged views on the left have slightly different orientations in order to show the detailed interactions better.
3.2. Structural comparisons and domain interactions in LdtMt2Δ130
The models of LdtMt2Δ130 are compared in Fig. 2 ▶(a) and the Cα root-mean-square (r.m.s.) deviations among them are given in Supplementary Table S2. The r.m.s. deviation between the apo and mercury-derivative models is small (0.40 Å for 246 Cα-atom pairs), indicating that the mercury binding did not cause a large overall structural change (Supplementary Table S2). However, the r.m.s. deviations between the models of the mercury derivative and of chains A and B of the meropenem complex are 1.34 and 1.82 Å for 263 Cα-atom pairs, respectively. Large r.m.s. deviations are observed in the N- and C-terminal regions (Leu141–His150 and Lys407–Ala408) and two internal regions: the β6–β7 loop of the NTD (Asn227–Asn242) and the active-site lid (residues His300–Asp323) (Fig. 2 ▶ a). The Cα r.m.s. deviation of the active-site lid is 3.95 Å for chain B of the meropenem-complexed model, with a maximum deviation of 6.01 Å occurring at Gly309. The covalent binding of meropenem induces a major conformational rearrangement of the active-site lid from the ‘open’ conformation to the ‘closed’ conformation (Fig. 2 ▶ a). The r.m.s. deviation between chains A and B of the meropenem-complexed model is 1.48 Å for 268 Cα-atom pairs (Fig. 2 ▶ a and Supplementary Table S2) and Cα r.m.s. deviations greater than 2.0 Å occur in the four regions mentioned above, with a maximum deviation of 8.89 Å at Pro148. The N-terminal residues around Pro148 are influenced by crystal packing in the meropenem complex. When the ten N-terminal residues (141–150) are excluded from the comparison, the r.m.s. deviation decreases to 0.82 Å, suggesting that the two chains of the meropenem complex represents largely identical states.
The Protein Interfaces, Surfaces and Assemblies (PISA) server (Krissinel & Henrick, 2007 ▶) revealed that the surface area buried at the interface between the NTD and the Ldt domain is 424, 431, 451 and 450 Å2 for the apo form, the mercury derivative and chains A and B of the meropenem complex, respectively. This indicates that the two domains themselves do not interact extensively with each other. However, the relative orientations of the two domains are virtually identical in all three models of LdtMt2Δ130 (Fig. 2 ▶ a). This is because the C-terminal tail holds them together. The C-terminal tail (coloured blue in Figs. 1 ▶ a–1c) interacts with both the Ldt domain and the NTD (Figs. 1 ▶ a and 2 ▶ b). The αC helix of the C-terminal tail interacts with one face of the large β-sheet of the NTD via hydrogen bonds and salt bridges (Fig. 2 ▶ b, upper panel). The aromatic rings of the tryptophan residues (Trp394, Trp398 and Trp401) in the C-terminal tail also stack tightly with the hydrophobic moieties of Tyr201, Arg211, Glu213 and Phe215 of the NTD, further stabilizing the interaction (Fig. 2 ▶ b, upper panel). The long β17–αC loop of the C-terminal tail forms a hydrogen-bonding network with the Ldt domain (Fig. 2 ▶ b, lower panel).
3.3. Analytical ultracentrifugation studies of LdtMt2Δ130
The largest surface area buried at the interface between two monomers of LdtMt2Δ130 within the crystals is 501 and 815 Å2 per monomer (4.5% and 6.6% of the monomer surface area) for the apo form and the mercury derivative (both in space group C2), respectively. Such monomer–monomer contacts can be regarded as crystallization artifacts. On the other hand, the largest surface area buried at the interface between the two tightly packed monomers of the meropenem complex is 1332 Å2 per monomer (10.6% of the monomer surface area), suggesting that such a large interface area may exist in solution (Supplementary Fig. S3a).
To test whether meropenem binding affects the oligomeric state of LdtMt2Δ130 in solution, we performed sedimentation-equilibrium studies under various experimental conditions. Supplementary Fig. S3(b) shows representative data and fits for monomer (1×) and dimer (2×) models of apo LdtMt2Δ130. The weighted r.m.s. errors for the monomer (1×) and dimer (2×) fits are 7.93 × 10−3 and 4.88 × 10−2, respectively, demonstrating the superiority of the 1× model. The residual plots shown in Supplementary Fig. S3(b) (top panel) also support our conclusion that apo LdtMt2Δ130 exists as monomers in solution. In Supplementary Fig. S3(c), sedimentation-equilibrium data and fits for the meropenem complex of LdtMt2Δ130 (after a 1 h reaction with a 100-fold molar excess of meropenem) are shown. The r.m.s. values for the monomer (1×) and dimer (2×) fits are 9.86 × 10−3 and 4.79 × 10−2, respectively. The r.m.s. values and residual plots (Supplementary Fig. S3c, top panel) indicate that meropenem-complexed LdtMt2Δ130 also exists as monomers in solution. Other heterogeneous or interactive models were also tested, but there was no indication of the possibility of their presence, indicating that meropenem binding has no effect on the monomeric state of LdtMt2Δ130 in solution. Therefore, we conclude that the apparent dimeric interface observed in the crystal structure of the meropenem complex arises from tight crystal packing.
3.4. LdtMt2 contains tandem immunoglobulin-like Big_5 domains
LdtMt2 possesses an additional N-terminal region (residues Met1–Asp251) which contains a putative transmembrane helix (Leu20–Ala42) in front of its catalytic Ldt domain (Fig. 1 ▶ b). Sequence analysis using the Prosite database (http://www.expasy.org/prosite) reveals that LdtMt2 appears to be a putative lipoprotein with Cys35 as a potential lipid-attachment site. Unexpectedly, our crystal structure of LdtMt2Δ130 reveals that His150–Gly250 of LdtMt2Δ130 are folded into a compact Ig-like domain (Fig. 3 ▶ a, inset; Bork et al., 1994 ▶), although it shows no detectable sequence similarity to eukaryotic Ig-fold proteins. A DALI structural-similarity search (Holm & Rosenström, 2010 ▶) reveals that the NTD of LdtMt2Δ130 structurally resembles the light chain of antibody Fab fragments such as the Mus musculus 6E1 Fab light chain (Jiang et al., 2003 ▶; PDB entry 1orq; r.m.s. deviation of 3.3 Å for 86 equivalent Cα positions, a Z-score of 7.2 and a sequence identity of 6%).
Figure 3.

Big_5 domain of Ldtc Mt2. (a) Topology diagrams of the Big_5 domain (His150–Gly250) of LdtMt2Δ130 (inset) and of four distinct subtypes of the Ig-like fold (modified from Bork et al., 1994 ▶): c-type (constant), v-type (variable), s-type (switched) and h-type (hybrid). The four-stranded structural core (strands b, c, e, and f; β2, β3, β5 and β6 of LdtMt2Δ130) common to all Ig-like domains (orange) is surrounded by structurally more variable strands (green). (b, c) Ribbon diagrams of the Big_5 domain (His150–Gly250) in the apo model of LdtMt2Δ130 (b) and meropenem-complexed LdtMt2Δ130 (c), and enlarged views of the β6–β7 loop (insets) coloured as in (a). The bound calcium ion and the residues around it (Asp232–Met237) are shown as a purple ball and as stick models, respectively, with a 2mF o − DF electron-density map (contoured at 1.5σ).
Ig folds have been identified in prokaryotic proteins and are known as bacterial immunoglobulin-like (Big) domains (Halaby et al., 1999 ▶). Big domains are present in proteins ranging from enzymes to chaperones (Halaby & Mornon, 1998 ▶). Among the topological subtypes of classical Ig-like domains (Bork et al., 1994 ▶), the NTD of LdtMt2Δ130 follows the topology of the C-type (constant) Ig fold, as the NTD of LdtMt2Δ130 does not possess an extra loop between strands β3 and β4 (Fig. 3 ▶ a). Thus, the Big domain in the NTD of LdtMt2Δ130 is structurally more similar to the constant region of the immunoglobulin light chain in the Fab fragment than the variable region (Fig. 3 ▶ a).
Following our structural analyses, we searched for other YkuD-family proteins that contain one or more Big domains such as Big_2 (Pfam accession No. PF02368), Big_3 (PF07523) or Big_5 (PF13205). The overall amino-acid sequence of LdtMt2 aligns well with those of proteins containing Big_5 domains, such as a membrane protein from Streptomyces sp. SPB74 (ExPASy accession No. B5GI65; overall sequence identity of 29%) and an YkuD-family protein from Nakamurella multipartita (ExPASy accession No. C8X6V6; overall sequence identity of 40%). Interestingly, the N. multipartita YkuD protein possesses two sequential Big_5 domains: one in residues 182–282 (corresponding to His150–Gly250 of LdtMt2) and one in residues 87–177 (corresponding to Asp56–Thr145 of LdtMt2). This clearly indicates that LdtMt2 also has two sequential Big_5 domains in front of its Ldt domain. In comparison, the minor l,d-transpeptidase in Mtb, LdtMt1, contains only a single Big_5 domain that shows 34% sequence identity to the second Big_5 domain of LdtMt2 but shows no sequence identity to the first Big_5 domain of LdtMt2. The first Big_5 domain of LdtMt2 may not be essential for catalytic activity, as LdtMt1 has been shown to be functional (Lavollay et al., 2008 ▶; Dubée, Triboulet et al., 2012 ▶). Our LdtMt2Δ130 structures reveal that the second Big_5 domain is rigidly held by the Ldt domain via the C-terminal tail. However, the C-terminal tail does not appear to be sufficiently long to interact with the first Big_5 domain. The C-terminal tail is also conserved in the above two proteins that contain Big_5 domains, suggesting that the observed domain arrangement of LdtMt2Δ130 may also be conserved and that the tandem Big_5 domains, in particular the second one, may play an important role in assisting the function of the catalytic Ldt domain.
Although the biological function of the Big_5 domains of LdtMt2 is unknown, various aspects of the general functions of other Big domains have been studied. For example, leptospiral immunoglobulin-like (Lig) proteins, which are surface-exposed proteins that belong to the Big_2 family, mediate host–pathogen interactions (Castiblanco-Valencia et al., 2012 ▶). The Big_2 domains of Lig proteins have been reported to bind Ca2+, suggesting possible involvement of Ca2+ binding in the function of proteins containing Big domains (Raman et al., 2010 ▶). Our crystals of apo LdtMt2Δ130 were obtained using a reservoir solution containing 50 mM calcium chloride, whereas the crystallization condition for the meropenem complex did not contain calcium ions. Interestingly, we observed extra electron density in close proximity to the β6–β7 loop in the structure of the apo form (Fig. 3 ▶ b), whereas no such electron density was observed at the same position in the meropenem complex (Fig. 3 ▶ c). The extra electron density is best interpreted as a Ca2+ ion bound to the Big_5 domain because the density peak is in close association with the backbone N atoms of Gly234 and Glu235 at distances of 2.33 and 2.78 Å, respectively, as well as a side-chain O atom of Asp232 at a distance of 2.93 Å (Figs. 3 ▶ b and 3 ▶ c). Gly234 and Gly236 appear to be crucial in allowing a sharp turn in the β6–β7 loop, which exhibits large r.m.s. deviations between the apo and meropenem-complex structures (Fig. 2 ▶ a). Asp232, Gly234 and Gly236 of LdtMt2 are semi-conserved in other Big_5-domain-containing proteins as (i) Glu235, Ala237 and Gly239 in the membrane protein from Streptomyces sp. SPB74, (ii) Asn264, Gly266 and Gly268 in the YkuD-family protein from N. multipartita and (iii) Glu73, Thr75 and Gly77 in another functional Mtb l,d-transpeptidase, LdtMt1.
3.5. l,d-Transpeptidase domain of LdtMt2, its covalent acylation by meropenem and a structural view
Our structures of LdtMt2Δ130 show that the C-terminal region of LdtMt2 (residues Asp251–Val378) is folded into a compact domain with the l,d-transpeptidase fold. Previously reported structural data on l,d-transpeptidases include the crystal structure of a C-terminal fragment (Ldtfm; residues 217–466) of E. faecium l,d-transpeptidase in the apo form (Biarrotte-Sorin et al., 2006 ▶), the crystal structure of the Lys117Ala/Gln118Ala double mutant of Bacillus subtilis YkuD protein (LdtBs) in the apo form (Bielnicki et al., 2006 ▶) and the solution structure of LdtBs in the apo and imipenem-bound forms (Lecoq et al., 2012 ▶). As expected, the two most similar structures detected using the DALI program (Holm & Rosenström, 2010 ▶) are (i) LdtBs (Bielnicki et al., 2006 ▶; PDB entry 1y7m; Z-score of 17.7, r.m.s. deviation of 4.3 Å and sequence identity of 17% for 131 equivalent Cα positions) and (ii) Ldtfm (Biarrotte-Sorin et al., 2006 ▶; PDB code 1zat, Z-score of 16.8, r.m.s. deviation of 6.5 Å and sequence identity of 20% for 151 equivalent Cα positions). The Z-scores are below 5 for other proteins. The Ldtfm structure consists of a domain (residues 217–336) with a novel mixed α/β fold and the l,d-transpeptidase catalytic domain (residues 337–466). LdtBs is encoded by the ykuD gene of B. subtilis and is one of three putative l,d-transpeptidases. Its l,d-transpeptidase catalytic domain (residues 55–164) is preceded by a LysM domain (residues 4–46). In the solution structure of imipenem-bound LdtBs the covalently bound imipenem ligand samples a wide range of orientations and does not adopt a well defined conformation, thus providing no detailed view of the ligand–protein interactions (Lecoq et al., 2012 ▶; De & McIntosh, 2012 ▶). Previously, it has been shown that the soluble fragment (residues Ala55–Ala408) of LdtMt2 containing the l,d-transpeptidase catalytic domain (residues Asp251–Val378) catalyzes the formation of 3→3 peptidoglycan cross-links with disaccharide–tetrapeptide monomers in vitro (Gupta et al., 2010 ▶). It has been shown that the l,d-transpeptidase activities of LdtMt2 and LdtMt1 can be inactivated by carbapenems, such as meropenem, which is effective against drug-resistant Mtb strains (Dubée, Triboulet et al., 2012 ▶; Hugonnet et al., 2009 ▶).
Using mass spectrometry, we observed the covalent binding of meropenem to LdtMt2Δ130 (Supplementary Fig. S4 and Table S1). After 20 min of incubating LdtMt2Δ130 with meropenem, both a first set of peaks corresponding to the mass of the covalently acylated LdtMt2Δ130–meropenem complex and a second set of peaks which were 44 Da smaller than the first set of peaks were present (Supplementary Figs. S4a and S4b and Table S1). After 3 d of incubation, only the second set of peaks remained (Supplementary Figs. S4a and S4c and Table S1). The covalent meropenem adduct corresponding to the second set of peaks could be produced by the subtraction of an acetaldehyde moiety (m/z = 44 Da), similarly to the previously proposed mechanism for meropenem-bound β-lactamase from Mtb (Hugonnet et al., 2009 ▶). These experiments indicate that meropenem-acylated LdtMt2Δ130 is stable for at least 3 d after β-lactam ring opening, demonstrating that the deacylation of meropenem-inactivated LdtMt2Δ130 is slow. In the case of Mtb LdtMt1, the hydrolysis rate was 3100 times slower than the acylation rate (Dubée, Triboulet et al., 2012 ▶).
To provide a detailed structural view of how meropenem inhibits LdtMt2, we have determined the crystal structure of LdtMt2Δ130 as a covalent complex with meropenem. In chain B of the meropenem-complex model, clear electron density for meropenem that is contiguous with the Sγ atom of Cys354 was observed (Supplementary Fig. S2), but the electron density was missing beyond the thioether S atom of meropenem. This is likely to reflect the free orientation of the terminal region of meropenem corresponding to the R 3 side chain of carbapenem (Supplementary Figs. S1 and S2). In chain A the covalently bound meropenem is observed up to the pyrroline ring and is less clear than in chain B (Supplementary Fig. S2). Therefore, we restrict our detailed discussion of meropenem–LdtMt2Δ130 interactions to chain B only.
The invariant Cys354 of LdtMt2 has been proposed to be the key catalytic residue in l,d-transpeptidase activity (Supplementary Fig. S5; Lecoq et al., 2012 ▶). Furthermore, we note that the disposition of three residues, His336 and Ser337 (both located on β15) and Cys354 (located on β16), is highly similar to the catalytic triad of a wide range of enzymes including serine proteases, esterases and β-lactamases (Fig. 4 ▶ a; Dodson & Wlodawer, 1998 ▶). Similar catalytic triads, H(S/G/D)x 16–19 C, are also present in the LdtBs and Ldtfm structures (Fig. 4 ▶ b and Supplementary Fig. S5), where x represents any amino acid and the strictly conserved residues are shown in bold. The main-chain carbonyl of Ser337 in LdtMt2, as well as the corresponding Gly127 in LdtBs and Asp422 in Ldtfm, acts as a hydrogen-bond acceptor. In our three structures of LdtMt2Δ130, the side chains of His336 and Cys354 are suitably positioned for a cooperative action with the main-chain carbonyl of Ser337 (marked by dashed red lines in Fig. 4 ▶ a). The N∊2 atom of His336 interacts with the Sγ atom of Cys354 with a distance of 4.09 Å, and the Nδ1 atom of His336 interacts with the main-chain carbonyl O atom of Ser337 with a distance of 2.86 Å. The side-chain conformation of the catalytic His336 is stabilized by an additional interaction between His336 N∊2 and Asn356 Oδ1 with a distance of 3.37 Å (Fig. 4 ▶ a).
Figure 4.

The active site of the Ldt domain and the substrate-binding sites. (a) Active-site superposition of the apo (orange), mercury-derivatized (magenta) and meropenem-complexed (chain B, cyan) LdtMt2Δ130. Dotted lines denote interactions: hydrogen bonds to the catalytic triad (red) and the oxyanion hole (green), Ser351 with the oxyanion hole (black), His336 with Asn356 (black) and His352 with Cys354 Sγ and the main-chain carbonyl O atom of His352 in the mercury-derivatized model (purple). The covalently bound meropenem adduct in the meropenem complex is shown as a stick model. (b) Active-site superposition of meropenem-complexed LdtMt2 (chain B, cyan), LdtBs (green) and Ldtfm (grey) in the same view as in (a). (c, d) Electrostatic surface representations of the predicted binding sites for the donor substrate in the open conformation of mercury-derivatized LdtMt2Δ130 (c) and the acceptor substrate in the closed conformation of meropenem-complexed LdtMt2Δ130 (d) coloured as in Fig. 1 ▶(d). The surfaces of the binding site are represented with constituent residues as stick models. The movement of the imidazole ring of His352 in the apo form (orange) and the mercury-derivatized form (magenta) is presented in stick models with dotted surfaces in (c). The peptide bond of the donor substrate (meso-DAP3-d-Ala4) is schematically modelled into the active site in the open conformation. The terminus of the acceptor substrate (meso-DAP3) is schematically modelled into the active site in the closed conformation, with red dotted lines depicting a plausible site for recognizing the terminal amine of meso-DAP3.
The structure of the meropenem complex of LdtMt2 represents the acylated-enzyme intermediate state (Fig. 4 ▶ a) in the proposed mechanism of l,d-transpeptidation (Supplementary Fig. S6) based on our structures and previous studies (Dodson & Wlodawer, 1998 ▶; Lecoq et al., 2012 ▶). In analogy with the proposed mechanism for LdtBs (Lecoq et al., 2012 ▶), we propose that His336 captures the Sγ proton of Cys354 to generate the nucleophilic thiolate that attacks the carbonyl C atom of the meropenem β-lactam ring, resulting in an acylated enzyme; His336 then releases its acidic proton to the N atom of the β-lactam ring (Supplementary Fig. S6). Upon nucleophilic attack by the thiolate of Cys354, the carbonyl C atom of the β-lactam ring of meropenem is covalently bound to the Sγ atom of Cys354. The bond distance between Cys354 Sγ and the carbonyl C atom of meropenem is 1.94 Å, which falls within the typical bond distances (1.81–2.55 Å) of carbon–sulfur single bonds (Fig. 4 ▶).
The so-called oxyanion hole stabilizes a tetrahedral enzyme–substrate intermediate (E-S1* in Supplementary Fig. S6) to increase the activity of l,d-transpeptidation. In LdtMt2, His352, Gly353 and Cys354 contribute to the formation of the oxyanion hole, in which the carbonyl group of the opened meropenem scissile bond is hydrogen-bonded to the backbone amide N atoms of His352, Gly353 and Cys354 with distances of 3.83, 3.27 and 2.83 Å, respectively (Fig. 4 ▶ a and Supplementary Fig. S6).
3.6. The active-site lid undergoes a large conformational change upon acylation by meropenem
The active-site lid (His300–Asp323) of Mtb LdtMt2 is much longer than those of LdtBs and Ldtfm (Supplementary Fig. S5). The active-site lid of Mtb LdtMt1 is equally long. Those of LdtBs and Ldtfm are truncated and appear to be too short to fully cover the active-site pocket of their l,d-transpeptidase domains. A comparison of our LdtMt2Δ130 structures demonstrates the conformational flexibility of the active-site lid (Figs. 2 ▶ a and 5 ▶). The active-site lid adopts an open conformation in the mercury-derivative structure (Fig. 5 ▶ b), whereas it is in a closed conformation in the meropenem complex (Fig. 5 ▶ a). This observation suggests that the open active-site lid before acylation by meropenem undergoes movement to close the active-site pocket upon acylation (Fig. 5 ▶ c). The flexible nature of the active-site lid is further supported by the apo structure, in which much of the lid is disordered. Upon closure of the active-site lid, the side chains of Tyr308 and Tyr318 move close to the active-site pocket (Fig. 5 ▶ c), with their Oη atoms shifting by 5.46 and 4.70 Å, respectively. The two tyrosines enclose tightly the covalently bound meropenem (Figs. 5 ▶ a and 6 ▶ b–6d).
Figure 5.

Conformational flexibility of the active-site lid in LdtMt2Δ130. (a, b) Three different views of the electrostatic potential surface diagrams of mercury-derivatized (a) and meropenem-complexed LdtMt2Δ130 (b). The closed conformation of the active-site lid in (b) reveals that meropem attached to catalytic Cys354 is accessible through three narrow paths (Paths A, B and C). (c) The active-site lids in meropenem-complexed (cyan) and mercury-derivatized (magenta) LdtMt2Δ130 are shown as ribbon models with the surface of the meropenem complex. Residues that show large shifts upon lid closure are shown as stick models; the movement is indicated by black dotted arrows. The two models are in the same orientation.
Figure 6.

Meropenem-inactivated LdtMt2Δ130. (a) Electron-density map (left) and a schematic diagram of interactions (right) of the covalently bound meropenem adduct with Cys354 in meropenem-complexed LdtMt2Δ130 (chain B). The OMIT mF o − DF c map (contoured at 2.5σ) for meropenem and the 2mF o − DF c map (contoured at 1.0σ) for Cys354 are coloured blue and yellow, respectively. Dotted lines denote interactions with LdtMt2Δ130 and the corresponding bond lengths are shown in Å. Variable regions (R 1, R 2 and R 3) of carbapenems are shaded in green, blue and red, respectively. (b) Interactions of the bound meropenem with Tyr318 (left) and Tyr308 (right). Dotted lines and the electron-density map are presented as in (a). (c) Surface representation (left) and ribbon diagram (right) of the active site of LdtMt2Δ130 enclosing the meropenem adduct viewed along Path B. The β14–β15 loop, the β15–β16 loop and the active-site lid, which surround the bound meropenem, are coloured plum, orange and green, respectively. (d) Surface representation (left) and ribbon diagram (right) of the active site in the meropenem-complexed LdtMt2Δ130 viewed along Path A presented as in (c).
In the open conformation before acylation, the Sγ atom of Cys354 is exposed to bulk solvent and appears to be readily accessible via two wide paths (Paths A and B in Fig. 5 ▶ a). In the closed conformation of the meropenem complex, meropenem attached covalently to Cys354 is accessible from the bulk solvent along three narrower paths which are formed by the closure of the active-site lid (Paths A, B and C in Fig. 5 ▶ b). Path B is occupied by the R 3 part of meropenem, indicating that Path B is likely to be the binding site for the donor substrate (S1; Fig. 4 ▶ c). The Sγ atom of Cys354 points toward the entrance to Path B in our three structures, suggesting that the meso-DAP3 part of the donor substrate (the peptide bond of meso-DAP3-d-Ala4; S1) may approach Cys354 along Path B (Fig. 4 ▶ c and ‘E + S1’ in Supplementary Fig. S6).
Moreover, the amine group of the side chain of the second substrate (meso-DAP3 of the acceptor peptide; S2) is expected to approach His336 of the catalytic triad to form a tetrahedral enzyme–substrate intermediate (‘AcylE+S2’ and ‘AcylE-S2*’ in Supplementary Fig. S6 and Fig. 4 ▶ d). Therefore, approach of the acceptor substrate for transpeptidation seems to only be allowed through Path A, in which the acylated Sγ of Cys354 is exposed and His336 lies on the surface of Path A (Figs. 4 ▶ d and 5 ▶ a). In addition, the surface of Path A is lined by Gly281, Lys282, Trp340 and Asn356 (Fig. 4 ▶ d). Gly281 and Lys282 are strictly conserved in the YkuD family, while Trp340 and Asn356 are semi-conserved (Supplementary Fig. S5). The strict conservation of Gly281 and Lys282 (boxed in brown in Supplementary Fig. S5) might be related to the possibility that the positive charge of Lys282 stabilizes the carboxyl group of the side chain of the acceptor substrate (S2) meso-DAP3 (Fig. 4 ▶ d). The invariant Lys282 of Mtb LdtMt2 may be critical in recognizing the acceptor peptide during the transpeptidation reaction that is catalyzed by l,d-transpeptidase (Supplementary Fig. S6).
3.7. Implications for anti-TB drug design
Our mass-spectrometric and crystal structure analyses revealed that meropenem is covalently bonded to the Sγ atom of Cys354 of Mtb LdtMt2. We have also shown that LdtMt2 is not recovered from the meropenem-inactivated acyl enzyme for at least 3 d after the acylation reaction. Even though it took three weeks from initial incubation with meropenem to the collection of X-ray diffraction data, the electron-density map clearly indicated the covalently bonded meropenem adduct, supporting the ability of meropenem to act as a suicide inhibitor of LdtMt2. Our structure of meropenem-acylated LdtMt2 sheds light on the design of improved anti-TB carbapenems by providing a detailed view of the interactions between meropenem and LdtMt2 (Fig. 6 ▶).
The meropenem C7 carbonyl group is bonded to Cys354 and forms hydrogen bonds to the backbone N atoms of His352, Gly353 and Cys354 in the oxyanion hole in chain B of the meropenem-inhibited model (Fig. 6 ▶ a). The side chains of two tyrosine residues, Tyr308 and Tyr318, from the active-site lid make hydrogen bonds to meropenem (Fig. 6 ▶ b). The ζ-hydroxyl group of Tyr308 interacts with the meropenem N1 atom and the thioether S atom of meropenem C3. In addition, the pyrroline ring of meropenem is stabilized by the aromatic ring of Tyr308 through hydrophobic interactions. The highly conserved Tyr318 interacts with His352 by π-stacking and forms hydrogen bonds to the C8 hydroxyl group, the N1 atom and the C2 carboxyl group of meropenem (Figs. 6 ▶ a and 6 ▶ b). These protein–meropenem interactions indicate that the two tyrosine side chains may undergo a large shift in their positions upon acylation by meropenem, thus triggering the observed conformational change in the active-site lid. The backbone carbonyl groups of Ser331 and His352, as well as the side chain of His352, also make hydrogen bonds to the bound meropenem.
Much of the R 3 part of meropenem beyond the thioether S atom (Supplementary Fig. S1) does not make tight interactions with the protein. This observation explains why the electron density is missing beyond the thioether S atom of the opened meropenem (for chain B). The surface of the potential binding site for the donor substrate (S1) or the R 3 side chain of carbapenem is mainly lined by hydrophobic residues, except for the hydroxyl groups from the side chains of Ser331, Thr287 and Tyr292 (Fig. 4 ▶ c). Based on the surface analysis of the meropenem-bound form, it appears that the R 3 side chain of carbapenems can be tailored to better accommodate the protein surface and to optimize their inhibitory activities towards LdtMt2 (Supplementary Fig. S7). This view is in agreement with previous findings that the R 3 side chain of the carbapenem modulates the kinetic constants for the inactivation reaction of LdtMt1 (Dubée, Triboulet et al., 2012 ▶) and Ldtfm (Dubée, Arthur et al., 2012 ▶). Moreover, inhibitor design towards the potential binding site of the second substrate (meso-DAP3 of the acceptor peptide; S2) could also be attempted, thereby blocking the final transpeptidation step of 3→3 cross-linking. The surface representation of the potential binding site for the acceptor substrate (S2; Fig. 4 ▶ d) provides the semi-conserved target residues, e.g. Lys282, Trp340 and Asn356, for the targeted inhibition of the transpeptidation step. As the key residues in the active sites of LdtMt2 and LdtMt1 are well conserved (Supplementary Fig. S5), a designed inhibitor is likely to inhibit both.
4. Conclusions
This study provides the first detailed structural view of the meropenem-inactivated l,d-transpeptidase domain of LdtMt2, which is predominantly expressed by Mtb. The l,d-transpeptidase domain of the minor l,d-transpeptidase in Mtb, LdtMt1, is expected to be highly similar in its structure as it shows 48% sequence identity and the key catalytic residues are conserved. Together with the previous kinetic studies (Dubée, Triboulet et al., 2012 ▶), our structural and mass-spectrometric analyses of the meropenem complex indicate that LdtMt2 is not readily recovered from the meropenem-acylated form. A large conformational change in the active-site lid of LdtMt2 upon acylation by meropenem to partially cover the bound meropenem may explain the long-lived acylated enzyme intermediate. This structural information will be valuable in further optimization of meropenem and will facilitate the structure-based discovery of new anti-TB carbapenem drugs.
Supplementary Material
PDB reference: LdtMt2, apo, 4gsq
PDB reference: mercury derivative, 4gsr
PDB reference: meropenem complex, 4gsu
Supplementary material file. DOI: 10.1107/S0907444912048998/be5223sup1.pdf
Acknowledgments
We thank the beamline staff for assistance during X-ray diffraction experiments at Pohang Light Source (beamlines BL-4A, BL-6C and BL-7A), SPring-8 (beamline BL26B1) and the Photon Factory (beamlines BL-1A, BL-5A, BL-17A, NE3A and NW12A). This work was supported by the Korea Ministry of Education, Science and Technology (MEST), the National Research Foundation (NRF) of Korea, Basic Science Outstanding Scholars Program, World-Class University Program (grant No. R31-10032) and the Innovative Drug Research Center for Metabolic and Inflammatory Disease (grant No. R11-2007-107-000000) to SWS, by the Korea Ministry of Health, Welfare and Family Affairs, Korea Healthcare Technology R&D Project (grant No. A092006) to SWS and BWH, and by NRF grants funded by the Korean government (MEST) to HSK (2012039930) and HJY (20110013663).
Footnotes
Supplementary material has been deposited in the IUCr electronic archive (Reference: BE5223). Services for accessing this material are described at the back of the journal.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Almeida Da Silva, P. E. & Palomino, J. C. (2011). J. Antimicrob. Chemother. 66, 1417–1430. [DOI] [PubMed]
- Biarrotte-Sorin, S., Hugonnet, J.-E., Delfosse, V., Mainardi, J.-L., Gutmann, L., Arthur, M. & Mayer, C. (2006). J. Mol. Biol. 359, 533–538. [DOI] [PubMed]
- Bielnicki, J., Devedjiev, Y., Derewenda, U., Dauter, Z., Joachimiak, A. & Derewenda, Z. S. (2006). Proteins, 62, 144–151. [DOI] [PMC free article] [PubMed]
- Bork, P., Holm, L. & Sander, C. (1994). J. Mol. Biol. 242, 309–320. [DOI] [PubMed]
- Brünger, A. T. (1992). Nature (London), 355, 472–475. [DOI] [PubMed]
- Castiblanco-Valencia, M. M., Fraga, T. R., Silva, L. B., Monaris, D., Abreu, P. A., Strobel, S., Józsi, M., Isaac, L. & Barbosa, A. S. (2012). J. Infect. Dis. 205, 995–1004. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Chiang, C.-Y., Centis, R. & Migliori, G. B. (2010). Respirology, 15, 413–432. [DOI] [PubMed]
- De, S. & McIntosh, L. P. (2012). Structure, 20, 753–754. [DOI] [PubMed]
- Dodson, G. & Wlodawer, A. (1998). Trends Biochem. Sci. 23, 347–352. [DOI] [PubMed]
- Dubée, V., Arthur, M., Fief, H., Triboulet, S., Mainardi, J.-L., Gutmann, L., Sollogoub, M., Rice, L. B., Ethève-Quelquejeu, M. & Hugonnet, J.-E. (2012). Antimicrob. Agents Chemother. 56, 3409–3412. [DOI] [PMC free article] [PubMed]
- Dubée, V., Triboulet, S., Mainardi, J.-L., Ethève-Quelquejeu, M., Gutmann, L., Marie, A., Dubost, L., Hugonnet, J.-E. & Arthur, M. (2012). Antimicrob. Agents Chemother. 56, 4189–4195. [DOI] [PMC free article] [PubMed]
- Dye, C. & Williams, B. G. (2010). Science, 328, 856–861. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- England, K., Boshoff, H. I., Arora, K., Weiner, D., Dayao, E., Schimel, D., Via, L. E. & Barry, C. E. III (2012). Antimicrob. Agents Chemother. 56, 3384–3387. [DOI] [PMC free article] [PubMed]
- Erdemli, S. B., Gupta, R., Bishai, W. R., Lamichhane, G., Amzel, L. M. & Bianchet, M. A. (2012). Structure, 20, 2013–2115. [DOI] [PMC free article] [PubMed]
- Gengenbacher, M. & Kaufmann, S. H. (2012). FEMS Microbiol. Rev. 36, 514–532. [DOI] [PMC free article] [PubMed]
- Goffin, C. & Ghuysen, J. M. (2002). Microbiol. Mol. Biol. Rev. 66, 702–738. [DOI] [PMC free article] [PubMed]
- Gupta, R., Lavollay, M., Mainardi, J.-L., Arthur, M., Bishai, W. R. & Lamichhane, G. (2010). Nature Med. 16, 466–469. [DOI] [PMC free article] [PubMed]
- Halaby, D. M. & Mornon, J.-P. (1998). J. Mol. Evol. 46, 389–400. [DOI] [PubMed]
- Halaby, D. M., Poupon, A. & Mornon, J.-P. (1999). Protein Eng. 12, 563–571. [DOI] [PubMed]
- Holm, L. & Rosenström, P. (2010). Nucleic Acids Res. 38, 545-549. [DOI] [PMC free article] [PubMed]
- Hugonnet, J.-E., Tremblay, L. W., Boshoff, H. I., Barry, C. E. III & Blanchard, J. S. (2009). Science, 323, 1215–1218. [DOI] [PMC free article] [PubMed]
- Jiang, Y., Lee, A., Chen, J., Ruta, V., Cadene, M., Chait, B. T. & MacKinnon, R. (2003). Nature (London), 423, 33–41. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol. 372, 774–797. [DOI] [PubMed]
- Labia, R., Barthélemy, M. & Peduzzi, J. (1985). Drugs Exp. Clin. Res. 11, 765–770. [PubMed]
- Lavollay, M., Arthur, M., Fourgeaud, M., Dubost, L., Marie, A., Veziris, N., Blanot, D., Gutmann, L. & Mainardi, J.-L. (2008). J. Bacteriol. 190, 4360–4366. [DOI] [PMC free article] [PubMed]
- Lecoq, L., Bougault, C., Hugonnet, J.-E., Veckerlé, C., Pessey, O., Arthur, M. & Simorre, J.-P. (2012). Structure, 20, 850–861. [DOI] [PubMed]
- Mainardi, J.-L., Hugonnet, J.-E., Rusconi, F., Fourgeaud, M., Dubost, L., Moumi, A. N., Delfosse, V., Mayer, C., Gutmann, L., Rice, L. B. & Arthur, M. (2007). J. Biol. Chem. 282, 30414–30422. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Raman, R., Rajanikanth, V., Palaniappan, R. U. M., Lin, Y.-P., He, H., McDonough, S. P., Sharma, Y. & Chang, Y.-F. (2010). PLoS One, 5, e14377. [DOI] [PMC free article] [PubMed]
- Terwilliger, T. C. (2003). Acta Cryst. D59, 38–44. [DOI] [PMC free article] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Veziris, N., Truffot, C., Mainardi, J.-L. & Jarlier, V. (2011). Antimicrob. Agents Chemother. 55, 2597–2600. [DOI] [PMC free article] [PubMed]
- Wietzerbin, J., Das, B. C., Petit, J. F., Lederer, E., Leyh-Bouille, M. & Ghuysen, J. M. (1974). Biochemistry, 13, 3471–3476. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: LdtMt2, apo, 4gsq
PDB reference: mercury derivative, 4gsr
PDB reference: meropenem complex, 4gsu
Supplementary material file. DOI: 10.1107/S0907444912048998/be5223sup1.pdf