

The crystal structures of two fragments of the l,d-transpeptidase from M. tuberculosis have been determined at 1.45 and 1.86 Å resolution. The extramembrane part of this enzyme consists of three domains: two domains related to the immunoglobulin fold and a catalytic domain belonging to the ErfK/YbiS/YhnG family at the C-terminus.
Keywords: Mycobacterium tuberculosis, cell wall, peptidoglycans, transpeptidases, antibiotics
Abstract
The transpeptidase LtdMt2 catalyzes the formation of the (3–3) cross-links characteristic of the peptidoglycan layer in the Mycobacterium tuberculosis cell wall. Bioinformatics analysis suggests that the extramembrane part of the enzyme consists of three domains: two smaller domains (denoted as A and B domains) and a transpeptidase domain (the C domain) at the C-terminus. The crystal structures of two fragments comprising the AB domains and the BC domains have been determined. The structure of the BC module, which was determined to 1.86 Å resolution using Se-SAD phasing, consists of the B domain with an immunoglobulin-related fold and the catalytic domain belonging to the ErfK/YbiS/YbnG fold family. The structure of the AB-domain fragment, which was solved by molecular replacement to 1.45 Å resolution, reveals that despite a lack of overall sequence identity the A domain is structurally very similar to the B domain. Combining the structures of the two fragments provides a view of the complete three-domain extramembrane part of LdtMt2 and shows that the protein extends at least 80–100 Å from the plasma membrane into the peptidoglycan layer and thus defines the maximal distance at which cross-links are formed by this enzyme. The LdtMt-related transpeptidases contain one or two immunoglobulin domains, which suggests that these might serve as extender units to position the catalytic domain at an appropriate distance from the membrane in the peptidoglycan layer.
1. Introduction
The cell wall of Mycobacterium tuberculosis is a complex multilayered structure with a unique architecture that protects the organism against both physical and chemical stress (Hett & Rubin, 2008 ▶). This barrier supports the survival of this pathogen in host macrophages and confers resistance to many antibiotics (Hett & Rubin, 2008 ▶; Gengenbacher & Kaufmann, 2012 ▶; Russell, 2011 ▶). The increasing number of multidrug-resistant (MDR-TB) and extensively drug-resistant (XDR-TB) strains of M. tuberculosis makes treatment using current strategies increasingly difficult (Cole & Riccardi, 2011 ▶). Since M. tuberculosis expresses a significant number of class A β-lactamases (Flores et al., 2005 ▶; Hugonnet & Blanchard, 2007 ▶), β-lactams are not considered to be a promising front-line medication against tuberculosis. However, interest in the treatment of tuberculosis by targeting the mycobacterial cell wall using a combination therapy of potent lactamase inhibitors of the clavulanate family and carbapenem-type β-lactams has been revived. For instance, the clavulanate–carbapenem combination has shown a bactericidal effect against dormant mycobacteria and drug-resistant strains and in a mouse tuberculosis model (Hugonnet et al., 2009 ▶; England et al., 2012 ▶).
In the complex mycobacterial cell wall, the peptidoglycan (PG) layer is responsible for rigidity and osmotic stability and provides the foundation of the cell envelope (Hett & Rubin, 2008 ▶). The mycolic acids and glycolipids that form a wax-like coat in the outer layer are covalently linked to arabinogalactan, which in turn is coupled to N-acetylmuramic acid units in the PG (Brennan & Crick, 2007 ▶). PG is a complex polymer built up of a glycan chain of alternating units of N-acetylglucosamine and N-acetylmuramic acid and short oligopeptide stems coupled to the N-acetylmuramic acid units via a lactate moiety. The peptide stems are cross-linked, connecting different glycan strands into a meshwork that builds up the PG layer surrounding the cell membrane (Vollmer et al., 2008 ▶; Meroueh et al., 2006 ▶). The integrity of the peptide cross-links is vital for bacteria and the enzymes involved in the formation of these linkages are targeted by β-lactam antibiotics. The amino-acid composition and cross-link architecture show large variations when comparing different bacterial species. M. tuberculosis displays a common peptide-stem sequence (l-Ala-d-γGlu-Dap-d-Ala) which is also found in Escherichia coli and Bacillus subtilis, but with characteristic modifications. These are the high level of amidation on the d-glutamate (d-Glu) and diaminopimelate (Dap) moieties and a high proportion (80%) of Dap–Dap (3–3) cross-links (Kumar et al., 2012 ▶; Lavollay et al., 2008 ▶). The peptide linkages are produced by periplasmic transpeptidases that are active in PG polymerization (Lovering et al., 2012 ▶). The common Dap-d-Ala (3–4) cross-links are formed by d,d-transpeptidases, which are also called penicillin-binding proteins (PBPs). In contrast, the Dap–Dap (3–3) cross-links are created by l,d-transpeptidases (Mainardi et al., 2005 ▶), which represent a different class of enzymes that are not related to the d,d-transpeptidases (Biarotte-Sorin et al., 2006 ▶; Bielnicki et al., 2006 ▶). l,d-Transpeptidases are also responsible for the coupling of nonconventional d-amino acids to PG peptide stems, which has been suggested to be a stress-induced cell-wall adaptation in Vibrio cholerae (Cava et al., 2011 ▶).
The M. tuberculosis H37Rv genome encodes five proteins that all contain a characteristic l,d-transpeptidase domain (Rv0116c, Rv0192, Rv0483, Rv1433 and Rv2518c). LdtMt2 (Rv2518c) has been reported to be expressed at the highest level and has been found to be essential for virulence. Inactivation of this gene led to an attenuated phenotype and to increased susceptibility to clavulanate/amoxicillin treatment in vitro and in a mouse tuberculosis model (Gupta et al., 2010 ▶). The importance of LdtMt2 in virulence makes it an attractive target for the development of inhibitors and novel antibiotics. Structurally characterized l,d-transpeptidases from B. subtilis (Bielnicki et al., 2006 ▶) and from Enterococcus faecium (Biarrotte-Sorin et al., 2006 ▶) define the architecture of the catalytic domain. However, both display an additional domain unrelated to the approximately 200-amino-acid long segment preceding the catalytic domain in LdtMt2 that may govern the positioning of the catalytic module in the PG layer of the cell wall.
Here, we report the three-dimensional structure of LdtMt2 (l,d-transpeptidase 2 from M. tuberculosis) based on the X-ray crystallographic structures of two fragments of LdtMt2 representing the extramembrane part of the protein (residue range 55–408). The structure analysis reveals that LdtMt2 folds into three domains: two domains in the N-terminal part, both of which show an immunoglobulin-related fold, and a C-terminal transpeptidase domain. This domain composition is different from the two-domain structure of the extramembrane part of LdtMt2 proposed recently (Erdemli et al., 2012 ▶). The crystal structures of the LdtMt2 constructs allow modelling of the full-length extramembrane part of the enzyme (residues 55–408), providing an estimate of the maximal distance of the catalytic site from the membrane and thereby the approximate distance at which 3–3 cross-links are formed in the PG layer. Mass-spectrometric analysis provides further evidence that LdtMt2 forms covalent adducts with the β-lactam antibiotics imipenem and ampicillin.
2. Materials and methods
2.1. Gene expression and protein purification
The expression construct for the complete extramembrane segment of LdtMt2 (UniProt code O53223; Rv2518c) included amino acids 34–408, lacking only the predicted transmembrane domain (residues 18–34) and the intracellular part of the polypeptide chain (residues 1–17). Additional constructs were based on the domain arrangement obtained from a bioinformatics analysis of the protein sequence (see §3). The construct used in structure determination comprising the two N-terminal domains A and B (the AB module) included residues 55–250, while the construct comprising the B and C domains (the BC module) encoded residues 149–408 (Fig. 1 ▶ a).
Figure 1.

(a) Domain arrangement in the extramembrane segment of the l,d-transpeptidase LdtMt2 from M. tuberculosis. TM, transmembrane helix. (b) Stereo cartoon of the structure of the BC module of LdtMt2. The catalytic domain is shown in grey and the B-domain in blue. The C-terminal extension that folds over the B domain is shown in green. The three tryptophan residues within this segment are shown as black sticks. (c) Stereo cartoon of the structure of the AB module of LdtMt2. The two immunoglobulin-like domains are shown in blue (B domain) and red (A domain).
The coding sequences for these constructs were amplified by PCR using Pfu Turbo polymerase (Stratagene) with M. tuberculosis H37Rv genomic DNA as the template. Each construct was expressed in the pNIC28Bsa4 vector (GenBank accession No. EF198106) with a cleavable N-terminal His6 tag (H2N-MHHHHHHSSGVDLGTENLYFQ*SM). Proteolytic tag removal by TEV protease (the cleavage position is indicated by an asterisk) resulted in recombinant proteins with two additional amino acids (Ser-Met) at the N-terminus. All proteins were expressed in E. coli BL21 (DE3) cells at 294 K in LB medium supplemented with 30 µg ml−1 kanamycin. Gene expression was induced by the addition of 0.1 mM IPTG at an OD600 of 0.5–0.6 and the cells were harvested 20 h post-induction. The pellets were washed in 0.3 M NaCl, 10 mM Tris–HCl pH 8.0 and the cell walls were disrupted by flash-freezing and treatment with lysozyme (40 µg ml−1) and DNase I (2 µg ml−1) followed by sonication. The recombinant proteins were purified by affinity chromatography using His-Pur Ni–NTA resin (Thermo Scientific) and were eluted with an increasing imidazole gradient. Subsequently, the buffer was exchanged to 0.15 M NaCl, 25 mM Tris–HCl pH 8.0 using a desalting column (HiPrep 26/10; Pharmacia Biotech). The His6 tag was removed by TEV protease cleavage with 5 µg TEV per milligram of recombinant protein at 293 K in the presence of 2 mM DTT. Cleaved proteins were separated from residual uncleaved proteins and TEV protease by a second Ni–NTA affinity step. Finally, a size-exclusion chromatography step was performed using a Superdex 200 column (Pharmacia Biotech) equilibrated with a buffer consisting of 0.15 M NaCl, 25 mM Tris–HCl pH 8.0. Pure protein samples were concentrated using a Vivaspin 20 centrifugal device (Sartorius, Göttingen, Germany) and the protein concentration was determined according to the Bradford assay using reagents from Thermo Scientific (Rockford, Illinois, USA) and a BSA standard. The samples were analyzed by SDS–PAGE and native PAGE (PHAST system, Pharmacia Biotech), aliquoted in the final buffer (0.15 M NaCl, 25 mM Tris–HCl pH 8.0), flash-frozen in liquid nitrogen and stored at 193 K. The selenomethionine-substituted BC-module construct was produced via the metabolic inhibition method (Doublié, 1997 ▶) and purification was carried out using the same steps as for the native protein.
2.2. Protein crystallization
Protein crystallization screening was carried out at protein concentrations of 17.5 mg ml−1 for the construct comprising the B and C domains and 23.2 mg ml−1 for the construct comprising the A and B domains by the vapour-diffusion method using a Mosquito Crystallization Robot (TTP LabTech Ltd, Melbourn, England) and the commercially available crystallization screens PACT, JCSG+, NeXtal Classics II Suite (Qiagen) and SaltRX (Hampton Research). Crystals of the AB module were obtained in sitting drops at 293 K by mixing 0.15 µl protein solution and 0.15 µl well solution [0.2 M ammonium sulfate, 0.1 M bis-tris–HCl pH 6.5, 25%(w/v) PEG 3350] and equilibrating against 50 µl well solution. The crystal used for data collection was flash-cooled in liquid nitrogen without using a cryoprotectant. The crystals of the BC module were grown in a hanging-drop format at 293 K by mixing 1 µl protein solution and 2 µl well solution (3 M sodium acetate pH 6.0, 0.1 M bis-tris propane–HCl pH 7.0) and equilibrating against 1.0 ml well solution. Crystals of the selenomethionine-substituted BC module were obtained in conditions identical to those for the native protein. The crystals of the BC module (native and selenomethionine-substituted) were cryoprotected by a short soak (20 s) in mother liquor supplemented with 30%(v/v) ethylene glycol prior to freezing. The crystals were frozen in nylon loops and stored in liquid nitrogen until data collection.
2.3. Data collection
X-ray data sets were collected to 1.45 Å resolution for the AB module and to 1.86 Å resolution for the BC module on beamline ID14-1 of the European Synchrotron Radiation Facility (ESRF), Grenoble, France. In addition, a data set was collected to 2.7 Å resolution from a crystal of the SeMet-substituted BC module on beamline ID29 at the ESRF at the Se edge at a wavelength of 0.97677 Å. All data sets were collected at 100 K. The X-ray data were processed and scaled using XDS (Kabsch, 2010 ▶; AB module) and MOSFLM (Leslie, 2006 ▶; BC module) and scaled with SCALA from the CCP4 suite (Winn et al., 2011 ▶).
The crystals of the AB module belonged to space group C2, with unit-cell parameters a = 114.4, b = 27.6, c = 68.0 Å, β =114.1°, and contained one molecule in the asymmetric unit with a solvent content of 47.8%. Crystals of the BC module (native and selenomethionine-substituted) belonged to space group I212121, with unit-cell parameters a = 116.0, b = 121.4, c = 123.3 Å, and contained two molecules in the asymmetric unit with a solvent content of 68%. The statistics for all data sets are summarized in Table 1 ▶.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| AB module (57250) | BC module (149408) | BC module (Se-SAD) | |
|---|---|---|---|
| PDB code | 4hu2 | 4huc | |
| Beamline | ID14-1, ESRF | ID14-1, ESRF | ID29, ESRF |
| Detector | ADSC Q210 | ADSC Q210 | Pilatus 6M |
| No. of images | 180 | 180 | 1800 |
| Oscillation interval () | 1.0 | 1.0 | 0.1 |
| Mosaicity () | 0.57 | 0.63 | 0.15 |
| Space group | C2 | I212121 | I212121 |
| Unit-cell parameters | |||
| a () | 114.4 | 116.0 | 116.1 |
| b () | 27.6 | 121.4 | 121.8 |
| c () | 68.0 | 123.3 | 124.5 |
| = () | 90.0 | 90.0 | 90.0 |
| () | 114.1 | 90.0 | 90.0 |
| Wavelength () | 0.93340 | 0.93340 | 0.97677 |
| Resolution () | 33.71.45 (1.531.45) | 58.01.86 (1.961.86) | 50.02.75 (2.902.75) |
| Total No. of reflections | 158723 (19668) | 514550 (50207) | 153521 (22186) |
| No. of unique reflections | 34254 (4636) | 71549 (9227) | 23295 (3369) |
| I/(I) | 21.6 (2.8) | 23.7 (3.1) | 13.7 (2.6) |
| Multiplicity | 4.6 (4.2) | 7.2 (5.4) | 6.6 (6.6) |
| Completeness (%) | 98.9 (94.1) | 98.1 (87.6) | 99.9 (99.9) |
| R merge (%) | 3.8 (48.9) | 6.5 (52.4) | 10.5 (82.2) |
| Wilson B value (2) | 17.6 | 22.3 | 74.9 |
| CC_anom | 0.423 (0.29) | ||
| Refinement | |||
| R cryst (%) | 16.2 | 16.5 | |
| R free (%) | 21.8 | 18.7 | |
| No. of atoms/B factor (2) | |||
| Overall | 1818/20.3 | 4570/24.9 | |
| Protein | 1499/17.8 | 4040/23.7 | |
| Ligands | 10/31.2 [sulfate] | 50/44.7 [acetate, Na+] | |
| Water | 309/32.1 | 480/32.8 | |
| R.m.s.d. from ideal geometry | |||
| Bond lengths () | 0.009 | 0.013 | |
| Bond angles () | 1.44 | 1.48 | |
| Ramachandran plot (%) | |||
| Residues in preferred regions | 98.3 | 95.9 | |
| Residues in allowed regions | 1.7 | 4.1 | |
| Outliers | 0 | 0 | |
2.4. Structure determination and refinement
The structure of the BC module was solved by Se-SAD phasing using the data sets obtained from crystals of the SeMet-substituted protein. The Se sites were identified and refined from the peak data set using SHELX (Sheldrick, 2008 ▶). Of the 12 selenium sites in the asymmetric unit, 11 were found by SHELX, with CC values of 47.2% (CCall) and 30.1% (CCweak). An almost complete model was built by ARP/wARP (Mooij et al., 2009 ▶) using the SeMet peak data set. From this point, the 1.86 Å resolution native data set was used to complete the model by iterative rounds of manual model building using Coot (Emsley et al., 2010 ▶) and crystallographic refinement with REFMAC5 (Murshudov et al., 2011 ▶) applying local NCS symmetry and TLS. The final model contained two chains of the BC module comprising protein residues 149–408, 12 acetate ions, two Na+ ions and 480 water molecules. The final crystallographic R and R free values were 16.5% and 18.7%, respectively (Table 1 ▶).
The structure of the AB module was solved by molecular replacement using Phaser (McCoy et al., 2007 ▶) with the B domain (149–250) derived from the BC module as the search model. Density modification using Parrot (Cowtan, 2010 ▶) was employed to improve the electron-density map, in particular for the unknown domain A at the N-terminus. The A domain was built manually using Coot (Emsley et al., 2010 ▶) and refined to 1.45 Å resolution by cycles of restrained refinement using REFMAC5 (Murshudov et al., 2011 ▶). The final structure comprised amino-acid residues 57–250, two sulfate ions and 309 water molecules; the crystallographic R and R free values were 16.2% and 21.8%, respectively (Table 1 ▶).
Both protein models were validated using Coot (Emsley et al., 2010 ▶) and MolProbity (Chen et al., 2010 ▶) to monitor the stereochemistry and model quality. A summary of the refinement statistics and model parameters is given in Table 1 ▶. Structural comparisons were carried out using the DALI algorithm (Holm & Rosenström, 2010 ▶; Holm & Park, 2000 ▶). Molecular contacts and interacting surfaces were analyzed using the PISA server (Krissinel & Henrick, 2007 ▶). Figures were produced using the program PyMOL (http://www.pymol.org). The coordinates of the AB module and BC module have been deposited in the Protein Data Bank under accession codes 4hu2 and 4huc, respectively.
2.5. Mass-spectrometric analysis of antibiotic binding
Samples of LdtMt2 (5 µg) were dispensed in water without antibiotics and with 2 mM imipenem (Sigma) or ampicillin (Duchefa Biochemie, Haarlem, The Netherlands) and incubated for 45 min at 295 K. Subsequently, the samples were diluted in 0.5 ml denaturing buffer [5%(v/v) acetonitrile, 0.1%(v/v) formic acid, 0.5 mM TCEP] and loaded onto a CapLC system and an ESI-Q-TOF quadrupole/orthogonal acceleration time-of-flight mass spectrometer (Waters Corp.) using the method described by Sundqvist et al. (2007 ▶). The spectra were combined and deconvoluted with the Maximum Entropy 1 algorithm from the MassLynx software suite (Waters Corp.) to obtain the molecular weights of the protein molecules and the protein–antibiotic adducts.
3. Results and discussion
3.1. Domain organization of LdtMt2, construct design and protein production
The lppS gene (Rv2518c) encodes an enzyme consisting of 408 amino acids denoted LdtMt2. The polypeptide chain contains a short intracellular segment (residues 1–17), a predicted transmembrane domain (residues 18–34) and a larger extramembrane part (residues 35–408). The amino-acid sequence of LdtMt2 was analyzed with respect to potential domain organization using transmembrane-domain prediction (Cserzö et al., 1997 ▶) and sequence comparisons by BLAST (Altschul et al., 1990 ▶) and ClustalW (Thompson et al., 1994 ▶). This resulted in a well defined prediction of the catalytic domain (residues 250–408). The sequence similarity of residues 150–250 (30–40% identity) to three other M. tuberculosis proteins (Rv0116c, Rv0483 and Rv1433) and several proteins derived from different corynebacteria genomes that also contain a predicted l,d-transpeptidase domain suggested that this fragment may be a separate domain. Furthermore, the sequence comprising residues 34–250 was subjected to domain-border analysis/prediction (http://www.tuat.ac.jp/~domserv/cgi-bin/DLP-SVM.cgi#Wahtis; Ebina et al., 2009 ▶) as well as to secondary-structure prediction by JPred (Cole et al., 2008 ▶) to define the domain borders. Finally, two likely starting points at the N-terminus were the end of the predicted transmembrane helix coinciding with a predicted lipoprotein-attachment site (Rahman et al., 2008 ▶) at position 34/35 and the beginning of the first predicted secondary-structure element at position 55. Taken together, these analyses suggested that the extramembrane part of the enzyme consists of three domains: two smaller domains (domain A, residues 34/55–146; domain B, residues 149–250) and a catalytic domain (domain C, residues 250–408) belonging to the ErfK/YbiS/YhnG family (Bielnicki et al., 2006 ▶; Fig. 1 ▶ a). Based on this analysis, three constructs were designed: a full-length construct (residues 34–408), the AB module (residue 55–146) and the BC module (residues 149–408).
The full-length extramembrane fragment was expressed in E. coli and purified to homogeneity. Based on size-exclusion chromatography, the protein is a monomer in solution. However, extensive crystallization screens did not result in crystal formation. The AB- and BC-module constructs also resulted in soluble protein and each construct behaved as a monomer in solution.
3.2. The structure of the BC module
The crystal structure of the BC module was solved to 1.86 Å resolution by Se-SAD phasing from crystals of selenomethionine-substituted protein. The electron density for the two polypeptide chains (residues 149–408) in the asymmetric unit is well defined (Fig. 2 ▶ a). Six surface residues were modelled in double conformations (Arg297 and Ile301 in chain A and Met237, His246, Arg297 and Lys370 in chain B). The model also contains 480 water molecules and 12 acetate ions, which mostly interact with surface residues. Two of the acetate ions are located in the active-site cleft of domain C. Finally, each polypeptide chain contains a bound metal ion. We modelled the bound metal as an Na+ ion based on the electron density (i.e. no remaining difference density after refinement), typical metal–ligand distances in the range 2.2–2.5 Å and the composition of the ligand sphere. The Na+ ion is located at the C-terminus of a surface helix and is coordinated by the carbonyl O atoms of residues 342 and 345 and by three water molecules.
Figure 2.

Parts of the refined 2F o − F c electron-density maps of LdtMt2 contoured at 1.5σ. (a) Stereoview of the electron-density map of the BC module in the vicinity of the active-site residue Cys354. (b) Stereoview of the electron-density map of the AB module in the hydrophobic core of the B domain.
The BC module consists of the catalytic domain of the typical ErfK/YbiS/YhnG fold and a smaller domain with an immunoglobulin-related fold (Fig. 1 ▶ b). The crystal asymmetric unit contains two molecules with a monomer–monomer interface of approximately 800 Å2, suggesting crystal-packing contacts rather than dimer formation. The structures of the two molecules in the asymmetric unit in the crystals of the BC module are essentially identical, displaying a Cα r.m.s.d. of 0.3 Å for 259 aligned residues. The overall structure of this module and the interactions of the two polypeptide chains in the asymmetric unit are very similar to a recently determined structure of a corresponding module of LdtMt2, albeit obtained from a different expression construct (residues 120–408 as judged from Fig. 1b in Erdemli et al., 2012 ▶; the atomic coordinates were not available in the PDB).
The core of the transpeptidase domain consists of a β-sandwich formed by two β-sheets packed together. Comparison of this domain with a related protein from E. faecium (PDB entry 1zat; Biarrotte-Sorin et al., 2006 ▶) using DALI results in a Z-score of 19.3 and a Cα r.m.s.d. of 1.9 Å for 120 aligned residues. A similar comparison with the B. subtilis homologue (PDB entry 1y7m; Bielnicki et al., 2006 ▶) gives a Z-score of 17.7 and a Cα r.m.s.d. of 1.6 Å for 109 aligned residues. The key residues of the catalytic site, Cys354 and His336, correspond to Cys442 and His421 in the E. faecium homologue. In LdtMt2 the active site is located under a lid formed by a pair of antiparallel β-strands and their connecting loop (residues 298–324) that protrude from the core of the catalytic domain (Fig. 3 ▶). This feature is not present in the B.subtilis enzyme and the tip of the lid is disordered in the E. faecium protein, making the active site solvent-accessible, while in LdtMt2 access is limited by the lid. The large hydrophobic residues of the lid (Tyr298, Tyr308 and Tyr318) and the residues surrounding the entrance to the active site (Tyr330, Phe334 and Trp340) contribute to the closure and to the exclusion of solvent, which may support efficient catalysis.
Figure 3.

The active site of LdtMt2. The lid covering the active site formed by a pair of antiparallel β-strands and their connecting loop (residues 298–324) is shown in red. The active-site residues Cys354 and His336 are depicted as green sticks. The bulky hydrophobic residues of the lid (Tyr298, Tyr308 and Tyr318) and its surroundings (Tyr330, Phe334 and Trp340), shown as red and orange sticks, respectively, limit access to and contribute to the closure of the active site.
Residues 382–408 form a C-terminal extension which is not present in the two characterized structural homologues. This sequence stretch runs along the C domain and contributes to the domain interface by forming additional contacts with the B domain (Fig. 1 ▶ b). The three tryptophan residues (Trp394, Trp398 and Trp401) in this segment form extensive stacking interactions at the domain interface and are likely to stabilize the relative orientation of the two domains. The pattern of tryptophan residues in this sequence stretch is retained in Rv0483 (LprQ), one of the homologues from the M. tuberculosis H37Rv genome.
As has been noted previously (Erdemli et al., 2012 ▶), residues 150–250 (here denoted domain B) fold into an antiparallel β-sandwich (Fig. 1 ▶ b) related to the immunoglobulin-like fold (see also below).
3.3. The structure of the AB module of LdtMt2
The structure of the segment comprising the A and B domains was solved to 1.45 Å resolution by molecular replacement using the B domain (residues 149–250) as the search model. The structure of the AB module includes residues 57–250, approximately half of the extramembrane part of LdtMt2, and 309 water molecules. Apart from the first two residues of the construct, the electron density for the polypeptide chain is well defined (Fig. 2 ▶ b). Seven residues, Val59, Val66, Asn121, Arg123, Thr138, Glu162 and His214, were modelled in double conformations. Each domain in the model contains a bound sulfate ion located on the protein surface. In domain A the sulfate is anchored to the protein through interactions with Asn95, Asn97, Arg99 and Arg122, whereas in the B domain the ligand forms hydrogen bonds to the side chains of Tyr201, Arg209 and Arg211.
The two domains are arranged in a V-shaped manner and both display a variant of the immunoglobulin fold built up by a β-sandwich of two antiparallel β-sheets (Figs. 1 ▶ c and 4 ▶). A structural classification of immunoglobulin-like β-sandwich domains defined four subclasses, s-type, h-type, c-type and v-type, based on strand connectivity and topology (Bork et al., 1994 ▶). Both immunoglobulin-like domains of LdtMt2 belong to the c-type Ig fold, with strand orders a–b–e–d and c–f–g in the two antiparallel β-sheets (Fig. 4 ▶).
Figure 4.
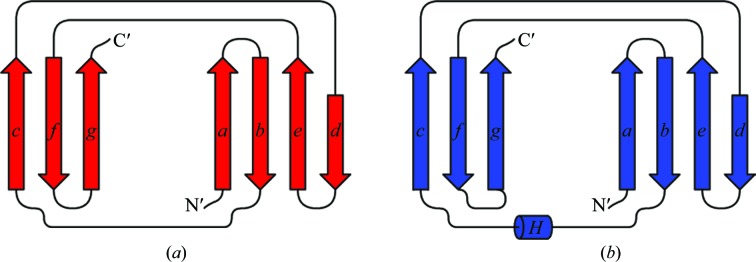
Topology diagrams of the immunoglobulin-like domains in LdtMt2. (a) Topology of the A domain. (b) Topology of the B domain.
The similarity between the A and B domains is reflected in the Cα r.m.s.d. of 2.7 Å based on 85 aligned residues. The main difference in their overall structure is the short α-helix (residues 180–187) found between β-strands β2 and β3, which is not present in domain A, and the loop connecting β6 and β7, which is substantially longer in the B domain (Figs. 4 ▶ and 5 ▶ a). The sequence identity derived from structural alignment of the two domains is 12% which does not provide strong evidence for gene duplication.
Figure 5.

(a) Superposition of the A domain (red) and B domain (blue) of LdtMt2. The major differences in the core fold (indicated by >) are the α-helix (residues 180–187) inserted between β-strands β2 and β3, which is not present in the A domain, and the longer loop connecting β6 and β7 in the B domain. (b) Superposition of domain A (red) from LdtMt2 with the homologous domain of the endo-β-1,4-mannanase from C. fimi (light blue; PDB entry 2x2y). Conserved residues of LdtMt2 are shown as yellow stick models. Five threonine side chains on the surface of the three-stranded β-sheet show a similar pattern in the mannanase and the A domain of LtdMt2 and are shown as blue sticks.
The domain interface accounts for 350 Å2 of buried surface area including 13 and ten buried residues from the A and B domains, respectively. The domain interface is formed by the loops connecting β-strands β1 and β2 and β-strands β5 and β6 in domain A and the loop connecting β-strands β6 and β7 in domain B. The two domains are connected by a short linker Ala149-His150 forming hydrogen bonds to the main chain of the B-domain residues involved in the interface (Gly236 and Phe237). The size of the AB-domain interface, the short linker and the fact that B factors of the linker residues are in the range 18–22 Å2 as in the remaining part of the structure suggest that the domain linker is not flexible and the orientation of the two domains is kept in this V-shaped form (Fig. 1 ▶ c). The structures of the B domain in the AB and BC modules are essentially identical and superposition gives an r.m.s.d. of 0.4 Å for 101 aligned residues, although the B-factor profiles for these domains are different in the two crystal forms, most likely owing to different crystal-packing interactions.
The two constructs used in the crystal structure analysis cover approximately the whole length of the extramembrane part of the polypeptide chain of LdtMt2 and the results presented here redefine the domain composition of this enzyme. A recent structure analysis described the extramembrane part of the protein as a two-domain structure extending from the membrane (Erdemli et al., 2012 ▶). The structure of the AB module, which covers a larger proportion of the N-terminal residues of the extramembrane part of the enzyme (residues 55–250) than in the previous analysis (residues 120–408), shows that this segment of LdtMt2 contains an additional domain, domain A, at the N-terminus (Fig. 1 ▶ c) which had not been recognized previously. The extramembrane part of the polypeptide chain of LdtMt2 is thus a three-domain entity, with two immunoglobulin-like domains rather than one and a C-terminal transpeptidase domain.
A structural comparison using DALI identified the closest structural homologues of the two immunoglobulin-like domains as the periplasmic copper-resistance protein from Pseudomonas syringae (PDB entry 2c9r; Zhang et al., 2006 ▶; Z-score of 10.2 and Cα r.m.s.d. of 2.4 Å) and the N-terminal domain of Halothermothrix orenii amylase (PDB entry 3bc9; Tan et al., 2008 ▶) and a similar domain of the endo-β-1,4-mannanase from Cellulomonas fimi (PDB entry 2x2y; Hekmat et al., 2010 ▶) with similar Z-scores (6.0–10.1) and Cα r.m.s.d. values (2.6–3.1 Å). Finally, the cell-wall-located S-layer protein from Geobacillus stearothermophilus (PDB entry 2ra1; Pavkov et al., 2008 ▶) aligned with a Z-score of 7.8 and a Cα r.m.s.d. value of 3.2 Å. Structure-based sequence alignments show that in the case of the B domain the sequence identities to these structural homologues are all below 10%, thus indicating no significant relationship at the amino-acid sequence level. However, the A domain shows a higher degree of amino-acid sequence conservation when compared with the copper-resistance protein from P. syringae (18.5%) and with the homologous domains of the mannanase (18.5%) and a glucodextranase from Arthrobacter globiformis (15.2%) (Mizuno et al., 2004 ▶). Functional analogy can be excluded in the case of the periplasmic copper-resistance protein, as the copper-binding residues are not present in LdtMt2. However, the similarity to the two domains that are present in the enzymes processing high-molecular-weight polysaccharides (mannanase and glucodextranase) could potentially be relevant as the PG backbone is a duplex of carbohydrate chains. In the multi-domain amylase enzyme from H. orenii it has been experimentally demonstrated that the small noncatalytic immunoglobulin-like domain mediates the interaction of the enzyme with the high-molecular-weight substrate (Tan et al., 2008 ▶). The residues identified as conserved between domain A and the corresponding domains from the polysaccharide-processing enzymes mannanase or glucodextranase are either part of the hydrophobic core or are located on the β-sheet surface (Fig. 5 ▶ b).
3.4. The extramembrane LdtMt2
As the two constructs include essentially the whole sequence of the extramembrane part of LdtMt2 and contain an overlapping domain (domain B), the structure of the full-length protein excluding the short intracellular and membrane-stretch parts can be reconstructed by superposition of the AB and the BC modules based on the B domains (Fig. 6 ▶). The three-domain structure of this transpeptidase can extend from the plasma membrane to a distance of at least 80 Å (plus the stretch of residues 34–54). The remaining four transpeptidase domain proteins in the M. tuberculosis H37Rv genome show differences in the length of the polypeptide chain. Rv0116c and Rv1433 both have a predicted transmembrane domain, but their extramembrane segment is shorter at approximately 250 residues and shows significant sequence identity (45 and 38%, respectively) to the BC module of LdtMt2. On the other hand, Rv0483 and Rv0192 appear to have a three-domain arrangement as in LdtMt2 as they not only show sequence homology to the B and C domains but also to the A domain (35% sequence identity for Rv0483 and 18% for Rv0192). The five l,d-transpeptidases in the M. tuberculosis genome thus can be divided into two groups with one or two immunoglobulin-like domains between the plasma membrane and the catalytic domain. The variation in the number of these domains positions the catalytic domain at different distances from the membrane, reflecting the multilayered structure of the peptidoglycan in the bacterial cell wall. These domains define the distance of the catalytic site and thus the site of formation of the 3–3 linkages in the PG relative to the membrane. Cross-link formation appears to occur at two levels carried out by the BC module-like l,d-transpeptidases (Rv0116c and Rv1433) and by the one domain longer LdtMt2-type (ABC module; Fig. 6 ▶) enzymes (Rv2518c, Rv0483 and Rv0192).
Figure 6.

Model of the complete extramembrane part of LdtMt2 from M. tuberculosis. The model was derived by superposition of the B domains common to the structures of the AB and BC modules. The maximal distance between the membrane and the catalytic site, where cross-link formation in the cell-wall peptidoglycan by LdtMt2 can occur, can be approximated to at least 80–100 Å.
3.5. Binding of β-lactam antibiotics to LdtMt2
The inhibitory effect of β-lactam-type antibiotics towards transpeptidases is based on the formation of a covalent adduct with the active-site serine residues in d,d-transpeptidases or cysteine residues in l,d-transpeptidases, as has been demonstrated for the l,d-transpeptidase from B. subtilis (Lecoq et al., 2012 ▶) and for LdtMt1 (Rv0116c), a homologue of LdtMt2 (Dubée et al., 2012 ▶). Incubation of the BC module or of the full-length extramembrane fragment (residues 34–408) of LdtMt2 from M. tuberculosis with the β-lactam-type antibiotics imipenem (299.3 Da, carbapenem class) and ampicillin (349.4 Da, penicillin class) led to the formation of covalent enzyme–antibiotic adducts as shown by ESI-MS (Table 2 ▶ and Supplementary Material1). The observed molecular masses of the extramembrane fragment (residues 34–408; 40 074.0 Da) and the BC module (residues 149–408; 28 482.0 Da) were very close to the calculated masses from the sequence: 40 073.8 and 28 481.8 Da, respectively (Supplementary Material). The mass difference between the samples before and after incubation with 2 mM ampicillin or 2 mM imipenem corresponds to the exact mass of the respective antibiotic (Table 2 ▶). Together with the previous demonstration of binding of imipenem and meropenem to LdtMt2 using ITC (Erdemli et al., 2012 ▶), these data indicate that the enzyme can recognize and bind a variety of β-lactam antibiotics, which also suggests that there is potential for the design of more specific inhibitors of LdtMt2.
Table 2. Covalent adducts formed by LdtMt2 from M. tuberculosis with the -lactam-type antibiotics imipenem and ampicillin.
| Protein construct, molecular mass (Da) | -Lactam, molecular mass (Da) | Detected mass (Da) | Mass difference (Da) |
|---|---|---|---|
| LdtMt2 periplasmic segment (34408), 40074.0 | Imipenem, 299.3 | 40374.6 | 300.6 |
| LdtMt2 BC module (149408), 28482.0 | Imipenem, 299.3 | 28780.9 | 298.9 |
| Ampicillin, 349.4 | 28831.7 | 349.7 |
Supplementary Material
PDB reference: LtdMt2, AB module, 4hu2
PDB reference: BC module, 4huc
Supporting information file. DOI: 10.1107/S0907444912049268/wd5198sup1.pdf
Acknowledgments
We acknowledge access to synchrotron radiation at beamlines 9-11 at MAX IV laboratory and ID14-1 and ID29 at the ESRF and thank the beamline staff for support. This work was funded by the Swedish Governmental Agency for Innovation Systems (VINNOVA).
Footnotes
Supplementary material has been deposited in the IUCr electronic archive (Reference: WD5198). Services for accessing this material are described at the back of the journal.
References
- Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. (1990). J. Mol. Biol. 215, 403–410. [DOI] [PubMed]
- Biarrotte-Sorin, S., Hugonnet, J.-E., Delfosse, V., Mainardi, J.-L., Gutmann, L., Arthur, M. & Mayer, C. (2006). J. Mol. Biol. 359, 533–538. [DOI] [PubMed]
- Bielnicki, J., Devedjiev, Y., Derewenda, U., Dauter, Z., Joachimiak, A. & Derewenda, Z. S. (2006). Proteins, 62, 144–151. [DOI] [PMC free article] [PubMed]
- Bork, P., Holm, L. & Sander, C. (1994). J. Mol. Biol. 242, 309–320. [DOI] [PubMed]
- Brennan, P. J. & Crick, D. C. (2007). Curr. Top. Med. Chem. 7, 475–488. [DOI] [PubMed]
- Cava, F., de Pedro, M. A., Lam, H., Davis, B. M. & Waldor, M. K. (2011). EMBO J. 30, 3442–3453. [DOI] [PMC free article] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Cole, C., Barber, J. D. & Barton, G. J. (2008). Nucleic Acids Res. 35, W197–W201. [DOI] [PMC free article] [PubMed]
- Cole, S. T. & Riccardi, G. (2011). Curr. Opin. Microbiol. 14, 570–576. [DOI] [PubMed]
- Cowtan, K. (2010). Acta Cryst. D66, 470–478. [DOI] [PMC free article] [PubMed]
- Cserzö, M., Wallin, E., Simon, I., von Heijne, G. & Elofsson, A. (1997). Protein Eng. 10, 673–676. [DOI] [PubMed]
- Doublié, S. (1997). Methods Enzymol. 276, 523–530. [PubMed]
- Dubée, V., Triboulet, S., Mainardi, J.-L., Ethève-Quelquejeu, M., Gutmann, L., Marie, A., Dubost, L., Hugonnet, J.-E. & Arthur, M. (2012). Antimicrob. Agents Chemother. 56, 4189–4195. [DOI] [PMC free article] [PubMed]
- Ebina, T., Toh, H. & Kuroda, Y. (2009). Biopolymers, 92, 1–8. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- England, K., Boshoff, H. I., Arora, K., Weiner, D., Dayao, E., Schimel, D., Via, L. E. & Barry, C. E. III (2012). Antimicrob. Agents Chemother. 56, 3384–3387. [DOI] [PMC free article] [PubMed]
- Erdemli, S. B., Gupta, R., Bishai, W. R., Lamichhane, G., Amzel, L. M. & Bianchet, M. A. (2012). Structure, 20, 2103–2115. [DOI] [PMC free article] [PubMed]
- Flores, A. R., Parsons, L. M. & Pavelka, M. S. (2005). Microbiology, 151, 521–532. [DOI] [PubMed]
- Gengenbacher, M. & Kaufmann, S. H. (2012). FEMS Microbiol. Rev. 36, 514–532. [DOI] [PMC free article] [PubMed]
- Gupta, R., Lavollay, M., Mainardi, J.-L., Arthur, M., Bishai, W. R. & Lamichhane, G. (2010). Nature Med. 16, 466–469. [DOI] [PMC free article] [PubMed]
- Hekmat, O., Lo Leggio, L., Rosengren, A., Kamarauskaite, J., Kolenova, K. & Stålbrand, H. (2010). Biochemistry, 49, 4884–4896. [DOI] [PubMed]
- Hett, E. C. & Rubin, E. J. (2008). Microbiol. Mol. Biol. Rev. 72, 126–156. [DOI] [PMC free article] [PubMed]
- Holm, L. & Park, J. (2000). Bioinformatics, 16, 566–567. [DOI] [PubMed]
- Holm, L. & Rosenström, P. (2010). Nucleic Acids Res. 38, W545–W549. [DOI] [PMC free article] [PubMed]
- Hugonnet, J.-E. & Blanchard, J. S. (2007). Biochemistry, 46, 11998–12004. [DOI] [PMC free article] [PubMed]
- Hugonnet, J.-E., Tremblay, L. W., Boshoff, H. I., Barry, C. E. III & Blanchard, J. S. (2009). Science, 323, 1215–1218. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol. 372, 774–797. [DOI] [PubMed]
- Kumar, P., Arora, K., Lloyd, J. R., Lee, I. Y., Nair, V., Fischer, E., Boshoff, H. I. & Barry, C. E. III (2012). Mol. Microbiol. 86, 367–381. [DOI] [PMC free article] [PubMed]
- Lavollay, M., Arthur, M., Fourgeaud, M., Dubost, L., Marie, A., Veziris, N., Blanot, D., Gutmann, L. & Mainardi, J.-L. (2008). J. Bacteriol. 190, 4360–4366. [DOI] [PMC free article] [PubMed]
- Lecoq, L., Bougault, C., Hugonnet, J.-E., Veckerlé, C., Pessey, O., Arthur, M. & Simorre, J.-P. (2012). Structure, 20, 850–861. [DOI] [PubMed]
- Leslie, A. G. W. (2006). Acta Cryst. D62, 48–57. [DOI] [PubMed]
- Lovering, A. L., Safadi, S. S. & Strynadka, N. C. (2012). Annu. Rev. Biochem. 81, 451–478. [DOI] [PubMed]
- Mainardi, J.-L., Fourgeaud, M., Hugonnet, J.-E., Dubost, L., Brouard, J.-P., Ouazzani, J., Rice, L. B., Gutmann, L. & Arthur, M. (2005). J. Biol. Chem. 18, 38146–38152. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Meroueh, S. O., Bencze, K. Z., Hesek, D., Lee, M., Fisher, J. F., Stemmler, T. L. & Mobashery, S. (2006). Proc. Natl Acad. Sci. USA, 103, 4404–4409. [DOI] [PMC free article] [PubMed]
- Mizuno, M., Tonozuka, T., Suzuki, S., Uotsu-Tomita, R., Kamitori, S., Nishikawa, A. & Sakano, Y. (2004). J. Biol. Chem. 279, 10575–10583. [DOI] [PubMed]
- Mooij, W. T., Cohen, S. X., Joosten, K., Murshudov, G. N. & Perrakis, A. (2009). Structure, 17, 183–189. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Pavkov, T., Egelseer, E. M., Tesarz, M., Svergun, D. I., Sleytr, U. B. & Keller, W. (2008). Structure, 16, 1226–1237. [DOI] [PubMed]
- Rahman, O., Cummings, S. P., Harrington, D. J. & Sutcliffe, I. C. (2008). World J. Microbiol. Biotechnol. 24, 2377–2382.
- Russell, D. G. (2011). Immunol. Rev. 240, 252–268. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Sundqvist, G., Stenvall, M., Berglund, H., Ottosson, J. & Brumer, H. (2007). J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 852, 188–194. [DOI] [PubMed]
- Tan, T.-C., Mijts, B. N., Swaminathan, K., Patel, B. K. & Divne, C. (2008). J. Mol. Biol. 378, 852–870. [DOI] [PubMed]
- Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994). Nucleic Acids Res. 22, 4673–4680. [DOI] [PMC free article] [PubMed]
- Vollmer, W., Blanot, D. & de Pedro, M. A. (2008). FEMS Microbiol. Rev. 32, 149–167. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Zhang, L., Koay, M., Maher, M. J., Xiao, Z. & Wedd, A. G. (2006). J. Am. Chem. Soc. 128, 5834–5850. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: LtdMt2, AB module, 4hu2
PDB reference: BC module, 4huc
Supporting information file. DOI: 10.1107/S0907444912049268/wd5198sup1.pdf