

Abstract
The fast development of software and hardware is notably helping in closing the gap between macroscopic and microscopic data. Using a novel theoretical strategy combining molecular dynamics simulations, conformational clustering, ab-initio quantum mechanics and electronic coupling calculations, we show how computational methodologies are mature enough to provide accurate atomistic details into the mechanism of electron transfer (ET) processes in complex protein systems, known to be a significant challenge. We performed a quantitative study of the ET between Cytochrome c Peroxidase and its redox partner Cytochrome c. Our results confirm the ET mechanism as hole transfer (HT) through residues Ala194, Ala193, Gly192 and Trp191 of CcP. Furthermore, our findings indicate the fine evolution of the enzyme to approach an elevated turnover rate of 5.47×106 s−1 for the ET between Cytc and CcP through establishment of a localized bridge state in Trp191.
Author Summary
We have developed a protocol capable of describing long-range electron transfer mechanisms at an atomic detailed level. We demonstrate the maturity of the computational techniques in obtaining a quantitative view of the Cytochrome c Peroxidase/Cytochrome c electron transfer process, known to be a significant challenge. In excellent agreement with experimental data, our results allow for the description of the electron transfer pathway, its mechanism and the electron transfer rate at a quantitative level. The overall protocol is free of parameterization and can be applied to any complex electron transfer process. Furthermore, the results reveal the fine enzyme evolution of this protein-protein complex to optimize its electron transfer rate by a localized bridge state.
Introduction
Electron transfer (ET) is a fundamental reaction in biochemistry [1], [2]. Its comprehensive elucidation is crucial for the understanding of biological function and the design of synthetic energy transduction systems. In this matter, the question of how intermediary medium controls the electron transfer process between two redox proteins remains an intriguing and challenging problem in biophysics and biochemistry [3]–[7]. In general, ET can be considered as a transition between two electronic states, donor (D) and acceptor (A). Following Marcus theory, the ET rate is determined by the electronic coupling between D and A (VDA), the reaction free energy (ΔG) and the reorganization energy (λ) needed to adopt the system from one state to the other:
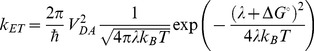 |
(1) |
Besides direct and bridge-mediated superexchange between donor and acceptor, ET can also occur through incoherent hopping including transiently populated electronic states localized on a bridge [8].
There have been many investigations on ET in enzymes [9]–[13], with several of them on ruthenium-modified proteins [14]–[28]. In terms of the theoretical study of ET in protein, the Pathways method developed by Beratan et al. has made considerable impact in the 90's [1], [29], while it got replaced by the combined QM-MD approach [30]–[33], being employed in this study. Of special interest is the ET in the Cytochrome c Peroxidase/Cytochrome c (CcP/Cytc) complex [9], [34]–[39]. In its catalytic cycle, CcP undergoes a two-electron oxidation by peroxide forming an oxo-ferryl intermediate called compound I (CpdI), which is then reduced by two distinct Cytc to firstly CpdII and finally the ferric resting state again (see Figure 1). The measurement of the pure ET rate (not including the complex formation) still bears difficulties due to instrumental dead-times. It was estimated to be greater than 2000 s−1 measured by stopped flow experiments on the ground state ET in the covalently linked complex, or rather 5.0×104 s−1 measured by flash photolysis methods using Ru-porphyrin photo-excited states of the noncovalent complex [38], [40]. Pelletier and Kraut proposed a σ-bond tunneling pathway from Cytc to Trp191 following the residues Ala194, Ala193 and Gly192 of CcP (shown in red in Figure 2), which is supported by several other studies [34], [36], [41], [42]. Furthermore, it is widely accepted that CpdI oxidizes Trp191 generating the radical intermediated Trp191+, which then is reduced by Cytc [43]–[48]. Consequently, the ET mechanism can be considered as hole transfer (HT).
Figure 1. Catalytic cycle of CcP.
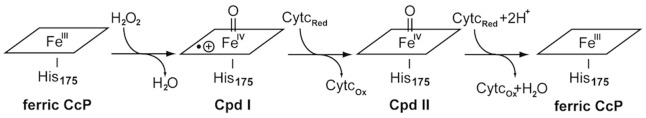
Figure 2. Electron transfer region of the CcP/Cytc complex.
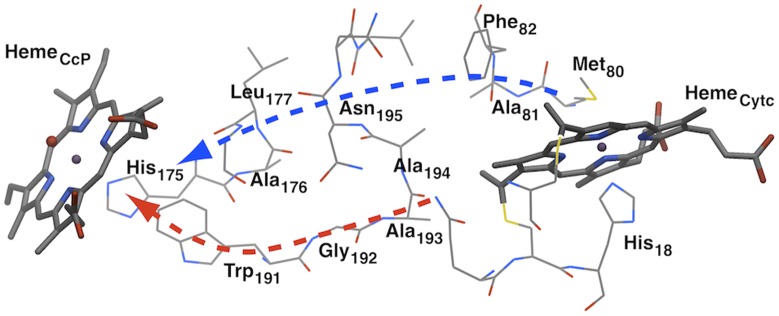
The ET pathway proposed by Pelletier and Kraut is shown in red, the ET pathway suggested by Siddarth is shown in blue.
Understanding the ET mechanism at an atomic detailed level is a crucial step towards rational enzyme engineering. In this paper we demonstrate how the evolution of computational methodologies allows today for a comprehensive study on complex protein-protein ET. Using a new theoretical strategy combining conformational sampling, a recently developed ET pathway search (QM/MM e-Pathway) [49], [50] and robust ab-initio quantum mechanics electronic coupling calculations, we perform a comprehensive case study of the ground state ET between CcP and its redox partner Cytc. In addition to the identification of the ET mechanism and pathway, this study provides a computational assessment of the ET rate constant. Our analysis confirms the underlying ET mechanism to be sequential hopping, with the Trp191 radical cation as an intermediate state. This finding provides a select example of enzyme evolution by means of breaking down slow processes into several faster ones, together with a fine-tuning of the ΔG and λ differences, thereby gaining total turnover rate.
Results/Discussion
The overall simulation protocol proposed here involves: 1) the conformational sampling of the protein-protein complex, 2) the analysis of possible electron transfer pathways, and 3) the computation of VDA, ΔG and λ to obtain the ET rate for the different pathways found in step 2.
Conformational sampling
Molecular dynamics techniques have experienced a significant advancement in the recent years. Both the development of better algorithms together with the availability of a larger number of processors make it easy to run tens of nanoseconds in few days on a small local cluster (16–32 processors). Furthermore, hundreds of nanoseconds or even microseconds are becoming a possibility with the usage of GPU clusters or specialized purpose machines [51], [52]. These long MD runs have also provided the necessary phase space exploration to further optimize the force fields [53].
For CcP/Cytc, the existence of a “rigid” engineered covalent cross-link protein-protein complex [38], with very similar activity and structure to the non-covalent (wild type) complex, points to the existence of a main ET conformation. Thus, a relatively short MD around the initial crystal should provide representative conformations. Nevertheless, we run a 30 ns MD trajectory that confirmed the stability of the protein-protein complex. Additionally the ET rate results (see below) confirm the properness of the conformational sampling.
Starting from the Pelletier and Kraut crystal structure [34] we performed 30 ns of molecular dynamics (MD) on the complex and extracted 10 snapshots within the first 2 ns (based on clustering of the ET region RMSD) for local sampling and 3 more snapshots at time steps 10 ns, 20 ns and 30 ns for nonlocal sampling. The RMSD for the heme groups as well as the superposition of all 14 conformations (crystal structure plus 13 snapshots from MD) is given in Figure S1 and S2 in Text S1. The overall results indicate clearly the stability of the complex and the lack of large fluctuations in the donor, acceptor and interphase region.
ET pathway
Following the conformational sampling, we identified the potentially important residues for the ET in each of the 14 snapshots. By using mixed quantum mechanics and molecular mechanics (QM/MM) techniques, it is now possible to track the electron pathway for complex biological systems. To this purpose, we developed the QM/MM e-Pathway, an iterative procedure capable of tracking the residues (molecular orbitals) with highest electron affinity in the transfer region (see Figure 3 and methods section).
Figure 3. Visual output of the QM/MM e-Pathway method applied on a single MD snapshot.
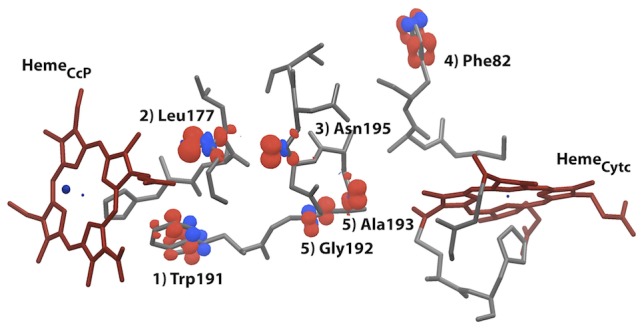
Molecular orbitals (spin densities of electron hole) are labeled by corresponding residue name and iteration of the approach. I.e. Trp191 being identified first, then excluded from QM region, such that Leu177 is identified second, etc. See methods and SI for details on the QM/MM e-Pathway approach.
In detail, the examined residues are Ala176, Leu177, Trp191, Gly192, Ala193, Ala194, Asn195 and Asn196 of CcP as well as Ala81 and Phe82 of Cytc, spanning the protein space between the donor and acceptor. The logo plot, given in Figure 4, summarizes the QM/MM e-Pathway calculations on all 14 snapshots, where the size of a digit d for residue r indicates the relative frequency of residue r being identified as hole acceptor at step d of the iterative approach. The total occurrence of a specific residue can deviate from 1.0 because multiple residues were sometimes identified within a single QM/MM e-Pathway step and we also stopped the search after 7 steps. The results clearly indicate that Trp191 is the first hole acceptor in 10 of the 14 conformations. Thus, from the QM/MM e-Pathway analysis, one can obtain qualitative mechanistic information. In this case, for example, it indicates the possibility of having Trp191 as the localized bridge state in a two-step, sequential hopping ET mechanism.
Figure 4. Logo plot summarizing the QM/MM e-Pathway calculations on all 14 snapshots, where the size of a digit d for residue r indicates the relative frequency of residue r being identified as hole acceptor at step d of the iterative approach.
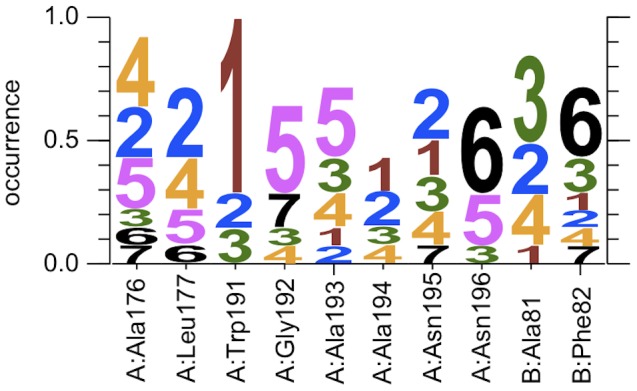
We denote a set of residues as the ET pathway for a specific snapshot, once a connecting chain of residues between the donor and acceptor is found through the stepwise application of the QM/MM e-Pathway approach (see supporting information). Our calculations result in 2 distinct ET pathways for the CcP/Cytc system. The first pathway path1 consists of residues Trp191, Gly192, Ala193 and Ala194 of CcP, and is assigned to 10 of the 14 conformations. The second pathway path2 consists of residues Ala176, Leu177, Ala194 and Asn195 of CcP, and Phe82 and Ala81 of Cytc, and is assigned to 4 of the 14 conformations. Comparison of our results with published data shows that path1 is identical to the ET pathway proposed by Pelletier and Kraut and confirmed by several studies [1], [34], [42]. There is less evidence for path2 to be involved in the ET between Cytc and CcP, yet it was proposed by Siddarth [54].
VDA, ΔG, and λ
The QM/MM e-Pathway approach only identifies important residues in the ET region in a qualitative manner. Therefore, we computed the ET rate for each possible pathway and mechanism in order to get a quantitative measure. As mentioned above, based on the QM/MM e-Pathway analysis we also investigated the hole localization on Trp191 and its role in the ET process. Thus, the following text is split into two parts: 1) single-step HT between Cytc and CcP and 2) sequential hopping HT with Trp191 as localized bridge.
Single step HT
In order to investigate the single-step HT, we calculated the electronic coupling between donor and acceptor for all 14 conformations based on 4 different setups: 1) direct electronic coupling of hemeCcP and hemeCytc only, 2) full bridge-mediated electronic coupling including the complete ET region, and bridge-mediated electronic coupling including residues of 3) path1 and 4) path2. The different setups are applied by including the corresponding residues in the QM region of the mixed QM/MM calculations. The rmsVDA values are given in Table 1 while the single VDA values for each snapshot can be found in the supporting information.
Table 1. Average distances d in Å, Electronic coupling rmsV in eV, ΔG° in eV, λ in eV and in s−1 calculated for HT between donor and acceptor (DA), donor and bridge (DB), and bridge and acceptor (BA), respectively.
| HT | Step | d | rmsV (direct) | rmsV (full) | rmsV (path1) | rmsV (path2) | ΔG° | λ | kET |
| 1-step | DA | 27.2 | 5.81×10−9 (0.68) | 3.04×10−6 (0.85) | 3.71×10−6 (0.28) | 3.99×10−7 (0.45) | −0.92 | 1.38 (0.85) | 8.99×104 |
| 2-step | DB | 21.2 | 1.35×10−7 (0.82) | 2.23×10−5 (0.66) | 1.81×10−5 (0.72) | −0.55 | 0.88 (0.74) | 5.47×106 | |
| BA | 7.1 | 1.47×10−2 (0.81) | −0.37 | 0.19 (0.73) | 1.78×1012 |
The electronic coupling is calculated applying QM setups direct, full, path1 and path2. kET is calculated by Marcus theory applying the respective highest electronic coupling of the system. Fluctuations are depicted through the coherence factor 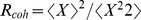 given in parentheses.
given in parentheses.
Clearly, the possible electron transfer between hemeCcP and hemeCytc is of bridge-mediated nature. Due to the large distance of about 27 Å between donor and acceptor, the direct electronic coupling is very weak with VDA(direct)∼10−9 eV. In contrast, the bridge-mediated electronic coupling lies within the range of 10−7 to 10−5 eV, indicating that the intermediary protein scaffold enhances the ET transfer between donor and acceptor. In detail, the electronic coupling based on the full and path1 transfer region are equally high, while the electronic coupling on path2 is one order of magnitude lower, with rmsVDA(path1) = 3.04×10−6 eV, rmsVDA(full) = 3.71×10−6 eV and rmsVDA(path1) = 3.99×10−7 eV, respectively. The results also show that the electronic coupling values fluctuate only moderately due to thermal movements of the protein, indicating that the single-step HT is not conformational gated.
In order to obtain the ET rate from Cytc to CcP by Marcus theory, the energies ΔG° and λ have to be determined. From QM calculation of separate species CpdI, CpdII, CytcOx and CytcRed in gas phase as well as implicit solvent, we obtained very similar values of ΔE = (E(CpdII)+E(CytcOx))−(E(CpdI)+E(CytcRed)) of −0.93 and −0.95 eV, respectively. In addition, we performed energy minimization of the initial and final states of the whole protein complex by QM/MM methodology, resulting into ΔE = −0.92 eV. These energies are in excellent agreement with the experimental value of ΔG° = −0.9±0.15 eV found for the reaction of CcPhorse with Cytc [55]. This result indicates that the entropic effects play only a minor role in this process, why we substitute ΔE by ΔG° for the rest of this study (see Methods section). We estimated the reorganization energy λ = 1.38 eV through the vertical energy difference between the different electronic states by applying the method described by Blumberger [56]. This value is comparable to λ = 1.5 eV experimentally determined by Conklin et al. on the CcP/Cytc system [55], and also lies within the range of λ = 1.18–1.80 eV computationally predicted for the intra-protein ET system Ru(bpy)2(im)His33 cyt c [56] and λ = 1.2 eV measured on Ru((NH3)4)LHis33-Zn-cyt c [57]. Using Marcus theory on the ET parameters obtained, the rate constant of the single-step ET between CcP and Cytc is calculated to be 8.99×104 s−1.
Two-step HT
The QM/MM e-pathway analysis indicated the importance of Trp191. Furthermore, we identified redox states localized on Trp191 within the energy window of the heme orbitals during the electronic coupling calculation of the single-step HT. It is also experimentally proven that Trp191 is able to localize a transient hole, forming a positive radical [47], [48], [58]. Thus, we split the ET process into two separate steps: donor-bridge (DB, Cytc to Trp191) and bridge-acceptor (BA, Trp191 to CcP) and calculated the electronic coupling for each of them.
The main step of the two-step process is HT from hemeCytc to Trp191. Its electronic coupling through residues of path1 is essentially the same as the full and significantly higher than the direct electronic coupling, thus indicating a bridge-mediated HT along path1, with rmsVDB(full) = 2.23×10−5 eV. The electronic coupling of the other step, Trp191 to hemeCcP, is very strong with rmsVBA(direct) = 1.47×10−2 eV, as Trp191 is in direct vicinity to hemeCcP. The rate of the total reaction is limited by the slowest step. Using QM/MM calculations as explained above, we estimated ΔG° = −0.55 eV and λ = 0.88 eV for the HT between Cytc and Trp191, resulting in kET = 5.47×106 s−1. Reduction of hemeCcP by Trp191 is very fast with ΔG° = −0.37 eV and λ = 0.19 eV, resulting in kET = 1.78×1012 s−1. Consequently, the rate-limiting step is HT between Cytc and Trp191 of CcP.
ET mechanism
Our results are in agreement with experiments in both the ET mechanism as well as the ET rate constant. From the literature it is known that CpdI oxidizes Trp191 generating the radical intermediated Trp191+, which then is reduced by Cytc [43]–[48]. Comparing our computed ET rates for the single- and two-step HT processes, it is clear that the electron hole is transferred between Cytc and CcP by sequential hopping with the intermediate state being Trp191+. Interestingly, when comparing the couplings and rates for the two-step and one-step processes, we observe a ∼60× larger rate constant for the two-step mechanism despite of only ∼6× larger electronic coupling. The reason for this is the relation between ΔG°, λ and kET depicted in the Marcus equation, with kET being maximal when λ = −ΔG°. The direct HT from Cytc to CcP has λ - (−ΔG°) = 0.46 eV, whereas the rate-limiting HT between Trp191 and Cytc of the two-step process has λ - (−ΔG°) = 0.33 eV only, thus enabling higher rate constant at similar electronic coupling. These findings indicate the fine evolution of the enzyme to approach an increased turnover rate for the ET between CcP and Cytc through establishment of the localizable bridge state Trp191+.
We computed the rate of the ground state ET transfer between Cytc and CcP to be 5.47×106 s−1. This result is in agreement with experimental ET rates to be greater than 2000 s−1, measured using stopped-flow methods on the ground state ET [38]. Although our results furthermore agree with ET rates measured to be greater than 5.0×106 s−1 by flash photolysis on photo-exited ruthenium-modified Cytc [40], these values should not be compared directly due to the different kind of ET states involved.
As indicated by the coherent numbers in Table 1 (or by the individual values for the different snapshots in the supporting information), the coupling and reorganization energies for the different snapshots are quite consistent. Taking into account the computed rate constant, in good agreement with the experimental values, the low fluctuations in VDA and λ, together with the small conformational changes along the MD, indicates a nongated ET mechanism. In terms of the reorganization energy, we are aware of the simplicity of our model and the importance of including protein electronic polarization into the calculations. Nevertheless, custom fit MD templates for both states RO and OR based on QM/MM optimization of the hemes plus ligated residues, as well as the rigidity of the interface and the fact that donor and acceptor are completely buried into protein and not accessible to solvent, lets us safely estimate correct reorganization energies as well as ΔG° through ΔE, as done in this study. However, the application of stated methodology to other ET systems might make specific adaptions necessary, such as more sampling or polarizable force fields to estimate λ and ΔG°.
In summary, we have performed a comprehensive mechanistic case study on the ET process in the CcP/Cytc complex. It shows that the maturity of computational techniques and a novel protocol combining conformational sampling, ET pathway mapping and calculation of the key ET parameters (ΔG°, V and λ), allows a detailed description of the underlying ET mechanism including the estimation of absolute rates of all relevant ET steps for this well-known system. Currently, there are efforts under way testing the approach in further systems in order to validate its general applicability. For the CcP/Cytc complex, our approach has confirmed a two-step nongated mechanism with residues Ala194, Ala193, Gly192 and Trp191 of CcP acting as the main ET pathway, with Trp191 serving as the transient hole acceptor at the first stage of the ET process. Most importantly, in the rate-limiting step of the two-step HT process, λ is found to be better matching -ΔG° than in the single-step HT, resulting in a much higher rate despite similar electronic coupling. This finding provides a select example of enzyme evolution by means of breaking down slow processes into several faster ones, thereby gaining total turnover rate.
Materials and Methods
All QM/MM calculations were performed with Schrodinger's QSite program [59]. The spin-unrestricted DFT method with the M06 functional [60] was applied for the QM/MM geometry optimization as well as for the analysis of ET mechanisms by using the QM/MM e-Pathway approach (see supporting information and references) [49], [50]. All setups included the 6–31G* basis set for main-group elements and the lacvp pseudo-potential for Fe. The DFT methodology does not include the dispersion corrections, which could slightly affect the geometries, However these corrections do not change the electron interaction in the system and therefore have no direct impact on the electronic coupling.
For our calculations, we chose the structure of the native CcP/Cytc complex derived by Pelletier and Kraut [34]. Protocols of the protein preparation, MD simulation and QM/MM energy minimization are given in the supporting information. In brief, the data of QM/MM calculations on both hemes in their reduced states were used to construct the diabatic states and derive the electronic coupling (see below). The resulting conformation is referred to as the crystal complex of CpdII/CytcRED since the proteins did not undergo conformational changes throughout the equilibration process. We performed 30 ns of molecular dynamics (MD) on the complex, applying GROMACS [61] together with the OPLS force field. Snapshots were extracted every 1 ps within the first 2 ns and furthermore at time steps 10 ns, 20 ns and 30 ns. Based on the RMSD of the ET region, we clustered all 2000 snapshots from the 2 ns trajectory by using the k-medoids algorithm [62] and chose the 10 resulting clustering modes as representatives. In particular, the selected conformations were taken at time steps 0, 162, 432, 990, 1083, 1356, 1527, 1764, 1814 and 1884. Together with the crystal complex and snapshots at 10, 20 and 30 ns, our study included 14 conformations.
ET pathways are identified through the recently developed QM/MM e-Pathway approach [49], [50], where the ET region between the donor and acceptor is described by QM and the remainder of the protein by MM level of theory. For details we refer to the supporting information. In brief, the iterative procedure starts by finding the first acceptor of the hole through localization of the spin density in the ET region, given by a single point calculation of the system having one electron missing and thus a doublet spin state. In the next iteration, the previously identified residue is excluded from the QM region, turning it into a classical residue. Therefore, an electronic description of this residue is no longer possible and the electron hole needs to find its next host. A set of residues is designated as an ET pathway, once a connecting chain of residues between the donor and acceptor is found through the stepwise application of the approach.
The electronic coupling values were calculated by the FCD method [24], [63]–[65] in combination with Koopmans' theorem [66]. Here, the properties of the adiabatic states get approximated through one-electron energies and the highest occupied molecular orbitals localized on the donor and acceptor sites in the corresponding neutral system. It is known, that DFT calculations of open-shell systems leads to artificial delocalization of the unpaired electron because of incomplete cancellation of the electron self-interaction [67], [68]. It means that excess charge delocalization in radical cations and radical anions computed by DFT may be considerably overestimated. It was shown, however, for different functionals that the excess charge distribution is well described by Kohn-Sham orbitals of neutral dimers [69]. An alternative promising approach is the construction of the diabatic Hamiltonian using charge-localized broken-symmetry states [70]. The performance of this scheme is still not well established. In particular, it was found that calculated values of electronic couplings may be extremely sensitive to the choice of the functional [71]. Another important point is the performance of the two-state model based on a unitary transformation of adiabatic states to diabatic states. A general consideration has been revealed that the coupling derived with the two-state FCD scheme accounts properly for both the direct and bridge mediated superexchange interaction of the donor and acceptor in donor-bridge-acceptor systems [72].
All rmsV values were computed following reference [73], applying 2 (nearly) degenerate states for the donor (ND) and acceptor (NA) site, respectively, and 1 for the bridge site. We note that ET from any initial state of the donor can occur to any of the NA final states of the acceptor (NA parallel ET processes); thus the rate computed with rmsV must be multiplied by NA. Alternatively, the effective electronic coupling can be defined as 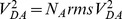 . We refer to direct coupling when the QM region consists solely of the donor and acceptor sites, and to bridge mediated coupling when the QM region also includes several specified bridging residues.
. We refer to direct coupling when the QM region consists solely of the donor and acceptor sites, and to bridge mediated coupling when the QM region also includes several specified bridging residues.
The ET rate was calculated using Marcus expression [74], where we estimated the driving force ΔG° as the energy change ΔE = E products - E reactants. QM geometry optimization of separated species CpdI, CpdII, CytcOx and CytcRed was performed in the gas phase as well as in the implicit solvent with the dielectric constant of 4.0 (to simulate the protein environment). In addition, we also computed the optimal geometry of the CpdI - CytcRed (reactant) and CpdII - CytcOx (product) state using the QM/MM approach. The neglected entropy contribution of ΔG° appears to be small; we did not find significant conformational changes accompanying the ET reaction. In order to estimate the reorganization energy λ, we ran 4 ns of MD on both electronic states RO and OR of each donor-acceptor pair DA, DB and BA, respectively. We calculated the vertical energy difference ΔEET = ERO – EOR for 5 randomly chosen snapshots from the MD simulation and estimated λ using
| (2) |
where the brackets indicate averages over snapshots of MD either for the RO or the OR states, as specified by the subscripts [56].
Supporting Information
Supporting information on preparation of the CcP/Cytc complex, MD simulation on the CcP/Cytc complex, QM/MM e-Pathway and ET parameters.
(DOCX)
Acknowledgments
Fruitful discussions with Harry B. Gray are thankfully acknowledged.
Funding Statement
Computational resources were provided by the Barcelona Supercomputing Center. Work was supported by startup funds from the Barcelona Supercomputer Center, by the Spanish Ministry of Education and Science to VG through the project CTQ2010-18123 as well as to AAV through the project CTQ2009-12346. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Beratan DN, Onuchic JN, Winkler JR, Gray HB (1992) Electron-Tunneling Pathways In Proteins. Science 258: 1740–1741. [DOI] [PubMed] [Google Scholar]
- 2. Langen R, Chang IJ, Germanas JP, Richards JH, Winkler JR, et al. (1995) Electron-Tunneling In Proteins - Coupling Through A Beta-Strand. Science 268: 1733–1735. [DOI] [PubMed] [Google Scholar]
- 3.Balzani V, Piotrowiak P, Rodgers MAJ, Mattay J, Astruc D, et al. (2001) Electron Transfer in Chemistry. Weinheim, Germany: Wiley-VCH.
- 4.Jortner J, Ratner MA (1997) Molecular Electronics. Oxford: Blackwell Scientific.
- 5.May V, Kühn O (2000) Charge and Energy Transfer Dynamics in Molecular Systems. Berlin: Wiley-VCH.
- 6.Jortner J, Bixon M (1999) Electron Transfer: From Isolated Molecules to Biomolecules. Advances in Chemical Physics. New York: Wiley.
- 7. Gray HB, Winkler JR (2003) Electron tunneling through proteins. Quarterly reviews of biophysics 36: 341–372. [DOI] [PubMed] [Google Scholar]
- 8. Berlin YA, Ratner MA (2005) Intra-molecular electron transfer and electric conductance via sequential hopping: Unified theoretical description. Radiation Physics and Chemistry 74: 124–131. [Google Scholar]
- 9. Nocek JM, Zhou JS, De Forest S, Priyadarshy S, Beratan DN, et al. (1996) Theory and Practice of Electron Transfer within Protein–Protein Complexes: Application to the Multidomain Binding of Cytochrome c by Cytochrome c Peroxidase. Chemical Reviews 96: 2459–2490. [DOI] [PubMed] [Google Scholar]
- 10. Ceccarelli M, Marchi M (2003) Simulation and Modeling of the Rhodobacter sphaeroides Bacterial Reaction Center II: Primary Charge Separation. Journal of Physical Chemistry B 107: 5630–5641. [Google Scholar]
- 11. Morozova OB, Hore PJ, Sagdeev RZ, Yurkovskaya AV (2005) Intramolecular Electron Transfer in Lysozyme Studied by Time-Resolved Chemically Induced Dynamic Nuclear Polarization. Journal of Physical Chemistry B 109: 21971–21978. [DOI] [PubMed] [Google Scholar]
- 12. Reece SY, Seyedsayamdost MR, Stubbe J, Nocera DG (2007) Direct Observation of a Transient Tyrosine Radical Competent for Initiating Turnover in a Photochemical Ribonucleotide Reductase. Journal of the American Chemical Society 129: 13828–13830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wittekindt C, Schwarz M, Friedrich T, Koslowski T (2009) Aromatic Amino Acids as Stepping Stones in Charge Transfer in Respiratory Complex I: An Unusual Mechanism Deduced from Atomistic Theory and Bioinformatics. Journal of the American Chemical Society 131: 8134–8140. [DOI] [PubMed] [Google Scholar]
- 14. Axup AW, Albin M, Mayo SL, Crutchley RJ, Gray HB (1988) Distance dependence of photoinduced long-range electron transfer in zinc/ruthenium-modified myoglobins. Journal of the American Chemical Society 110: 435–439. [Google Scholar]
- 15. Gehlen JN, Daizadeh I, Stuchebrukhov AA, Marcus RA (1996) Tunneling matrix element in Ru-modified blue copper proteins: pruning the protein in search of electron transfer pathways. Inorganica Chimica Acta 243: 271–282. [Google Scholar]
- 16. Kawatsu T, Kakitani T, Yamato T (2002) Destructive Interference in the Electron Tunneling through Protein Media. Journal of Physical Chemistry B 106: 11356–11366. [Google Scholar]
- 17. Prytkova TR, Kurnikov IV, Beratan DN (2005) Ab Initio Based Calculations of Electron-Transfer Rates in Metalloproteins. Journal of Physical Chemistry B 109: 1618–1625. [DOI] [PubMed] [Google Scholar]
- 18. Shih C, Museth AK, Abrahamsson M, Blanco-Rodriguez AM, Di Bilio AJ, et al. (2008) Tryptophan-Accelerated Electron Flow Through Proteins. Science 320: 1760–1762. [DOI] [PubMed] [Google Scholar]
- 19.Schuster GB (2004) Long-Range Charge Transfer in DNA (I and II): Topics in Current Chemistry. Berlin, New York: Springer-Verlag.
- 20. Arkin MR, Stemp EDA, Holmlin RE, Barton JK, Hörmann A, et al. (1996) Rates of DNA-Mediated Electron Transfer Between Metallointercalators. Science 273: 475–480. [DOI] [PubMed] [Google Scholar]
- 21. Lewis FD, Liu X, Miller SE, Wasielewski MR (1999) Electronic Interactions between π-Stacked DNA Base Pairs and Diphenylacetylene-4,4′-dicarboxamide in Hairpin DNA. Journal of the American Chemical Society 121: 9746–9747. [Google Scholar]
- 22. Lewis FD, Liu X, Liu J, Miller SE, Hayes RT, et al. (2000) Direct measurement of hole transport dynamics in DNA. Nature 406: 51–53. [DOI] [PubMed] [Google Scholar]
- 23. Lewis FD, Kalgutkar RS, Wu Y, Liu X, Liu J, et al. (2000) Driving Force Dependence of Electron Transfer Dynamics in Synthetic DNA Hairpins. Journal of the American Chemical Society 122: 12346–12351. [Google Scholar]
- 24.Rösch N, Voityuk AA (2004) Quantum Chemical Calculation of Donor–Acceptor Coupling for Charge Transfer in DNA. Long-Range Charge Transfer in DNA II. pp. 37–72.
- 25. Voityuk AA (2005) Electronic Couplings in DNA π-Stacks: Multistate Effects. Journal of Physical Chemistry B 109: 17917–17921. [DOI] [PubMed] [Google Scholar]
- 26. Zheng J, Kang YK, Therien MJ, Beratan DN (2005) Generalized Mulliken–Hush Analysis of Electronic Coupling Interactions in Compressed π-Stacked Porphyrin–Bridge–Quinone Systems. Journal of the American Chemical Society 127: 11303–11310. [DOI] [PubMed] [Google Scholar]
- 27. Blancafort L, Voityuk AA (2006) CASSCF/CAS-PT2 Study of Hole Transfer in Stacked DNA Nucleobases. Journal of Physical Chemistry A 110: 6426–6432. [DOI] [PubMed] [Google Scholar]
- 28. Félix M, Voityuk AA (2008) Parameters For Excess Electron Transfer in DNA. Estimation Using Unoccupied Kohn–Sham Orbitals and TD DFT. Journal of Physical Chemistry A 112: 9043–9049. [DOI] [PubMed] [Google Scholar]
- 29. Beratan DN, Onuchic JN, Betts JN, Bowler BE, Gray HB (1990) Electron tunneling pathways in ruthenated proteins. Journal of the American Chemical Society 112: 7915–7921. [Google Scholar]
- 30. Keinan S, Venkatramani R, Balaeff A, Beratan DN (2010) Is MD Geometry Sampling Sufficient for Nucleobase Electronic Structure Analysis of ET Reactions? Comparing Classical MD and QM/MM Methods. Journal of Physical Chemistry C 114: 20496–20502. [Google Scholar]
- 31. Siriwong K, Voityuk AA (2008) pi stack structure and hole transfer couplings in DNA hairpins and DNA. A combined QM/MD study. Journal of Physical Chemistry B 112: 8181–8187. [DOI] [PubMed] [Google Scholar]
- 32. Beratan DN, Skourtis SS, Balabin IA, Balaeff A, Keinan S, et al. (2009) Steering Electrons on Moving Pathways. Accounts of chemical research 42: 1669–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Balabin IA, Beratan DN, Skourtis SS (2008) Persistence of Structure Over Fluctuations in Biological Electron-Transfer Reactions. Physical Review Letters 101: 158102–158106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pelletier H, Kraut J (1992) Crystal structure of a complex between electron transfer partners, cytochrome c peroxidase and cytochrome c. Science 258: 1748–1755. [DOI] [PubMed] [Google Scholar]
- 35. Pappa HS, Tajbaksh S, Saunders AJ, Pielak GJ, Poulos TL (1996) Probing the Cytochrome c Peroxidase–Cytochrome c Electron Transfer Reaction Using Site Specific Cross-Linking. Biochemistry 35: 4837–4845. [DOI] [PubMed] [Google Scholar]
- 36. Rosenfeld RJ, Hays A-MA, Musah RA, Goodin DB (2002) Excision of a proposed electron transfer pathway in cytochrome c peroxidase and its replacement by a ligand-binding channel. Protein Science 11: 1251–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Erman JE, Vitello LB (2002) Yeast cytochrome c peroxidase: mechanistic studies via protein engineering. Biochimica Et Biophysica Acta (BBA) - Protein Structure and Molecular Enzymology 1597: 193–220. [DOI] [PubMed] [Google Scholar]
- 38. Guo M, Bhaskar B, Li H, Barrows TP, Poulos TL (2004) Crystal structure and characterization of a cytochrome c peroxidasecytochrome c sitespecific crosslink. Proceedings of the National Academy of Sciences of the United States of America 101: 5940–5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kang SA, Marjavaara PJ, Crane BR (2004) Electron transfer between cytochrome c and cytochome c peroxidase in single crystals. Journal Of The American Chemical Society 126: 10836–10837. [DOI] [PubMed] [Google Scholar]
- 40. Geren L, Hahm S, Durham B, Millett F (1991) Photoinduced electron transfer between cytochrome c peroxidase and yeast cytochrome c labeled at Cys 102 with (4-bromomethyl-4′-methylbipyridine)-[bis(bipyridine)]-ruthenium2+. Biochemistry 30: 9450–9457. [DOI] [PubMed] [Google Scholar]
- 41. Liu R-Q, Hahm S, Miller M, Durham B, Millett F (1995) Photooxidation of Trp-191 in cytochrome c peroxidase by ruthenium-cytochrome c derivatives. Biochemistry 34: 973–983. [DOI] [PubMed] [Google Scholar]
- 42. Hays Putnam A-MA, Lee Y-T, Goodin DB (2009) Replacement of an Electron Transfer Pathway in Cytochrome c Peroxidase with a Surrogate Peptide. Biochemistry 48: 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Musah RA, Goodin DB (1997) Introduction of Novel Substrate Oxidation into Cytochrome c Peroxidase by Cavity Complementation: Oxidation of 2-Aminothiazole and Covalent Modification of the Enzyme. Biochemistry 36: 11665–11674. [DOI] [PubMed] [Google Scholar]
- 44. Musah RA, Jensen GM, Rosenfeld RJ, McRee DE, Goodin DB, et al. (1997) Variation in Strength of an Unconventional C–H to O Hydrogen Bond in an Engineered Protein Cavity. Journal of the American Chemical Society 119: 9083–9084. [Google Scholar]
- 45. Sivaraja M, Goodin DB, Smith M, Hoffman BM (1989) Identification by ENDOR of Trp191 as the free-radical site in cytochrome c peroxidase compound ES. Science 245: 738–740. [DOI] [PubMed] [Google Scholar]
- 46. Miller MA, Vitello L, Erman JE (1995) Regulation of Interprotein Electron Transfer By Trp 191 of Cytochrome c Peroxidase. Biochemistry 34: 12048–12058. [DOI] [PubMed] [Google Scholar]
- 47. Barrows TP, Bhaskar B, Poulos TL (2004) Electrostatic Control of the Tryptophan Radical in Cytochrome c Peroxidase. Biochemistry 43: 8826–8834. [DOI] [PubMed] [Google Scholar]
- 48. Seifert JL, Pfister TD, Nocek JM, Lu Y, Hoffman BM (2005) Hopping in the Electron-Transfer Photocycle of the 1∶1 Complex of Zn–Cytochrome c Peroxidase with Cytochrome c. Journal of the American Chemical Society 127: 5750–5751. [DOI] [PubMed] [Google Scholar]
- 49. Guallar V, Wallrapp F (2008) Mapping protein electron transfer pathways with QM/MM methods. Journal of the Royal Society, Interface 5: 233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wallrapp F, Masone D, Guallar V (2008) Electron Transfer in the P450cam/PDX Complex. The QM/MM e-Pathway. Journal of Physical Chemistry A 112: 12989–12994. [DOI] [PubMed] [Google Scholar]
- 51. Harvey MJ, Giupponi G, De Fabritiis G (2009) ACEMD: Accelerating Biomolecular Dynamics in the Microsecond Time Scale. Journal of Chemical Theory and Computation 5: 1632–1639. [DOI] [PubMed] [Google Scholar]
- 52. Klepeis JL, Lindorff-Larsen K, Dror RO, Shaw DE (2009) Long-timescale molecular dynamics simulations of protein structure and function. Current Opinion in Structural Biology 19: 120–127. [DOI] [PubMed] [Google Scholar]
- 53. Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, et al. (2010) Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 78: 1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Siddarth P (1994) An artificial intelligence search for key residues in protein electron transfer systems. Journal of photochemistry and photobiology A: Chemistry 82: 117–121. [Google Scholar]
- 55. Conklin KT, McLendon G (1988) Free energy effects on biological electron transfer: reactions of iron(IV) cytochrome c peroxidase (ES) with metallocytochromes c. Journal of the American Chemical Society 110: 3345–3350. [Google Scholar]
- 56. Blumberger J (2008) Free energies for biological electron transfer from QM/MM calculation: method, application and critical assessment. Physical chemistry chemical physics : PCCP 10: 5651–5667. [DOI] [PubMed] [Google Scholar]
- 57. Meade TJ, Gray HB, Winkler JR (1989) Driving-force effects on the rate of long-range electron transfer in ruthenium-modified cytochrome c. Journal Of The American Chemical Society 111: 4353–4356. [Google Scholar]
- 58. Mauro JM, Fishel LA, Hazzard JT, Meyer TE, Tollin G, et al. (1988) Tryptophan-191. fwdarw. phenylalanine, a proximal-side mutation in yeast cytochrome c peroxidase that strongly affects the kinetics of ferrocytochrome c oxidation. Biochemistry 27: 6243–6256. [DOI] [PubMed] [Google Scholar]
- 59.Columbia University. (2010) QSite. 5.6 ed. New York, NY: Schrödinger, LLC.
- 60. Truhlar DG, Zhao (2008) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other function. Theoretical Chemistry Accounts 120: 215–241. [Google Scholar]
- 61. Hess B, Kutzner C, van der Spoel D, Lindahl E (2008) GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. Journal of Chemical Theory and Computation 4: 435–447. [DOI] [PubMed] [Google Scholar]
- 62.Theodoridis S, Koutroumbas K (2006) Pattern Recognition. 3rd edition. Orlando, FL, USA: Academic Press, Inc.
- 63. Voityuk AA, Rösch N (2002) Fragment charge difference method for estimating donor-acceptor electronic coupling: Application to DNA pi-stacks. Journal of Chemical Physics 117: 5607–5616. [Google Scholar]
- 64. Wallrapp F, Voityuk A, Guallar V (2009) Solvent Effects on Donor–Acceptor Couplings in Peptides. A Combined QM and MD Study. Journal of Chemical Theory and Computation 5: 3312–3320. [DOI] [PubMed] [Google Scholar]
- 65. Wallrapp F, Voityuk A, Guallar V (2010) Temperature Effects on Donor–Acceptor Couplings in Peptides. A Combined QM and MD Study. Journal of Chemical Theory and Computation 6: 3241–3248. [DOI] [PubMed] [Google Scholar]
- 66. Newton MD (1991) Quantum chemical probes of electron-transfer kinetics: the nature of donor-acceptor interactions. Chemical Reviews 91: 767–792. [Google Scholar]
- 67. Cohen AJ, Mori-Sanchez P, Yang WT (2008) Insights into current limitations of density functional theory. Science 321: 792–794. [DOI] [PubMed] [Google Scholar]
- 68. Dreuw A, Head-Gordon M (2005) Single-reference ab initio methods for the calculation of excited states of large molecules. Chemical reviews 105: 4009–4037. [DOI] [PubMed] [Google Scholar]
- 69. Felix M, Voityuk AA (2011) DFT Performance for the Hole Transfer Parameters in DNA pi Stacks. International journal of quantum chemistry 111: 191–201. [Google Scholar]
- 70. Kaduk B, Kowalczyk T, Van Voorhis T (2012) Constrained Density Functional Theory. Chemical reviews 112: 321–370. [DOI] [PubMed] [Google Scholar]
- 71. Pavanello M, Neugebauer J (2011) Modelling charge transfer reactions with the frozen density embedding formalism. Journal of Chemical Physics 135: 234103. [DOI] [PubMed] [Google Scholar]
- 72. Voityuk AA (2012) Electronic coupling for charge transfer in donor-bridge-acceptor systems. Performance of the two-state FCD model. Physical Chemistry Chemical Physics 14: 13789–13793. [DOI] [PubMed] [Google Scholar]
- 73. Voityuk AA (2010) Electron transfer between [4Fe-4S] clusters. Chemical Physics Letters 495: 131–134. [Google Scholar]
- 74. Marcus RA, Sutin N (1985) Electron transfers in chemistry and biology. Biochimica Et Biophysica Acta 811: 265–322. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information on preparation of the CcP/Cytc complex, MD simulation on the CcP/Cytc complex, QM/MM e-Pathway and ET parameters.
(DOCX)