

Abstract
Background
Ventricular tachycardia (VT) is the second most common cause of death in patients with Duchenne muscular dystrophy (DMD). Recent studies have implicated enhanced sarcoplasmic reticulum (SR) Ca2+ leak via ryanodine receptors (RyR2) as a cause of VT in the mdx mouse model of DMD. However, the signaling mechanisms underlying induction of SR Ca2+ leak and VT are poorly understood.
Objective
To test whether enhanced CaMKII phosphorylation of RyR2 underlies SR Ca2+ leak and induction of VT in mdx mice.
Methods
Programmed electrical stimulation (PES) was performed on anesthetized mice, and confocal imaging of calcium release events in isolated ventricular myocytes.
Results
PES revealed inducible VT in mdx mice, which was inhibited by CaMKII inhibition or mutation S2814A in RyR2. Myocytes from mdx mice exhibited more Ca2+ sparks and Ca2+ waves compared with wild type (WT) mice, in particular at faster pacing rates. Arrhythmogenic Ca2+ waves were inhibited by CaMKII but not PKA inhibition. Moreover, mutation S2814A but not S2808A in RyR2 suppressed spontaneous Ca2+ waves in myocytes from mdx mice.
Conclusion
CaMKII blockade and genetic inhibition of RyR2-S2814 phosphorylation prevent VT induction in a mouse model of DMD. In ventricular myocytes from mdx mice, spontaneous Ca2+ sparks and Ca2+ waves can be suppressed by CaMKII inhibition or mutation S2814A in RyR2. Thus, inhibition of CaMKII-induced SR Ca2+ leak might be a new strategy to prevent arrhythmias in patients with DMD without heart failure.
Keywords: Cardiac arrhythmias, Ca2+/calmodulin kinase II, mouse model, ryanodine receptor, Duchenne muscular dystrophy, ventricular tachycardia
INTRODUCTION
Duchenne muscular dystrophy (DMD) is the most common type of muscular dystrophy with an incidence of 1 in 3,500 male births.1 Although the most common cause of death is respiratory failure, 90% of the patients manifest evidence of cardiac disease at the time of death.1 Overall, a quarter of DMD patients die from cardiac causes, half of which are due to lethal ventricular tachyarrhythmias (VT). At present, there are no effective treatments to prevent these lethal ventricular arrhythmias due to a lack of understanding of the underlying mechanisms.
In DMD, the absence of dystrophin cause abnormal stress-induced entry of Ca2+ into the cells in turn leading to diastolic sarcoplasmic reticulum (SR) Ca2+ release events.2, 3 Ryanodine receptor type 2 channels (RyR2) are intracellular Ca2+ release channels on the SR membrane responsible for Ca2+ release associated with excitation-contraction coupling. We and other laboratories have recently provided evidence of defective RyR2 function in the mdx mouse, a mouse model of DMD.2–4 Leakage of Ca2+ from the SR due to defective RyR2 regulation may lead to depletion of SR Ca2+ stores and reduced systolic SR Ca2+ release associated with contractile impairments in mdx mice. Moreover, diastolic SR Ca2+ release via RyR2 may promote arrhythmias in mdx mice, but the exact molecular mechanisms underlying RyR2 dysfunction remain incompletely understood.4
The open probability of RyR2 can be modulated by binding of accessory subunits (e.g., calmodulin, calsequestrin, FKBP12.6) and post-translational modifications (e.g., phosphorylation, nitrosylation, oxidation).5, 6 It has been demonstrated that RyR2 activity is modulated by phosphorylation of at least two residues, namely S2808, primarily by protein kinase A (PKA),7, 8 and S2814, primarily by Ca2+/calmodulin-dependent protein kinase II (CaMKII).9 Whereas PKA is activated by beta-adrenergic stimulation, CaMKII can be activated via the beta-adrenergic pathway, high [Ca2+]i level and oxidative stress.10, 11 Therefore, it is possible that elevated diastolic Ca2+ level in cardiomyocytes from mdx mice activate or potentiate activation of CaMKII.12, 13 Moreover, elevated levels of oxidative stress in hearts of mdx mice might promote CaMKII activation.14, 15 An alternative hypothesis is that CaMKII becomes activated due to increases in heart rates or elevations of beta-adrenergic levels.9 Recently, we demonstrated that constitutive CaMKII hyperphosphorylation of RyR2 promotes diastolic SR Ca2+ leak and induction of VT in mice with pressure overload-induced heart failure.16 In this paper, we examined whether CaMKII activation and downstream phosphorylation of RyR2, and the ensuing diastolic SR Ca2+ leak, are determinants of ventricular arrhythmogenesis in the mdx mouse.
METHODS
Animals
Animal studies were performed according to protocols approved by the Institutional Animal Care and Use Committee conforming to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH Publication No. 85–23, revised 1996).
Programmed electrical stimulation
Atrial and ventricular intracardiac ECG were recorded using a 1.1 F octapolar electrode catheter (EPR-800, Millar Instruments, Houston, Texas) inserted into the right ventricle via the right jugular vein, as described previously.17
Ventricular myocyte Ca2+ imaging
Mouse ventricular myocytes were isolated and loaded with Ca2+ sensitive dye Fluo-4 AM as described previously.16, 18 After pacing at 1-Hz (10 V) for 2 minutes, only rod-shaped myocytes showing clear striation and normal contractility were selected for further experiments. Once steady-state Ca2+ transient was observed after 20 second of pacing at either 1-Hz or 3-Hz, pacing was stopped for 60 seconds and spontaneous Ca2+ release events and Ca2+ sparks were counted. Steady state SR Ca2+ content was estimated by rapid application of 10 mM caffeine after pacing.
Western blot analyses
Heart lysates were prepared from flash-frozen mouse hearts and Western blot analyses were performed as described previously.19
RESULTS
Inhibition of CaMKII prevents ventricular tachycardia in mdx mice
Prior studies have demonstrated an increased propensity towards cardiac arrhythmias in mdx mice, a commonly used small animal model of DMD.4, 20 To determine the mechanisms underlying ventricular arrhythmogenesis in mdx mice, we performed programmed electrical stimulation (PES) in anesthetized mice. Under baseline (non-paced) conditions, there were no significant differences in cardiac conduction and repolarization parameters at 4 months of age comparing WT and mdx mice (Table S1). Intracardiac pacing revealed similar right ventricular effective refractory periods (Table S1). Moreover, none of the mice exhibited spontaneous ectopic beats or episodes of ventricular arrhythmia during baseline recordings.
Next, PES was performed to assess VT inducibility. Whereas none of the WT mice exhibited sustained VT (0 of 8), 50% of the mdx mice developed an episode of VT following PES (7 of 14, P<0.05 vs WT, Fig. 1). In recent studies, we demonstrated a role for CaMKII in determining the susceptibility to both atrial and ventricular arrhythmias in mice.8, 16 Following administration of CaMKII inhibitor KN93, VT inducibility in mdx mice was significantly decreased (2 of 14, P < 0.05 vs mdx). These data were confirmed in mdx mice crossed with transgenic mice that overexpress CaMKII-inhibitory peptide AC3I in the heart.21 Genetic, cardiac-restricted inhibition of CaMKII also suppressed VT induction in mdx mice (0 of 12 developed VT, P <0.05 vs mdx). To determine whether CaMKII phosphorylation of RyR2 was involved in VT generation, mdx mice were crossed to RyR2-S2814A mice in which CaMKII phosphorylation site S2814 has been inactivated. PES revealed that mdx:S2814A mice were also protected from VT induction (1 of 10 positive, P < 0.05 vs mdx). Finally, S2814A mice did not exhibit any arrhythmias (0 of 7, not shown). The duration of ventricular tachycardia following PES was also quantified, since this represents a good index of arrhythmia propensity. Figure 1C reveals that the average duration of VT in mdx mice was 0.8 (IQR: 0 to 3.2) seconds, significantly longer than in WT mice (0, 0 to 0 seconds; P<0.05 compared with mdx). Moreover, the duration of VT episodes was significantly shorter in mice treated with KN93, mdx:AC3I, or mdx:S2814A mutant mice (Fig. 1C). Thus, the PES studies revealed that CaMKII and, specifically, CaMKII phosphorylation site S2814 on RyR2 are involved in arrhythmogenesis in mdx mice. Thus, the PES studies revealed that CaMKII and, specifically, CaMKII phosphorylation site S2814 on RyR2 are involved in arrhythmogenesis in mdx mice.
Figure 1. Genetic inhibition of CaMKII phosphorylation of RyR2 prevents induction of ventricular tachycardia (VT) in mdx mice.

(A) Representative surface ECG tracings revealing ventricular tachycardia (VT) in an mdx mouse following intracardiac pacing (indicated by arrows). (B) Bar graph showing quantification of the incidence of pacing-induced sustained VT in WT and mdx mice. (C) Boxplot showing quantification of the duration of pacing induced sustained VT in WT and mdx mice. Pharmacologic (KN93) or genetic inhibition of CaMKII (AC3I peptide), as well as mutation S2814A on RyR2, prevented VT induction in mdx mice (B, C). *p<0.05 vs. WT. #p<0.05 vs. mdx.
Rapid pacing activates CaMKII and S2814 phosphorylation on RyR2
In prior studies, we demonstrated that PES activates CaMKII and leads to increased S2814 phosphorylation on RyR2 in mouse atria.8 To determine whether PES also activates CaMKII in the ventricle, heart samples were flash frozen immediately after rapid ventricular pacing. Western blot analysis revealed enhanced CaMKII autophosphorylation at T287 (Fig. 2A). The level of T287 autophosphorylation was increased similarly in WT and mdx mice (Fig. 2B). To assess the effects of activated CaMKII on RyR2 as a downstream target, S2814 phosphorylation was determined using a phospho-epitope specific antibody. S2814 phosphorylation was increased similarly in WT and mdx mice following rapid pacing (Fig. 2C, D). By contrast, phosphorylation of another major site (S2808) was unaltered following pacing (Fig. 2E, F). These data in conjunction with the PES data suggest that rapid pacing-induced activation of CaMKII associated with S2814 phosphorylation on RyR2 promotes VT induction in mdx mice.
Figure 2. Pacing-induced CaMKII activation and increased S2814 phosphorylation on RyR2 in WT and mdx mice.

Representative Western blots and bar graphs with quantification of averaged data showing (A, B) Increased CaMKII autophosphorylation levels at T287 following intracardiac pacing in both WT and mdx mice. (C, D) Increased RyR2 phosphorylation levels at S2814 following intracardiac pacing in both WT and mdx mice. (E, F) Unaltered RyR2 phosphorylation levels at S2808 following pacing. Each bar represents averaged data from 4–5 mice. * p<0.05.
Mutations S2814A and S2808A in RyR2 reduce Ca2+ spark frequency in mdx mice at 1-Hz
To compare the effects of RyR2 phosphorylation sites S2814 and S2808 on Ca2+ handling in mdx mice, we crossed mdx mice with RyR2-S2814A (see above) and RyR2-S2808A knockin mice.3, 22 Western blot analysis revealed that expression levels of RyR2 and CaMKII were similar in WT, mdx, mdx:S2814A, and mdx:S2808A mice (Fig. S1). Moreover, at basal heart rates RyR2 and CaMKII phosphorylation levels were similar in all 4 genotypes (Fig. S1). As male mdx mice have been shown to develop dilated cardiomyopathy starting at around 9 months of age,3 we conducted all experiments around 4 months of age to exclude potential confounding effects of altered contractile function. Echocardiography revealed similar ejection fractions in all 4 genotypes of mice (Table S2).
Ventricular myocytes were isolated from WT, mdx, mdx:S2814A, and mdx:S2808A mice. First, the Ca2+ spark frequency (CaSpF) was measured immediately following a 20-s pacing train at 1-Hz (Fig. 3A). The CaSpF was significantly elevated in myocytes from mdx mice (3.7 ± 0.9 sparks/1003m/s) compared with WT mice (1.2 ± 0.5; P<0.01, Fig. 3B). In contrast, compared with mdx mice, CaSpF was significantly decreased in myocytes from both mdx:S2814A (1.2 ± 0.3 sparks/1003m/s; P<0.01) and mdx:S2808A mice (1.9 ± 0.4 sparks/1003m/s; P<0.05), suggesting that inhibition of phosphorylation at either S2814 or S2808 site prevents diastolic Ca2+ release in mdx mice at slow heart rates (Fig. 3B).
Figure 3. Mutations S2814A and S2808A in RyR2 reduce Ca2+ spark frequency (CaSpF) leak in mdx mice at 1-Hz.

(A) Representative line scans showing spontaneous Ca2+ sparks in ventricular myocytes from wildtype (WT), mdx, mdx:S2814A and mdx:S2808A cardiomyocytes following a 20-s 1 Hz pacing train. Bar graph showing (B) CaSpF and (C) frequency of spontaneous Ca2+ waves (SCaW), during the 1-minute pause. **p<0.01 vs. WT. #p<0.05, ##p<0.01 vs. mdx.
Coalescence of multiple Ca2+ sparks into spontaneous Ca2+ waves (SCaWs) can trigger cellular depolarizations associated with arrhythmias.23 Following a conditioning pacing train at 1-Hz, myocytes from all groups of mice exhibited a low incidence of SCaWs. Compared with WT cardiomyocytes, there was a trend towards a higher incidence of SCaWs in mdx cardiomyocytes (0.3 ± 0.3 and 0.9 ± 0.4 events/min, respectively; P=0.08, Fig. 3C). Similarly, although there seemed to be a reduction in SCaWs in mdx:S2814A and mdx:S2808A cardiomyocytes, it did not reach statistical significance (Fig. 3C).
Alterations in SR Ca2+ stores can affect diastolic Ca2+ leak.24 Specifically, it has been suggested that spontaneous Ca2+ release occurs due to ‘store overload-induced Ca2+ release’ (SOICR).24 Therefore, we determined the loading of SR Ca2+ stores in mdx mice to identify the potential contribution of SOICR. Cardiomyocytes from mdx mice had significantly lower SR Ca2+ stores in comparison with cardiomyocytes from WT mice, excluding a pro-arrhythmic role of SOICR in mdx cardiomyocytes (WT: 4.4 ± 0.4 vs mdx: 2.6 ± 0.3 F/F0, P<0.01, Fig. 4A, B). Similarly, the reduction in CaSpF was accompanied by an increase in SR Ca2+ stores in mdx:S2814A (4.9 ± 0.5 F/F0) and mdx:S2808A (4.7 ± 0.3 F/F0), further confirming that the increased CaSpF in mdx mice was due to aberrant RyR2 activity and not SOICR (both P<0.001 vs. mdx; see Fig. 4).
Figure 4. Reduced SR Ca2+ stores in mdx ventricular myocytes at 1-Hz.
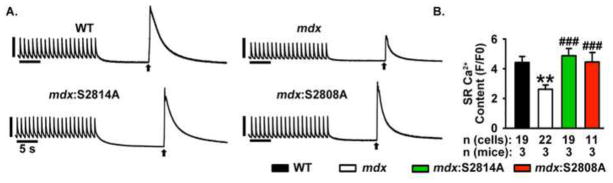
(A) Representative [Ca2+]i recordings from ventricular myocytes during 1-Hz pacing train, followed by caffeine-evoked SR Ca2+ dump, and (B) SR Ca2+ content, in ventricular myocytes from WT, mdx, and mdx:S2814A mice. Numbers in bar graphs represent number of cardiomyocytes obtained from 3–5 mice in each group. * p<0.01, ### P<0.001.
Mutation S2814A but not S2808A in RyR2 reduces Ca2+ sparks frequency in mdx mice at 3-Hz
Next, we studied Ca2+ sparks properties at a faster pacing rate to identify changes in Ca2+ handling that could explain the pacing-induced arrhythmia induction in vivo. CaSpF and detailed properties of Ca2+ sparks were assessed in ventricular myocytes from all 4 mice groups at 3-Hz pacing frequency (Fig. 5A–E). CaSpF was significantly higher in mdx (6.5 ± 1.0 sparks/100μm/s) compared with WT cardiomyocytes (3.2 ± 0.6; P<0.05, Fig. 5B). In contrast, CaSpF in mdx:S2814A cardiomyocytes was similar to WT cardiomyocytes (4.7 ± 0.8 sparks/100μm/s), but significantly higher in mdx:S2808A cardiomyocytes (6.9 ± 1.1 sparks/100μm/s, P<0.05 Fig. 5B). Moreover, cardiomyocytes from mdx mice had a larger Ca2+ spark mass as reflected by higher full width half maximum and longer full duration half maximum, in comparison with cardiomyocytes from WT mice (Fig. 5C–E). Thus, the S2814A mutation in RyR2 restored all Ca2+ spark properties to normal, whereas the S2808A mutation failed to do so (Fig. 5C–F). Moreover, there was a more pronounced enhancement in Ca2+ spark mass in mdx mice compared with WT myocytes at 3-Hz, compared with similar measurements made at 1-Hz (Fig. S2).
Figure 5. Mutation S2814A but not S2808A in RyR2 normalizes Ca2+ spark frequency (CaSpF) and mass in mdx mice at 3 Hz.

(A) Representative line scans showing spontaneous Ca2+ sparks in ventricular myocytes from wildtype (WT), mdx, mdx:S2814A and mdx:S2808A cardiomyocytes following a 20-s 3 Hz pacing train. Bar graph showing (B) CaSpF, (C) Spark amplitude, (D) Full diameter half maximum (FDHM), and (E) Full width half maximum (FWHM), during the 1-minute pause. *p<0.05, ***p<0.001 vs. WT. ##p<0.01, ###p<0.001 vs. mdx.
Inhibition of CaMKII and RyR2-S2814 phosphorylation suppress Ca2+ waves in mdx ventricular myocytes
Next, we examined the determinants of SCaWs in ventricular myocytes paced at 3-Hz. After pacing at 3-Hz for 20 seconds, ventricular myocytes from mdx mice exhibited a higher incidence of SCaWs in comparison with myocytes from WT mice (7.0 ± 0.4 vs. 1.0 ± 0.7 events/min; P<0.001, Fig. 6A, B). In contrast, cardiomyocytes from mdx:S2814A mice had a significantly lower incidence of SCaWs compared with cardiomyocytes from mdx mice (2.4 ± 0.7 events/min, Fig. 6B). Similar to the Ca2+ sparks data at 3-Hz, cardiomyocytes from mdx:S2808A mice had a significantly higher SCaWs frequency, in comparison with cardiomyocytes from WT mice (5.4 ± 1.2 events/min; P<0.05 vs. WT). These data suggest that only inhibition of S2814 but not S2808 can normalize Ca2+ spark properties and reduce SCaW propensity in mdx mice. We also measured the level of SR Ca2+ store loading at the 3-Hz pacing frequency (Fig. 6C). These data revealed that the elevated SCaW frequency in mdx mice led to reduced Ca2+ accumulation into the SR during diastole.
Figure 6. Mutation S2814A but not S2808A in RyR2 reduces spontaneous Ca2+ waves (SCaW) in mdx mice at 3 Hz.

(A) Representative line scan images and [Ca2+]i tracings showing spontaneous Ca2+ waves in ventricular myocytes from mdx mice following a 20-s 3-Hz pacing train. Scale bars: 2 F/F0 (vertical) and 5 seconds (horizontal). (B) Bar graph showing incidence of SCaWs following pacing train. (C) Bar graph showing SR Ca2+ load at 3 Hz. (D) Line graph showing the increase in SCaW frequency when pacing rate was increased from 1-Hz to 3-Hz. The proportional increase in SCaWs was greater in mdx and mdx:S2808A mice, compared to WT and mdx:S2814A mice. *p<0.05, **p<0.01, ***p<0.001 vs. WT. ###p<0.001 vs. mdx.
We next plotted the increase in SCaW frequency that occurred when the pacing rate was accelerated from 1-Hz to 3-Hz (Fig. 6D). This graph reveals that there was a modest increase in SCaW in ventricular myocytes from WT mice. In contrast, myocytes from mdx mice exhibited a much greater rate-dependent increase in SCaW numbers. This relative increase appeared to be blunted in mdx:S2814, but not mdx:S2808A mice. These findings are consistent with the pacing-induced ventricular tachycardias observed in mdx mice following PES, and the absence of VTs in mdx:S2814A mice (see Fig. 1).
We have demonstrated above that inhibition of CaMKII phosphorylation of RyR2 can prevent ventricular arrhythmias at least in the mdx mouse model of DMD. Specific inhibition of CaMKII phosphorylation of RyR2 may have limited translatability. Thus, we also assessed whether pharmacological inhibition of CaMKII could suppress ScaW in myocytes from mdx mice (Fig. 7). After an initial pacing at 3-Hz in Tyrode’s solution, the frequency of SCaWs was assessed. Next, CaMKII inhibitor KN-93 or PKA inhibitor H-89 was added, respectively. The incidence of SCaWs after inhibitor addition, normalized to SCaW frequency before inhibitor application, was assessed for each cell. Averaged data revealed that KN-93 almost completely inhibited SCaWs in mdx myocytes, whereas H-89 had little effect on SCaWs. To determine the potential importance of S2814 phosphorylation of RyR2 relative to CaMKII phosphorylation of other downstream targets, we performed similar experiments on myocytes from mdx:S2814D mice, in which RyR2 is constitutively phosphorylated at S2814D.16 These studies revealed that at baseline the SCaW frequency was higher in mdx:S2814D mice compared to mdx mice. Moreover, CaMKII inhibition using KN-93 did not reduced SCaW frequency in mdx:S2814D mice, suggesting that increased S2814 phosphorylation on RyR2 is an important contributor to SR Ca2+ waves and thus arrhythmogenesis in mdx mice.
Figure 7. CaMKII inhibition suppresses SCaW in mdx ventricular myocytes at 3-Hz.
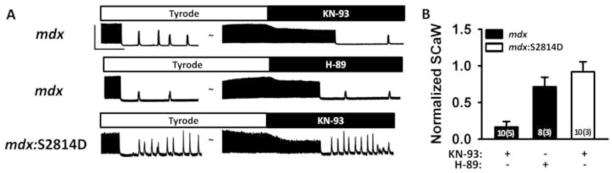
(A) Representative tracings showing spontaneous Ca2+ waves (SCaW) following 3-Hz pacing. CaMKII inhibitor KN-93 (1 μM) or PKA inhibitor H-89 (1 μM) were added subsequently, followed by another observation period for SCaWs. Scale bars: 2 F/F0 (vertical) and 20 seconds (horizontal). (B) Bar graph showing SCaW frequency after inhibitor administration normalized to SCaW frequency in Tyrode.
DISCUSSION
Previous studies have demonstrated an important role for defective RyR2 regulation and the resulting abnormal intracellular Ca2+ release in the pathogenesis of VT in mdx mice, a mouse model of DMD.4 Here we demonstrated that RyR2 is dysregulated in mdx mice prior to the development of overt structural heart disease. Although RyR2 phosphorylation is not enhanced in young mdx mice, ventricular myocytes are prone to defective Ca2+ release events following rapid pacing. Pharmacological inhibition of CaMKII but not PKA normalized diastolic SR Ca2+ leak in mdx mice, and genetic inhibition of S2814 but not S2808 phosphorylation on RyR2 prevented the induction of proarrhythmogenic Ca2+ waves in mdx mice. Our data revealed that SR Ca2+ load was markedly reduced in mdx mice, excluding a role of store overload-induced Ca2+ release as an arrhythmia mechanism. Together these data suggest that CaMKII and in particular CaMKII phosphorylation of RyR2 at S2814 can amplify SR Ca2+ leak, resulting in cellular Ca2+ waves and induction of VT in mice with DMD.
Development of ventricular arrhythmias depends on CaMKII phosphorylation of RyR2
Ventricular tachyarrhythmias in patients with DMD may be caused by a combination of mechanisms, including conduction abnormalities, fibrosis and fatty replacement of the myocardium. In addition, enhanced triggered activity due to abnormal myocyte Ca2+ homeostasis could promote arrhythmias in patients with DMD.25 Whereas it should be noted that patients with DMD typically develop ventricular arrhythmias in the presence of structural heart disease, we focused our experimental studies on young mice (< 6 months of age) to elucidate arrhythmia mechanisms in the absence of potentially confounding structural heart disease.25
Our data demonstrate that VT induction in mdx mice can be prevented by inhibition of CaMKII-dependent phosphorylation of S2814 on RyR2. At the cellular level, inhibition of CaMKII but not PKA suppressed abnormal Ca2+ waves, which are known to correlate with arrhythmogenesis at the whole animal level. The discordance between the effects of CaMKII and PKA phosphorylation of RyR2 in terms of their arrhythmogenic potential might seem surprising especially as both events enhance diastolic SR Ca2+ leak. However, there are differences in the functional effects of S2814 and S2808 phosphorylation. For example, CaMKII activity is governed by the frequency of local [Ca2+]i levels, and may thus be upregulated at faster heart rates.26 Our data suggest that baseline CaMKII levels in the hearts of mdx mice were not elevated, specifically in terms of level of T287 auto-phosphorylation (Fig. S1). However, increased diastolic SR Ca2+ leak could promote arrhythmias in mdx mice when the leak is amplified by CaMKII at faster heart rates.4 In contrast, our findings suggest that PKA phosphorylation of S2808 on RyR2 is not involved in cellular arrhythmogenesis in mdx mice. Finally, we recently demonstrated that genetic inhibition of CaMKII phosphorylation of RyR2 prevents VT in mice with non-dystrophic heart failure by normalization of diastolic SR Ca2+ leak, suggesting that similar mechanisms might have broader implications for arrhythmia mechanisms, especially in the setting of cardiomyopathy.16
Changes in RyR2 posttranslational regulation in dystrophic cardiomyopathy
It is possible that posttranslational mechanisms other than phosphorylation also contribute to defective RyR2 regulation in dystrophic cardiomyopathy. Bellinger et al. demonstrated in skeletal muscles that changes in RyR1 S-nitrosylation precede changes in phosphorylation.27 There might also be excessive ROS production in the hearts of young mdx mice, which could be inhibited by transgenic expression of neuronal nitric oxide synthase in the mdx mice.28, 29 Thus, it appears that RyR2 can be activated directly by S-nitrosylation, independent of phosphorylation. In addition, oxidation of RyR2 could directly or indirectly (via oxidation of CaMKII) promote activation of the channel. These mechanisms might explain the presence of increased diastolic SR Ca2+ leak in hearts of young mdx mice in the absence of elevated RyR2 phosphorylation levels.3, 4 In this study, we also found that baseline CaMKII phosphorylation of RyR2 at S2814 was not elevated in the hearts of young mdx mice. Nevertheless, our data suggest that mdx mice are predisposed to develop VT, and that activation of CaMKII could act as a ‘second hit’ to initiate ventricular arrhythmias.
Requirement of a ‘double hit ‘for induction of arrhythmias in mdx mice
We observed rescue from inducible VT and normalization of SR Ca2+ handling in cardiomyocytes from mdx mice as a result of pharmacologic inhibition of CaMKII or genetic inhibition of S2814 phosphorylation on RyR2. Although mdx mice at this age exhibit increased diastolic SR Ca2+ leak,3 they do not develop spontaneous arrhythmias, and that rapid cardiac pacing was required to induce VT.4. Similar findings have been reported previously in a mouse model of atrial fibrillation, in which a combination of a gain-of-function mutation in RyR2 and CaMKII activation were required to induce arrhythmias.8 Moreover, it has become increasingly apparent that RyR2 is among the major targets of CaMKII that increase arrhythmogenesis due to triggered activity.16 This notion is supported by recent findings that RyR2 stabilizers can reduce arrhythmogenesis in mdx mice.4, 30 Thus, even though mdx mice are predisposed toward the development of VT as evident by increased diastolic SR Ca2+ leak, a second hit – which could be CaMKII-mediated phosphorylation of RyR2 - is required to induce the arrhythmias.
Study limitations
Patients with DMD typically do not exhibit VT until they develop structural heart disease.1 Therefore, further studies are required to examine the role of CaMKII phosphorylation of RyR2 in the pathogenesis of VT after DMD patients (or animal models) develop heart failure. It is possible that the mechanisms we described are only relevant to VT induced by changes in heart rate or due to beta-adrenergic stimulation in patients, since no spontaneous VT episodes were observed in mdx mice. Our cellular studies were performed at 1 Hz and 3 Hz, rates that are slower than in vivo heart rates. It is well recognized that isolated myocytes lose the ability to electrically activate at fast rates (8–10 Hz) once isolated, especially when they are studied at room temperature. However, our data did reveal a striking rate-dependence of SR Ca2+ waves, which correlated nicely with the in vivo arrhythmia susceptibility studies. Finally, further validation in a large animal model or in patients with DMD would be important to examine the potential therapeutic potential of CaMKII inhibitors, which are currently in development.
CONCLUSIONS
DMD is the most common cause of muscular dystrophy and is associated with a substantial number of deaths due to lethal VT. Currently, there is no specific treatment to either prevent or treat these arrhythmias. Our study suggests that CaMKII-mediated phosphorylation of RyR2 promotes VT in DMD and represents a promising therapeutic target. The reduction of SR Ca2+ leak via RyR2 may constitute a novel strategy to treat VT in DMD patients by means of pharmacological inhibition of either CaMKII or RyR2.4, 9 Additional studies in larger mammals are required to confirm the therapeutic potential of this target.31
Supplementary Material
Acknowledgments
S.A. was supported by American Heart Association SCA predoctoral fellowship (2010-2012) and fellowship from the Alkek foundation from Baylor College of Medicine. N.L. was supported by AHA SCA postdoctoral fellowship (2010-2012), and AHA beginning grant-in-aid (2012-2014). X.H.T.W. is a W.M. Keck Foundation Distinguished Young Scholar in Medical Research, and is supported by NIH grants HL089598 and HL091947, and Muscular Dystrophy Association grant #69238. M.E.A. is funded by NIH grants HL 079031, HL 096652, HL 113001, and HL 070250. This work was also funded by the Foundation Leducq Alliance for CaMKII Signaling (M.E.A., X.H.T.W.).
Abbreviations
- CaMKII
calcium/calmodulin-dependent protein kinase II
- CaSpF
Ca2+ sparks frequency
- DMD
Duchenne muscular dystrophy
- PKA
Protein kinase A
- RyR2
Type 2 ryanodine receptors
- SCaW
Spontaneous Ca2+ wave
- SOICR
Store overload-induced Ca2+ release
- SR
Sarcoplasmic reticulum
- VT
Ventricular tachycardia
Footnotes
Conflicts of Interest: None of the authors have a conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Groh WJ, Zipes DL. Neurological disorders and cardiovascular disease. In: Bonow RO, Mann DL, Zipes DL, Libby P, editors. Braunwald’s heart disease. A textbook of cardiovascular medicine. Philadephia: Saunders; 2011. pp. 1916–1919. [Google Scholar]
- 2.Jung C, Martins AS, Niggli E, Shirokova N. Dystrophic cardiomyopathy: amplification of cellular damage by Ca2+ signalling and reactive oxygen species-generating pathways. Cardiovascular research. 2008;77:766–773. doi: 10.1093/cvr/cvm089. [DOI] [PubMed] [Google Scholar]
- 3.Sarma S, Li N, van Oort RJ, Reynolds C, Skapura DG, Wehrens XH. Genetic inhibition of PKA phosphorylation of RyR2 prevents dystrophic cardiomyopathy. Proc Natl Acad Sci U S A. 2010;107:13165–13170. doi: 10.1073/pnas.1004509107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fauconnier J, Thireau J, Reiken S, et al. Leaky RyR2 trigger ventricular arrhythmias in Duchenne muscular dystrophy. Proc Natl Acad Sci U S A. 2010;107:1559–1564. doi: 10.1073/pnas.0908540107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dobrev D, Wehrens XH. Calmodulin kinase II, sarcoplasmic reticulum Ca2+ leak, and atrial fibrillation. Trends in cardiovascular medicine. 2010;20:30–34. doi: 10.1016/j.tcm.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wehrens XH, Lehnart SE, Marks AR. Intracellular calcium release and cardiac disease. Annual review of physiology. 2005;67:69–98. doi: 10.1146/annurev.physiol.67.040403.114521. [DOI] [PubMed] [Google Scholar]
- 7.Marx SO, Reiken S, Hisamatsu Y, et al. PKA phosphorylation dissociates FKBP12. 6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 8.Chelu MG, Sarma S, Sood S, et al. Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J Clin Invest. 2009;119:1940–1951. doi: 10.1172/JCI37059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wehrens XH, Lehnart SE, Reiken SR, Marks AR. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res. 2004;94:e61–70. doi: 10.1161/01.RES.0000125626.33738.E2. [DOI] [PubMed] [Google Scholar]
- 10.Hilliard FA, Steele DS, Laver D, et al. Flecainide inhibits arrhythmogenic Ca2+ waves by open state block of ryanodine receptor Ca2+ release channels and reduction of Ca2+ spark mass. J Mol Cell Cardiol. 2010;48:293–301. doi: 10.1016/j.yjmcc.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.MacQuaide N, Dempster J, Smith GL. Measurement and modeling of Ca2+ waves in isolated rabbit ventricular cardiomyocytes. Biophysical journal. 2007;93:2581–2595. doi: 10.1529/biophysj.106.102293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allen DG, Whitehead NP, Yeung EW. Mechanisms of stretch-induced muscle damage in normal and dystrophic muscle: role of ionic changes. J Physiol. 2005;567:723–735. doi: 10.1113/jphysiol.2005.091694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fanchaouy M, Polakova E, Jung C, Ogrodnik J, Shirokova N, Niggli E. Pathways of abnormal stress-induced Ca2+ influx into dystrophic mdx cardiomyocytes. Cell Calcium. 2009;46:114–121. doi: 10.1016/j.ceca.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prosser BL, Ward CW, Lederer WJ. X-ROS signaling: rapid mechano-chemo transduction in heart. Science. 2011;333:1440–1445. doi: 10.1126/science.1202768. [DOI] [PubMed] [Google Scholar]
- 15.Erickson JR, Joiner ML, Guan X, et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–474. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Oort RJ, McCauley MD, Dixit SS, et al. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation. 2010;122:2669–2679. doi: 10.1161/CIRCULATIONAHA.110.982298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li N, Wehrens XH. Programmed electrical stimulation in mice. Journal of visualized experiments: JoVE. 2010;39:1730. doi: 10.3791/1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Oort RJ, Garbino A, Wang W, et al. Disrupted junctional membrane complexes and hyperactive ryanodine receptors after acute junctophilin knockdown in mice. Circulation. 2011;123:979–988. doi: 10.1161/CIRCULATIONAHA.110.006437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sood S, Chelu MG, van Oort RJ, et al. Intracellular calcium leak due to FKBP12. 6 deficiency in mice facilitates the inducibility of atrial fibrillation. Heart Rhythm. 2008;5:1047–1054. doi: 10.1016/j.hrthm.2008.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Branco DM, Wolf CM, Sherwood M, Hammer PE, Kang PB, Berul CI. Cardiac electrophysiological characteristics of the mdx (5cv) mouse model of Duchenne muscular dystrophy. J Interv Card Electrophysiol. 2007;20:1–7. doi: 10.1007/s10840-007-9168-z. [DOI] [PubMed] [Google Scholar]
- 21.Zhang R, Khoo MS, Wu Y, et al. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med. 2005;11:409–417. doi: 10.1038/nm1215. [DOI] [PubMed] [Google Scholar]
- 22.Lehnart SE, Wehrens XH, Reiken S, et al. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell. 2005;123:25–35. doi: 10.1016/j.cell.2005.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li N, Wang T, Wang W, et al. Inhibition of CaMKII Phosphorylation of RyR2 Prevents Induction of Atrial Fibrillation in FKBP12. 6 Knockout. Mice Circ Res. 2012;110:465–470. doi: 10.1161/CIRCRESAHA.111.253229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang D, Xiao B, Yang D, et al. RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR) Proc Natl Acad Sci U S A. 2004;101:13062–13067. doi: 10.1073/pnas.0402388101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Groh WJ. Arrhythmias in the muscular dystrophies. Heart Rhythm. 2012 doi: 10.1016/j.hrthm.2012.06.038. [DOI] [PubMed] [Google Scholar]
- 26.Vinogradova TM, Zhou YY, Bogdanov KY, et al. Sinoatrial node pacemaker activity requires Ca(2+)/calmodulin-dependent protein kinase II activation. Circ Res. 2000;87:760–767. doi: 10.1161/01.res.87.9.760. [DOI] [PubMed] [Google Scholar]
- 27.Bellinger AM, Reiken S, Carlson C, et al. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat Med. 2009;15:325–330. doi: 10.1038/nm.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tidball JG, Wehling-Henricks M. The role of free radicals in the pathophysiology of muscular dystrophy. J Appl Physiol. 2007;102:1677–1686. doi: 10.1152/japplphysiol.01145.2006. [DOI] [PubMed] [Google Scholar]
- 29.Wehling-Henricks M, Jordan MC, Roos KP, Deng B, Tidball JG. Cardiomyopathy in dystrophin-deficient hearts is prevented by expression of a neuronal nitric oxide synthase transgene in the myocardium. Hum Mol Genet. 2005;14:1921–1933. doi: 10.1093/hmg/ddi197. [DOI] [PubMed] [Google Scholar]
- 30.Wehrens XH, Lehnart SE, Reiken SR, et al. Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science. 2004;304:292–296. doi: 10.1126/science.1094301. [DOI] [PubMed] [Google Scholar]
- 31.Collins CA, Morgan JE. Duchenne’s muscular dystrophy: animal models used to investigate pathogenesis and develop therapeutic strategies. Int J Exp Pathol. 2003;84:165–172. doi: 10.1046/j.1365-2613.2003.00354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.