

Abstract
Background
Heart failure is characterised by impaired function and disturbed Ca2+ homeostasis. Transgenic increases in inhibitor-1 activity have been shown to improve Ca2 cycling and preserve cardiac performance in the failing heart. The aim of this study is to evaluate the effect of activating the inhibitor (I-1c) of protein phosphatase 1 (I-1) through gene transfer on cardiac function in a porcine model of heart failure induced by myocardial infarction (MI).
Methods and Results
Myocardial infarction was created by a percutaneous, permanent left anterior descending artery (LAD) occlusion in Yorkshire Landrace swine (n = 16). One month post MI, pigs underwent intracoronary delivery of either recombinant adeno-associated virus type 9 (rAAV9) carrying I-1c (n = 8) or saline (n = 6) as control. One month post MI creation animals exhibited severe heart failure demonstrated by decreased ejection fraction (46.4±7.0% vs. sham 69.7±8.5%) and impaired (dP/dt)max as well as (dP/dt)min. Intracoronary injection of AAV9.I-1c prevented further deterioration of cardiac function and led to a decrease of scar size.
Conclusions
In this preclinical model of heart failure, overexpression of I-1c by intracoronary in vivo gene transfer, preserved cardiac function and reduced scar size.
Keywords: I-1c, AAV9, SERCA2a, heart failure, myocardial infarction
One of the key abnormalities in both human and experimental HF is a defect in sarcoplasmic reticulum (SR) function, resulting in abnormal intracellular Ca2+ handling 1–4. Deficient SR Ca2+ uptake during relaxation has been identified in failing hearts from both humans and animal models and has been associated with a decrease in the activity of the SR Ca2+-ATPase (SERCA2a), which is at least partially due to enhanced phospholamban (PLN) inhibition5–7. Restoring SERCA2a levels or reducing PLN inhibition has been shown to improve function, metabolism and/or survival in a number of experimental models of heart failure5. Targeting SERCA2a in experimental models of heart failure by somatic gene transfer has proven effective in rescuing contractile function while at the same time decreasing ventricular arrhythmias and restoring energetics.
This has led to the initiation and the recent completion of a first-in-man phase 1 clinical trial of gene therapy for heart failure, using adeno-associated type (AAV) -1 vector carrying SERCA2a 8. The safety profile of AAV gene therapy along with the positive biological signals obtained from this phase 1 trial has led to the initiation of a phase 2 trial of AAV1.SERCA2a in NYHA class III/IV patients9. More recently, several studies have shown that by constitutively activating the inhibitor of protein phosphatase 1 (I-1) within the failing heart, there is improvement of SR Ca2+-handling, contractility and most importantly, reversal of adverse remodelling by directly decreasing fibrosis and cardiac hypertrophy 10–14. In this study, we have chosen to focus on the expression of the constitutively active, truncated form of Inhibitor-1, the I-1c protein, because it has the ability to enhance contractility while at the same time decreasing fibrosis in small animal models of heart failure12. Overexpression of I-1c in the heart results in significant enhancement of phospholamban (PLN) phosphorylation and cardiac function and abrogates the negative effects of aortic constriction in this model. In fact, beyond the rescue of contractile function, overexpression of I-1c resulted in a significant decrease in fibrosis12. However there have been conflicting reports that I-1c overexpression in a murine model may result in amplification of the beta-adrenergic system and worsening of function when subjected to specific stresses 15.
Therefore the specific aim of the study is to evaluate the effects of long term I-1c expression in a porcine model of heart failure. We assessed the dynamic changes in ventricular volumes, and contractility following gene transfer with AAV9.I-1c in a porcine model of volume overload and infarct after the development of heart failure.
Methods
Animal care and all procedures were approved by local institutional committees and were performed in accordance with the “Principles of Laboratory Animal Care by the National Society for Medical Research and the Guide for the Care and Use of Laboratory Animals” (NIH Publication No. 86-23, revised 1985).
AAV9.I-1c construction and characterization
There are two components to AAV9.I-1c. The first is an active I-1 (GenBank: AAL48321.1) transgene (AA 1-65 with T35D), and the second is the vector, AAV9, which delivers the gene selectively to the heart after intracoronary administration. We used a well characterized, human, constitutively active I-1 (AA 1-65 with T35D) gene that has been truncated to remove the S67 and T75 PKC sites. This truncated gene and the protein it encodes for is designated I-1c. The rationale for removing the PKC sites on I-1 is that their phosphorylation increases PP1 activity, resulting in decreased SERCA and SR Ca-transport and attenuated contractility 16. Because PKC activity is increased in heart failure 17, it is desirable to remove these sites on I-1. In addition to removal of the PKC sites, the threonine35 PKA-phosphorylation site of the I-1c gene is constitutively active by replacing threonine with aspartic acid. AAV9 vectors carrying I-1c under control of the CMV promoter were produced using the two-plasmids protocol described by Zolotukhin et al 18 with the following modifications. 293-T cells (ATCC, Manassas, VA) were grown in triple flasks for 24 h (DMEM, 10% FBS) prior to adding the calcium phosphate precipitate. After 72 hours, the virus was purified from benzonase-treated cell crude lysates over an iodixanol density gradient (Optiprep, Greiner Bio-One Inc., Longwood, FL), followed by heparin-agarose type I affinity chromatography (Sigma-Aldrich Inc., St. Louis, MO). Finally viruses were concentrated and formulated into lactated Ringer’s solution (Baxter Healthcare Corporation, Deerfield, IL) using a Vivaspin 20 Centrifugal concentrators 50K MWCO (Vivascience Inc., Carlsbad, CA), and stored at −80°C. Vector stock biochemical purity (>95%) was assessed by silver staining after electrophoresis. Genome containing particles (gcp) were determined by a real-time PCR approach (LightCycler, Roche Diagnostics) using the SYBR™ Green Taq ReadyMix™ (SIGMA, Saint-Louis, MO) and primers CMV-F and CMV-R.
Animal studies and myocardial infarction model
This study, using female Yorkshire landrace pigs (~ 20 kg body weight) was approved by the Institutional Animal Care and Use Committee. All procedures were performed under anaesthesia with inhaled isoflurane (0.5–2%). All animals enrolled in the study were pre-screened for neutralizing antibodies and had a maximum of 8-fold dilution for detection as described by Rapti et al19. For MI generation, we introduced an 8F sheeth into the femoral artery and cannulated the left anterior descending artery (LAD) with an 7F hockey stick guiding catheter (Cordis Infiniti, Johnson & Johnson). After injecting 100 µg nitroglycerin and obtaining a baseline coronary angiogram, we placed a platinum embolic coil (0.035 in, 40 mm length, 5×3-mm diameter, Cook Medical Inc.) using a 4F AR catheter (Cordis Infiniti, Johnson & Johnson) into the LAD after the takeoff of the first diagonal branch, thus occluding 2/3 of the LAD tributary, determined by coronary angiography. \ Fourteen animals were randomized to receive either control intracoronary injection with saline or AAV9.I-1c, three animals were subject to a sham procedure.
Intracoronary antegrade injection of AAV9.I-1c
The detailed injection procedure has been described previously20. Briefly, the solution for intracoronary injection was prepared by adding 2.5 × 1012 AAV9.I-1c vector genomes (vg) to a mixture of 10 mL of normal saline and 10 mL blood. Via the arterial sheath, the left coronary artery (LCA) was cannulated with a 5F guiding catheter (Cordis Infiniti, Johnson & Johnson). Two hundered micrograms of nitroglycerin were injected into the coronary arteries to enhance coronary perfusion. Two coronary guide wires were introduced in left anterior descending artery (LAD) and the left circumflex artery (LCX) to stabilise the position of the guiding catheter during the injection. A programmable precision infusion pump (Harvard Apparatus) was used for the injection. Fifteen ml of the viral solution were injected into the proximal part of the LCA at 1 ml/min followed immediately by a 5 ml flush of saline/blood mixture. Afterwards the left coronary artery catheters were removed and a 5F Hockey stick guiding catheter (Cordis Infiniti, Johnson & Johnson) was placed in the right coronary artery. Here 5 ml of viral solution were injected into the Right Coronary Artery (RCA) at 1 ml/min followed immediately by a 5 ml flush of saline/blood mixture. On the day of gene delivery, some animals (four control, five AAV9.I-1c) underwent subcutaneous implantation of a Medtronic (Minneapolis, MN ) Reveal insertable loop recorder at the T4 level to monitor them for arrhythmic events.
Assessment of myocardial function and structure
We assessed myocardial function and structure at baseline (i.e. before MI generation), 1 month after myocardial infarction (i.e. before application of the virus), and 2 months after application (the three month time point). Echocardiography was performed obtaining apical images at 10-degree intervals with a 5MHz TTE probe (Vivid 7, GE), rotated by software and gated to ECG. Digital images were analysed on a workstation with custom software. Two stable and well-defined consecutive cardiac cycles were acquired digitally for each measurement.
For hemodynamic catheterization, we accessed the femoral artery and vein with 8F sheaths and placed a 7F Millar Micro-Tip catheter system (Millar Instruments Inc.) into the aorta, the left ventricle. We determined the following parameters: systolic pressure, end-diastolic pressure, peak LV pressure rate of rise (dP/dt)max and Tau value (time constant of isovolumic relaxation); (dP/dt)max/P was calculated as (dP/dt)max/(systolic – end-diastolic pressure). The mean of at least 3 consecutive cardiac cycles was calculated for each measurement.
We performed coronary angiography at the one-month, and three-month time points using an Integris H5000 single-plane fluoroscopy system (Philips Medical Systems). All images were acquired and analysed by an investigator blinded to the study arm. Just prior to sacrifice, a dobutamine stress test was performed to assess the hemodynamic alteration and the potential arrhythmogenicity in response to increased β-stimulation. Intravenous dobutamine infusion was started at 5 µg/kg/min and gradually increased up to 40 µg/kg/min. Continuous pressure and electrocardiographic recording were obtained during the stress test.
The animals were euthanized by i.v. injection of EuthasolR (pentobarbital, phenytoin, 1ml/4.5kg), removed the hearts, resected the right ventricle, and cut the left ventricle into 6 slabs of the same thickness. We visualised viable myocardium by staining 5 of these slices with 2,3,5-Triphenyltetrazolium chloride and quantified scar volume using unbiased stereology.
Real Time qPCR detection of I-1C mRNA
Total RNA was isolated with TRIzol reagent (Invitrogen) followed by DNase I treatment (Qiagen). Total RNA was reverse transcribed using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems, ABI) according to the manufacturer’s protocol. Quantitative PCR reactions were performed with Power SYBR Green Master Mix using an ABI Prism 7500 Real Time PCR System. The real time qPCR assay detects a 125 bp sequence unique to AAV9.I-1C and the number of copies of AAV9.I-1C was quantified using serial dilutions of a plasmid containing the target sequences as standards. Primers for the I-1C were designed with the Primer Express software based on the human protein sequence (NP 006732.3; Forward: CGTGCCCCTGCTGGAA Reverse: TCCGGTCCTCGTCGATCTC). Relative quantitation is the I-1c copies detected relative the number of lowest number of I-1copies detected in the treatment group. Since the actual number of cycles required for detection was relatively, high, determination of vector genomes per gram of tissue may not be justified.
Western blot
Cardiac whole homogenate samples (50 μg) in SDS sample buffer were run on an SDS-PAGE gradient gel(4–20%, Bio-rad), and proteins in the gel were transferred to NC membrane (Bio-Rad). The membrane was blocked with 5% skim milk and incubated overnight with an antibody directed against I-1c (provided by Dr. Kranias) and GAPDH (Sigma). The membrane was then incubated with an anti-rabbit IgG secondary antibody conjugated to horseradish peroxidase (Cell Sinaling) and developed using SuperSignal West Pico Chemiluminescent Sustrate (Thermo scientific).
Statistical analyses
Investigators quantified observations independently from one another and in a blinded manner. Numeric data are presented as mean ± SD unless indicated otherwise. Pairwise statistical comparisons were performed statistically between Saline vs. I1-C and Sham vs. I1 C groups using Graph Pad software. Within-group comparisons were carried out using the paired t-test and between-group comparisons were carried out using the two-independent sample Student t-test. P-values are two-sided and there was no adjustment for multiple comparisons due to the nature of the study. The α-value was set at 0.05.
Results
Twenty-one pigs underwent myocardial infarction as described below and fourteen survived one month following myocardial infarction. During the first month, the pigs died mainly from ventricular arrhythmias or sudden cardiac death. Eight pigs received AAV9.I-1c while six pigs received saline. (Figure 1) The eight pigs receiving AAV9.I-1c survived while one of the pigs in the saline group died. The pigs were then sacrificed 2 months following gene transfer. Three animals were subject to sham procedure only.
Figure 1. Study design.
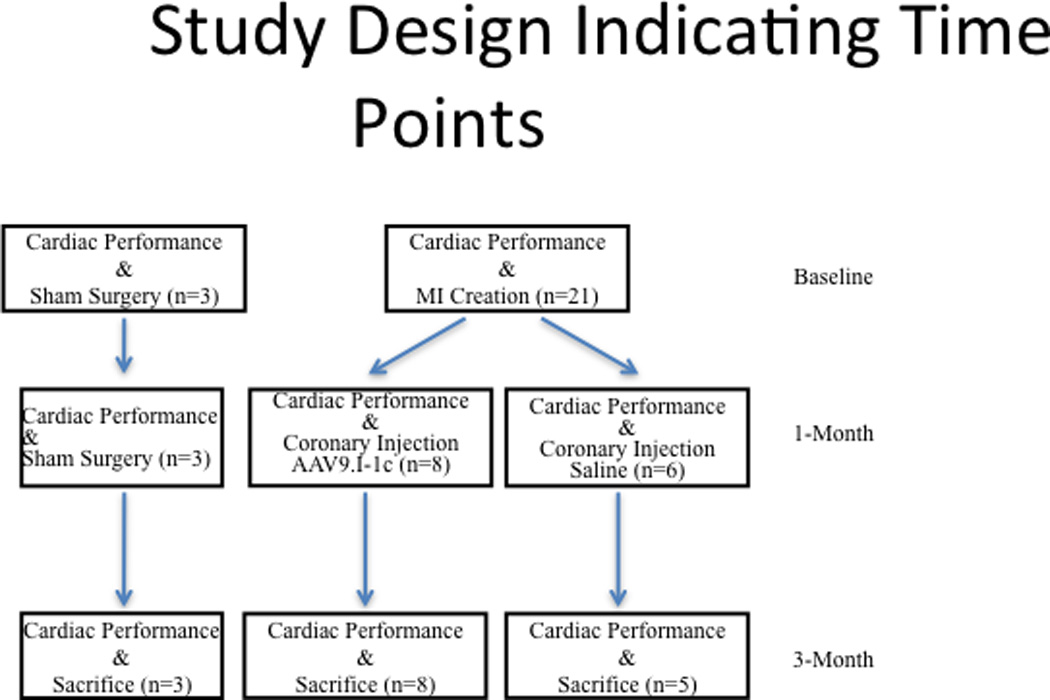
Twenty one pigs underwent myocardial infarction and fourteen survived one month following myocardial infarction. Eight pigs received AAV9.I-1c while six pigs received saline. All animals receiving AAV9.I-1c survived until sacrifice after 3months, while there was one death in the control group. Referenced time points are indicated on the right.
We used a pig model of MI, which consists of placement of an occlusive coil in the LAD. Chronic heart failure models of swine are suitable preclinical models, as they share anatomical and pathophysiological similarities to humans. One month following infarct generation, AAV9.I-1c or saline was administered intracoronary. We determined the effect on cardiac function at baseline, time of gene transfer (one-month) and at three months.
Cardiac output (CO) increased in all groups one month post MI creation. (Figure 2) In the control group only receiving intracoronary saline cardiac output deteriorated in the following two months (6.32 ± 1.9 to 5.0 ± 0.96 L/min at three month) indicating severe progression of heart failure. In contrast CO in animals treated with AAV9.I-1c was preserved and did not significantly differ from sham procedure animals (7.1 ± 1.1 vs. 7.2 ± 0.5 L/min).
Figure 2. Cardiac output.
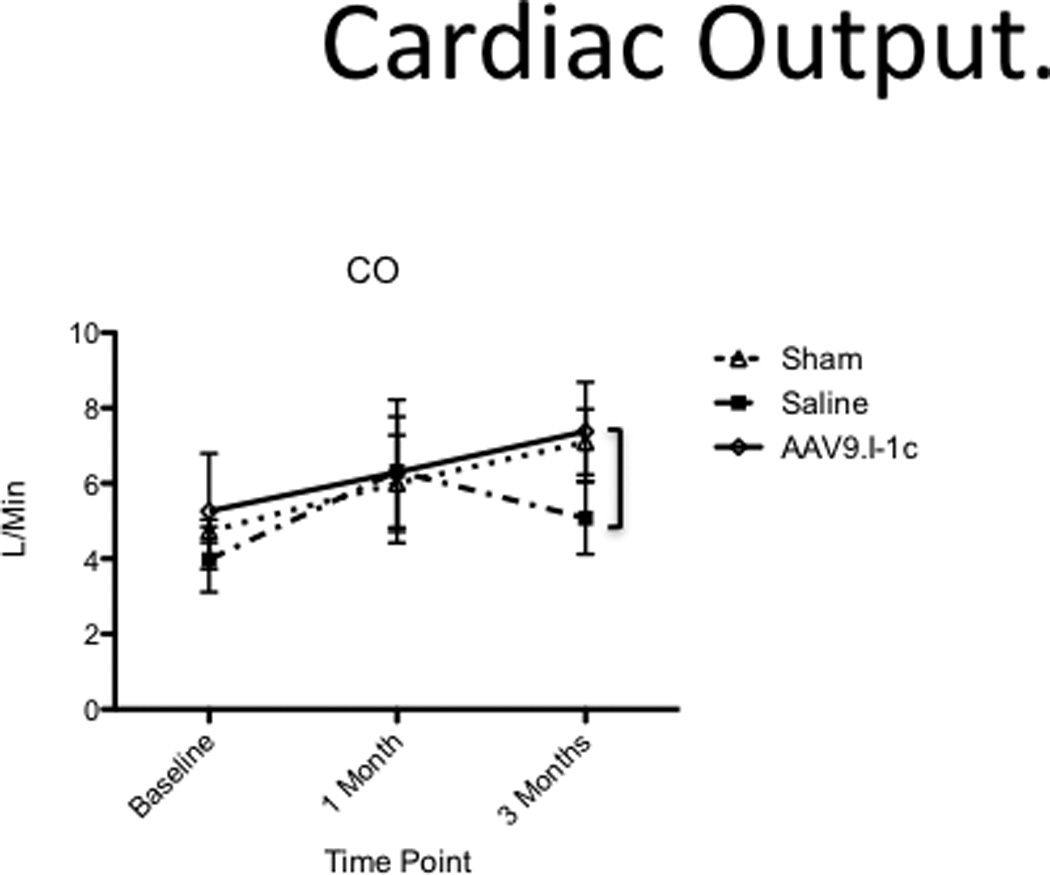
Cardiac output was measured before MI creation, at the time of gene transfer and before sacrifice at three months.
Likewise the ejection fraction (EF) remained stable after animals were treated with AAV9.I-1c 1 month post MI (from 46.4 ± 5.8 to 50.5 ± 3.5% at 3 months) while continuously deteriorating in control animals from 44% to 37.4% at 3 months.(Figure 3) In parallel hemodynamic catheterization showed increased (dP/dt)max/P, indicating improved myocardial contractility (Figure 4, Table 1). While sham animals showed almost no change over time (+1.6 ± 3.6), animals with heart failure that received saline had a pronounced decrease of systolic function measured by (dP/dt)max/P (−41.7 ± 14.2). In contrast, gene therapy with AAV9.I-1c preserved function with a smaller decrease of −4.9 ± 19.8 (P < 0.05 to saline). The time constant of isovolumic relaxation (tau) was measured to assess diastolic left ventricular function. Diastolic dysfunction was prominent after 1 month and remained impaired after 3 months in animals that underwent MI and received saline injection (+1.3 ± 18.8). While gene transfer of I-1c average tau change (−13.3 ± 27.0 s) suggests improved diastolic dysfunction in the treated animals after one month, although it failed to reach statistical significance when compared to saline.
Figure 3. Ejection fraction.
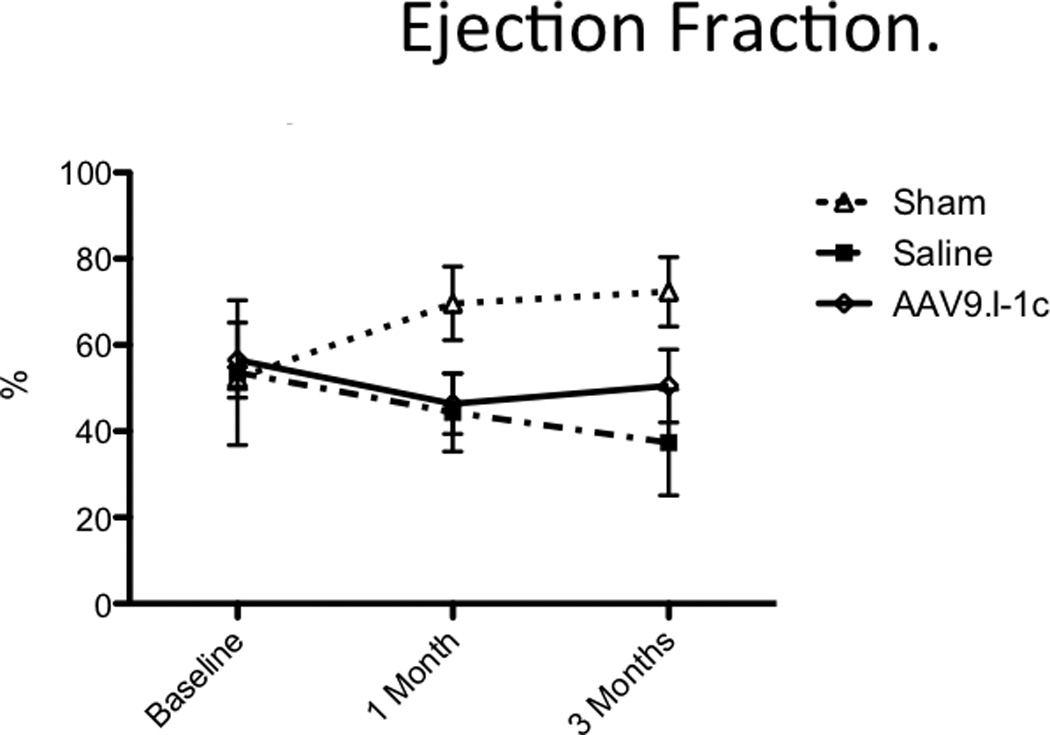
Global functional analysis shows long-term improvements in AAV9.I-1c-treated animals as determined by ejection fraction.
Figure 4. Differences in the adjusted peak LV pressure rate of rise and the time constant isovolumic relaxation.
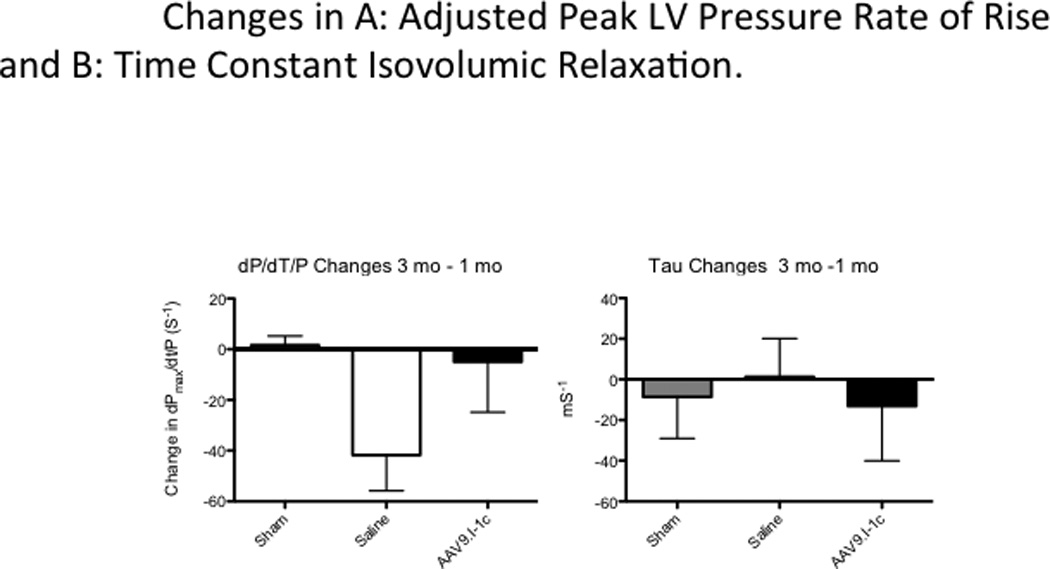
(dP/dt)max as a measure of cardiac contractility was normalized to the average Pressure: (dP/dt)max /P.
Table 1.
Hemodynamic Data
| Control (n=6) | AAV.I-1c (n=8) | |||||
|---|---|---|---|---|---|---|
| Baseline | 1 Month | 3 Months | Baseline | 1 Month | 3 Months | |
| (dP/dt)max (mmHg/s) | 1441 ± 131 | 1914 ± 310 | 1266 ± 357 | 1316 ± 426 | 1635 ± 396 | 1675 ± 325* |
| EDP (mmHg) | 11.8 ± 2.0 | 10.9 ± 0.9 | 11.9 ± 2.0 | 12.4 ± 1.1 | 12.3 ± 1.51 | 12.6 ± 1.9 |
| SBP (mmHg) | 97.8 ± 14.3 | 104.6 ± 20.1 | 87.8 ± 56.4 | 87.2 ± 11.1 | 98.2 ± 13.8 | 87.8 ± 40.8 |
| HR (bpm) | 91.4 ± 19.1 | 98.2 ± 10.8 | 61.1 ± 33.5 | 87.3 ± 9.4 | 81.8 ± 12.9 | 69.7 ± 31.8 |
At Baseline, 1 month and 3 months after myocardial infarction cardiac catherization was performed, left ventricular peak pressure rate of rise (dP/dt)max, end diastolic pressure (EDP), systolic blood pressure (SBP), and heart rate (HR). The average of three measurements was used for analysis. Results are given as mean ± SD.
P < 0.05 vs. control group.
At the end of the study period fractional shortening (FS), determined by echocardiography, was significantly higher in I-1c treated animals (25.3 ± 5.2 %) than in control animals (18.8 ± 6.1 %, P < 0.05, Table 2), consistent with improved contractility. In summary, the presented data suggest that gene therapy with I-1c may induce functional improvements after myocardial infarction in swine.
Table 2.
Echocardiographic Data
| Control (n=6) | AAV.I-1c (n=8) | |||||
|---|---|---|---|---|---|---|
| Baseline | 1 Month | 3 Months | Baseline | 1 Month | 3 Months | |
| IVSTd (mm) | 0.7 ± 0.07 | 0.4 ± 0.05# | 0.42 ± 0.12# | 0.7 ± 0.06 0 | .38 ± 0.05# | 0.43 ± 0.14# |
| FS (%) | 36.4 ± 13.2 | 22.8 ± 5.9# | 18.8 ± 6.1# | 30.3 ± 5.5 | 26.3 ± 7.0 | 25.3 ± 5.2* |
| SV (mL) | 53.8 ± 14.5 | 76.0 ± 18.9# | 82.4 ± 26.0 # | 50.6 ± 11.2 | 60.9 ± 23.7 | 101.6 ± 23.7# |
| ESV (mL) | 41.6 ± 15.7 | 97.5 ± 34.8# | 146.6 ± 64.6# | 36.5 ± 4.7 | 75.2 ± 24.7# | 108.3 ± 59.4# |
| EDV (mL) | 91.4 ± 20.2 | 174.3 ± 44.7# | 229 ± 61.0# | 87.3 ± 11.4 | 143.5 ± 40.9# | 222.6 ± 59.8# |
| E/Ea | 0.05 ± 0.004 | 0.06 ± 0.007 | 0.07 ± 0.013 | 0.05 ± 0.010 | 0.06 ± 0.012 | 0.05 ± 0.019 |
At Baseline, 1 month and 3 months after myocardial infarction, interventricular septum thickness (IVSTd), fractional shortening (FS), stroke volume (SV), end-systolic volume (ESV), end-diastolic volume (EDV) and E/Ea were measured at the mid ventricular level. The average of three measurements was used for analysis. Results are given as mean ± SD.
P < 0.05 vs. control group;
P < 0.05 vs. baseline.
In order to evaluate the arrhythmogenicity of I-1c gene transfer to the porcine myocardium, four of the control animals and five of the treatment group animals were implanted with loop recorders for longer term monitoring. No events due to ventricular tachycardia (VT) were detected in the two-month period from the time of injection, to sacrifice. Furthermore, we subjected the animals to a dobutamine challenge prior to sacrifice. In both arms of the study, we detected two ventricular tachycardia (VT) events in each group. Therefore, while the animal model itself might be considered arrhythmogenic, the AAV9. I-1c delivery did not appear to enhance this effect.
Adjusted for body weight, I-1c overexpressing animals had lower average LV muscle mass (2.9 ± 0.08 mg/kg vs. 3.1 ± 0.05 mg/kg, p = 0.11) relative to the saline group (Figure 5) but these differences did not reach statistical significance. In addition, the right ventricular muscle mass was also not statistically different from the control group.
Figure 5. Heart Weight to Body Weight Ratio.
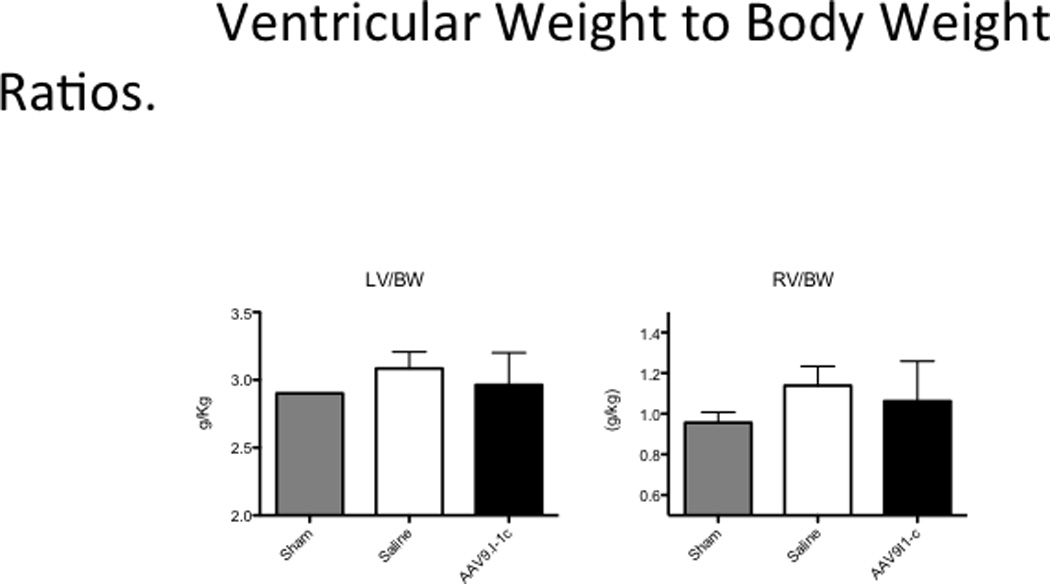
Weight of the left ventricle was determined after explanation at sacrifice and the ratio to animal’s body weight was calculated.
Examination of slices (4–6mm) of explanted hearts by vital staining with tetrazolium chloride (TTC), which highlights scar area in white and viable tissue in pink (Figure 6), revealed that hearts of I-1c group had significantly smaller fibrotic scar areas at 2 months after intracoronary gene transfer.
Figure 6. Left Ventricular Scar Size.
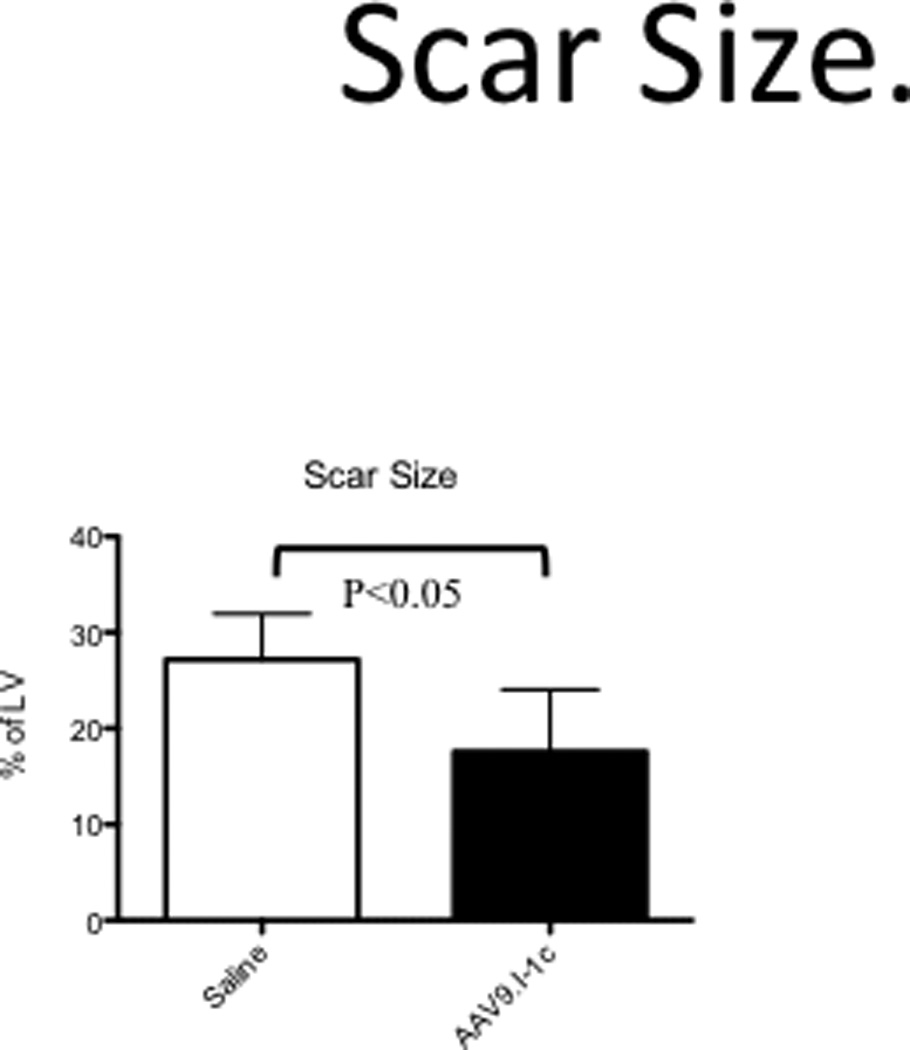
Quantification of scar volume by tetrazolium chloride (TTC) staining.
To evaluate the effectiveness of our gene transfer method we examined the expression of I-1c by RTPCR and western blotting. As shown in Figure 7a, I-1c expression was present in left ventricular tissue (anterior wall) from animals that had received AAV9.I-1c while Figure 7b shows the presence of I-1c genomes.
Figure 7. Expression of I-1c.
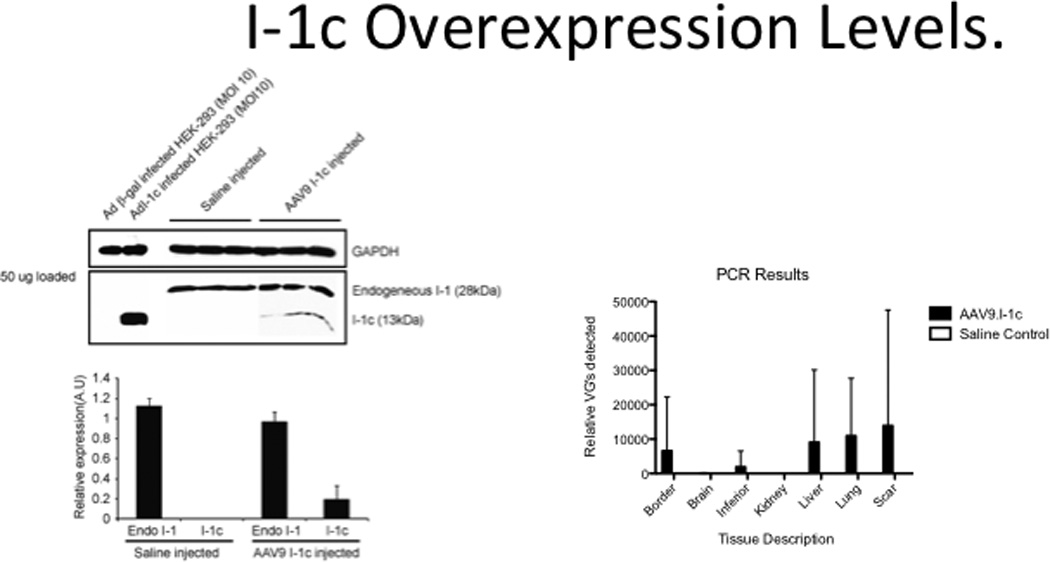
a) Western blot for I-1 and I-1c in left ventricular tissues following gene transfer of I-1c or injection with saline. b) RTPCR of I-1c in left ventricular tissues following gene transfer of I-1c. Border: Viable tissue adjacent to scar tissue. Inferior: Myocardium supplied by RCA at mid-papillary muscle level.
Discussion
There are several key conclusions from our porcine model. First, intracoronary gene transfer of I-1c leads to expression of the constitutive protein as indicated by our qPCR and Western Blot analyses with a significant amount of variability. While the endogenous total I-1 remains relatively high in both animal groups studied, the western blot analysis clearly demonstrates about 20% additional inhibitor present due to gene transfer. Thus, our data suggest the translational potential of I-1c therapy for the failing heart in a clinically relevant large animal ischemia injury model. This adds another potential option to consider in addition to the now well-documented SERCA2a therapy now being evaluated in clinical trials21. It is notable that I-1c therapy could be considered either as an alternative, or as a potential enhancement to other proven therapies.
Though the I-1c protein levels are significantly higher in our treatment group, these values need to be kept in context of the constitutive I-1 levels including both phosphorylated and un-phosphorylated forms. It is notable that I-1c imparts constitutive inhibition while the endogenous I-1 could be phosphorylated rendering it inactive against protein phosphatase. El-Armouche et al. showed that the phosphorylated I-1 levels in the failing human heart22 are elevated with concomitant decreases in phosphorylated phospholamban22, 23, with these overall changes negatively impacting calcium handling. Our goal was to alter the balance between phosphorylated and un-phosphorylated 28kD I-1 by overexpression of the constitutively active I-1c. Our data suggest that the expression levels achieved positively impacted cardiac function. These results support the conclusion that even modest transfection of I-1c can have beneficial effects to the failing heart.
Our biodistribution studies clearly demonstrated that though expression levels are variable, there is clear transfection of I-1C in the treatment group myocardial border zone, scar tissue as well as a the more remote inferior zone. Furthermore, we did detect some off target transfection. The variability in transfection and expression levels can be due to a number of sources, including the inherent intra-animal variability we experience in our porcine models that at least partially recapitulates the variability seen in the clinic. Notably, there can be significant variability in infarct size and in coronary artery architecture of the porcine heart24 which is also expected in the human heart25. Thus, while the observed variability increases the uncertainty in determining differences between experimental groups, it does serve to represent some of the variability expected in translation to clinical trials.
There are several measurements of cardiac performance that suggest overall cardio-protection from the progression of heart failure or potentially reverse remodelling. There is preservation in the native increase in cardiac output compared to the sham group as shown in Figure 2. Systolic function recovers from a negative trajectory following model creation since there is a levelling off of the EF decline determined in the control group as shown in Figure 3. In the treatment group, ejection fraction measures suggests improved cardiac performance. Furthermore, the scar size is significantly smaller in the treated animals relative to the controls. Taken together, these results support the potential of I-1c gene transfer to heart failure patients.
PLN has been shown to be the major regulator of basal contractility, a key mediator of the inotropic and lusitropic effects of β-agonists in the mammalian heart Dysregulation of the PLN phosphorylation state is one of the insults contributing to the depressed SR Ca2+-transport function in heart failure. Indeed, the SR protein phosphatase-1 activity is increased in patients with end-stage heart failure, which coupled with the down regulation of β-receptors10, 12, 13, 26 would contribute to decreased PLN phosphorylation and increased inhibition of SERCA2a’s Ca2+ affinity. Thus, regulation of the phosphatase activity in SR may provide another mechanism by which the cell achieves increases in PLN phosphorylation resulting in increases in basal contractile parameters and their responses to β-agonists10, 12, 13, 26. β-adrenergic stimulation results in cAMP-dependent increased phosphorylation of the I-1 protein, effectively inhibiting the SR phosphatase activity resulting in stimulatory effects through “unopposed” PLN phosphorylation27, 28.
However, in heart failure, the I-1 is dephosphorylated due to decreased β-adrenergic drive and this results in diminished inhibition of the type 1 phosphatase and dephosphorylation of PLN29. Ablation of I-1 resulted in increased protein phosphatase activity, which was associated with decreases in PLN phosphorylation status and thus, increased inhibition of SERCA2a’s Ca2+ affinity and myocardial contractility. Overexpression of the constitutively active I-1 had opposite effects than its ablation10. Phosphorylation at Ser67 of I-1 may be associated with diminished inhibitor activity and increased PP1 activity. Indeed, our preliminary studies in human failing hearts show that the phosphorylation of Ser67 in I-1 is increased (55%) and PLN phosphorylation (pSer16; pThr17) is decreased, in agreement with previous reports on enhanced PP1 activity. Furthermore, we have recently identified an additional PKC phosphorylation site on I-1, Thr7513. Expression of a constitutively phosphorylated I-1 at either S67D or T75D in isolated rat cardiomyocytes is associated with significantly depressed contractile performance.
Given these the in vitro and in vivo findings, potent and specific inhibition of PP1 activity might improve cardiac function and arrest the progression of hypertrophy and subsequent heart failure10,30.
These experiments suggest that I-1 is a “molecular inotrope” that regulates cardiac β-adrenergic responses through PLN phosphorylation. Indeed, overexpression of the active truncated I-1 in vivo was associated with enhanced cardiac function and protection against pressure-overload hypertrophy12. In addition, gene delivery of this activated molecule in failing rat hearts resulted in rescue of the depressed cardiac function and partial restoration of cardiac remodeling. Importantly, the effects of I-1 in vivo appeared to be mediated by specific alterations only in PLN phosphorylation while there were no alterations in either ryanodine receptor or troponin I phosphorylation levels and the activity of the L-type Ca2+-channel remained unaltered12.
More recently, an inducible transgenic mouse model was generated that enabled temporally controlled expression of active I-131. Active I-1 expression in the adult heart elicited significant enhancement of contractile function, associated with preferential PLN phosphorylation and enhanced SR Ca2+-transport. During ischemia / reperfusion-induced injury, active I-1 expression augmented contractile function and recovery. Further examination revealed that the infarct region and apoptotic as well as necrotic injuries were significantly attenuated by enhanced I-1 activity. These cardioprotective effects were associated with suppression of the endoplasmic reticulum stress response.
The protective role of I-1c was challenged by recent reports from El Armouche and colleagues who found that I-1 deletion exhibited less susceptibility to acute isoprenaline-induced deaths, associated with a lesser extent of ventricular arrhythmias and also protected against chronic isoprenaline-induced hypertrophy, dilatation and fibrosis 22, 30. More recently the same group found that mice with conditional cardiomyocyte-restricted expression of I-1c on an I-1–deficient background exhibited enhanced cardiac contractility but exaggerated contractile dysfunction and ventricular dilation upon catecholamine infusion15. It is not clear why different groups have such different results however different genetic backgrounds (FVB/N vs C57Bl/6J) are known to alter morphology, function, and survival in transgenic mice.
For this reason, our studies in a relevant pre-clinical model of heart failure are quite important to specifically address the issue of I-1c overexpression on contractile measures. Using gene transfer methodologies targeting the heart our studies clearly showed that I-1c overexpression improved contractile function, mitigated the adverse remodelling post –MI and led to a decrease in infarct size. We did not observe any negative effects of I-1c overexpression in this pre-clinical model of heart failure.
There are some limitations to this study that can be addressed in subsequent studies to further assess the benefits of I-1c gene transfer as a therapeutic approach to heart failure. While we have chosen the 2.5 × 1012 VG dose in our treatment groups, it is unclear what the optimal dose is and furthermore, if there would be any negative effects at elevated doses. Therefore, determination of dose dependence on cardiac performance would be beneficial to determining the safety and efficacy. While we have clearly demonstrated myocardial transfection of I-1c, a more comprehensive myocardial distribution pattern study would be beneficial so that we could better understand correlation of I-1c levels to cardiac function. In addition, while our study design included evaluating the arrhythmogenicity of gene transfer by dobutamine challenges just prior to sacrifice, determination of PLN phosphorylation state was precluded. Now that we have determined that there are no significant increases in rates of arrhythmia, future more extensive studies will focus on determining the PLN phosphorylation as well as more quantitative determination of I-1c mRNA. In addition, while some studies have demonstrated long-term efficacy21 on AAV administration, our three month study may not be predictive of long term I-1c gene delivery effects of will ultimately be, especially regarding the potential immunogenicity of this approach. We have taken the precaution of screening our animals for neutralizing antibodies to mitigate this effect. Therefore, longer-term follow-up would be beneficial. In Addition, scar sizes before gene therapy were not realized in this study. Measurement by MRI will be beneficial in future studies. Finally, while we have shown statistically significant differences between the AAV9.I-1c and control groups, future studies would benefit from groups with higher populations, thus enhancing our ability to measure more subtle cardiac performance changes and alterations in signalling pathways.
Conclusion
In this porcine model of myocardial infarction, we found that once left ventricular dysfunction was established for one month, AAV9.I-1c injection into the coronary arteries induced improvements that reversed the adverse remodelling of the left ventricle,which could be detected two months later. The beneficial effects were specifically on contractility (dP/dt/P) and ejection fraction. In addition, there was evidence that I-1c overexpression halted infarct expansion.
Congestive heart failure is a major cause of morbidity and mortality in the United States. While progress in conventional treatments is making steady and incremental gains to reduce heart failure mortality, there is a critical need to explore new therapeutic approaches. Gene therapy was initially applied in the clinical setting for inherited monogenic disorders. It is now apparent that gene therapy has broader potential in diseases such as congestive heart failure. Improvement in our understanding of the molecular mechanisms of heart failure, along with the development of novel and safer vectors for gene delivery, have led to the identification of novel targets that are difficult to manipulate pharmacologically but may be more amenable to gene therapy. Over the past few years calcium cycling abnormalities and specifically deficiencies in sarcoplasmic reticulum calcium uptake have been hallmarks of advanced heart failure. The complex of SERCA2a-phospholamban-Protein phosphatase 1 has been difficult to target pharmacologically. However the encouraging results from the CUPID trial in which AAV1.SERCA2a gene transfer was found to be safe and demonstrated benefit in clinical outcomes, symptoms, functional status, NT-proBNP and cardiac structure in a phase 2 study, has once again validated calcium cycling as being an important target for heart failure treatment. For this reason, I-1c with its additional benefits is emerging as an important and valid target for the treatment of heart failure. In this research report, we describe positive impacts of I1-c delivery using the AAV9 vector to a clinically relevant large animal model of heart failure.
Acknowledgements
We thank Dorothee Ladage, MD, PhD, for assistance with statistical analyses as well as Lauren Leonardson, Catherine McMahon and James Lough for excellent technical assistance (all Mount Sinai School of Medicine).
Sources of Funding
DL was supported by the German Research Foundation (DFG). This work is supported by Leducq Foundation through the Caerus network (RJH & EGK), by NIH R01 HL093183, HL088434, HL071763, HL080498, HL083156, and P20HL100396 (RJH) and HL26057 and HL64018 (EGK).
Abbreviations
- EF
ejection fraction
- LV
left ventricle / ventricular
- LAD
left anterior descending artery
- MI
myocardial infarction
- HF
heart failure
- dPdt
peak left ventricular pressure rate
- Tau
time constant of isovolumic relaxation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
EGK, RJS and RJH are scientific co-founders of NanoCor Therapeutics. NanoCor is planning to commercialize I-1c for future therapeutic use.
References
- 1.Hasenfuss G, Pieske B. Calcium cycling in congestive heart failure. J Mol Cell Cardiol. 2002;34:951–969. doi: 10.1006/jmcc.2002.2037. [DOI] [PubMed] [Google Scholar]
- 2.Hasenfuss G, Reinecke H, Studer R, Pieske B, Meyer M, Drexler H, Just H. Calcium cycling proteins and force-frequency relationship in heart failure. Basic Res Cardiol. 1996;91 Suppl 2:17–22. doi: 10.1007/BF00795357. [DOI] [PubMed] [Google Scholar]
- 3.Gwathmey JK, Copelas L, MacKinnon R, Schoen FJ, Feldman MD, Grossman W, Morgan JP. Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ Res. 1987;61:70–76. doi: 10.1161/01.res.61.1.70. [DOI] [PubMed] [Google Scholar]
- 4.Gwathmey JK, Hajjar RJ. Intracellular calcium related to force development in twitch contraction of mammalian myocardium. Cell Calcium. 1990;11:531–538. doi: 10.1016/0143-4160(90)90029-t. [DOI] [PubMed] [Google Scholar]
- 5.Kranias EG, Hajjar RJ. Modulation of cardiac contractility by the phospholamban/serca2a regulatome. Circ Res. 2012;110:1646–1660. doi: 10.1161/CIRCRESAHA.111.259754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Florea S, Anjak A, Cai WF, Qian J, Vafiadaki E, Figueria S, Haghighi K, Rubinstein J, Lorenz J, Kranias EG. Constitutive phosphorylation of inhibitor-1 at ser67 and thr75 depresses calcium cycling in cardiomyocytes and leads to remodeling upon aging. Basic Res Cardiol. 2012;107:279. doi: 10.1007/s00395-012-0279-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen G, Zhou X, Florea S, Qian J, Cai W, Zhang Z, Fan GC, Lorenz J, Hajjar RJ, Kranias EG. Expression of active protein phosphatase 1 inhibitor-1 attenuates chronic beta-agonist-induced cardiac apoptosis. Basic Res Cardiol. 2010;105:573–581. doi: 10.1007/s00395-010-0106-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hajjar RJ, Zsebo K, Deckelbaum L, Thompson C, Rudy J, Yaroshinsky A, Ly H, Kawase Y, Wagner K, Borow K, Jaski B, London B, Greenberg B, Pauly DF, Patten R, Starling R, Mancini D, Jessup M. Design of a phase 1/2 trial of intracoronary administration of aav1/serca2a in patients with heart failure. J Card Fail. 2008;14:355–367. doi: 10.1016/j.cardfail.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 9.Jaski BE, Jessup ML, Mancini DM, Cappola TP, Pauly DF, Greenberg B, Borow K, Dittrich H, Zsebo KM, Hajjar RJ. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (cupid trial), a first-in-human phase 1/2 clinical trial. J Card Fail. 2009;15:171–181. doi: 10.1016/j.cardfail.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carr AN, Schmidt AG, Suzuki Y, del Monte F, Sato Y, Lanner C, Breeden K, Jing SL, Allen PB, Greengard P, Yatani A, Hoit BD, Grupp IL, Hajjar RJ, DePaoli-Roach AA, Kranias EG. Type 1 phosphatase, a negative regulator of cardiac function. Mol Cell Biol. 2002;22:4124–4135. doi: 10.1128/MCB.22.12.4124-4135.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky R, Kimball TF, Lorenz JN, Nairn AC, Liggett SB, Bodi I, Wang S, Schwartz A, Lakatta EG, DePaoli-Roach AA, Robbins J, Hewett TE, Bibb JA, Westfall MV, Kranias EG, Molkentin JD. Pkc-alpha regulates cardiac contractility and propensity toward heart failure. Nat Med. 2004;10:248–254. doi: 10.1038/nm1000. [DOI] [PubMed] [Google Scholar]
- 12.Pathak A, del Monte F, Zhao W, Schultz JE, Lorenz JN, Bodi I, Weiser D, Hahn H, Carr AN, Syed F, Mavila N, Jha L, Qian J, Marreez Y, Chen G, McGraw DW, Heist EK, Guerrero JL, DePaoli-Roach AA, Hajjar RJ, Kranias EG. Enhancement of cardiac function and suppression of heart failure progression by inhibition of protein phosphatase 1. Circ Res. 2005;96:756–766. doi: 10.1161/01.RES.0000161256.85833.fa. [DOI] [PubMed] [Google Scholar]
- 13.Rodriguez P, Mitton B, Nicolaou P, Chen G, Kranias EG. Phosphorylation of human inhibitor-1 at ser67 and/or thr75 attenuates stimulatory effects of protein kinase a signaling in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2007;293:H762–H769. doi: 10.1152/ajpheart.00104.2007. [DOI] [PubMed] [Google Scholar]
- 14.Chen G, Zhou X, Nicolaou P, Rodriguez P, Song G, Mitton B, Pathak A, Zachariah A, Fan GC, Dorn GW, 2nd, Kranias EG. A human polymorphism of protein phosphatase-1 inhibitor-1 is associated with attenuated contractile response of cardiomyocytes to beta-adrenergic stimulation. FASEB J. 2008;22:1790–1796. doi: 10.1096/fj.07-097428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wittkopper K, Fabritz L, Neef S, Ort KR, Grefe C, Unsold B, Kirchhof P, Maier LS, Hasenfuss G, Dobrev D, Eschenhagen T, El-Armouche A. Constitutively active phosphatase inhibitor-1 improves cardiac contractility in young mice but is deleterious after catecholaminergic stress and with aging. J Clin Invest. 2010;120:617–626. doi: 10.1172/JCI40545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodriguez P, Mitton B, Waggoner JR, Kranias EG. Identification of a novel phosphorylation site in protein phosphatase inhibitor-1 as a negative regulator of cardiac function. The Journal of biological chemistry. 2006;281:38599–38608. doi: 10.1074/jbc.M604139200. [DOI] [PubMed] [Google Scholar]
- 17.Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky R, Kimball TF, Lorenz JN, Nairn AC, Liggett SB, Bodi I, Wang S, Schwartz A, Lakatta EG, DePaoli-Roach AA, Robbins J, Hewett TE, Bibb JA, Westfall MV, Kranias EG, Molkentin JD. Pkc-alpha regulates cardiac contractility and propensity toward heart failure. Nature medicine. 2004;10:248–254. doi: 10.1038/nm1000. [DOI] [PubMed] [Google Scholar]
- 18.Zolotukhin S, Byrne BJ, Mason E, Zolotukhin I, Potter M, Chesnut K, Summerford C, Samulski RJ, Muzyczka N. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Therapy. 1999;6:973–985. doi: 10.1038/sj.gt.3300938. [DOI] [PubMed] [Google Scholar]
- 19.Rapti K, Louis-Jeune V, Kohlbrenner E, Ishikawa K, Ladage D, Zolotukhin S, Hajjar RJ, Weber T. Neutralizing antibodies against aav serotypes 1, 2, 6, and 9 in sera of commonly used animal models. Mol Ther. 2012;20:73–83. doi: 10.1038/mt.2011.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ishikawa K, Ladage D, Tilemann L, Fish K, Kawase Y, Hajjar RJ. Gene transfer for ischemic heart failure in a preclinical model. J. Vis. Exp. 2011;51 doi: 10.3791/2778. pii: 2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaski BE, Jessup ML, Mancini DM, Cappola TP, Pauly DF, Greenberg B, Borow K, Dittrich H, Zsebo KM, Hajjar RJ. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (cupid trial), a first-in-human phase 1/2 clinical trial. Journal of cardiac failure. 2009;15:171–181. doi: 10.1016/j.cardfail.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.El-Armouche A, Wittkopper K, Degenhardt F, Weinberger F, Didie M, Melnychenko I, Grimm M, Peeck M, Zimmermann WH, Unsold B, Hasenfuss G, Dobrev D, Eschenhagen T. Phosphatase inhibitor-1-deficient mice are protected from catecholamine-induced arrhythmias and myocardial hypertrophy. Cardiovasc Res. 2008;80:396–406. doi: 10.1093/cvr/cvn208. [DOI] [PubMed] [Google Scholar]
- 23.Huang B, Wang S, Qin D, Boutjdir M, El-Sherif N. Diminished basal phosphorylation level of phospholamban in the postinfarction remodeled rat ventricle: Role of beta-adrenergic pathway, g(i) protein, phosphodiesterase, and phosphatases. Circ Res. 1999;85:848–855. doi: 10.1161/01.res.85.9.848. [DOI] [PubMed] [Google Scholar]
- 24.Weaver ME, Pantely GA, Bristow JD, Ladley HD. A quantitative study of the anatomy and distribution of coronary arteries in swine in comparison with other animals and man. Cardiovasc Res. 1986;20:907–917. doi: 10.1093/cvr/20.12.907. [DOI] [PubMed] [Google Scholar]
- 25.Cademartiri F, La Grutta L, Malago R, Alberghina F, Meijboom WB, Pugliese F, Maffei E, Palumbo AA, Aldrovandi A, Fusaro M, Brambilla V, Coruzzi P, Midiri M, Mollet NR, Krestin GP. Prevalence of anatomical variants and coronary anomalies in 543 consecutive patients studied with 64-slice ct coronary angiography. Eur Radiol. 2008;18:781–791. doi: 10.1007/s00330-007-0821-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen J, Petranka J, Yamamura K, London RE, Steenbergen C, Murphy E. Gender differences in sarcoplasmic reticulum calcium loading after isoproterenol. Am J Physiol Heart Circ Physiol. 2003;285:H2657–H2662. doi: 10.1152/ajpheart.00557.2003. [DOI] [PubMed] [Google Scholar]
- 27.Ahmad Z, Green FJ, Subuhi HS, Watanabe AM. Autonomic regulation of type 1 protein phosphatase in cardiac muscle. The Journal of biological chemistry. 1989;264:3859–3863. [PubMed] [Google Scholar]
- 28.Gupta RC, Neumann J, Watanabe AM, Lesch M, Sabbah HN. Evidence for presence and hormonal regulation of protein phosphatase inhibitor-1 in ventricular cardiomyocyte. Am J Physiol. 1996;270:H1159–H1164. doi: 10.1152/ajpheart.1996.270.4.H1159. [DOI] [PubMed] [Google Scholar]
- 29.Neumann J, Eschenhagen T, Jones LR, Linck B, Schmitz W, Scholz H, Zimmermann N. Increased expression of cardiac phosphatases in patients with end-stage heart failure. J Mol Cell Cardiol. 1997;29:265–272. doi: 10.1006/jmcc.1996.0271. [DOI] [PubMed] [Google Scholar]
- 30.El-Armouche A, Rau T, Zolk O, Ditz D, Pamminger T, Zimmermann WH, Jackel E, Harding SE, Boknik P, Neumann J, Eschenhagen T. Evidence for protein phosphatase inhibitor-1 playing an amplifier role in beta-adrenergic signaling in cardiac myocytes. FASEB J. 2003;17:437–439. doi: 10.1096/fj.02-0057fje. [DOI] [PubMed] [Google Scholar]
- 31.Nicolaou P, Rodriguez P, Ren X, Zhou X, Qian J, Sadayappan S, Mitton B, Pathak A, Robbins J, Hajjar RJ, Jones K, Kranias EG. Inducible expression of active protein phosphatase-1 inhibitor-1 enhances basal cardiac function and protects against ischemia/reperfusion injury. Circ Res. 2009;104:1012–1020. doi: 10.1161/CIRCRESAHA.108.189811. [DOI] [PMC free article] [PubMed] [Google Scholar]