

Abstract
Background
The ascomycete fungus Ceratocystis cacaofunesta is the causal agent of wilt disease in cacao, which results in significant economic losses in the affected producing areas. Despite the economic importance of the Ceratocystis complex of species, no genomic data are available for any of its members. Given that mitochondria play important roles in fungal virulence and the susceptibility/resistance of fungi to fungicides, we performed the first functional analysis of this organelle in Ceratocystis using integrated “omics” approaches.
Results
The C. cacaofunesta mitochondrial genome (mtDNA) consists of a single, 103,147-bp circular molecule, making this the second largest mtDNA among the Sordariomycetes. Bioinformatics analysis revealed the presence of 15 conserved genes and 37 intronic open reading frames in C. cacaofunesta mtDNA. Here, we predicted the mitochondrial proteome (mtProt) of C. cacaofunesta, which is comprised of 1,124 polypeptides - 52 proteins that are mitochondrially encoded and 1,072 that are nuclearly encoded. Transcriptome analysis revealed 33 probable novel genes. Comparisons among the Gene Ontology results of the predicted mtProt of C. cacaofunesta, Neurospora crassa and Saccharomyces cerevisiae revealed no significant differences. Moreover, C. cacaofunesta mitochondria were isolated, and the mtProt was subjected to mass spectrometric analysis. The experimental proteome validated 27% of the predicted mtProt. Our results confirmed the existence of 110 hypothetical proteins and 7 novel proteins of which 83 and 1, respectively, had putative mitochondrial localization.
Conclusions
The present study provides the first partial genomic analysis of a species of the Ceratocystis genus and the first predicted mitochondrial protein inventory of a phytopathogenic fungus. In addition to the known mitochondrial role in pathogenicity, our results demonstrated that the global function analysis of this organelle is similar in pathogenic and non-pathogenic fungi, suggesting that its relevance in the lifestyle of these organisms should be based on a small number of specific proteins and/or with respect to differential gene regulation. In this regard, particular interest should be directed towards mitochondrial proteins with unknown function and the novel protein that might be specific to this species. Further functional characterization of these proteins could enhance our understanding of the role of mitochondria in phytopathogenicity.
Keywords: Cacao wilt disease, Ceratocystis cacaofunesta, Mitogenomics, Mitochondrial proteome, Fungal virulence
Background
Ceratocystis cacaofunesta is an ascomycete fungus (Class Sordariomycetes, Order Microascales) that causes wilt disease in cacao (Theobroma cacao). This fungus is a member of the Latin American clade of the Ceratocystis fimbriata complex [1,2], a taxonomic group that includes species with high genetic variability and wide host ranges [3,4]. These species cause canker and wilt diseases in many economically important crops, such as Coffea arabica and Eucalyptus spp.[5]. Engelbrecht and Harrington [6] proposed that host specialization may influence speciation in this group.
C. cacaofunesta is indigenous to Central and South America [1,2]. In Brazil, this pathogen was first reported in the Amazon region [7]. In the 1990s, C. cacaofunesta was introduced to the southern region of Bahia, which is the largest Brazilian cacao-producing state [8]. This fungus is able to penetrate cacao trees through stem wounds that are caused either by insects (natural vectors) or through infected cutting tools [9]. Unlike other cacao diseases that primarily affect branches and fruits, such as Witches’ broom disease and Frosty pod, wilt disease is a systemic infection that damages the entire plant. The fungus enters its host through the secondary xylem, resulting in the formation of deep spots and leading to the obstruction of water and nutrient transport [10]. Consequently, the plant turns yellow and then brown; the infection culminates in the wilting and sudden death of the tree. This disease is responsible for reductions in the cacao population in plantation areas, which has resulted in great economic losses in the affected regions. Extensive damages have also been reported in Trinidad [11], Venezuela and Colombia [12].
Several studies have attempted to characterize the genetic variation, aggressiveness and host specialization of different populations of C. fimbriata complex including C cacaofunesta[6,13-15]. However, little is known at the molecular level regarding this fungus and its interaction with cacao. Notably, no genomic data are available for any members of the Ceratocystis genus. In 2011, our group initiated the C. cacaofunesta Genome Project (http://www.lge.ibi.unicamp.br/ceratocystis), with the goal of understanding the mechanisms that underlie the interactions between C. cacaofunesta and Theobroma cacao in the development of wilt disease. We initially focused on the study of C. cacaofunesta mitochondria for two reasons: (i) their potential role in fungal pathogenesis and (ii) the relevance of this organelle as a target for fungicides [16].
In addition to their canonical function as an energy-producing compartment, mitochondria are involved in multiple cellular processes [17]. These organelles play important roles in calcium homeostasis [18], the biosynthesis of iron-sulfur clusters [19], lipid and amino acid metabolism, aging and the signaling of programmed cell death [20]. Due to the importance of these processes, mitochondrial dysfunctions cause serious consequences for the cell and, ultimately, the entire organism. Functionally compromised mitochondria are associated with senescence in non-pathogenic fungi, such as Podospora anserina[21] and N. crassa[22].
Mitogenomics has become a useful tool for evolutionary studies [23], and the continuous advances in this field have contributed to the understanding of the diverse topology, organization and structure of mtDNA in fungi [24]. However, as mtDNA encodes approximately 1% of the mitochondrial proteome, scant information concerning the roles of mitochondria in the metabolism, development and lifestyle of organisms has been gleaned from exclusive analyses of the mitochondrial genome. Fungal mtDNA generally contains 14 genes that encode hydrophobic subunits of the respiratory chain complexes, two genes that encode ribosomal RNAs (small and large subunits) and genes that encode a full set of tRNAs [25]. The other 99% of the mitochondrial proteome is encoded by nuclear DNA and imported into the mitochondria [26]. Nuclear-encoded mitochondrial proteins (NMP) are produced by cytosolic ribosomes and are targeted to the proper mitochondrial subcompartment [27-29].
To understand mitochondrial function, studies using genomic, transcriptomic and proteomic approaches have been performed in several types of organisms, such as fungi [30], plants [31] and humans [32]. The first mitochondrial analysis using integrative proteomic and genomic approaches in fungi was conducted in S. cerevisiae[33]. By overcoming the individual limitations of each technique, this integrative study produced a powerful tool for the prediction of mitochondrial processes in yeast. The S. cerevisiae mitochondrial proteome contains an estimated 1,000 proteins, of which 851 were identified using proteomic assays [34]. In N. crassa, proteomic studies have led to the identification of 438 mitochondrial proteins [35]. Proteomics has previously been used to illuminate central processes in phytopathogenic fungi [36,37]. Certain more recent studies have analyzed of mycelial, conidiospores and haustoria proteomes [37-41], fungal secretomes and proteomes that are associated with fungal virulence [42]. However, the mitochondrial proteomes of phytopathogenic fungi have not been explored.
In the present study, we performed a global analysis of C. cacaofunesta mitochondria using an integrative approach (mitogenomics, transcriptomics and proteomics). We predicted the C. cacaofunesta mitochondrial proteome (mtProt), including mitochondrial-encoded proteins and mitochondrial proteins encoded in the nuclear genome (NMP). Moreover, 27% of predicted mtProt was validated by experimental proteome analysis (LC-MS/MS). We focused on comparisons with available mitochondrial proteomes from non-pathogenic model fungi to increase our knowledge on the role of mitochondria in pathogenicity. The present study is the first partial genomic analysis of a Ceratocystis species to be published. Additionally, to the best of our knowledge, this study is pioneering in that it presents a global analysis of the mitochondrial proteome of a phytopathogenic fungus using this integrative approach.
Results and discussion
C. cacaofunesta mitochondrial genome structure
The complete mitochondrial genome of Ceratocystis cacaofunesta was assembled with 2,153-fold coverage as a single circular molecule comprising 103,147 bp (Figure 1). It is the fourth largest published fungal mitochondrial genome, and the second largest among the Sordariomycetes, after that of Chaetomium thermophylum (127 kb) [43]. However, the differences in the sizes of fungal mitochondrial genomes are not correlated with differences in their number of conserved genes [44,45]. Therefore, C. cacaofunesta mtDNA contains 15 genes that encode the conserved proteins NADH dehydrogenase subunits 1 to 6 and 4 L (nad1 to nad6 and nad4 L); cytochrome c oxidase subunits I, II and III (cox1, cox2 and cox3); ATP synthase subunits 6, 8 and 9 (atp6, atp8 and atp9); apocytochrome b (cob); and the ribosomal protein S3 (rps3), (Figure 1). With the exception of rps3, all of these conserved genes are involved in oxidative phosphorylation and ATP synthesis. Genes that encode the small and large rRNA subunits (rns and rnl, respectively) were also identified. The rps3 gene is located within a group I intron (small RNAs that have ability to self-splice from RNA transcripts) of the rnl gene, as has been described for many other Sordariomycetes [46,47]. It has been proposed that the structure of one gene (e.g., rps3) within another (e.g., rnl) guarantees that they are co-transcribed and that the stoichiometry of the two components is adequate for ribosome biogenesis [46]. In addition to rRNA and conserved coding genes, the tRNAscan-SE program [48] identified 31 putative tRNAs for the 20 standard amino acids and a possible suppressor tRNA (amber tRNA). All of the tRNAs, rRNAs and protein-coding genes are oriented in the same direction (clockwise in Figure 1). Also, the conserved genes are organized in the same four synthetics units described for Sordariomycetes[49], except by minor changes in trn genes distribution. The gene pair nad2-nad3 is partially overlapping as the nad2 ORF extends 49 nucleotides into the nad3 reading frame (Figure 1). It has been proposed that the preservation of syntenic units could play a functional role, enabling the polycistronic transcription of these genes [49].
Figure 1.

Map of C. cacaofunesta mitochondrial DNA. The numbers along the outermost circle are the DNA coordinates in kb, beginning at 12 o’clock and continuing clockwise. The scales for the GC content (in %, light blue histogram) and the GC skew (dimensionless, black curve) are at 12 o’clock in the innermost circle. Conserved coding genes (reds), pseudogenes (red dots), tRNAs and rRNAs (green) are shown in the outermost circle. Introns (light red) and intronic ORFs (red) are shown in the middle circle. The intronic ORFs of each gene are numbered as oi (intron number). The domains of each intronic ORF are indicated as LL (for LAGLIDADG) or G (for GIY-YIG). Similar intronic ORFs (with more than 50% coverage and identity) are linked by yellow ribbons.
The annotation of the C. cacaofunesta mt genome revealed that introns form 48.7% of this DNA molecule, explaining its large size. The invasion of mitochondrial genes by group I introns is a primary reason for the wide variation in fungal mitochondrial genome sizes (Additional file 1) [50]. A total of 37 mitochondrial group I introns, with an average size of 1,535 bp, were identified in the conserved coding and rRNA genes. These introns are distributed within conserved genes throughout the C. cacaofunesta mitochondrial genome, with cox1 harboring the largest number of these elements (10 introns) (Figure 1, Additional file 1). Cox1 has been described as a reservoir for mitochondrial group I introns in fungi, harboring as many as 18 of these elements in Agaricus bisporus[51]. Of the total of mitochondrial group I interns, 36 encoded homing endonuclease genes (HEG) (Figure 1, Additional file 1). The dynamic properties of these introns suggest that they can be moved into other regions of the genome and between the genomes of phylogenetically distant species [52].
Although the presence of mitochondrial plasmids is a common feature of Sordariomycetes, such as G. zeae[53], N. crassa[54]and P. anserine[55], no integrated or free linear mitochondrial plasmids were identified in the C. cacaofunesta mtDNA. However, a pseudogene with similarity to the RNA polymerase (rpo) of the P. anserina mitochondrial plasmid pAL2-1(e-value: 4e-92, 100% of query coverage and 57% of positives matches) was identified downstream of the cox1 gene (Figure 1, labeled, “pseudo rpo”). The presence of this pseudogene with plasmidial origin suggests an ancestral plasmid insertion in this region. We found no evidence of inverted repeats flanking this pseudogene.
The analysis of the C. cacaofunesta mtDNA also revealed an average of 26.8% G+C residues, which were uniformly distributed throughout the sequence (Figure 1, blue histogram). The intronic ORFs exhibited a slightly lower GC content (25.2%) than did the conserved genes (26.5%). Moreover, a GC skew analysis was performed, and the shift points of the GC skew graphs were consistent with the loci of the origin (org) and termination (ter) of replication in bacteria [56] and certain fungi [57].
Predicted mitochondrial proteome and annotation
Considering that approximately 99% of the mitochondrial proteins are encoded in the nucleus [25] and that we have the complete genome sequences of C. cacaofunesta, we estimated the total mitochondrial proteome of C. cacaofunesta based on the numbers of mitochondrial- and nuclear-encoded proteins.
The use of these combined approaches generated a predicted mitochondrial proteome for C. cacaofunesta that consisted of 1,124 polypeptides, with 1,072 NMP and 52 mitochondrial-encoded proteins. Of the total proteins, 584 were identified using the predictor softwares (TargetP and WoLF PSORT), 309 were identified only by homology analysis and 179 were identified using both methods (Figure 2, Venn diagram). The predicted C. cacaofunesta mtProt has a similar size to the mtProt that was estimated for S. cerevisiae (approximately 1,000 polypeptides) [58]; whereas the A. thaliana mtProt is estimated to contain approximately 850 polypeptides [31], and the mouse mtProt contains about 1,500 polypeptides [59].
Figure 2.

A Flowchart of the mitochondrial proteome prediction (blue) and experimental proteome (green). The complete set of predicted mitochondrial proteins is surrounded by a black dotted line.
The predicted mtProt of C. cacaofunesta was annotated using AutoFACT [60] to summarize the results of the BLAST [61] searches of the NR/NCBI, KEGG [62] and UniRef90 databases [63]. An independent search of the CDD (conserved domain) database [63] was also performed. The Blast2GO [64] program was used to produce Gene Ontology (GO) classification (see Material and Methods). Table 1 summarizes the main results of annotation. From the predicted MtProt, 38 codifying genes result in no hits in AutoFACT. These genes were collectively annotated as hypothetical unconserved genes. Three hundred forty-nine other genes were similar only with genomic sequences and/or predict hypothetical proteins; these genes were therefore annotated as hypothetical conserved genes. Lastly, the remaining 737 genes are similar to at least one other known (described) gene in the databases that were used for AutoFACT annotation and were therefore considered to be known conserved genes. The GO annotation assigned 724 proteins to at least one level of ontology (411 proteins were assigned to cellular component, 619 to molecular function and 599 to biological process). A total of 190 enzymes were identified by Blast2GO, from which, 138 were distributed in 78 metabolic pathways. The most represented metabolic pathway were nitrogen metabolism (41 enzymes) and oxidative phosphorylation (40 enzymes). Additional file 2 shows the entire catalogue of predicted C. cacaofunesta mitochondrial proteins and their respective annotation.
Table 1.
Summary of MtProt annotation
| Gene category | Predicted MtProt | Experimental MtProt |
|---|---|---|
| Hypothetical unconserved |
38 |
1 |
| Hypothetical conserved |
349 |
83 |
| Known conserved |
737 |
220 |
| Total | 1,124 | 304 |
Transcriptomic analysis
A global analysis of the C. cacaofunesta transcriptome was performed using large-scale mRNA sequencing (RNA-Seq), as described in the corresponding section of Methods. This technique is widely used to analyze gene expression and to validate gene predictions [65-68]. This methodology was recently used to analyze mitochondrial gene expression [69,70]. Approximately 55 million of reads were generated in two biological replicates (CER1 and CER2). As expected, only a very small number of reads mapped to mitochondrial genes (400 from CER1 and 600 from CER2) (Additional file 2A). The RNAseq methodology used here requires a mRNA polyA tail, but RNAs that are transcribed from fungal mtDNAs generally lack this feature. Polyadenylation of mitochondrial mRNA appears to be restricted to higher eukaryotes [71]. Therefore, the 1,000 RNAseq reads aligned with C. cacaofunesta mtDNA most likely represents artifacts.
Genes with RPKM < 1 were considered to not be expressed. Among the total reads that mapped to nuclear genes 21.6% (approximately 12 million) align with genes that encode mitochondrial proteins (5.6 and 6.2 million from CER1 and CER2, respectively). This result indicates that the average expression of genes that encode NMPs is approximately 2.18 times greater than the expression of the other nuclear genes (mean RPKMs of 73.00 and 33.36, respectively) (Figure 3). Of the 1,072 NMP genes, only 10 presented RPKM values of less than 1, and 398 had RPKM values greater than 100 (Additional file 2B). More highly expressed genes are involved in protein synthesis and oxidative phosphorylation, which is consistent with the high rates of fungal growth in the culture conditions. Approximately 99% of the NMP codifying genes are expressed. RNAseq also aids in the identification of new genes. Based on our prediction, a total of 38 novel putative genes encode mitochondrial proteins. However, RNAseq analysis indicated that only 33 of these are expressed (RPKM > 1) (Additional file 2B).Transcriptomic information consists of the complete set of transcripts and their abundance in the cell in specific physiological conditions [36]. Therefore, the transcriptome of fungi in vitro may be quite different when compared with the transcriptome of fungi during its interaction with its host. Although we are unable to make conclusions regarding expression profile of C. cacaofunesta genes during pathogenesis based on these data, the transcriptome analysis of C. cacaofunesta mitochondrial genes in vitro provides valuable information with respect to validating gene prediction, including the discovery of 33 novel putative genes.
Figure 3.
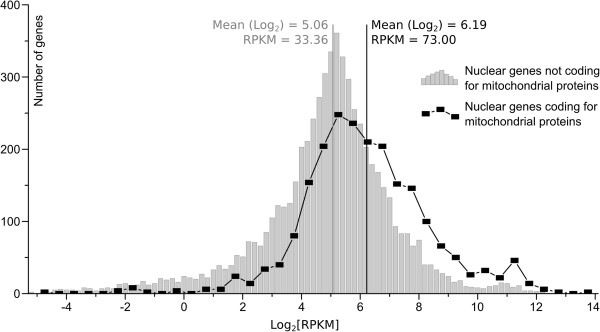
The distribution of RPKM values (in log2 scale) in mitochondrial (black line) and non-mitochondrial (gray histogram) genes. The transcription of the mitochondrial genes was 2.18 x higher (on average) than that of the non-mitochondrial genes.
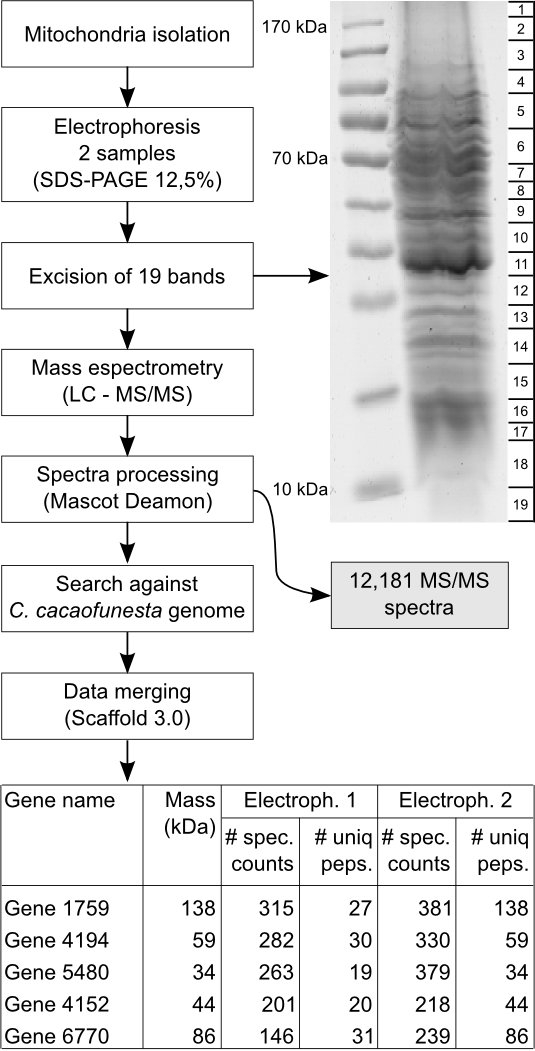
Experimental mitochondrial proteome
The isolated mitochondrial proteome was electrophoretically separated on a 12.5% SDS-PAGE gel, and 19 bands were excised from the gel and subjected to LC-MS/MS analysis in the corresponding section (Additional file 3). A total of 12,181 MS/MS spectra were generated and confidently assigned to 476 unique proteins with statistically significant (p < 0.05) Mascot-based scores and a 9.1% FDR (Additional file 4, Sheet 4A). Among the 476 proteins that were identified using LC-MS/MS, the 5 most abundant proteins presented more than 140 spectral counts in both samples (Additional file 4, Sheet 4A). The size of these proteins ranged from 34 to 138 kDa, and there was not a molecular weight bias in their abundance.
Among the total proteins that were identified using LC-MS/MS, 64% (304) were contained within the predicted mtProt (Figure 2). This result indicates that approximately 27% of the predicted mtProt was experimentally validated. Accordingly, the mitochondrial proteome analysis by LC-MS/MS is capable of identifying 23%-40% of the known mitochondrial proteins but does not capture low-abundance proteins [59], except for proteins that are produced under specific circumstances [72]. Experimental mitochondrial proteome analysis of N. crassa has identified 473 proteins that correspond to 169 unique genes; these represent 20% of the total mtProt that has been described for this species [73].
Of the 304 predicted mitochondrial proteins that were validated by LC-MS/MS, we identified 5 mitochondrial-encoded conserved proteins (ATP6, COB, COX1, COX2 and NAD2) and an intronic ORF (oi5cox2). Table 2 lists a subset of the experimentally identified NMP proteome that was associated with important metabolic process. According with transcriptome analysis, the set of experimentally identified proteins reflects the metabolic status of the fungal cell in rich growth conditions.
Table 2.
Proteins identified by LC-MS/MS involved in the respiration metabolic process
| Methabolic Process | Description | Number |
|---|---|---|
| Pyruvate dehydrogenase complex |
Pyruvate Dehydrogenase (lipoamide) alpha |
1 |
| Pyruvate Dehydrogenase (lipoamide) beta |
1 |
|
| Pyruvate Dehydrogenase Kinase |
1 |
|
| Dihydrolipoamide S-acetyltransferase |
1 |
|
| Total |
4 |
|
| Tricarboxylic acid cycle |
2-methylcitrate dehydratase |
2 |
| Citrate Synthase |
2 |
|
| Isocitrate Dehydrogenase1 |
1 |
|
| Isocitrate Dehydrogenase 2 |
1 |
|
| Isocitrate dehydrogenase |
1 |
|
| Homoisocitrate dehydrogenase |
1 |
|
| Isocitrate lyase |
1 |
|
| Total |
9 |
|
| Oxidative phosphorylation |
Complex I |
13 |
| Complex II |
3 |
|
| Complex III |
3 |
|
| Complex IV |
2 |
|
| Complex V |
5 |
|
| Total | 25 |
Remarkably, we identified 83 conserved hypothetical proteins and 1 novel protein in the experimental proteome that exhibited putative mitochondrial localization (Additional file 4, Sheet 4B). We believe that these results are an important contribution because they give the first experimental evidence of the existence of these putative mitochondrial proteins. The functional characterization of these proteins and their association with particular mitochondrial pathways is a great challenge but could certainly improve our understanding of this organelle.
We also identified 172 proteins that were not predicted to be mitochondrial based on our analysis (Figure 2, Additional file 4, Sheet 4C). Manual annotation was used to elucidate the composition of this group of proteins. Total number of mitochondrial proteins identified using LC-MS/MS could reach 388 (304 from prediction, and 84 from manual annotation), representing 81% of the total experimental proteome. This result indicates that the enriched-mitochondrial preparation of C. cacaofunesta was performed with high efficiency. However, the standard methodology chosen here was automatic annotation due to the high stringency of the parameters; hence, the 84 proteins that were classified as putative mitochondrial based on manual annotation were not included in the final catalogue of mitochondrial proteins that are proposed here. The experimental proteome was used to validate a fraction of predicted proteome, focusing on the identification of hypothetical and novel proteins.
One of the main contributions of this study is the experimental identification of two groups of predicted mitochondrial proteins with no assigned function: (i) proteins for which there was no experimental evidence (hypothetical proteins) and (ii) novel proteins. Table 1 shows the total of proteins of groups (i) and (ii) based both on the predicted and the experimental proteomes. Of the 349 predicted hypothetical proteins, 83 were identified by mass spectrometry, as mentioned above. With respect to group (ii), the predicted mtProt identified 38 novel putative proteins. However, RNAseq confirmed the expression of 33 of them, and one protein was identified experimentally. The 84 hypothetical proteins that were identified by mass spectrometry were reclassified as conserved unknown function (Additional file 4, Sheet 4B). The S. cerevisiae mitochondrion is the best-understood and characterized molecularly [24,29,33,72]. However, approximately 19% of the identified proteins remain have no known function [29]. The predicted C. cacaofunesta mtProt contains approximately 34% of proteins with unknown function. These data suggest that we are far from fully understanding the function of this organelle, a fact that reflects its functional plasticity. Further functional characterization of these proteins may enable a better understanding of mitochondrial function. It is important to highlight that there are more than 65 known Sordariomycetes genomes published, and we suggest that the 38 predicted novel genes (33 expressed) may be specific to this species. Moreover, it is very likely that these proteins are shared with other species from the Ceratocystis fimbriata complex, a hypothesis that will investigated with the upcoming availability of other Ceratocystis genomes. Our results provide a framework for examining the involvement of novel proteins in mitochondrial pathways.
The functional annotation of the C. cacaofunesta mitochondrial proteome and a comparison with those of S. cerevisiae and N. crassa
We focused on the GO annotation to perform a global function analysis of the predicted C. cacaofunesta proteome. Putative functions were assigned to the predicted mtProt using GO and were manually grouped into 12 categories according to biological process. Only the amino acid, carbohydrate and lipid metabolism categories were queried using specific GO IDs: GO:0006519, GO:0005975 and GO:0006629, respectively. The remaining 9 categories included several related GO IDs (Additional file 5). The GO distribution is shown in Figure 4, together with the GO distributions of N. crassa and S cerevisiae mtProt. The largest GO category is “other” (27.6%), which was expected given that it contained proteins from 26 small categories (Additional file 6, Sheet 6A). The second-most represented category is “genome maintenance and transcription”, which includes 21.7% of all of the predicted mitochondrial proteins. Based on sequence, approximately 19% of the mtProt are related to protein metabolism, and 18.7% of them are assigned to “transport of metabolites”. This latter category includes ATP-binding cassette (ABC) transporters, ion transport families, protein transporters and other metabolite transporters. More than 15.2% of the proteins were directly involved in energy metabolism, including parts of the oxidative phosphorylation machinery (OXPHOS), members of the tricarboxylic acid cycle (TCA), and the pyruvate dehydrogenase complex (PDH). Among underrepresented categories were the following: (i) response to stimulus and (ii) signaling, which included 5.5% and 4.0% of all of the predicted mitochondrial proteins, respectively (Figure 4). At 3.0%, lipid metabolism was the smallest category represented in the C. cacaofunesta mtProt.
Figure 4.
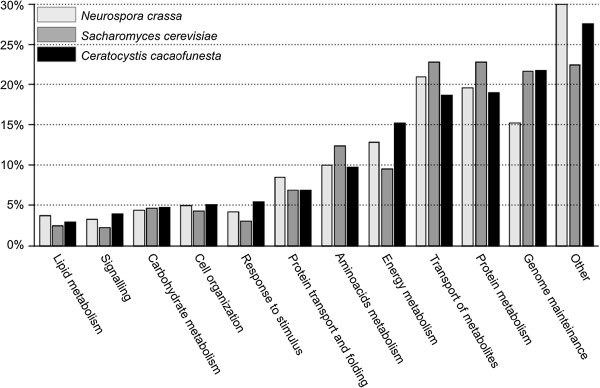
Distribution of Gene Ontology (GO) categories for the predicted mitochondrial genes of N. crassa (light gray), S. cerevisiae (dark gray) and C. cacaofunesta (black).
We used the same GO categorization criteria as above to annotate the S. cerevisiae and N. crassa mitochondrial proteomes [72,73] to compare their global functional profiles to that from C. cacaofunesta (Figure 4 and Additional file 6, Sheet 6B and 6C). Due to the redundancy of GO categories associated with a same protein, it is difficult to perform a statistical analysis of these data. However, Figure 4 clearly shows that there is a similar pattern to the distribution of mitochondrial proteins in different GO functional categories among the three species. Considering the important role of mitochondrial function to basic cell metabolism, the conservation of core mitochondrial functions is expected [74]. However, it is known that the expansion or reduction in the size of individual protein families, and hence individual functions, plays a specific role in lifecycle of the organism. In phytopathogenic fungi, differences in protein families have been associated with the importance of the related functions in plant-pathogen interactions [5]. Considering that C. cacaofunesta is a phytopathogenic fungus and that S. cerevisiae and N. crassa are non-pathogenic, it would be plausible to expect more differences between these species regarding the distribution of proteins in functional categories that are related to transport of metabolites, signaling, and defense [75]. However, no such major differences were identified. According the most recent publication on the subject, certain categories that were represented in this analysis, such as signaling, energy metabolism, lipid metabolism, protein transport and protein folding, have similar protein percentages as those that have been described for the S. cerevisiae mtProt [29]. These results suggest that the involvement of the mitochondria in virulence and pathogenicity may rely on specific proteins and/or in the convergence in time and space of different components.
The same GO categorization was performed using the experimental proteome. For this purpose, the 388 proteins with potential mitochondrial localizations that had been identified using LC-MS/MS were subjected to a GO analysis (Additional file 6, Sheet 6A). The results shown in Figure 5 revealed that the categories most strongly represented in the experimental proteome were transport of metabolites (23.9%), energy metabolism (20.4%) and protein metabolism (19.7%). The classifications of the experimental proteins reflect the C. cacaofunesta culture conditions (malt, yeast extract and agar). The fungus grows rapidly, suggesting an intense metabolism, including the synthesis of enzymes and the import of proteins and metabolites. Moreover, the availability of glucose as the primary carbon source likely enhanced the expression of genes that are involved in energy metabolism. Vodisch and coworkers (2011) examined the mtProt of Aspergillus nidulans as a part of the global fungal proteome [76]. These authors observed a clear relationship between the culture conditions and the enriched classes of experimentally identified proteins.
Figure 5.
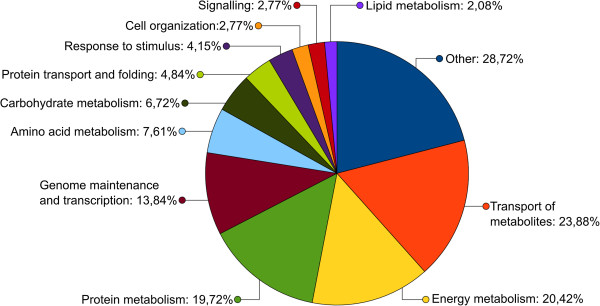
Distribution of Gene Ontology (GO) categories for the C. cacaofunesta nuclear genes encoding the mitochondrial proteins that were identified using LC-MS/MS.
Notably, the global analysis of the C. cacaofunesta mitochondrial function that was based on functional classification (GO) of the experimentally identified proteins (Figure 5) reflects the functional grouping of the full set of predicted mitochondrial proteins (Figure 4). Considering that we performed mass spectrometry only for fungi in cultured conditions, which appears to be far from mimicking fungal growth in nature, the similarity of functions between the predicted and experimental proteomes reinforces the importance of the core mitochondrial functions. Additionally, this similarity suggests that the mitochondrial role in the adaptation of the fungus to certain growth conditions, including the environment of its host, depends of specific sets of proteins, pathways and/or differential gene regulation rather than on major changes in mitochondrial function.
C. cacaofunesta global proteome survey
It is well established that mitochondrial function is important for the virulence and pathogenicity of fungi [16,77]. Here, we demonstrated that the global proteome compositions of S. cerevisiae, N. crassa and C. cacaofunesta do not contain major differences. However, of these three fungi, C. cacaofunesta is the only pathogenic fungus, which prompted us to perform a more detailed investigation of its proteome composition.
Early stages of necrotrophic infections, like those caused by C. cacaofunesta, are associated with host cell death, the production of secondary metabolites and the accumulation of reactive oxygen species (ROS) [78,79]. Mitochondrial proteins that are associated with detoxification may play an important role in the success of pathogen colonization. We therefore analyzed the predicted mtProt of C. cacaofunesta to identify proteins that may play important roles in pathogenicity and sensitivity/resistance to fungicides.
We focused the search on proteins that have been previously related to virulence, specifically ATP-binding cassette (ATP) transporters, aldehyde dehydrogenase, alternative NADH dehydrogenase, alternative oxidase, the mitochondrial carrier FOW1, glutathione S-transferase and hypoxia-related protein. Figure 6 shows a schematic summary of a subset of the predicted proteins in the mtProt of C. cacaofunesta with potential involvement in the pathogenicity of this fungus.
Figure 6.
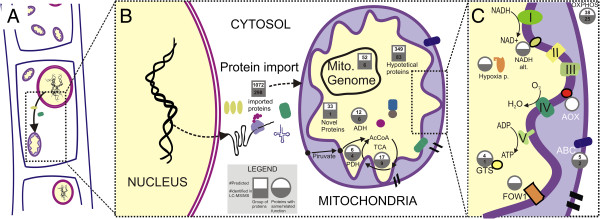
Schematic summary of the C. cacaofunesta mitochondrial proteome survey. (A) Detailed representation of a hypha, including the nucleus and the mitochondria. (B) Nuclear – mitochondrial communication represented by the protein import (predicted/identified by LC-MS/MS). In the mitochondria are represented proteins involved in pyruvate dehydrogenase complex (PDH) and tricarboxylic acids cycle (TCA). Also, are represented aldehyde dehydrogenase (ADH); hypothetical proteins; novel proteins and proteins encoded in mtDNA. (C) Zoom of the mitochondrial subcompartments. Proteins involved in oxidative phosphorylation (OXPHOS) and polypeptides with probable role in pathogenicity are depicted. Cytochrome-dependent respiratory chain components (complexes I to V); alternative respiratory pathway including alternative NADH (NADH alt) and alternative oxidase (AOX); glutathione S-transferases (GST); ATP-binding cassette transporters (ABC) and protein homologous to mitochondrial carrier of Fussarium oxysporum (FOW1).
The C. cacaofunesta predicted mtProt contains 5 proteins classified as ABC transporters. All of the genes codifying these proteins were expressed under the tested conditions, with average RPKM values of 140.6 and 1,463.7. Mass spectrometry identified 2 of this class of transporters (Additional file 2, Sheet 2B; Additional file 4, Sheet 4A). The ABC transporter of a Nectria haematococca (NhABC1) proved to be an important virulence factor that contributes to the N. haematococca tolerance of phytoalexin during its interaction with the host [79]. Furthermore, ABC transporters have been associated with fungicide resistance [80].
Aldehyde dehydrogenase (ADH) has also been implicated in the pathogenicity of fungi. ADH is regarded as a detoxification enzyme due to its role in the metabolism of intermediates and exogenous aldehydes [81]. An analysis of adh expression in Clasdosporium fulvum, both in vitro and in vivo, during its interactions with tomato indicated that the expression of this gene was associated with infection and starvation conditions [82]. Interestingly, the predicted C. cacaofunesta mtProt survey identified 12 ADH-encoding genes with confirmed expression (RPKMs > 10), and 6 ADH proteins were identified by LC-MS/MS. Moreover, glutathione S-transferases (GSTs) are a large family of proteins that are involved in cellular detoxification processes during oxidative stress responses [83] and may be involved in fungicide resistance [84]. Mao and coworkers (2012) suggested a link between the number of GST-encoding genes in the fungal genome and its lifecycle [85]. The predicted C. cacaofunesta mtProt contains 4 proteins that are homologous to GST. The RNA-Seq analysis indicated varying average expression levels, with RPKM values of as high as 150; one of those proteins was identified in the experimental proteome (Additional file 2, Sheet 2B; Additional file 4, Sheet 4B).
Moreover, components of the alternative respiratory chain have been related to fungal pathogenicity, specifically alternative NADH dehydrogenase [86] and alternative oxidase (AOX) [87]. Studies in which alternative NADH dehydrogenase was blocked in Aspergillus niger demonstrated the role of this protein in protection against oxidative stress [88]. C. cacaofunesta has one encoded alternative NADH dehydrogenase gene with RPKM values > 60; this gene was also identified in the experimental proteome (Additional file 2, Sheet 2B; Additional file 4, Sheet 4B). Moreover, the predicted mtProt included a putative AOX. This enzyme has been implicated in fungal protection against oxidative stress, pathogenicity and fungicide resistance [89,90]. C. cacaofunesta AOX was not identified in the experimental proteome, but the gene aox exhibited detectable expression, with a RPKM value of approximately 88 (Additional file 2, Sheet 2B; Additional file 4, Sheet 4A). Recently, Thomazella and coworkers (2012) provided strong evidence for the involvement of AOX in maintaining the infective phase of the basidiomycete Moniliophthora perniciosa[91].
The deletion of a Fusarium oxysporum mitochondrial carrier-encoding gene (fow1) caused the loss of its ability to infect plant tissues [92]. A putative FOW1 protein was identified in the C. cacaofunesta predicted mtProt. The RNA-Seq data indicated that the FOW1 homologue in C. cacaofunesta was highly expressed (with an RPKM value of 536), and its product was identified in the experimental proteome (Additional file 2, Sheet 2B; Additional file 4, Sheet 4A).
It has been reported that the ability of certain fungi to withstand different levels of hypoxia is a critical virulence factor in terms of fungal pathogenicity in mammals [93]. Hypoxia-related genes have been identified as important virulence factors for Aspergillus fumigatus and Candida albicans given that they are exposed to low oxygen concentrations during infection [93,94]. Oxygen levels in the secondary xylem, the plant tissue that is infected by C. cacaofunesta, are as low as are observed in human tissues that are infected by these fungal pathogens, reaching levels less than 5% [93,95]. This observation suggests C. cacaofunesta should possess mechanism with which to adapt to hypoxia. In this regard, the predicted C. cacaofunesta mtProt contains one protein that has a conserved hypoxia response domain. This gene was expressed in in vitro conditions, with an RPKM value as high as 259 and its product was also identified in the experimental proteome (Additional file 2, Sheet 2B; Additional file 4, Sheet 4A). Although hypoxia response mechanisms are more complex and involve a series of regulatory pathways [96], these results open the way for the exploration of the role of C. cacaofunesta mitochondria in fungal phytopathogens responses to hypoxia.
Due to inherent limitations for each of the techniques that were used to predict the subcellular localization of proteins, we chose to perform a stringent integrative approach to predict the C. cacaofunesta mtProt. We therefore must state that the list of mitochondrial proteins proposed here as the predicted mitochondrial proteome of C. cacaofunesta may not correspond to the full set of mitochondrial polypeptides. This mtProt survey is a preliminary analysis due to the relatively small number of proteins discussed but reveals how these data can be exploited in future studies. The presented genome and estimated proteome data provide a starting point for the improved characterization of C. cacaofunesta mitochondrial function in two ways: (i) facilitating the identification of proteins that may be relevant to the lifestyle of this fungus and (ii) expanding the literature that relates to studies of mitochondrial function and evolution to the discovery of new mitochondrial genes. In this regard, the identification of both conserved proteins with unknown function and novel proteins is particularly relevant. The comprehensive characterization of these proteins opens up exciting new opportunities for understanding the role of mitochondria in phytopathogenicity. Due to the important role of this organelle in resistance/susceptibility to fungicides, this study could aid in the identification of more effective strategies by which to the control this fungus.
Conclusions
In the present study, we performed a global analysis of C. cacaofunesta mitochondria using an integrative approach (mitogenomics, transcriptomics and proteomics). We predicted the C. cacaofunesta mitochondrial proteome, including 52 mitochondrial encoded proteins and 1,072 proteins encoded in the nuclear genome (NMP). All of the NMP (except for 10 genes) were transcribed in mycelia that were grown in vitro; approximately 27% of these genes were also identified by mass spectrometry. RNAseq analysis allowed to detected expression of 33 novel putative genes. The comparison of the global analysis of mitochondrial function between C. cacaofunesta, S. cerevisiae and N. crassa revealed that, in addition to the known involvement of mitochondria in the pathogenesis of fungi, the differences in the lifestyle of these organisms (pathogenic and no-pathogenic) are not accompanied by major differences in the functional composition of their mitochondrial proteomes. Therefore, the relevance of this organelle in different fungi lifestyle could lie in a particular set of proteins and/or in a different pattern of gene regulation. In this regard, LC-MS analysis validated the existence of 84 proteins with unknown function, of which one is probably specific to this species and likely to the Ceratocystis species complex. Further functional characterization of these putative mitochondrial proteins could improve our understanding of the mitochondria’s role in fungal pathogenesis.
Methods
Biological material and nucleic acid isolation
Dr. Tomas Harrington, from the Iowa State University Department of Plant Pathology, generously donated Ceratocystis cacaofunesta strain C1593. This strain was isolated in 1999 from infected cacao trees located on a farm in the district of Uruçuca, Bahia, Brazil. In our laboratory, C. cacaofunesta cultures were maintained on 2% malt, yeast extract and agar (MYEA) plates inside a BOD chamber at 28°C. For liquid cultures, 20 to 30 agar blocks (1 mm2) that were removed from the solid cultures were inoculated into flasks that contained 100 mL of malt media and were cultivated at 28°C for 7 days, under constant agitation at 150 rpm.
The mycelia were separated from the media using filtration, were washed twice with sterile distilled water and were frozen with liquid nitrogen. The samples were ground to a fine powder in a mortar and were processed for DNA isolation as previously described [97]. The RNA isolation was performed using the RNeasy Plant Mini Kit (Qiagen, Valencia, CA, USA), according to the manufacturer’s protocol. All of the isolated genetic materials were qualitatively analyzed using denaturing formaldehyde/agarose gel electrophoresis and were quantified using a NanoDropTM 1000 spectrophotometer (Thermo Scientific).
Genome sequencing and assembly
The C. cacaofunesta mitochondrial genome was sequenced as part of the C. cacaofunesta Genome Project (http://www.lge.ibi.unicamp.br/ceratocystis). The DNA was sequenced on a Genome Analyzer IIx platform (Illumina) at the University of North Carolina, Chapel Hill High-Throughput Sequencing Facility. The whole-genome shotgun strategy was used to produce 76-bp paired-end reads (400-bp insert size) and 36-bp mate-pair reads (3-kb insert size).
The paired-end reads were assembled into longer scaffolds using de novo assembler VELVET 1.0.12 software [98] with a k-mer parameter of 69, which maximizes the length of the mitochondrial DNA (mtDNA) contig. The mtDNA was identified through comparisons with C. cacaofunesta scaffolds and Gibberella zeae mtDNA (NC_009493), resulting in the identification of a single mtDNA contig. To verify the topology of the mtDNA, we used the paired-end and mate-pair reads that had been aligned with the mtDNA using the SOAP2 aligner [99]. The reads that mapped to the borders of the contig were located at an expected distance from their respective pairs.
The complete mtDNA sequence was deposited into the GenBank database and is available under the accession number JX185564.
mtDNA annotation
C. cacaofunesta mtDNA was inspected by tBLASTn searches to identify 14 known conserved coding genes using their Gibberella zeae mtDNA orthologous as the query (Accession NC_009493). The rps3 gene was predicted using the C. fimbriata rps3 sequence available at GenBank (Accession FJ895616.1). Open Reading Frame Finder (ORF Finder) was used to search for intronic and hypothetical ORFs using Genetic code 4 (mold mitochondrial). Alternative start codons were allowed for predicted intronic ORFs when the initial ATG site resulted in an incomplete LAGLIDADG or GIY-YIG domain as indicated by a BLASTp search. The set of tRNAs was identified using tRNAscan-SE [48]. Genes encoding both small and large rRNA subunits were identified through comparisons with orthologous Gibberella zeae sequences using BLAST2seq (BLASTn).
mtDNA GC content and GC skew
The GC content, local GC content and cumulative GC skew of the C. cacaofunesta mtDNA were calculated using customized Perl scripts (available upon request). For the local GC content and cumulative GC skew, a sliding window of over 5,000 bp with a 500-bp range was used.
Predicted mitochondrial proteome
The putative genes encoding NMPs were identified using two strategies: ab initio prediction and sequence comparison. For the ab initio prediction, SignalP 3.0 software [100] was used to analyze all of the predicted nuclear proteins to identify those with a low probability (≤ 50%) of containing a signal peptide. Using the SignalP results as the input, the WoLF PSORT [101] and TargetP [100] programs were used to classify putative mitochondrial proteins. Within the TargetP output, only the proteins that were identified with a high probability of encoding mitochondrial genes (mTP ≥ 50% and other ≤ 50%) were considered. The proteins that were identified by both programs (TargetP and WoLF PSORT) constituted the final ab initio protein dataset. For the sequence comparison approach, we built a database that contained 1,583 known fungal mitochondrial proteins (Additional file 7; 741 proteins from S. cerevisiae and 842 from N. crassa). After the C. cacaofunesta nuclear encoded proteins were compared against this database using BLASTp (with an e-value cutoff of 1e-5), the comparative protein dataset was obtained from the BLAST results by applying query coverage (≥ 70%) and similarity (≥ 70%) filters. The ab initio and comparative protein datasets were merged to generate the final NMP dataset. The sequences of the nuclear-encoded mitochondrial proteins from C. cacaofunesta are provided in Additional file 8.
The automatic annotation of the NMPs was performed using the NR/NCBI, KEGG [62] and UniRef90 databases (BLASTp with an e-value cutoff of 1e-5) [61] and summarized using the AutoFACT program [60]. The CDD/Pfam database was used to identify the conserved domains. The Blast2GO program [64] was used to perform the gene ontology classification (BLASTp, with an e-value cutoff of 1e-5 on Generic GO Slim; the first 50 hits were considered). The proteins with identified biological functions according to GO Slim were manually grouped into 12 classes: amino acid metabolism, carbohydrate metabolism, cell organization, energy metabolism, genome maintenance and transcription, lipid metabolism, protein metabolism, protein transport and folding, response to stimulus, signaling, transport of metabolites and other. The proteins that were classified into multiple categories were considered to be members of both categories, with the exception of proteins that were classified as both “other” and another category; these were excluded from the “other” category and remained in the specific category. Thus, the “other” category includes proteins of known function that were not included in any of the above-described categories. The GO Slim biological processes that clustered in each category are shown in Additional file 5. Similar analyses were performed for the S. cerevisiae and N. crassa mitochondrial proteomes to compare them with the C. cacaofunesta data (Additional file 6, Sheets 6A, 6B and 6C).
Preparation of RNA-seq libraries and transcriptome analysis
RNA from C. cacaofunesta liquid cultures was extracted using the RNeasy Plant mini kit (Qiagen). Approximately 5 μg total RNA were used to prepare RNA-seq libraries following the procedures described by the manufacturer (Illumina). Briefly, the mRNA was purified using Sera-mag oligo (dT) beads and subsequently used for cDNA synthesis. Double-stranded cDNA was subjected to end-repair, A-tailing, adapter ligation and PCR amplification. The libraries were quantified using a Qubit fluorometer (Invitrogen), and quality control was performed using the Experion automated electrophoresis system (Bio-Rad). Two independent biological replicates were used for transcriptome sequencing. Each sample was sequenced in one lane of an Illumina Genome Analyzer IIx sequencer. Approximately 27 and 29 million single-end reads of 36 bp were produced for the CER1 and CER2 libraries, respectively.
The SOAP2 aligner [99] was used to align the RNA-Seq reads from mycelia that were grown in vitro with predicted genes that encode nuclear-encoded mitochondrial proteins. The program was configured to allow for as many as two mismatches, to discard sequences that contained “N”s and to return all optimal alignments. The expression level of each gene was estimated as an RPKM (reads per kilobase of exon per million reads mapped) value [102].
Mitochondrial isolation and proteomic assays
Mitochondrial isolation
The mitochondrial isolation was performed according to the protocol that is described by Sorensen and coworkers [103], with minor modifications. C. cacaofunesta was cultured for seven days in standard MYEA growth medium. Subsequently, the fungal mycelia were washed in cold water and homogenized using a bead beater in cold extraction buffer that contained 330 mM sucrose, 10 mM Tris–HCl, 1 mM EDTA, 1% PVP, 0.1% BSA and 0.3 mM PMSF (pH 7.5). The homogenates were filtered and centrifuged at 1,500 g for 15 min to remove the cellular debris. The resulting supernatants were centrifuged at 15,500 g for 20 min. The samples were rinsed twice in wash buffer (330 mM sucrose, 10 mM Tris–HCl, 1 mM EDTA, pH 7.2), and the final mitochondrial preparation was used to perform the proteomic assays.
The identification of proteins using LC-MS/MS
Enzymatic in-gel digestion for mass spectrometry analysis
A total of 20 μg of the concentrated mitochondrial proteome was obtained as described above and was separated using one-dimensional SDS-PAGE electrophoresis. Nineteen bands were excised from the gel and subjected to in-gel trypsin digestion, as described previously [104], with modifications.
The resulting peptide solution was dried in a SpeedVac concentrator and resuspended in 100 μL of 0.1% formic acid. An aliquot of 4.5 μL was separated using C18 (75 μm × 100 mm) RP-nanoUPLC (nanoACQUITY, Waters) coupled with a Q-Tof Ultima mass spectrometer (Waters) with a nano-electrospray source. The flow rate was 600 nL/min, and the gradient was 2-90% acetonitrile in 0.1% formic acid over 45 min. The instrument was operated in the “top three” mode, in which one MS spectrum was acquired, followed by an MS/MS analysis of the three most intense peaks [105].
The spectra were acquired using MassLynx v.4.1 software, and the raw data files were converted to a peak list format (mgf) using the Mascot Distiller v.2.3.2.0, 2009 software (Matrix Science Ltd.) without summing the scans, allowing for a label-free analysis (Additional file 9). The files were then searched against the Ceratocystis cacaofunesta database (7,321 entries – 7,269 nuclear proteins, and 52 mitochondrial proteins) using Mascot engine v.2.3.01 (Matrix Science Ltd.). Carbamidomethylation was used as a fixed modification, methionine oxidation as a variable modification, one missed trypsin cleavage and a tolerance of 0.1 Da for both precursor and fragment ions. For the protein quantitation, the .dat files from the Mascot output were analyzed using Scaffold Q+ (version 3_00_03, Proteome Software), and quantitative values (normalized spectral counts) were obtained [106,107]. For the endogenous peptide identification, methionine oxidation was set as a variable modification, with a tolerance of 0.1 Da for both the precursor and fragment ions. Only peptides with a minimum of 5 amino acid residues and significance (p < 0.05) based on the Mascot-based scores were considered in the results.
Abbreviations
ABC tranporter: ATP binding cassette (family of membrane transport proteins); ADH: Alcohol dehydrogenase; AOX: Alternative oxidase; ATP: Adenosine triphosphate; BLAST: Basic local alignment search tool; BSA: Bovine serum albumin; CDD: Conserved domains database; COB: Cytochrome b oxidade; COX: Cytochrome c oxidase; CS: Citrate synthase; EDTA: Ethylenediaminetetraacetic acid; FOW1: Fusarium oxysporum mitochondrial carrier; GO: Gene ontology; GST: Glutathione S-transferase; IDH: Isocitrate dehydrogenases (IDH1 and IDH2); KEGG: Kyoto encyclopedia of genes and genomes; NAD: Nicotinamide adenine dinucleotide (electron donor); NADH: Reduced B-nicotinamide adenine dinucleotide; NCBI: National Center for Biotechnology Information; NMP: Nuclear-encoded mitochondrial proteins; NR: Non-redundant GenBank database; ORF: Open reading frame; OXPHOS: Proteins of oxidative phosphorylation machinery; PDH: Proteins of the pyruvate dehydrogenase complex (; PMSF: Phenylmethylsulfonyl fluoride; PVP: Polyvinylpyrrolidone; rnl: Large RNA subunit; rns: Small RNA subunit; RPKM: Reads per kilobase of exons per million; rpo: RNA polymerase; SDS-PAGE: Sodium dodecyl sulfate polyacrylamide gel electrophoresis; TCA: Tricarboxylic acid cycle; Tris–HCl: Tris (hydroxymethyl) aminomethane hydrochloride; UFP: Unknown function protein
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ABA: preparation of the manuscript and data analysis; LCN, MFC, ROV: bioinformatics analysis and revision of manuscript; BVO, DPTT: collaboration in sampling the material and the revision of manuscript; RAT: collaboration in bioinformatics analysis; AFPL: collaboration in the generation of LC-MS/MS data; PJPLT, PM: sequencing of the libraries by RNAseq, and collaboration in data analysis; LWM: collaboration in sampling the material; GAGP, OGC: coordination of the molecular and bioinformatics analysis, preparation and organization of the manuscript. All of the authors have read and approved the final version of the manuscript.
Supplementary Material
All intronic ORFs sequences.
Table of all genes predicted in this work and its RPKMs and its annotations (RPKMs, NR, KEGG, CDD, Autofact, Biological process, Molecular function and Cellular component, Predicted by BlastP, Predicted by TargetP/WolfpSort, Identified by LC-MS/MS). Sheet 2A: mitochondrial genes. Sheet 2B: predicted nuclear genes.
Scheme of experimental method.
{kind=link}
Table of all LC-MS/MS.data. Sheet 4A: all proteins identified by LC-MS/MS. Sheet 4B: new annotation proposed to hypothetical proteins. Sheet 4C: possible cellular location of proteins not predicted.
Table grouping of biological process categories identified by GOslim.
Table of mitochondrial predicted genes annotation by GOslim.
Amino acid sequences of fungal mitochondrial proteins.
Amino acid sequences of all proteins analyzed in this work.
Table mzML of mass spectrometry data.
Contributor Information
Alinne Batista Ambrosio, Email: alinne@lge.ibi.unicamp.br.
Leandro Costa do Nascimento, Email: leandro@lge.ibi.unicamp.br.
Bruno V Oliveira, Email: bvo@lge.ibi.unicamp.br.
Paulo José P L Teixeira, Email: paulo@lge.ibi.unicamp.br.
Ricardo A Tiburcio, Email: tiburcio@lge.ibi.unicamp.br.
Daniela P Toledo Thomazella, Email: danitt@lge.ibi.unicamp.br.
Adriana F P Leme, Email: adriana.paesleme@lnbio.org.br.
Marcelo F Carazzolle, Email: mcarazzo@lge.ibi.unicamp.br.
Ramon O Vidal, Email: ramon.vidal@gmail.com.
Piotr Mieczkowski, Email: piotr_mieczkowski@med.unc.edu.
Lyndel W Meinhardt, Email: lyndel.meinhardt@ars.usda.gov.
Gonçalo A G Pereira, Email: goncalo@unicamp.br.
Odalys G Cabrera, Email: odalys@lge.ibi.unicamp.br.
Acknowledgements
The authors would like to acknowledge to Dr. Tomas Harrington, from the Iowa State University Department of Plant Pathology, who generously donated Ceratocystis cacaofunesta strain C1593. This work was supported by research funding of Fundação de Amparo à Pesquisa do Estado de São Paulo, grant number 2009/50119-9.
References
- Baker CJ, Harrington TC, Krauss U, Alfenas AC. Genetic variability and host specialization in the latin american clade of ceratocystis fimbriata. Phytopathology. 2003;93(10):1274–1284. doi: 10.1094/PHYTO.2003.93.10.1274. [DOI] [PubMed] [Google Scholar]
- Johnson JA, Harrington TC, Engelbrecht CJ. Phylogeny and taxonomy of the North American clade of the ceratocystis fimbriata complex. Mycologia. 2005;97(5):1067–1092. doi: 10.3852/mycologia.97.5.1067. [DOI] [PubMed] [Google Scholar]
- Barnes I, Gaur A, Burgess T, Roux J, Wingfield BD, Wingfield MJ. Microsatellite markers reflect intra-specific relationships between isolates of the vascular wilt pathogen Ceratocystis fimbriata. Mol Plant Pathol. 2001;2(6):319–325. doi: 10.1046/j.1464-6722.2001.00080.x. [DOI] [PubMed] [Google Scholar]
- Engelbrecht CJ, Harrington TC, Steimel J, Capretti P. Genetic variation in eastern North American and putatively introduced populations of Ceratocystis fimbriata f. platani. Mol Ecol. 2004;13(10):2995–3005. doi: 10.1111/j.1365-294X.2004.02312.x. [DOI] [PubMed] [Google Scholar]
- Soanes DM, Alam I, Cornell M, Wong HM, Hedeler C, Paton NW, Rattray M, Hubbard SJ, Oliver SG, Talbot NJ. Comparative genome analysis of filamentous fungi reveals gene family expansions associated with fungal pathogenesis. PLoS One. 2008;3(6):e2300. doi: 10.1371/journal.pone.0002300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelbrecht CJ, Harrington TC. Intersterility, morphology and taxonomy of Ceratocystis fimbriata on sweet potato, cacao and sycamore. Mycologia. 2005;97(1):57–69. doi: 10.3852/mycologia.97.1.57. [DOI] [PubMed] [Google Scholar]
- Bastos CNE HC. Ocorrencia de Ceratocystis fimbriata. Acta Amazonica. 1978;8(4):543–544. [Google Scholar]
- Bezerra JL. Ceratocystis fimbriata causing death of budded cocoa seedlings in Bahia, Brazil. Incoped Newsletter. 1997;1 [Google Scholar]
- Malaguti G. Ceratostomella fimbriata en el cacao de Venezuela. Acta cientif venezol. 1952;3(3):94–97. [Google Scholar]
- Harrington TC. Host specialization and speciation in the American wilt pathogen Ceratocystis fimbriata. Fitopatol Bras. 2000;25S:262–263. [Google Scholar]
- Spence JL. Preliminary observations on a wilt condition of cocoa. J Agric Soc Trinidad and Tobago. 1958;58(3):349–359. [Google Scholar]
- Thorold CA. Diseseas of cocoa. XII. Oxford: Clarendon Press; 1975. p. 423. [Google Scholar]
- Engelbrecht CJ, Harrington TC, Alfenas A. Ceratocystis wilt of cacao-a disease of increasing importance. Phytopathology. 2007;97(12):1648–1649. doi: 10.1094/PHYTO-97-12-1648. [DOI] [PubMed] [Google Scholar]
- Ferreira MA, Harrington TC, Alfenas AC, Mizubuti ES. Movement of genotypes of Ceratocystis fimbriata within and among Eucalyptus plantations in Brazil. Phytopathology. 2011;101(8):1005–1012. doi: 10.1094/PHYTO-01-11-0015. [DOI] [PubMed] [Google Scholar]
- Harrington TC, Thorpe DJ, Alfenas AC. Genetic variation and variation in aggressiveness to native and exotic hosts among Brazilian populations of Ceratocystis fimbriata. Phytopathology. 2011;101(5):555–566. doi: 10.1094/PHYTO-08-10-0228. [DOI] [PubMed] [Google Scholar]
- Shingu-Vazquez M, Traven A. Mitochondria and fungal pathogenesis: drug tolerance, virulence, and potential for antifungal therapy. Eukaryot Cell. 2011;10(11):1376–1383. doi: 10.1128/EC.05184-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz G. Mitochondria: beyond oxidative phosphorylation. Biochim Biophys Acta. 1995;1271(1):123–126. doi: 10.1016/0925-4439(95)00018-Y. [DOI] [PubMed] [Google Scholar]
- Gunter TE, Yule DI, Gunter KK, Eliseev RA, Salter JD. Calcium and mitochondria. FEBS Lett. 2004;567(1):96–102. doi: 10.1016/j.febslet.2004.03.071. [DOI] [PubMed] [Google Scholar]
- Muhlenhoff U, Richhardt N, Gerber J, Lill R. Characterization of iron-sulfur protein assembly in isolated mitochondria. A requirement for ATP, NADH, and reduced iron. J Biol Chem. 2002;277(33):29810–29816. doi: 10.1074/jbc.M204675200. [DOI] [PubMed] [Google Scholar]
- Eisenberg T, Buttner S, Kroemer G, Madeo F. The mitochondrial pathway in yeast apoptosis. Apoptosis. 2007;12:1011–1023. doi: 10.1007/s10495-007-0758-0. [DOI] [PubMed] [Google Scholar]
- Lorin S, Dufour E, Sainsard-Chanet A. Mitochondrial metabolism and aging in the filamentous fungus Podospora anserina. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1757;5-6:604–610. doi: 10.1016/j.bbabio.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Court DA, Griffiths AJF, Kraus SR, Russell PJ, Bertrand H. A new senescence-inducing mitochondrial linear plasmid in field-isolated <i>Neurospora crassa strains from India. Curr Genet. 1991;19(2):129–137. doi: 10.1007/BF00326294. [DOI] [PubMed] [Google Scholar]
- Torriani SF, Goodwin SB, Kema GH, Pangilinan JL, McDonald BA. Intraspecific comparison and annotation of two complete mitochondrial genome sequences from the plant pathogenic fungus Mycosphaerella graminicola. FG & B. 2008;45(5):628–637. doi: 10.1016/j.fgb.2007.12.005. [DOI] [PubMed] [Google Scholar]
- Solieri L. Mitochondrial inheritance in budding yeasts: towards an integrated understanding. Trends Microbiol. 2010;18(11):521–530. doi: 10.1016/j.tim.2010.08.001. [DOI] [PubMed] [Google Scholar]
- Wiedemann N, Frazier AE, Pfanner N. The protein import machinery of mitochondria. J Biol Chem. 2004;279(15):14473–14476. doi: 10.1074/jbc.R400003200. [DOI] [PubMed] [Google Scholar]
- Gray MW, Lang BF. Transcription in chloroplasts and mitochondria: a tale of two polymerases. Trends Microbiol. 1998;6(1):1–3. doi: 10.1016/S0966-842X(97)01182-7. [DOI] [PubMed] [Google Scholar]
- Dolezal P, Likic V, Tachezy J, Lithgow T. Evolution of the molecular machines for protein import into mitochondria. Science. 2006;313(5785):314–318. doi: 10.1126/science.1127895. [DOI] [PubMed] [Google Scholar]
- Neupert W, Herrmann JM. Translocation of proteins into mitochondria. Annu Rev Biochem. 2007;76:723–749. doi: 10.1146/annurev.biochem.76.052705.163409. [DOI] [PubMed] [Google Scholar]
- Schmidt O, Pfanner N, Meisinger C. Mitochondrial protein import: from proteomics to functional mechanisms. Nat Rev Mol Cell Biol. 2010;11(9):655–667. doi: 10.1038/nrm2959. [DOI] [PubMed] [Google Scholar]
- Costa GG, Cabrera OG, Tiburcio RA, Medrano FJ, Carazzolle MF, Thomazella DP, Schuster SC, Carlson JE, Guiltinan MJ, Bailey BA. The mitochondrial genome of Moniliophthora roreri, the frosty pod rot pathogen of cacao. Fungal Biol. 2012;116(5):551–562. doi: 10.1016/j.funbio.2012.01.008. [DOI] [PubMed] [Google Scholar]
- David Day A, Heazlewood J, Julian Tonti-Filippini S, Alexander Gout M, David Day A, James W, Harvey Millar A. Experimental analysis of the arabidopsis mitochondrial proteome highlights signaling and regulatory components, provides assessment of targeting prediction programs, and indicates plant-specific mitochondrial proteins. Plant Cell. 2004;16(1):241–256. doi: 10.1105/tpc.016055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer Tim R, Neph S, Dinger Marcel E, Crawford J, Smith Martin A, Shearwood A-Marie J, Haugen E, Bracken Cameron P, Rackham O, Stamatoyannopoulos John A. The human mitochondrial transcriptome. Cell. 2011;146(4):645–658. doi: 10.1016/j.cell.2011.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokisch H, Scharfe C, Camp DG II, Xiao W, David L, Andreoli C, Monroe ME, Moore RJ, Gritsenko MA, Kozany C. Integrative analysis of the mitochondrial proteome in yeast. PLoS Biol. 2004;2(6):e160. doi: 10.1371/journal.pbio.0020160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinders J, Zahedi RP, Pfanner N, Meisinger C, Sickmann A. Toward the complete yeast mitochondrial proteome: multidimensional separation techniques for mitochondrial proteomics. J Proteome Res. 2006;5(7):1543–1554. doi: 10.1021/pr050477f. [DOI] [PubMed] [Google Scholar]
- Doyle C, Donaldson M, Morrison E, Saville B. Ustilago maydis transcript features identified through full-length cDNA analysis. Mol Genet Genomics. 2011;286(2):143–159. doi: 10.1007/s00438-011-0634-z. [DOI] [PubMed] [Google Scholar]
- Tan K-C, Ipcho SVS, Trengove RD, Oliver RP, Solomon PS. Assessing the impact of transcriptomics, proteomics and metabolomics on fungal phytopathology. Mol Plant Pathol. 2009;10(5):703–715. doi: 10.1111/j.1364-3703.2009.00565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Fernandez R, Prats E, Jorren-Novo JV. Proteomics of plant pathogenic fungi. J Biomed Biotechnol. 2010;2010:1–36. doi: 10.1155/2010/932527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yajima W, Kav NNV. The proteome of the phytopathogenic fungus Sclerotinia sclerotiorum. Proteomics. 2006;6(22):5995–6007. doi: 10.1002/pmic.200600424. [DOI] [PubMed] [Google Scholar]
- Fernández-Acero FJ, Jorge I, Calvo E, Vallejo I, Carbú M, Camafeita E, López JA, Cantoral JM, Jorrín J. Two-dimensional electrophoresis protein profile of the phytopathogenic fungus Botrytis cinerea. Proteomics. 2006;6(S1):S88–S96. doi: 10.1002/pmic.200500436. [DOI] [PubMed] [Google Scholar]
- Medina M. Genomes, phylogeny, and evolutionary systems biology. Proc Natl Acad Sci U S A. 2005;102(Suppl 1):6630–6635. doi: 10.1073/pnas.0501984102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey D, Zhang Z, Saalbach G, Thordal-Christensen H. A proteomics study of barley powdery mildew haustoria. Proteomics. 2009;9(12):3222–3232. doi: 10.1002/pmic.200800645. [DOI] [PubMed] [Google Scholar]
- Xu J, Saunders CW, Hu P, Grant RA, Boekhout T, Kuramae EE, Kronstad JW, DeAngelis YM, Reeder NL, Johnstone KR. Dandruff-associated Malassezia genomes reveal convergent and divergent virulence traits shared with plant and human fungal pathogens. Proc Natl Acad Sci. 2007;104(47):18730–18735. doi: 10.1073/pnas.0706756104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amlacher S, Sarges P, Flemming D, van Noort V, Kunze R, Devos Damien P, Arumugam M, Bork P, Hurt E. Insight into structure and assembly of the nuclear pore complex by utilizing the genome of a eukaryotic thermophile. Cell. 2011;146(2):277–289. doi: 10.1016/j.cell.2011.06.039. [DOI] [PubMed] [Google Scholar]
- Paquin B, Franz Lang B. The mitochondrial DNA of allomyces macrogynus: the complete genomic sequence from an ancestral fungus. J Mol Biol. 1996;255(5):688–701. doi: 10.1006/jmbi.1996.0056. [DOI] [PubMed] [Google Scholar]
- Turmel M, Lemieux C, Burger G, Lang BF, Otis C, Plante I, Gray MW. The complete mitochondrial DNA sequences of nephroselmis olivacea and pedinomonas minor: Two radically different evolutionary patterns within green algae. The Plant Cell Online. 1999;11(9):1717–1729. doi: 10.1105/tpc.11.9.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethuraman J, Majer A, Friedrich NC, Edgell DR, Hausner G. Genes within genes: multiple LAGLIDADG homing endonucleases target the ribosomal protein S3 gene encoded within an rnl group I intron of ophiostoma and related taxa. Mol Biol Evol. 2009;26(10):2299–2315. doi: 10.1093/molbev/msp145. [DOI] [PubMed] [Google Scholar]
- Sethuraman J, Majer A, Iranpour M, Hausner G. Molecular evolution of the mtDNA encoded rps3 gene among filamentous ascomycetes fungi with an emphasis on the ophiostomatoid fungi. J Mol Evol. 2009;69(4):372–385. doi: 10.1007/s00239-009-9291-9. [DOI] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantou MP, Kouvelis VN, Typas MA. The complete mitochondrial genome of the vascular wilt fungus Verticillium dahliae: a novel gene order for Verticillium and a diagnostic tool for species identification. Curr Genet. 2006;50(2):125–136. doi: 10.1007/s00294-006-0079-9. [DOI] [PubMed] [Google Scholar]
- Pantou MP, Kouvelis VN, Typas MA. The complete mitochondrial genome of fusarium oxysporum: insights into fungal mitochondrial evolution. Gene. 2008;419(1–2):7–15. doi: 10.1016/j.gene.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Ferandon C, Moukha S, Callac P, Benedetto JP, Castroviejo M, Barroso G. The Agaricus bisporus cox1 gene: the longest mitochondrial gene and the largest reservoir of mitochondrial group i introns. PLoS One. 2010;5(11):e14048. doi: 10.1371/journal.pone.0014048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevalier BS, Stoddard BL. Homing endonucleases: structural and functional insight into the catalysts of intron/intein mobility. Nucleic Acids Res. 2001;29(18):3757–3774. doi: 10.1093/nar/29.18.3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgenson JE, Bowden RL, Zeller KA, Leslie JF, Alexander NJ, Plattner RD. A genetic map of Gibberella zeae (Fusarium graminearum) Genetics. 2002;160(4):1451–1460. doi: 10.1093/genetics/160.4.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galagan JE, Calvo SE, Borkovich KA, Selker EU, Read ND, Jaffe D, FitzHugh W, Ma LJ, Smirnov S, Purcell S. The genome sequence of the filamentous fungus Neurospora crassa. Nature. 2003;422(6934):859–868. doi: 10.1038/nature01554. [DOI] [PubMed] [Google Scholar]
- Osiewacz HD, Hermanns J, Marcou D, Triffi M, Esser K. Mitochondrial DNA rearrangements are correlated with a delayed amplification of the mobile intron (plDNA) in a long-lived mutant of Podospora anserina. Mutat Res. 1989;219(1):9–15. doi: 10.1016/0921-8734(89)90036-2. [DOI] [PubMed] [Google Scholar]
- Blattner FR, Plunkett G 3rd, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF. The complete genome sequence of Escherichia coli K-12. Science. 1997;277(5331):1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- Andersson SG, Kurland CG. Ancient and recent horizontal transfer events: the origins of mitochondria. APMIS Suppl. 1998;84:5–14. doi: 10.1111/j.1600-0463.1998.tb05641.x. [DOI] [PubMed] [Google Scholar]
- Schmitt S, Prokisch H, Schlunck T, Camp DG 2nd, Ahting U, Waizenegger T, Scharfe C, Meitinger T, Imhof A, Neupert W. Proteome analysis of mitochondrial outer membrane from Neurospora crassa. Proteomics. 2006;6(1):72–80. doi: 10.1002/pmic.200402084. [DOI] [PubMed] [Google Scholar]
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong S-E, Walford GA, Sugiana C, Boneh A, Chen WK. A Mitochondrial Protein Compendium Elucidates Complex I Disease Biology. Cell. 2008;134(1):112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koski LB, Gray MW, Lang BF, Burger G. AutoFACT: an automatic functional annotation and classification tool. BMC Bioinforma. 2005;6:151. doi: 10.1186/1471-2105-6-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis S, Madden TL. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004;32(suppl 2):W20–W25. doi: 10.1093/nar/gkh435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A, Birney E, Durbin R, Eddy SR, Howe KL, Sonnhammer EL. The Pfam protein families database. Nucleic Acids Res. 2000;28(1):263–266. doi: 10.1093/nar/28.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- Wilhelm BT, Landry J-R. RNA-Seq—quantitative measurement of expression through massively parallel RNA-sequencing. Methods. 2009;48(3):249–257. doi: 10.1016/j.ymeth.2009.03.016. [DOI] [PubMed] [Google Scholar]
- Au KF, Jiang H, Lin L, Xing Y, Wong WH. Detection of splice junctions from paired-end RNA-seq data by SpliceMap. Nucleic Acids Res. 2010;38(14):4570–4578. doi: 10.1093/nar/gkq211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard H, Schulz MH, Sultan M, Nürnberger A, Schrinner S, Balzereit D, Dagand E, Rasche A, Lehrach H, Vingron M. Prediction of alternative isoforms from exon expression levels in RNA-Seq experiments. Nucleic Acids Res. 2010;38(10):e112. doi: 10.1093/nar/gkq041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers M, Thomas J, Reddy A, Ben-Hur A. SpliceGrapher: detecting patterns of alternative splicing from RNA-Seq data in the context of gene models and EST data. Genome Biol. 2012;13(1):R4. doi: 10.1186/gb-2012-13-1-r4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres TT, Dolezal M, Schlötterer C, Ottenwälder B. Expression profiling of Drosophila mitochondrial genes via deep mRNA sequencing. Nucleic Acids Res. 2009;37(22):7509–7518. doi: 10.1093/nar/gkp856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neira-Oviedo M, Tsyganov-Bodounov A, Lycett GJ, Kokoza V, Raikhel AS, Krzywinski J. The RNA-Seq approach to studying the expression of mosquito mitochondrial genes. Insect Mol Biol. 2011;20(2):141–152. doi: 10.1111/j.1365-2583.2010.01053.x. [DOI] [PubMed] [Google Scholar]
- Chang JH, Tong L. Mitochondrial poly(A) polymerase and polyadenylation. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms. 1819;9-10:992–997. doi: 10.1016/j.bbagrm.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sickmann A, Reinders J, Wagner Y, Joppich C, Zahedi R, Meyer HE, Schonfisch B, Perschil I, Chacinska A, Guiard B. The proteome of Saccharomyces cerevisiae mitochondria. Proc Natl Acad Sci U S A. 2003;100(23):13207–13212. doi: 10.1073/pnas.2135385100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeping A, DeAbreu D, DiBernardo M, Collins RA. Gel-based mass spectrometric and computational approaches to the mitochondrial proteome of Neurospora. Fungal Genet Biol. 2011;48(5):526–536. doi: 10.1016/j.fgb.2010.11.011. [DOI] [PubMed] [Google Scholar]
- Scheffler IE. Mitochondria make a come back. Adv Drug Deliv Rev. 2001;49(1–2):3–26. doi: 10.1016/s0169-409x(01)00123-5. [DOI] [PubMed] [Google Scholar]
- Idnurm A, Howlett BJ. Pathogenicity genes of phytopathogenic fungi. Mol Plant Pathol. 2001;2(4):241–255. doi: 10.1046/j.1464-6722.2001.00070.x. [DOI] [PubMed] [Google Scholar]
- Vödisch M, Scherlach K, Winkler R, Hertweck C, Braun H-P, Roth M, Haas H, Werner ER, Brakhage AA, Kniemeyer O. Analysis of the aspergillus fumigatus proteome reveals metabolic changes and the activation of the pseurotin a biosynthesis gene cluster in response to hypoxia. J Proteome Res. 2011;10(5):2508–2524. doi: 10.1021/pr1012812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo S, Jain M, Xie X, Sheth SA, Chang B, Goldberger OA, Spinazzola A, Zeviani M, Carr SA, Mootha VK. Systematic identification of human mitochondrial disease genes through integrative genomics. Nat Genet. 2006;38(5):576–582. doi: 10.1038/ng1776. [DOI] [PubMed] [Google Scholar]
- Mengiste T. Plant immunity to necrotrophs. Annu Rev Phytopathol. 2012;50(1):267–294. doi: 10.1146/annurev-phyto-081211-172955. [DOI] [PubMed] [Google Scholar]
- Coleman JJ, White GJ, Rodriguez-Carres M, VanEtten HD. An ABC transporter and a cytochrome P450 of nectria haematococca MPVI Are virulence factors on Pea and Are the major tolerance mechanisms to the phytoalexin pisatin. Mol Plant Microbe Interact. 2010;24(3):368–376. doi: 10.1094/MPMI-09-10-0198. [DOI] [PubMed] [Google Scholar]
- de Waard MA. Significance of ABC transporters in fungicide sensitivity and resistance. Pestic Sci. 1997;51(3):271–275. doi: 10.1002/(SICI)1096-9063(199711)51:3<271::AID-PS642>3.0.CO;2-#. [DOI] [Google Scholar]
- Jacoby WBaZ DM. The enzymes of detoxification. J Biol Chem. 1990;256:20715–20718. [PubMed] [Google Scholar]
- Coleman M, Henricot B, Arnau J, Oliver RP. Starvation-induced genes of the tomato pathogen cladosporium fulvum are also induced during growth in planta. Mol Plant Microbe Interact. 1997;10(9):1106–1109. doi: 10.1094/MPMI.1997.10.9.1106. [DOI] [PubMed] [Google Scholar]
- Mariani D, Mathias C, da Silva C, Herdeiro RS, Pereira R, Panek A, Eleutherio E, Pereira MD. Involvement of glutathione transferases, Gtt1and Gtt2, with oxidative stress response generated by H2O2 during growth of Saccharomyces cerevisiae. Redox Rep. 2008;13(6):246–254. doi: 10.1179/135100008X309028. [DOI] [PubMed] [Google Scholar]
- Cohen E, Gamliel A, Katan J. Glutathione and glutathione-S-transferase in fungi: Effect of pentachloronitrobenzene and 1-chloro-2,4-dinitrobenzene; Purification and characterization of the transferase from Fusarium. Pestic Biochem Physiol. 1986;26(1):1–9. doi: 10.1016/0048-3575(86)90056-8. [DOI] [Google Scholar]
- Mao P, Meshul CK, Thuillier P, Goldberg NRS, Reddy PH. CART peptide is a potential endogenous antioxidant and preferentially localized in mitochondria. PLoS One. 2012;7(1):e29343. doi: 10.1371/journal.pone.0029343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph-Horne T, Hollomon DW. Functional diversity within the mitochondrial electron transport chain of plant pathogenic fungi. Pest Manag Sci. 2000;56(1):24–30. doi: 10.1002/(SICI)1526-4998(200001)56:1<24::AID-PS71>3.0.CO;2-Y. [DOI] [Google Scholar]
- Avila-Adame C, Köller W. Disruption of the alternative oxidase gene in magnaporthe grisea and its impact on host infection. Mol Plant Microbe Interact. 2002;15(5):493–500. doi: 10.1094/MPMI.2002.15.5.493. [DOI] [PubMed] [Google Scholar]
- Voulgaris I, O'Donnell A, Harvey LM, McNeil B. Inactivating alternative NADH dehydrogenases: enhancing fungal bioprocesses by improving growth and biomass yield? Sci Rep. 2012;2(322):1–9. doi: 10.1038/srep00322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miguez M, Reeve C, Wood PM, Hollomon DW. Alternative oxidase reduces the sensitivity of Mycosphaerella graminicola to QOI fungicides. Pest Manag Sci. 2004;60(1):3–7. doi: 10.1002/ps.837. [DOI] [PubMed] [Google Scholar]
- Kaneko I, Ishii H. Effect of azoxystrobin on activities of antioxidant enzymes and alternative oxidase in wheat head blight pathogens Fusarium graminearum and Microdochium nivale. J Gen Plant Pathol. 2009;75(5):388–398. doi: 10.1007/s10327-009-0178-9. [DOI] [Google Scholar]
- Thomazella DPT, Teixeira PJPL, Oliveira HC, Saviani EE, Rincones J, Toni IM, Reis O, Garcia O, Meinhardt LW, Salgado I. The hemibiotrophic cacao pathogen Moniliophthora perniciosa depends on a mitochondrial alternative oxidase for biotrophic development. New Phytol. 2012;194(4):1025–1034. doi: 10.1111/j.1469-8137.2012.04119.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue I, Namiki F, Tsuge T. Plant colonization by the vascular wilt fungus fusarium oxysporum requires FOW1, a gene encoding a mitochondrial protein. The Plant Cell Online. 2002;14(8):1869–1883. doi: 10.1105/tpc.002576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warn PA, Sharp A, Guinea J, Denning DW. Effect of hypoxic conditions on in vitro susceptibility testing of amphotericin B, itraconazole and micafungin against Aspergillus and Candida. J Antimicrob Chemother. 2004;53(5):743–749. doi: 10.1093/jac/dkh153. [DOI] [PubMed] [Google Scholar]
- Adachi H, Shimizu K, Hattori H, Tanaka R, Chibana H, Takagi Y, Tomita Y, Kanbe T. Genotyping of candida albicans by fragment analysis of microsatellites combined with 25S rDNA and RPS-based strategies. Nippon Ishinkin Gakkai Zasshi. 2009;50(3):167–174. doi: 10.3314/jjmm.50.167. [DOI] [PubMed] [Google Scholar]
- Spicer R, Holbrook NM. Within-stem oxygen concentration and sap flow in four temperate tree species: does long-lived xylem parenchyma experience hypoxia? Plant Cell Environ. 2005;28(2):192–201. doi: 10.1111/j.1365-3040.2004.01262.x. [DOI] [Google Scholar]
- Grahl N, Cramer RA. Regulation of hypoxia adaptation: an overlooked virulence attribute of pathogenic fungi? Medical Mycology. 2010;48(1):1–15. doi: 10.3109/13693780902947342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondego J, Carazzolle M, Costa G, Formighieri E, Parizzi L, Rincones J, Cotomacci C, Carraro D, Cunha A, Carrer H. A genome survey of moniliophthora perniciosa gives new insights into Witches' broom disease of cacao. BMC Genomics. 2008;9(1):548. doi: 10.1186/1471-2164-9-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, McEwen GK, Margulies EH, Birney E. Pebble and rock band: heuristic resolution of repeats and scaffolding in the velvet short-read de novo assembler. PLoS One. 2009;4(12):e8407. doi: 10.1371/journal.pone.0008407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Yu C, Li Y, Lam T-W, Yiu S-M, Kristiansen K, Wang J. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25(15):1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- Emanuelsson O, Brunak S, von Heijne G, Nielsen H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protocols. 2007;2(4):953–971. doi: 10.1038/nprot.2007.131. [DOI] [PubMed] [Google Scholar]
- Horton P, Park K-J, Obayashi T, Fujita N, Harada H, Adams-Collier CJ, Nakai K. WoLF PSORT: protein localization predictor. Nucleic Acids Res. 2007;35(suppl 2):W585–W587. doi: 10.1093/nar/gkm259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Meth. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Sorensen M, Sanz A, Gómez J, Pamplona R, Portero-Otín M, Gredilla R, Barja G. Effects of fasting on oxidative stress in rat liver mitochondria. Free Radic Res. 2006;40(4):339–347. doi: 10.1080/10715760500250182. [DOI] [PubMed] [Google Scholar]
- Hanna SL, Sherman NE, Kinter MT, Goldberg JB. Comparison of proteins expressed by Pseudomonas aeruginosa strains representing initial and chronic isolates from a cystic fibrosis patient: an analysis by 2-D gel electrophoresis and capillary column liquid chromatography–tandem mass spectrometry. Microbiology. 2000;146(10):2495–2508. doi: 10.1099/00221287-146-10-2495. [DOI] [PubMed] [Google Scholar]
- Paes Leme AF, Sherman NE, Smalley DM, Sizukusa LO, Oliveira AK, Menezes MC, Fox JW, Serrano SMT. Hemorrhagic activity of HF3, a snake venom metalloproteinase: insights from the proteomic analysis of mouse skin and blood plasma. J Proteome Res. 2011;11(1):279–291. doi: 10.1021/pr2006439. [DOI] [PubMed] [Google Scholar]
- Escalante T, Rucavado A, Pinto AFM, Terra RMS, Gutiérrez JM, Fox JW. Wound exudate as a proteomic window to reveal different mechanisms of tissue damage by snake venom toxins. J Proteome Res. 2009;8(11):5120–5131. doi: 10.1021/pr900489m. [DOI] [PubMed] [Google Scholar]
- Eming SA, Koch M, Krieger A, Brachvogel B, Kreft S, Bruckner-Tuderman L, Krieg T, Shannon JD, Fox JW. Differential proteomic analysis distinguishes tissue repair biomarker signatures in wound exudates obtained from normal healing and chronic wounds. J Proteome Res. 2010;9(9):4758–4766. doi: 10.1021/pr100456d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
All intronic ORFs sequences.
Table of all genes predicted in this work and its RPKMs and its annotations (RPKMs, NR, KEGG, CDD, Autofact, Biological process, Molecular function and Cellular component, Predicted by BlastP, Predicted by TargetP/WolfpSort, Identified by LC-MS/MS). Sheet 2A: mitochondrial genes. Sheet 2B: predicted nuclear genes.
Scheme of experimental method.
Table of all LC-MS/MS.data. Sheet 4A: all proteins identified by LC-MS/MS. Sheet 4B: new annotation proposed to hypothetical proteins. Sheet 4C: possible cellular location of proteins not predicted.
Table grouping of biological process categories identified by GOslim.
Table of mitochondrial predicted genes annotation by GOslim.
Amino acid sequences of fungal mitochondrial proteins.
Amino acid sequences of all proteins analyzed in this work.
Table mzML of mass spectrometry data.