

The epigenetic regulator MeCP2 is critical for normal brain function. One puzzle has been the mechanism of the age-dependent relationship between MeCP2 levels and MECP2 3′ UTR length, especially the predominance of the long 3′ UTR of MECP2 in human fetal brains. Here Han et al. show that miR-483-5p, an intragenic microRNA of the imprinted gene IGF2, regulates MeCP2 in fetal brains through the extended 3′ UTR. This study provides new insight into the post-transcriptional regulation of MeCP2 during human fetal development.
Keywords: MeCP2, human fetal brain, 3′ UTR, miR-483-5p, HDAC4, TBL1X
Abstract
Proper neurological function in humans requires precise control of levels of the epigenetic regulator methyl CpG-binding protein 2 (MeCP2). MeCP2 protein levels are low in fetal brains, where the predominant MECP2 transcripts have an unusually long 3′ untranslated region (UTR). Here, we show that miR-483-5p, an intragenic microRNA of the imprinted IGF2, regulates MeCP2 levels through a human-specific binding site in the MECP2 long 3′ UTR. We demonstrate the inverse correlation of miR-483-5p and MeCP2 levels in developing human brains and fibroblasts from Beckwith-Wiedemann syndrome patients. Importantly, expression of miR-483-5p rescues abnormal dendritic spine phenotype of neurons overexpressing human MeCP2. In addition, miR-483-5p modulates the levels of proteins of the MeCP2-interacting corepressor complexes, including HDAC4 and TBL1X. These data provide insight into the role of miR-483-5p in regulating the levels of MeCP2 and interacting proteins during human fetal development.
Methyl CpG-binding protein 2 (MeCP2) is a transcriptional regulator of gene expression that is critical for normal brain function (Chahrour and Zoghbi 2007; Guy et al. 2011). Loss-of-function mutations in MECP2 cause Rett syndrome as well as a spectrum of phenotypes ranging from autism to intellectual disabilities and mood disorders (Amir et al. 1999; Chahrour and Zoghbi 2007). Duplications and triplications spanning MECP2 also cause progressive neurological disorders characterized by autism, motor abnormalities, and seizures (Van Esch et al. 2005; del Gaudio et al. 2006; Ramocki et al. 2009). These findings argue that the levels of MeCP2 must be tightly regulated to ensure normal neurological function. Importantly, it is increasingly clear that maintenance of MeCP2 levels throughout life is critical, as revealed by recent reports demonstrating that inducible adult knockout of Mecp2 recapitulated the germline-null phenotypes (McGraw et al. 2011; Cheval et al. 2012; Nguyen et al. 2012). In human brains, MeCP2 protein level is repressed during the fetal stages and elevated during postnatal development (Balmer et al. 2003), which could be critical for the timely regulation of expression of hundreds of downstream genes (Chahrour et al. 2008). Alternative polyadenylation of MECP2 generates transcripts with differential 3′ untranslated region (UTR) length (Coy et al. 1999; Shahbazian et al. 2002), and interestingly, the repression of MeCP2 production in human fetal brains correlates with the predominant use of the exceptionally long (∼8.5-kb) MECP2 3′ UTR (Coy et al. 1999; Balmer et al. 2003). The direct relationship between these two events, however, has never been explained. In cancer cells, oncogenes have long-to-short 3′ UTR shifts, which lead to overexpression of oncogene-encoded proteins by escaping the effects of microRNA (miRNA) binding to the 3′ UTR (Mayr and Bartel 2009). Therefore, predominance of the long 3′ UTR of MECP2 in human fetal brains could render these transcripts more sensitive to miRNA-mediated repression and thereby keep MeCP2 protein levels low until the proper developmental stage. We hypothesized that there could be fetal brain-enriched miRNAs targeting human MECP2 and that the identification of such miRNAs would provide insight into the regulation of MeCP2 protein levels in human fetal brains.
In this study, we identified miR-483-5p, an intragenic miRNA of the imprinted gene IGF2, as a human-specific regulator of MeCP2 levels. Using fibroblasts from patients with imprinting defects leading to overexpression of miR-483-5p, we demonstrated that the increase in miR-483-5p leads to reduction of MeCP2 levels in human cells. In addition, we showed the enrichment of miR-483-5p in human fetal brains and the inverse correlation between miR-483-5p and MECP2 levels in human brains. We also showed that expression of miR-483-5p in hippocampal neurons rescues the abnormal dendritic spine phenotype caused by overexpression of human MeCP2. Finally, we discovered that miR-483-5p actually regulates the levels of other chromatin remodeling proteins that interact with MeCP2. This study provides interesting insight into the post-transcriptional regulation of MeCP2 and some of its interactors during human brain development.
Results and Discussion
Given the paucity of miRNAs that have been demonstrated to regulate human MECP2 and the fact that none of them are enriched in human fetal brains (Klein et al. 2007; Kuhn et al. 2010), we sought to identify additional MECP2-regulating miRNAs in the hope of discovering some that might play a critical role during fetal development. To this end, we used a newly developed bioinformatic tool—coexpression meta-analysis of miRNA targets (CoMeTa)—that predicts and ranks human miRNA target mRNAs based on their expression relationships (Gennarino et al. 2012). CoMeTa predicted six miRNAs for human MECP2 (Fig. 1A), and we tested the effects of each miRNA on endogenous MeCP2 levels in human medulloblastoma-derived DAOY cells. This analysis showed that the top-ranked miRNA, miR-483-5p, significantly reduced MeCP2 protein levels (Fig. 1B). Moreover, real-time quantitative RT–PCR (qRT–PCR) using primers against protein-coding exons—thus detecting both MECP2 transcripts with short and long 3′ UTRs—showed that miR-483-5p also decreased MECP2 mRNA levels (Fig. 1C). Consistent with the overexpression results, inhibition of endogenous miR-483-5p increased MeCP2 protein levels in DAOY cells (Fig. 1D). Furthermore, miR-483-5p decreased the luciferase activity of a construct containing most of the human MECP2 long 3′ UTR (1–7293), suggesting that the 3′ UTR is responsible for the down-regulation of MECP2 levels (Fig. 1E).
Figure 1.

miR-483-5p regulates human MeCP2 levels. (A) Six putative miRNAs and their ranks for human MECP2 predicted by CoMeTa. (B) Representative Western blot image shows that, among the six candidate miRNAs, overexpression of miR-483-5p reduced human MeCP2 protein levels by ∼80% compared with the cel-miR-67 transfection control. siRNA against MECP2 (si-MECP2) was used as a positive control. (C) qRT–PCR analysis shows that miR-483-5p reduced MECP2 mRNA levels. si-MECP2 was used as a positive control. (D) Inhibitor against miR-483-5p increased MeCP2 protein levels compared with the cel-miR-67 inhibitor control. (E) Luciferase activity of a construct containing the human MECP2 3′ UTR (1–7293) was down-regulated by miR-483-5p. miR-1302 was used as a negative control. (RL) Renilla luciferase; (FL) firefly luciferase. (*) P < 0.05; (**) P < 0.01.
We identified two putative miR-483-5p-binding sites (413–420 and 4090–4097) in the long 3′ UTR of human MECP2 (Fig. 2A). To test which putative miR-483-5p-binding site is authentic, we performed additional luciferase assays in HEK293T cells, which showed that mutagenesis of the second binding site (4090–4097), but not the first site (413–420), affected the efficiency of the down-regulation of luciferase activity by miR-483-5p (Fig. 2B). This suggests that only the second site (4090–4097) is a functional target site for miR-483-5p. Since this functional target site (4090–4097) is located outside the length of the short 3′ UTR (132 nucleotides [nt]) (Fig. 2A), we reasoned that miR-483-5p should regulate specifically the long, but not the short, 3′ UTR of MECP2. To validate this, we designed two additional sets of primers for qRT–PCR. One selectively detects the long 3′ UTR, and the other recognizes both short and long 3′ UTRs (thus total) of MECP2 (Fig. 2A). It was impossible to design primers that detect only the short 3′ UTR, since the long 3′ UTR contains the same sequence of short 3′ UTR. Consistent with the previous result (Fig. 1C), MECP2 mRNA levels measured by both primer sets were decreased by miR-483-5p expression in DAOY cells (Fig. 2C). Notably, the fold changes measured by each primer set were very similar (long 3′ UTR, 0.56; total MECP2, 0.62), suggesting that the decrease of the long 3′ UTR accounts for the change in the total MECP2 level. These results support that miR-483-5p regulates specifically the long 3′ UTR of MECP2.
Figure 2.

miR-483-5p regulates human MECP2 levels through the human-specific binding site in the MECP2 long 3′ UTR. (A) Two putative miR-483-5p-binding sites in the human MECP2 3′ UTR are shown. The colored numbers indicate positions of binding sequences in the 3′ UTR. Base pairs between miR-483-5p and the MECP2 3′ UTR are indicated by vertical lines. The red colored “GUC” was changed to “CAG” in mutant luciferase constructs. Red and blue arrows indicate the target sites of qRT–PCR primers used in C. (B) Schematic diagram shows luciferase constructs containing each putative miR-483-5p-binding site with or without mutation. Luciferase assay with the constructs shows that only the mutation in the second binding site (4090–4097) changed the effect of miR-483-5p. miR-1302 was used as a negative control. (C) qRT–PCR analysis with primers targeting the long 3′ UTR or short and long 3′ UTRs shows that miR-483-5p expression decreased MECP2 mRNA levels by down-regulating specifically the long 3′ UTR. (D) The construct with a longer MECP2 3′ UTR showed lower expression compared with that with a shorter MECP2 3′ UTR, and expression was further decreased by miR-483-5p in DAOY cells. (E) Alignment of MECP2 3′ UTRs from different species (primates and rodents) for the miR-483-5p-binding site. (F) Alignment of miR-483-5p seed sequences from different species. (G) Overexpression of miR-483-5p reduced MeCP2 protein levels in human SK-N-SH cells but had no effect in mouse Neuro2a cells. (H) The relative expression levels of the MECP2 long 3′ UTR were similar in SK-N-SH cells and Neuro2a cells. (I, left panel) The chimpanzee MECP2 sequence has a single-nucleotide difference from human family in the miR-483-5p-binding region. (Right panel) Luciferase assay with a “G-to-C” mutant construct (3514–4561 of the 3′ UTR) confirmed human specificity of miR-483-5p binding to the MECP2 3′ UTR. (J) When overexpressed, miR-483-5p reduced MeCP2 protein levels more efficiently than miR-132 and miR-155 in DAOY cells. In all experiments, cel-miR-67 was used as a transfection control. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001.
To test our initial hypothesis that the long 3′ UTR is more sensitive to miRNA-mediated down-regulation and thereby is causally associated with lower MeCP2 levels, we compared luciferase activity of constructs with either long (1–7293) or short (1–1600) human MECP2 3′ UTR in DAOY cells. The luciferase activity associated with the long 3′ UTR was significantly lower than that of the short form, and expression of miR-483-5p further decreased the activity of the long, but not the short, 3′ UTR (Fig. 2D), which confirms the specific effect of miR-483-5p on the long 3′ UTR of MECP2.
Unlike most known and functionally established miRNA-binding sites, the miR-483-5p-binding site (4090–4097) in the human MECP2 3′ UTR is not evolutionarily conserved, while the seed sequence of miR-483-5p itself is conserved from mice to humans (Fig. 2E,F). Consistent with this finding, miR-483-5p reduced endogenous MeCP2 levels only in human SK-N-SH cells but not in mouse Neuro2a cells (Fig. 2G). The relative expression level of the MECP2 long 3′ UTR (long 3′ UTR/total MECP2) was similar in SK-N-SH and Neuro2a cells (Fig. 2H), suggesting that the minimal effect of miR-483-5p on MeCP2 levels in Neuro2a cells was not due to the lower expression of long 3′ UTR of MeCP2 but to the lack of miR-483-5p-binding sequence in the MeCP2 long 3′ UTR. Interestingly, the miR-483-5p-binding site is conserved in the Denisovan, a Paleolithic species of human family (Meyer et al. 2012), but not in the chimpanzee MECP2 gene, which differs by 1 nt in the binding region (Fig. 2I). This single-nucleotide change blocked the ability of miR-483-5p to down-regulate MeCP2 levels (Fig. 2I), suggesting that the miR-483-5p-mediated regulation of MeCP2 levels is indeed human-specific. We also found that miR-483-5p was more effective in down-regulating human MeCP2 levels than miR-132 and miR-155, the two known miRNAs targeting MECP2 mRNA through evolutionarily conserved binding sites (Fig. 2J; Klein et al. 2007; Kuhn et al. 2010).
miR-483-5p is located in the second intron of IGF2, and the level of miR-483-5p correlates with IGF2 expression (Ma et al. 2011). IGF2 is an imprinted gene that is expressed almost exclusively from the paternal allele in many tissues. Misregulation of imprinting on the maternal IGF2 allele can activate IGF2 transcription from the maternal allele, causing Beckwith-Wiedemann Syndrome (BWS; Online Mendelian Inheritance in Man [OMIM] 130650) (Weksberg et al. 2010). To confirm the negative regulation of MeCP2 by miR-483-5p, we analyzed human fibroblasts from BWS patients carrying an epimutation (gain of methylation) at the imprinting control region 1 (ICR1) that results in biallelic IGF2 expression. We found that IGF2 and miR-483-5p levels were increased, while MeCP2 protein levels were decreased in the BWS fibroblasts (Fig. 3A,B). Notably, miR-483-5p and MeCP2 protein levels were inversely correlated in each control/BWS pair (Fig. 3B).
Figure 3.

Inverse correlation between miR-483-5p and MeCP2 levels in human BWS fibroblasts and developing human brain samples. (A) qRT–PCR analysis shows elevated expression of miR-483-5p and its host gene, IGF2, in the BWS human fibroblasts compared with the control human fibroblasts. (B) MeCP2 protein levels were decreased in BWS human fibroblasts. The colored numbers indicate fold changes of MeCP2 (blue) and miR-483-5p (red) in each cell pair. The histogram at the right shows the average of MeCP2 fold changes from the three cell pairs. (C) qRT–PCR analysis of the miR-483-5p and miR-132 levels in human fetal and postnatal brains. (D,E) The correlation between expression levels of MECP2 and miR-483-5p (D) or miR-132 (E) in human brain (cortex) samples from different ages ([green dots] fetal samples; [red dots] postnatal samples) measured by qRT–PCR. GAPDH and RNU48 were used to normalize the expression of genes in qRT–PCR. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001.
Given that IGF2 is a growth factor that is highly expressed in fetal life (Gicquel and Le Bouc 2006), miR-483-5p could be the miRNA repressing MECP2 expression in human fetal brains. To test this hypothesis, we measured the levels of miR-483-5p and miR-132 (Klein et al. 2007) in different human cortex samples obtained at fetal and postnatal stages. Interestingly, those two miRNAs showed differential expression depending on the developmental stage. While miR-132 was increased in postnatal stages, miR-483-5p was enriched in fetal brains then down-regulated postnatally (Fig. 3C). Consistent with this observation, the levels of miR-483-5p inversely correlated with MECP2 levels in human cortex, whereas the levels of miR-132 did not (Fig. 3D,E). These results suggest that miR-483-5p could be important in repressing MeCP2 levels, especially during fetal stages, while miR-132 could function in fine-tuning MeCP2 levels during postnatal stages (Klein et al. 2007). We also measured the levels of miR-483-3p, the opposite strand miRNA generated from the same precursor miRNA of miR-483-5p. Interestingly, in contrast to miR-483-5p, the expression of miR-483-3p was increased in postnatal stages, suggesting that these two miRNAs are differentially expressed at least in developing human brains (Supplemental Fig. 1A,B). Moreover, in luciferase assays, miR-483-3p did not change the activity of a construct with the MECP2 long 3′ UTR (Supplemental Fig. 1C), suggesting that miR-483-5p is the only miRNA regulating MECP2 levels expressed from the IGF2 locus.
In cultured mouse hippocampal neurons, miR-483-5p reduced the luciferase activity of a construct with the human MECP2 3′ UTR, suggesting that miR-483-5p could target human MECP2 in neurons (Supplemental Fig. 2A). We also cultured hippocampal neurons from MECP2 transgenic mice (Tg1) that express, in addition to the endogenous mouse Mecp2, the human MECP2 (Collins et al. 2004) with the long 3′ UTR, including the functional miR-483-5p-binding site (4090–4097) (Supplemental Fig. 2B,C). As described previously (Collins et al. 2004), the MeCP2 protein level was about twofold higher in Tg1 neurons compared with wild-type neurons (Fig. 4A). In the Tg1 neurons, expression of miR-483-5p restored MeCP2 to the wild-type level and rescued abnormal dendritic spine phenotype (Fig. 4A,B; Supplemental Fig. 2D). However, miR-483-5p did not affect either MeCP2 levels or dendritic spines in wild-type mouse neurons, confirming its specificity to human MECP2 (Fig. 4A,B).
Figure 4.
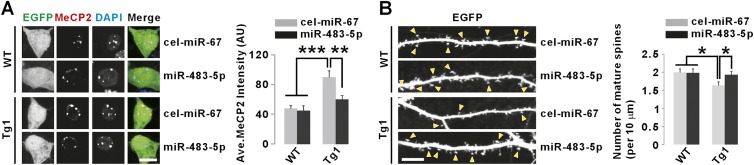
Overexpression of miR-483-5p restores MeCP2 protein levels and rescues abnormal mature spine density in neurons from MECP2 Tg1 mice. (A) Representative images of MeCP2 immunostaining in neuronal cell bodies show that miR-483-5p reduced MeCP2 protein levels in cultured hippocampal neurons from Tg1 mice without changing those in wild-type (WT) neurons. DAPI signals were not affected by miR-483-5p (data not shown). (Ave.) Average; (AU) arbitrary unit. Bar 10 μm. (B) Confocal images of dendritic spines show that miR-483-5p rescued reduced mature spine density in cultured hippocampal neurons from Tg1 mice. The yellow arrowheads indicate mature spines. Bar, 10 μm. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001.
A single miRNA or group of miRNAs can regulate a specific biological pathway by modulating the expression of various proteins participating in the same pathway (Gennarino et al. 2012). To understand the major functional pathway regulated by miR-483-5p, we ran co-operational level (COOL) analysis (Gennarino et al. 2012) followed by gene ontology (GO) analysis for miR-483-5p. COOL analysis clusters the putative targets of specific miRNA based on the highest degree of coexpression (Gennarino et al. 2012). Interestingly, the COOL and GO analysis for miR-483-5p showed enrichment of chromatin-related functions (Fig. 5A; Supplemental Table 1), which is reminiscent of the function of MeCP2 itself (Nan et al. 1997; Jones et al. 1998). To identify authentic miR-483-5p targets among the genes in those biological categories, we initially screened the effect of miR-483-5p overexpression on the levels of the putative targets using qRT–PCR in DAOY cells (data not shown). Those genes with a >25% decrease in levels were further validated with inhibitor of miR-483-5p and si-MECP2 (Fig. 5B). Through this approach, we found that human CNOT6, MAML1, RBM14, HDAC4, and TBL1X could be additional targets of miR-483-5p (Fig. 5B). To test whether miR-483-5p directly binds to the 3′ UTRs of the five genes, we performed luciferase assays. miR-483-5p decreased the activities of luciferase reporters fused to the wild-type, but not the binding site mutant, 3′ UTR (Fig. 5C; Supplemental Fig. 3). These results suggest that, in addition to MECP2, those five genes are directly regulated by miR-483-5p.
Figure 5.

miR-483-5p regulates the levels of additional targets in the MeCP2-interacting corepressor complexes. (A) Enriched CoMeTa COOL biological categories and their P-values predicted for miR-483-5p. (B) Validation of miR-483-5p putative targets in DAOY cells by qRT–PCR. The effect of si-MECP2 was analyzed to exclude secondary changes caused by MeCP2 down-regulation upon miR-483-5p expression (CITED2 and ARID5B). Green colored genes are more likely targets. MEF2C (a gene negatively regulated by MeCP2) levels were measured as a positive control (orange colored). GAPDH was used to normalize the expression of genes (black dotted line). (C) Luciferase assays with the constructs containing 3′ UTRs of indicated genes show down-regulation of the expression by miR-483-5p. Mutations in the miR-483-5p-binding sites inhibited the effect of miR-483-5p on the expression. Mutated sites are described in Supplemental Figure 3. (D) Diagram shows interactions among the miR-483-5p targets (HDAC4, TBL1X, and MeCP2). Solid lines indicate direct interactions. (E) Overexpression and inhibition experiments show that miR-483-5p regulates protein levels of HDAC4 and TBL1X in DAOY cells. (F) HDAC4 and TBL1X protein levels were decreased in BWS human fibroblasts carrying an epimutation (gain of methylation) at ICR1. (G,H) The inverse correlation between expression levels of miR-483-5p and HDAC4 (G) or TBL1X (H) in human brain (cortex) samples from different ages measured by qRT–PCR. In all experiments, cel-miR-67 was used as a transfection control. (*) P < 0.05; (**) P < 0.01.
Next, we tried to further understand the functional relationship among the authentic miR-483-5p targets. Since protein interaction could reveal the direct functional relationships, we searched a database (Ingenuity Pathway Analysis) to find any protein interaction among CNOT6, MAML1, RBM14, HDAC4, TBL1X, and MeCP2. This analysis revealed the interactions among MeCP2, HDAC4, and TBL1X, which are mediated by corepressor complexes (Fig. 5D). We could not find any interaction with CNOT6, MAML1, and RBM14. MeCP2-dependent regulation of gene expression involves recruitment of corepressors such as NCoR and SIN3A to the MeCP2-bound genomic locus (Guy et al. 2011). Indeed, HDAC4 directly interacts with these corepressors, and TBL1X is a subunit of the NCoR/SMRT corepressor complex (Fischle et al. 2002; Yoon et al. 2003), suggesting their functional association with MeCP2. Accordingly, we further characterized the relationship between miR-483-5p and expression of HDAC4 and TBL1X. We found that miR-483-5p modulates HDAC4 and TBL1X protein levels in DAOY cells (Fig. 5E). Consistently, their protein levels were also decreased in BWS fibroblasts (Fig. 5F). Finally, similar to MECP2, both HDAC4 and TBL1X levels were inversely correlated with miR-483-5p in human brain samples (Fig. 5G,H). Together, these results suggest that miR-483-5p can regulate the abundance of MeCP2 and MeCP2-interacting corepressor complexes in human fetal brains (Fig. 5D).
Interestingly, we found that the miR-483-5p-binding sites in the 3′ UTRs of human CNOT6, MAML1, RBM14, HDAC4, and TBL1X are conserved only in primates but not in rodents (Supplemental Fig. 3), which is reminiscent of the human-specific binding site in the MECP2 long 3′ UTR. This finding prompted us to analyze the conservation of miR-483-5p-binding sites in a large number of putative targets to help us understand the conserved or species-specific function of miR-483-5p. We analyzed the conservation of 223 miR-483-5p-binding sites in the 3′ UTRs of 169 putative target genes predicted by CoMeTa (see the Supplemental Material for details). Interestingly, only ∼5% (12 out of 223) of the binding sites in nine genes (NRXN3, ELK1, HOXC11, SAMD4A, PRPF4B, FHL1, TYRO3, CFLAR, and KIAA0913) were conserved from humans to rodents (Supplemental Fig. 4A,B; Supplemental Table 2). In contrast to the primate-specific putative targets of miR-483-5p (∼95% of putative targets), GO analysis of the nine genes with conserved binding sites from humans to rodents revealed other functional categories that are not enriched for chromatin-related functions (Supplemental Fig. 5A,B).
In spite of the well-established fact that precise MeCP2 levels are critical for normal brain function (Chao and Zoghbi 2012), the molecular mechanism underlying MeCP2 level regulation is poorly understood. miRNA-mediated regulation has been considered one of the potential mechanisms based on the exceptionally long MECP2 3′ UTR and broad implications of miRNAs in many biological processes, including neuronal development and function (Kosik 2006; Saba and Schratt 2010). Therefore, identification of miR-483-5p provides important evidence supporting the critical role of miRNAs for regulating MeCP2 levels.
MeCP2 levels are regulated spatiotemporally in human brains. Each cell type requires a specific level of MeCP2, which gradually increases throughout development (Shahbazian et al. 2002; Samaco et al. 2004). Considering this complexity, there could be multiple regulatory components of MeCP2, and those components should function coordinately to control MeCP2 levels precisely. In this study, we showed that miR-483-5p and miR-132 are differentially expressed in human fetal and postnatal brains. MeCP2 levels are repressed during the fetal stage, at which time the expression of miR-483-5p is high. As brain and synaptic connections are mature and MeCP2 levels become relatively stable, miR-132, which is expressed in mature neurons, could fine-tune MeCP2 levels in an activity-dependent manner (Klein et al. 2007; Nudelman et al. 2010).
To our knowledge, miR-483-5p is the first example of a human-specific regulator of MeCP2 levels. We clearly demonstrate the human specificity of miR-483-5p using MECP2 Tg1 neurons as well as luciferase assays. Although the human-specific interaction between miR-483-5p and the MECP2 3′ UTR was unexpected, our additional data shed some light on this and support the significance of the interaction. First, miR-483-5p is enriched in human fetal brains as compared with postnatal brains, and this expression pattern matches with the predominant use of the MECP2 long 3′ UTR, the target of miR-483-5p. To date, there has been no insight into why human fetal brains predominantly express the long 3′ UTR of MECP2 and how it correlates with low levels of MeCP2 proteins during fetal development. The identification of miR-483-5p helps shed light on this process. Consistent with the biological relevance of our finding, we discovered and verified additional targets of miR-483-5p (HDAC4 and TBL1X) that function in the MeCP2-interacting corepressor complexes. Altogether, this supports the role of miR-483-5p in regulating MeCP2 function. Notably, the miR-483-5p-binding sites in the 3′ UTR of human HDAC4 and TBL1X are conserved only in primates.
It is also notable that miR-483-5p is an intragenic miRNA residing in an imprinted gene, IGF2. Indeed, we demonstrated that miR-483-5p levels were increased, while MeCP2 levels were down-regulated in the BWS human fibroblasts. Based on this finding, our study may potentially link some human phenotypes associated with abnormal imprinting (BWS) and a mild decrease in MeCP2 levels. Interestingly, a higher prevalence of autism spectrum disorders in BWS patients has been reported (Kent et al. 2008).
Our study suggests that human fetal brains coordinately express the two regulatory components of MECP2—the long 3′ UTR and miR-483-5p—to repress its levels until proper maturation of the brain. Identification of such an exquisite mechanism of regulation of a protein and its cofactors and revealing that this mechanism is limited to higher primates underscore the importance of tightly controlling MeCP2 levels in human brains throughout their lifetime.
Materials and methods
Real-time qRT–PCR
DAOY cells in six-well plates were transfected with 70 pmol of miRNA duplex or siRNA or 200 pmol of miRNA hairpin inhibitor using a Dharmafect reagent. After 2 d, total RNA was extracted using a miRNeasy minikit (Qiagen), and 1 μg of total RNA was used to synthesize cDNA by Quantitect reverse transcription kit (Qiagen). The same protocol was applied to the human fibroblasts. For human brains, total RNA was extracted from ∼50 mg of each sample, and cDNA was synthesized as DAOY cells. Mature miR-483-5p, miR-483-3p, and miR-132 were detected using TaqMan miRNA assays (Applied Biosystems). The primers used in qRT–PCR reactions are described in the Supplemental Material.
Other information on materials and methods is described in the Supplemental Material.
Acknowledgments
We are grateful to R. Person and A. Beaudet for sharing human brain samples, and H. Kang for helping on the protein interaction network. We thank M. Sardiello, C. Schaaf, C. McGraw, J. Holder, B. Cochran, and the members of the H.Y.Z. laboratory for comments and discussions on the manuscript. This project was supported by The Howard Hughes Medical Institute (H.Y.Z.), NIH grants (5R01NS057819 to H.Y.Z; R01-DA023999 and R01-NS038296 to P.R.; and NIH/NIAAA K99AA018387 to K.H.-T.), and the Baylor Intellectual and Developmental Disabilities Research Center (P30HD024064) Administrative and Confocal cores from the Eunice Kennedy Shriver National Institute of Child Health and Human Development. C.S.R. is supported by a post-doctoral grant from the Wenner-Gren Foundations, Sweden.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.207456.112.
References
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY 1999. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23: 185–188 [DOI] [PubMed] [Google Scholar]
- Balmer D, Goldstine J, Rao YM, LaSalle JM 2003. Elevated methyl-CpG-binding protein 2 expression is acquired during postnatal human brain development and is correlated with alternative polyadenylation. J Mol Med (Berl) 81: 61–68 [DOI] [PubMed] [Google Scholar]
- Chahrour M, Zoghbi HY 2007. The story of Rett syndrome: From clinic to neurobiology. Neuron 56: 422–437 [DOI] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY 2008. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320: 1224–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao HT, Zoghbi HY 2012. MeCP2: Only 100% will do. Nat Neurosci 15: 176–177 [DOI] [PubMed] [Google Scholar]
- Cheval H, Guy J, Merusi C, De Sousa D, Selfridge J, Bird A 2012. Postnatal inactivation reveals enhanced requirement for MeCP2 at distinct age windows. Hum Mol Genet 21: 3806–3814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins AL, Levenson JM, Vilaythong AP, Richman R, Armstrong DL, Noebels JL, David Sweatt J, Zoghbi HY 2004. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet 13: 2679–2689 [DOI] [PubMed] [Google Scholar]
- Coy JF, Sedlacek Z, Bachner D, Delius H, Poustka A 1999. A complex pattern of evolutionary conservation and alternative polyadenylation within the long 3′-untranslated region of the methyl-CpG-binding protein 2 gene (MeCP2) suggests a regulatory role in gene expression. Hum Mol Genet 8: 1253–1262 [DOI] [PubMed] [Google Scholar]
- del Gaudio D, Fang P, Scaglia F, Ward PA, Craigen WJ, Glaze DG, Neul JL, Patel A, Lee JA, Irons M, et al. 2006. Increased MECP2 gene copy number as the result of genomic duplication in neurodevelopmentally delayed males. Genet Med 8: 784–792 [DOI] [PubMed] [Google Scholar]
- Fischle W, Dequiedt F, Hendzel MJ, Guenther MG, Lazar MA, Voelter W, Verdin E 2002. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol Cell 9: 45–57 [DOI] [PubMed] [Google Scholar]
- Gennarino VA, D'Angelo G, Dharmalingam G, Fernandez S, Russolillo G, Sanges R, Mutarelli M, Belcastro V, Ballabio A, Verde P, et al. 2012. Identification of microRNA-regulated gene networks by expression analysis of target genes. Genome Res 22: 1163–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gicquel C, Le Bouc Y 2006. Hormonal regulation of fetal growth. Horm Res 65: 28–33 [DOI] [PubMed] [Google Scholar]
- Guy J, Cheval H, Selfridge J, Bird A 2011. The role of MeCP2 in the brain. Annu Rev Cell Dev Biol 27: 631–652 [DOI] [PubMed] [Google Scholar]
- Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP 1998. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet 19: 187–191 [DOI] [PubMed] [Google Scholar]
- Kent L, Bowdin S, Kirby GA, Cooper WN, Maher ER 2008. Beckwith Weidemann syndrome: A behavioral phenotype–genotype study. Am J Med Genet B Neuropsychiatr Genet 147B: 1295–1297 [DOI] [PubMed] [Google Scholar]
- Klein ME, Lioy DT, Ma L, Impey S, Mandel G, Goodman RH 2007. Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA. Nat Neurosci 10: 1513–1514 [DOI] [PubMed] [Google Scholar]
- Kosik KS 2006. The neuronal microRNA system. Nat Rev Neurosci 7: 911–920 [DOI] [PubMed] [Google Scholar]
- Kuhn DE, Nuovo GJ, Terry AV Jr, Martin MM, Malana GE, Sansom SE, Pleister AP, Beck WD, Head E, Feldman DS, et al. 2010. Chromosome 21-derived microRNAs provide an etiological basis for aberrant protein expression in human Down syndrome brains. J Biol Chem 285: 1529–1543 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ma N, Wang X, Qiao Y, Li F, Hui Y, Zou C, Jin J, Lv G, Peng Y, Wang L, et al. 2011. Coexpression of an intronic microRNA and its host gene reveals a potential role for miR-483-5p as an IGF2 partner. Mol Cell Endocrinol 333: 96–101 [DOI] [PubMed] [Google Scholar]
- Mayr C, Bartel DP 2009. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138: 673–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGraw CM, Samaco RC, Zoghbi HY 2011. Adult neural function requires MeCP2. Science 333: 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, Kircher M, Gansauge MT, Li H, Racimo F, Mallick S, Schraiber JG, Jay F, Prufer K, de Filippo C, et al. 2012. A high-coverage genome sequence from an archaic Denisovan individual. Science 338: 222–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan X, Campoy FJ, Bird A 1997. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 88: 471–481 [DOI] [PubMed] [Google Scholar]
- Nguyen MV, Du F, Felice CA, Shan X, Nigam A, Mandel G, Robinson JK, Ballas N 2012. MeCP2 is critical for maintaining mature neuronal networks and global brain anatomy during late stages of postnatal brain development and in the mature adult brain. J Neurosci 32: 10021–10034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nudelman AS, DiRocco DP, Lambert TJ, Garelick MG, Le J, Nathanson NM, Storm DR 2010. Neuronal activity rapidly induces transcription of the CREB-regulated microRNA-132, in vivo. Hippocampus 20: 492–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramocki MB, Peters SU, Tavyev YJ, Zhang F, Carvalho CM, Schaaf CP, Richman R, Fang P, Glaze DG, Lupski JR, et al. 2009. Autism and other neuropsychiatric symptoms are prevalent in individuals with MeCP2 duplication syndrome. Ann Neurol 66: 771–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saba R, Schratt GM 2010. MicroRNAs in neuronal development, function and dysfunction. Brain Res 1338: 3–13 [DOI] [PubMed] [Google Scholar]
- Samaco RC, Nagarajan RP, Braunschweig D, LaSalle JM 2004. Multiple pathways regulate MeCP2 expression in normal brain development and exhibit defects in autism-spectrum disorders. Hum Mol Genet 13: 629–639 [DOI] [PubMed] [Google Scholar]
- Shahbazian MD, Antalffy B, Armstrong DL, Zoghbi HY 2002. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum Mol Genet 11: 115–124 [DOI] [PubMed] [Google Scholar]
- Van Esch H, Bauters M, Ignatius J, Jansen M, Raynaud M, Hollanders K, Lugtenberg D, Bienvenu T, Jensen LR, Gecz J, et al. 2005. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet 77: 442–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weksberg R, Shuman C, Beckwith JB 2010. Beckwith-Wiedemann syndrome. Eur J Hum Genet 18: 8–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon HG, Chan DW, Huang ZQ, Li J, Fondell JD, Qin J, Wong J 2003. Purification and functional characterization of the human N-CoR complex: The roles of HDAC3, TBL1 and TBLR1. EMBO J 22: 1336–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]