

The absence of hypocretinergic (Hcrt) neurons causes narcolepsy, possibly due to an autoimmune response. Using TRAP lines for translational profiling, Dalal et al. identify a suite of actively translated genes in Hcrt neurons and validate a new set of Hcrt neuronal markers as candidate targets. Functional studies show that mice deficient in transcription factor Lhx9 are hypersomnolent. Thus, Lhx9 may play a role in the development of a subset of Hcrt neurons to affect normal sleep behavior.
Keywords: translational profiling, hypocretin, orexin, bacTRAP, Lhx9, narcolepsy
Abstract
Hypocretin (orexin; Hcrt)-containing neurons of the hypothalamus are essential for the normal regulation of sleep and wake behaviors and have been implicated in feeding, anxiety, depression, and reward. The absence of these neurons causes narcolepsy in humans and model organisms. However, little is known about the molecular phenotype of these cells; previous attempts at comprehensive profiling had only limited sensitivity or were inaccurate. We generated a Hcrt translating ribosome affinity purification (bacTRAP) line for comprehensive translational profiling of all ribosome-bound transcripts in these neurons in vivo. From this profile, we identified >6000 transcripts detectably expressed above background and 188 transcripts that are highly enriched in these neurons, including all known markers of the cells. Blinded analysis of in situ hybridization databases suggests that ∼60% of these are expressed in a Hcrt marker-like pattern. Fifteen of these were confirmed with double labeling and microscopy, including the transcription factor Lhx9. Ablation of this gene results in a >30% loss specifically of Hcrt neurons, without a general disruption of hypothalamic development. Polysomnography and activity monitoring revealed a profound hypersomnolence in these mice. These data provide an in-depth and accurate profile of Hcrt neuron gene expression and suggest that Lhx9 may be important for specification or survival of a subset of these cells.
Narcolepsy is a profound disorder of sleep regulation, characterized by excessive daytime sleepiness, sleep attacks, cataplexy, and sleep-onset REM (rapid eye movement) periods. Studies indicate that narcolepsy, especially when accompanied by cataplexy, is due to the absence of hypocretinergic neurons and signaling (Nishino et al. 2000; Thannickal et al. 2000; Bourgin et al. 2008). The hypocretins (Hcrts; also called orexins) are peptidergic neurotransmitters used by a discrete population of neurons in the lateral hypothalamus (de Lecea et al. 1998; Sakurai et al. 1998). In model organisms, disruption of the gene encoding the Hcrt peptides or a Hcrt receptor (Chemelli et al. 1999; Lin et al. 1999) or, alternatively, the ablation of the neurons producing Hcrt (Gerashchenko et al. 2001; Hara et al. 2001; Zhang et al. 2007) recapitulates features of narcolepsy. In humans, there are two variations of the disorder: narcolepsy with cataplexy (NC) and narcolepsy without cataplexy (NwoC). NC is strongly linked and associated with the HLA DQB1*0602 allele, a T-cell receptor locus, and P2RY11, a receptor that can regulate immune cell survival (Hallmayer et al. 2009; Kornum et al. 2011). This strongly suggests involvement of the immune system in the etiology of NC, while the involvement in NwoC is less clear.
There is an interest in identifying a comprehensive profile of proteins specifically found in Hcrt neurons for three reasons. First, there is a long-standing hypothesis that epitopes found specifically in Hcrt neurons may serve to direct the autoimmune-mediated destruction of these cells (Lim and Scammell 2010). Previous work suggests that Trib2 may be one such epitope (Cvetkovic-Lopes et al. 2010; Kawashima et al. 2010; Toyoda et al. 2010), although this did not replicate in more recent patients with narcolepsy following H1N1 (Dauvilliers et al. 2010). Second, there is an interest in understanding the suit of receptors and channels specifically expressed by these neurons, as these may serve as drug targets for modulating sleep even in healthy individuals. Third, there is an interest in understanding the transcription factors that may serve to specify these neurons as both candidates for rare variant analyses in humans with early onset narcolepsy or NwoC and tools to direct the differentiation of Hcrt neurons from patient cells. In the present study, we used translating ribosome affinity purification (TRAP) (Doyle et al. 2008; Heiman et al. 2008; Dougherty et al. 2010) to generate a comprehensive translational profile of Hcrt neurons. We then confirmed these results with independent methods and pursued functional studies of one transcription factor, Lhx9, that may be important for the normal development of a subset of Hcrt neurons and normal sleep behavior.
Results
Translational profiling of Hcrt neurons
To apply the TRAP methodology to Hcrt neurons, we generated mouse lines targeting Hcrt-producing cells (Hcrt∷eGFP-RpL10a) (Fig. 1A). Successful targeting was confirmed using confocal immunofluorescent microscopy for eGFP and Hcrt antibodies (Fig. 1B) as well as in situ hybridization (ISH) for Hcrt transcript and GFP (data not shown). Both methods revealed GFP expression in >85% of neurons with detectable Hcrt expression and no instances of eGFP-RpL10a expression in Hcrt-negative cells. There was no eGFP-RPL10a expression elsewhere in the adult brain.
Figure 1.

Characterization of a Hcrt bacTRAP line. (A) Immunohistochemistry for Hcrt1 (orexin A) in a wild-type (WT) mouse brain (left panel) shows the same pattern of expression as eGFP antibodies on Hcrt∷eGFP-RpL10a mice (middle panel). (Right panel) Both label scattered cells with neuronal morphology in the lateral hypothalamus. (B) Confocal immunofluorescence on the Hcrt∷eGFP-RpL10a mouse line reveals >85% of Hcrt-positive neurons (red; middle panel) are eGFP-RpL10a-positive (green; left panel). No Hcrt-negative, eGFP-RpL10a-positive cells were seen anywhere in brain. Bar, 50 μm.
We then conducted replicate TRAP assays on four independent pools of adult mice. Biological replicates showed good reproducibility, with the minimum Pearson's correlation >0.96 across all comparisons (Fig. 2A). Compared with total RNA from the diencephalon, mRNA purified from Hcrt neurons showed a substantial enrichment of many transcripts, including known markers of these cells (Fig. 2B).
Figure 2.

TRAP purification of Hcrt cell mRNA. (A) Scatter plot of mRNA levels from microarray data from replicate Hcrt neuron TRAP assays. Log10 expression values. (B) Scatter plot of mRNA levels from microarray data from Hcrt neuron TRAP assays (X-axis, average of four arrays), compared with whole hypothalamic RNA, shows robust enrichment (>16-fold) of known Hcrt neuron-expressed genes (blue) and depletion (<0.5-fold) of negative controls (red, markers of glia). Multiple probe sets per gene are labeled when available. (C) Scatter plot of Pabp arrays (downloaded from Gene Expression Omnibus [GEO]: GSE17617; average of n = 9 arrays per group) shows a larger number of probe sets enriched more than twofold, including some glial genes. Most of the marker probe sets showed no signal. (D) Scoring rubric for Allen Brain Atlas analysis. (E) Blinded scoring of ISH patterns reveals a significant enrichment within the TRAP gene list for transcripts with Hcrt marker-like expression (scores of 1–3, P < 3 × 10−10, χ2 test). Each list normalized to the total number of scorable transcripts on the list (n = 32–67).
Identification of transcripts enriched in the Hcrt neuron translational profile
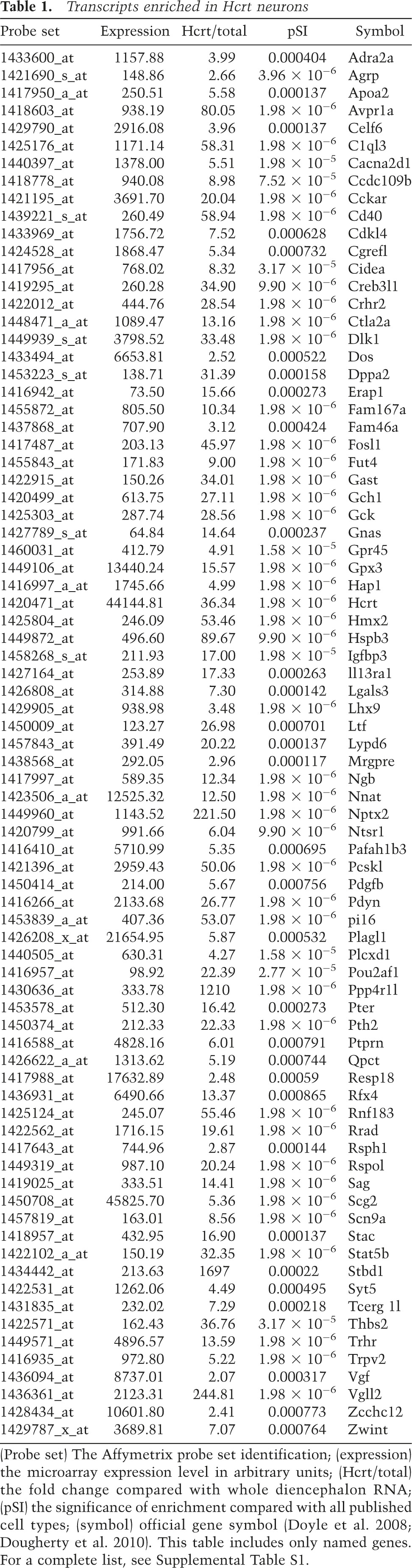
To systematically identify a set of RNAs specifically enriched in Hcrt neurons compared with cell types across the nervous system, we combined the results of three statistical filters: We removed probe sets with low expression, selected for those with more than twofold enrichment in Hcrt neurons compared with total diencephalic RNA, and selected those significantly specific to Hcrt neurons (pSI < 0.01) when compared with a previously collected set of 26 neural samples (Doyle et al. 2008) as described (Dougherty et al. 2010). This identified 220 probe sets, representing products from 188 genes (Table 1; Supplemental Table S1), and included a significant overlap with the few known Hcrt cell markers Hcrt, Nptx2, Pdyn, and Igfpb3 (P < 10 × 10−100, χ2 test) (Reti et al. 2002; Crocker et al. 2005; Honda et al. 2009).
Table 1.
Transcripts enriched in Hcrt neurons
Comparison with previous studies and validation of microarray results
Previously, others have used different techniques to profile Hcrt neurons: using affinity purification of RNAs bound to a tagged polyA-binding protein (Pabp) expressed in Hcrt neurons (Cvetkovic-Lopes et al. 2010) or screening for transcripts lost from hypothalamic RNA when Hcrt neurons were ablated (Honda et al. 2009). Careful comparison with these studies using statistical criteria matched to each publication indicates that our data are more accurate than Pabp (Fig. 2C–E) and are consistent with Honda et al. (2009) but with an order of magnitude more sensitivity.
As Hcrt neurons are relatively rare and thus contribute little RNA to a total RNA sample analyzed from whole hypothalamus, the strategy taken by Honda et al. (2009) would only be predicted to detect only very abundant and specific transcripts from these cells as well as changes in other cells responding to the loss of the Hcrt neurons. They discovered 53 probe sets depleted more than threefold, which overlapped with the known markers of these cells (P < 10 × 10−100, χ2 test). Thus, the approach was accurate although likely not comprehensive. Matching statistical criteria to the previous studies, TRAP would detect 534 probe sets more than threefold enriched. TRAP probe sets significantly overlap with that of Honda et al. (2009) (P < 7 × 10−7, χ2 test) and the known markers of these cells (P < 10 × 10−100, χ2 test). In contrast, the Pabp microarrays detected 1709 probe sets as enriched in Hcrt neurons more than threefold, although there was no significant overlap with either our analysis, that of Honda et al. (2009), or the known markers of these cells (all P > 0.1, χ2 test) (see also Fig. 2C).
For a systematic and independent confirmation of our data, we also examined the Allen Brain Atlas database of mouse ISH patterns (Lein et al. 2007) for our 188 most enriched transcripts (Table 1; Supplemental Table S1). Blinded scorers simultaneously examined the top 188 transcripts from Pabp and 188 transcripts randomly selected from the microarray. Due to the scattered nature of Hcrt neuron expression (Fig. 1B), it is impossible to unambiguously identify Hcrt neurons from single-label ISH. However, all Hcrt neuron-enriched genes should show a pattern of enrichment in scattered cells of the lateral hypothalamus. Therefore, we scored for this “Hcrt-like pattern,” (HLP) using as guides the Allen ISH patterns of Hcrt, Pdyn, Nptx2, and Igfbp3—markers that are known to have varying degrees of specificity for these cells (Fig. 2D).
Of the 188 TRAP transcripts, 65 had coronal ISH patterns with some detectable signal. Of these, 37 (59%) had an expression pattern clearly consistent with a robust enrichment in Hcrt neurons, analogous to positive controls Pdyn and Nptx2. Compared with the frequency of HLPs from the 188 randomly selected transcripts, the odds of this enrichment occurring by chance are slim (P < 3 × 10−10, χ2 test). Seven seemed to nearly match the exact pattern of Hcrt expression (Fam46a, Creb3I1, Gng2, Lhx9, Tmed3, Rfx4, and Pcbd1) in the hypothalamus (Supplemental Fig. S1). Of the 65 scored, only two (Agrp and Bend5) had patterns of expression that would seem to exclude them from Hcrt neurons entirely. The remaining genes were more broadly expressed in hypothalamic cells. These would not necessarily be considered to contradict the microarray results: A difference of twofold may not be readily discernible by ISH. Also ISH and TRAP may differ because of differences between transcription and translation, and occasionally ISH from the Allen Brain Atlas are simply inaccurate. For example, Lhx9 shows strong ubiquitous brain expression in the Allen Brain Atlas at most ages, in contrast to other reports (Bertuzzi et al. 1999; Retaux et al. 1999; Nakagawa and O'Leary 2001; Gray et al. 2004). Nonetheless, even using only the 188 transcripts derived from our most stringent criteria, TRAP increased by an order of magnitude the number of known Hcrt neuron-enriched genes. The Honda et al. (2009) data did not have a comparable number of transcripts to score, but of their scorable ISH patterns, they showed nearly the same pattern of enrichment as the TRAP data (57% with scores of 1–3). In contrast, the transcripts from the Pabp pulldown were no more likely than chance to be expressed in a HLP (P > 0.15, χ2 test) (Fig. 2E).
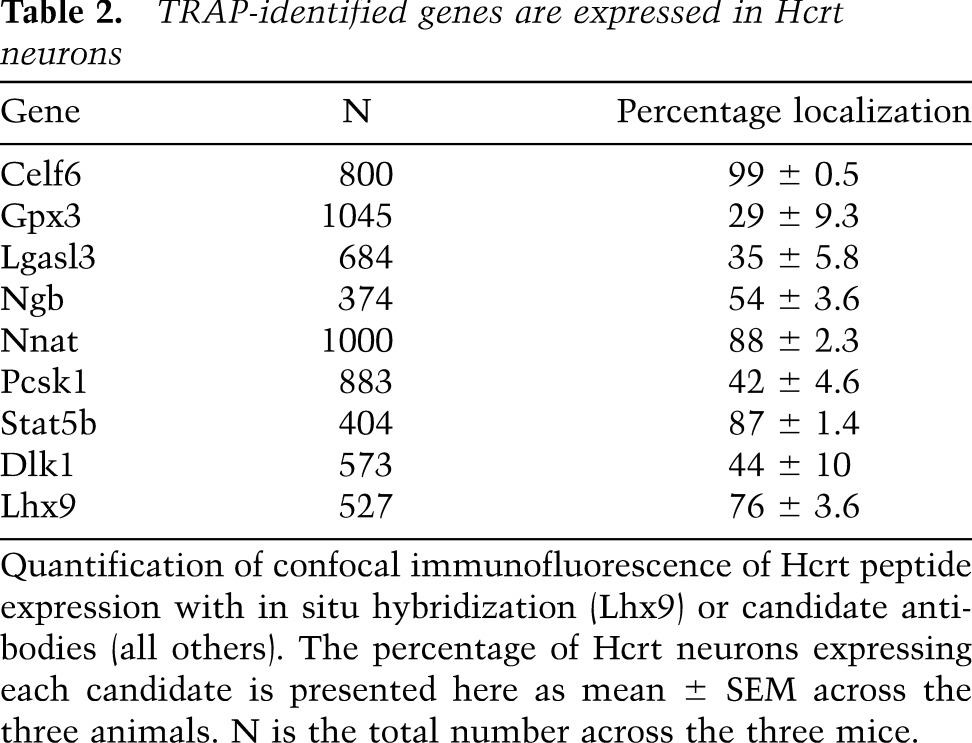
We further confirmed colocalization with Hcrt peptide for 15 of our 188 genes by confocal immunofluorescence and ISH (Fig. 3; Table 2). This suggests that the microarray results are predicting expression correctly in a substantial portion of the cases. Celf6 and Nnat labeled nearly all Hcrt neurons, but the Hcrt neurons were clearly only a subset of the hypothalamic cells expressing these proteins. This is similar to what has been seen with traditional markers of these cells, Pdyn and Nptx2. From these results, however, we also identified a previously unreported molecular diversity within these neurons. For example, both Pcsk1 and Ngb only labeled subsets of the Hcrt neurons at the protein level, in addition to labeling adjacent nuclei. Finally, probes of two genes, Stat5b and especially Rfx4, had nearly perfect overlap with Hcrt neurons, with the latter having expression only within these neurons.
Figure 3.

TRAP accurately predicts gene expression in Hcrt neurons. (A) Confocal immunofluorescence for positive control anti-Nptx2 (green) and anti-Hcrt (red) antibodies shows Hcrt is expressed in a subset of Npt2x cells, consistent with their known expression patterns. Colocalization of nine novel Hcrt neuron gene products identified as enriched in Hcrt neurons (Table 1) shows substantial overlap (Table 2). Lhx9 protein was expressed in a subset of Hcrt neurons, and a subset of Lhx9 cells were positive for Hcrt. (B) In situ hybridization for potential novel markers of Hcrt neurons (scored as a 1 or 2 in Supplemental Table S1), identifies six genes highly enriched in Hcrt neurons. (C) Genes identified as expressed but not substantially enriched by TRAP (Supplemental Table S2) also show robust expression in Hcrt neurons. Note the molecular diversity of Hcrt neurons in their expression of calcium-binding proteins Calb1 and Calb2 (white arrows point to triple expression of Calb1, Calb2, and Hcrt; yellow arrows point to double expression of Calb1 or Calb2 and Hcrt; cyan arrows point to single expression of Calb1). All of the images are captured at 40× magnification.
Table 2.
TRAP-identified genes are expressed in Hcrt neurons
Finally, to determine whether the data continued to be informative even beyond the top 188 genes, we also stained with two robust antibodies: Calb1 and Calb2. These were also predicted to have expression in Hcrt neurons, although only very modest enrichment (1.4-fold and 1.2-fold). We detected robust labeling in most Hcrt neurons, although again there was an interesting and previously unreported molecular diversity (Fig. 3C).
Correlating the Hcrt neuron profile with function
An unbiased examination of our transcripts (Supplemental Table S1) using two gene categorization tools (DAVID [Database for Annotation, Visualization, and Integrated Discovery] and BiNGO [Biological Network for Gene Ontology]) highlights several features that make these cells distinct. There is a substantial enrichment for genes containing signal peptides, such as peptide neurotransmitters, suggesting that these cells have an usually robust repertoire of secreted molecules (UniprotKB term “Signal,” P < 1.6 × 10−5, as calculated by DAVID with Benjamini-Hochberg [B-H] correction). More surprisingly, there seem to be intact pathways enriched in these neurons related to cytokine signaling (for example, gene ontology [GO] terms “regulation of immune effector process,” and “regulation of cytokine production,” both P < 5 × 10−5, BiNGO hypergeometic test with B-H correction), driven by the genes H2-M3, Cd59A, Stat5b, Tlr2, Cd40, Il13ra1, Panx1, and Apoa2, and an unusually high level of Caspase1. It is possible that something about these transcripts renders Hcrt neurons particularly vulnerable to destruction in NC. For example, the presence of Caspase1, which perhaps normally serves some role in protein secretion in these cells (Keller et al. 2008), could render them vulnerable to pyroptosis (Miao et al. 2011) after infection.
Our analysis also identifies a set of receptors that makes clear predictions about what type of neurotransmitters or pharmacological agents these cells should be especially responsive to, including serotonin (Htr1a and Htr1f) and other monoamines (Adra2a); neurotensin (Ntsr1); arginine vassopressin (Avpr1a); growth hormone (Ghr); thyrotropin-releasing hormone (Trhr); parathyroid hormone (Pth2r); cholecystokinin (Cckar); and an orphan receptor and potentially novel drug target, Gpr45. The predictions about the monoamines, Avp, Ntsr, Trh, and Cck are supported by the literature (Bayer et al. 2005; Tsujino et al. 2005; Kumar et al. 2007; Hara et al. 2009), while the others are novel. Finally, we identify a novel set of transcription factors—including Elk3, Hmx2, Fosl1, Creb3l1, Rfx4, Stat5b, and Lhx9—that may be important for the specification of these cells.
Lhx9 is necessary for a subset of Hcrt neurons in vivo
To determine whether any of the Hcrt neuron transcripts that we discovered here may have some functional significance for these cells, we examined Lhx9 knockout mice. Previously, two groups have generated deletions of Lhx9 in mice. One group identified an essential role of this transcription factor in formation of the gonads (Birk et al. 2000), while the other focused on the role of Lhx9 in conjunction with Lhx2 in commissural relay neuron axonal guidance in the embryonic development of the spinal cord (Wilson et al. 2008). To our knowledge, no one has studied the functional consequences of Lhx9 deletion on hypothalamic development.
To determine whether Lhx9 may contribute to development or axonogenesis of Hcrt neurons, we quantified the distribution and density of Hcrt axons by immunohistochemistry from serial sections of adult brains in Lhx9 knockout mice (Wilson et al. 2008) and wild-type littermate controls (n = 4). In the absence of Lhx9, Hcrt fibers continued to project to all of their targets with a normal pattern of innervation (Peyron et al. 1998) but a somewhat decreased amount of fiber density. Throughout the brain, we found that targets had lost 20%–40% of Hcrt innervation (P < 0.05, t-tests, in three regions counted: cortex, paraventricular thalamus, and septum). This could be due to a failure of either axonogenesis (Wilson et al. 2008) or specification of Hcrt neurons themselves. Therefore, we counted all Hcrt neuron cell bodies in the hypothalamus. There was a 39% (±5%) decrease of Hcrt cell number in the Lhx9 knockouts (P < 0.05 × 10−5, one-tailed t-test).
This loss of Hcrt neurons could be due to a broad failure of brain development, as has been previously described for Ebf2 knockouts (De La Herran-Arita et al. 2011). However, careful characterization of our Lhx9 knockout and wild-type mice demonstrated no consistent differences in brain size, weight, or structure. To determine whether this loss of neurons is due to a general decrease in the number of cells in the hypothalamus, we also examined two other populations of neurons in the hypothalamus that were not predicted to be influenced by Lhx9 expression: dopaminergic neurons, labeled by antibodies to tyrosine hydroxylase (Th), and melanin-concentrating hormone (Mch) neurons, labeled with antibodies to Mch. For this, we examined an independent set of wild-type and Lhx9 knockout mice with two-color immunofluorescence for Hcrt and Mch or Hcrt and Th and counted all Hcrt-, Mch-, or Th-positive cells in any sections of hypothalamus containing Hcrt neurons. We again observed a 33% decrease of Hcrt cells in Lhx9 knockout mice (P < 0.05, t-test, n = 3) with no decrease in Th- or Mch-positive neurons on the same sections. Finally, to determine whether this absence of Hcrt neurons may be due to a failure of development or whether they are lost in adulthood, we counted Hcrt neurons in 28-d-old Lhx9 knockout and wild-type mice. This is the earliest feasible time point for this experiment, as Hcrt peptide does not reach adult levels until after the end of the juvenile period (Yamamoto et al. 2000). The 33% decrease was also detectable by this time (data not shown), consistent with an early loss or failure of specification or maturation of the Hcrt expression rather than a degeneration in adult mice. mRNA expression studies of this transcript in wild-type mice at postnatal day 28 (P28) (Fig. 3) suggest that nearly all Hcrt neurons may express some amount of Lhx9 transcript, although protein was only detectable in a subset of these cells and was too dim to reliably quantify.
Loss of Lhx9 leads to a profound perturbation of sleep behavior
The human disorder of narcolepsy is characterized by a profound dysregulation of sleep. NC is accompanied in >90% of patients by a complete loss of Hcrt peptide, as assayed from the cerebral spinal fluid (CSF) of living patients (Nishino et al. 2000) or immunohistochemistry in deceased (Thannickal et al. 2000). However, NwoC is more variable with regards to Hcrt. Post-mortem studies have shown that NwoC is accompanied by only a partial loss of Hcrt neurons (Thannickal et al. 2009). We sought to determine whether Lhx9 knockout mice also have abnormal sleep physiology.
Five Lhx9 knockout and five wild-type littermates were implanted with electrodes to permit continuous monitoring of the sleep–wake cycle. Lhx9 knockout mice showed no evidence of cataplexy (Fig. 4A) but did demonstrate a profound hypersomnolence, with a 20% decrease in time spent awake across a 24-h recording period and a corresponding increase in NREM (non-REM) sleep (Fig. 4B,C). REM sleep was not affected. Circadian rhythm appears intact in these animals, with the normal increase in wakefulness seen during the dark period (Fig. 4C). Thus, these mice that have a loss of a subset of Hcrt neurons also recapitulate a portion of the narcolepsy phenotype, hypersomnolence, in the absence of REM abnormalities or cataplexy. Submaximal doses of Hcrt receptor antagonists (Morairty et al. 2012) or partial ablations of the Hcrt neuron population (Gerashchenko et al. 2001) can produce a hypersomnolence similar to what we observed here. Thus, the most parsimonious explanation of our data is that Lhx9 is important for a subset of Hcrt neurons and that this subset is essential for normal sleep behavior.
Figure 4.

Genetic ablation of Lhx9 results in hypersomnolence. (A) Representative hypnograms for a single Lhx9 knockout (KO) and wild-type (WT) mouse over a 12-h active period show a lack of sleep-onset REM periods but an apparent increase in NREM sleep in the knockout mouse. (B, top panel) Quantification of minutes per hour spent in REM and NREM sleep and wakefulness for Lhx9 knockout and wild-type mice reveals a significant increase in NREM sleep at the expense of wakefulness (n = 5 per group, two-tailed t-test). (Bottom panel) There was no difference in bout duration of wakefulness and NREM sleep between Lhx9 knockout and wild-type mice (n = 5 per group, two-tailed t-test). (C) Both Lhx9 knockout and wild-type mice show normal circadian differences in activity between night and day (n = 5 per group, two-tailed t-test) with no significant difference between genotypes. (*) P < 0.05; (**) P < 0.01. (***) P < 0.001. Values represent mean ± SEM.
However, this is but one of two alternative hypotheses regarding the mechanism of Hcrt loss in Lhx9 knockout mice. Either Lhx9 is essential in early fate specification or survival of a subset of Hcrt neurons or Lhx9 simply promotes expression of the Hcrt gene. We next conducted a set of experiments to address these alternative mechanisms.
Lhx9 does not regulate Hcrt promoter activity in vitro
To determine whether Lhx9 is a direct regulator of the known Hcrt promoter (pHcrt), we cloned a fragment of the Hcrt promoter, including the conserved regions previously reported to provide Hcrt neuron-specific activity in vivo and in vitro (Moriguchi et al. 2002; Adamantidis et al. 2007; Silva et al. 2009), and conducted luciferase assays in a neuroblastoma cell line overexpressing Lhx9 (Supplemental Fig. S2A). Lhx9 did not induce activity from this promoter and indeed led to a very modest suppression of luciferase activity, providing strong evidence that Lhx9 is not a direct positive regulator of this genomic region. However, this experiment does not preclude the possibility that Lhx9 positively regulates Hcrt expression through a different genomic region, regulates Hcrt expression indirectly through a second transcription factor, or requires a cofactor not expressed in the neuroblastoma cell line. Finally, although it has been used experimentally in this cell line before (Silva et al. 2009), pHcrt has very little activity in vitro in any reported cell line, precluding analyses of the ability of Lhx9 to robustly suppress Hcrt expression. To test these possibilities, we generated a high-titer Lhx9 lentivirus and injected it stereotactically into the lateral hypothalamus in a set of two experiments.
Lhx9 does not positively or negatively regulate Hcrt expression in vivo
We first injected wild-type mice unilaterally in the hypothalamus with either Lhx9-producing (n = 3) or GFP-producing (n = 4) viruses and, 8 d later, processed them for immunofluorescence for Lhx9, GFP, and Hcrt peptide. Confocal fluorescence microscopy confirmed expression of exogenous Lhx9 and GFP in the injected sides and accurate targeting to the region of Hcrt neurons in six of seven injected animals. Excluding the one off-target GFP animal, we counted all Hcrt neurons in all animals on both injected and uninjected sides. There was no significant difference between injected and uninjected sides for either Lhx9 or GFP or between Lhx9- and GFP-injected animals (Supplemental Fig. S2B). These data demonstrate that Lhx9 is not a direct or indirect transcriptional regulator of Hcrt expression in adult animals in vivo, at least in wild-type animals. However, the possibility remained that Lhx9 could rescue the Hcrt neuron number and/or behavioral disruptions seen in adult Lhx9 knockout mice. Therefore, in our second experiment, we first validated a more high-throughput method for phenotyping the behavior of our animals and then attempted to rescue the knockout phenotype with bilateral Lhx9 injection into the lateral hypothalamus.
Replacement of Lhx9 in the lateral hypothalamus does not rescue disruption of behavior or Hcrt neuron number
Quantification of time at rest and horizontal ambulations using 24-h activity monitoring has previously been shown to provide reliable estimates of sleep and wakefulness (Pack et al. 2007). The rest and activity patterns of Lhx9 knockout (n = 9) and wild-type (n = 6) mice were evaluated over a 48-h period in automated chambers using photobeam breaks. The hypersomnolence demonstrated by the Lhx9 knockout mice during EEG recording was replicated during activity monitoring. Lhx9 knockout mice exhibited a 20% increase in time at rest (minutes per hour) compared with wild-type mice (P < 0.05, ANOVA) (Fig. 5A) as well as normal increases in time at rest during the light period relative to the dark period, indicating intact circadian rhythms (P < 0.05, paired t-test) (Fig. 5B). Complementary to the sleep behavior results, a 57% decrease in average activity per hour was displayed by the Lhx9 knockout mice compared with wild-type mice (P < 0.01, ANOVA) (Fig. 5C). The decreases in activity were observed across both the light and dark periods (P < 0.05, repeated measures ANOVA) (Fig. 5D). These results support the EEG data and confirm that Lhx9-null mice with a loss of Hcrt neurons demonstrate a profound hypersomnolence.
Figure 5.

Disturbances in behavior and Hcrt expression of Lhx9 knockout (KO) mice are not rescued by adult reinstatement of Lhx9 expression. During 48-h activity monitoring, Lhx9 knockout mice exhibited a 20% increase in time at rest, an estimate of sleep, compared with wild-type (WT) mice (P < 0.05, ANOVA) (A), while both groups demonstrated intact circadian differences in time at rest between the light and dark periods (P < 0.05, paired t-test) (B). (C) Lhx9 knockout mice also demonstrated a 57% decrease in ambulatory activity, an estimate of wakefulness, relative to wild-type mice (P < 0.01, ANOVA). (D) The ambulatory differences were maintained across the light and dark periods (P < 0.05, repeated measures ANOVA). (E) Confocal microscopy revealed double-immunofluorescent labeling of GFP with orexin within Hcrt neurons, confirming successful targeting of hypocretinergic regions. (F) Representative example of Lhx9 expression in the targeted region of the hypothalamus in Lhx9 knockout mice. The white asterisk represents a labeling artifact at injection sites. (G,H) No differences were observed in time at rest or ambulatory activity between pre- and post-injection activity monitoring for Lhx9-injected Lhx9 knockout mice. In addition, time at rest and ambulatory activity was comparable during post-injection activity monitoring for both the Lhx9-injected Lhx9 knockout mice and the GFP-injected Lhx9 knockout mice (repeated measures ANOVA). (I) No difference was noted in the number of Hcrt neurons between Lhx9-injected and GFP-injected animals. (*) P < 0.05; (**) P < 0.01. Values represent mean ± SEM.
Following the first 48-h activity monitoring session, a lentiviral vector was bilaterally injected into the Hcrt neurons of Lhx9 knockout mice to produce expression of either Lhx9 (n = 5) or GFP (n = 4). Sixteen days following injection, successful targeting was confirmed through confocal microscopy of immunofluorescence-labeled sections, showing GFP and Lhx9 expression within the Hcrt neuron-containing lateral hypothalamus (Fig. 5E) with successful Lhx9 expression in the targeted areas in four of five mice (Fig. 5F) and GFP in four of four mice. Immediately prior to these neuroanatomical studies, the behavior of these mice was evaluated again over a 48-h period to determine whether reinstatement of Lhx9 expression in adult lateral hypothalamus could rescue the perturbations observed in sleep and wakefulness behaviors. While the Lhx9 expression was successfully rescued within the lateral hypothalamus, the behavioral phenotype was not. During post-injection activity monitoring, the Lhx9-injected Lhx9 knockout mice did not demonstrate a change in rest or ambulatory activity levels from those observed during preinjection activity monitoring (P > .05, repeated measures ANOVAs) (Fig. 5G,H). The Lhx9-injected Lhx9 knockout mice also demonstrated levels of rest and activity comparable with that of the GFP-injected Lhx9 knockout controls during both pre- and post-injection activity monitoring. In addition, staining and quantification indicated that Lhx9-injected Lhx9 knockout animals showed no rescue of Hcrt neuron number (P > 0.05, two-tailed t-test) (Fig. 5I).
Overall, we could not rescue either Hcrt neuron number or Lhx9 knockout sleep behavior, which suggests that these are related phenomena and that the perturbations in sleep and wakefulness behavior are due to a loss of cells during development rather than simple dysregulation of Hcrt promoter activity. Our current data do not address whether the cell loss is due to a cell-autonomous failure of specification or a secondary consequence of either pathfinding defects or loss of a distally acting signal. These hypotheses will need to be tested in the future with direct genetic approaches, including cell-specific deletion of Lhx9.
Regardless of the outcome of those studies, we discovered a genetic manipulation that results in a profound perturbation of sleep behavior. It is also of interest that a genome-wide association study of human narcoleptics (Hallmayer et al. 2009) identified a SNP on chromosome 1 with a suggestive association to narcolepsy (P < 1.04 × 10−7) that is 96 kb upstream of Lhx9 (HG19 assembly, as accessed December 2012, University of California at Santa Cruz Genome Browser, http:/www.genome.ucsc.edu). Thus, there remains the possibility that Lhx9 may be an important regulator of sleep in humans as well.
Discussion
We described here a mouse line permitting the in vivo profiling of Hcrt neurons and identified a set of 188 transcripts robustly enriched in these neurons (Supplemental Table S1) in addition to thousands more moderately enriched or nominally expressed (Supplemental Table S2), including many additional channels and receptors. Our validation studies suggest that our methods have a high true positive rate (at least 59%) and low false positive rate (∼5%). There are a small number of genes previously reported in Hcrt neurons (such as Foxa2) for which the arrays simply showed no signal in any sample. This suggests that there are some false negatives in our data, which may be ameliorated in future studies using RNA sequencing (RNA-seq). Also, TRAP does have some low level of nonspecific background, as indicated by the presence of signal from some glial genes (Fig. 2B, red circles). We used the fold change from these glial genes to establish a fold change threshold (0.47) below which we cannot be certain whether a gene is expressed in Hcrt neurons or not. The proposed narcolepsy autoantigen Trib2 (Lim and Scammell 2010) falls below this threshold. This suggests that if it is expressed in healthy Hcrt neurons, it is at a much lower level in these cells than surrounding tissue.
Our anatomical analysis of these molecular results largely supported the array data but also discovered a surprising molecular heterogeneity in these neurons, with several gene products expressed only in a subset of these neurons (Dlk1, Gpx3, Pcsk1, Lgals3, and Ngb). It remains to be determined whether these represent truly functionally distinct subsets of the Hcrt population or are merely transiently expressed and reflect the state of particular neurons at the time of perfusion, analogous to the classically activity-induced protein cFos. Among those assayed, Stat5b and Rfx4 showed the highest concordance rate while remaining largely specific to Hcrt neurons and thus could potentially serve as alternate markers for the population in future studies. These would be of great interest to examine in human post-mortem tissue from narcoleptic patients.
Additionally, we noted an unusual concordance between the expression of our candidate genes in Hcrt neurons and in the axonal targets of Hcrt neurons: For example, both Ngb and Lhx9 proteins were dimly expressed in Hcrt neurons but more robustly expressed in their targets in the laterodorsal tegmentum and paraventricular thalamus, respectively. This hints that target-mediated signals may in some way be contributing to the gene expression of Hcrt neurons and will be an interesting direction for future investigation.
Finally, while we emphasized here the clear significance of the Hcrt system to narcolepsy, it is essential to mark the accumulating evidence that these neurons may also play a role in anxiety, depression, and reward in addition to their roles in sleep and feeding (Borgland and Labouebe 2010; Sakurai and Mieda 2011). Thus the data we provide here may serve to both better inform these studies and provide an important tool to profile Hcrt neurons under a variety of conditions.
Materials and methods
Animal research committees
All procedures involving animals were approved by the appropriate institutional animal care and use committees.
Generation of mice
A bacterial artificial chromosome (BAC), RP23-258L05, was modified as described (Doyle et al. 2008) to insert the eGFP-RPL10a transgene at the translation start site of the Hcrt gene. Successful modification was confirmed with Southern blot, and lack of gross rearrangement of the BAC was checked by BAC fingerprinting. Modified BAC DNA was purified with CsCl gradient centrifugation, dialyzed into injection buffer, and injected into FVB mouse eggs. Eggs were transplanted into pseudopregnant Swiss Webster dams, and the resultant pups were screened with tail tip PCR for eGFP to identify founders. Each founder was crossed to C57/bl6j wild-type mice, and F1 progeny were genotyped and then processed for anatomy as described below.
Immunofluorescence microscopy
Mice were killed and then perfused with phosphate-buffered saline (PBS) followed by 4% paraformaldehyde in PBS. Brains were dissected, cryoprotected in 30% sucrose PBS solution, and then frozen and sectioned serially on a Leica cryostat into PBS and 0.1% sodium azide for storage at 4°C. Floating sections were blocked in 5% normal donkey serum, 0.25% Triton, and PBS for 30 min and then incubated overnight with primary antibodies in the same buffer. All primary antibodies are detailed in Supplemental Table S3. Sections were then washed three times with PBS and then incubated for 60–90 min with the appropriate Alexa fluorophore-conjugated secondary antibodies and nuclear dyes (DAPI and or TOPO-3-Iodide, Life Technologies). Images were acquired on a LSM 510 Zeiss confocal microscope (Fig. 1) or a Perkin Elmer UltraView Vox spinning-disk confocal on a Zeiss Axiovert microscope (all other confocal figures).
Immunohistochemistry
Mice were processed as above. For anti-Orx-A DAB immunohistochemistry, brains were processed using multibrain technology by Neuroscience Associates as described (Doyle et al. 2008) using orexin A antibody (1:10,000; Calbiochem) and a nickel-enhanced DAB substrate for HRP. Sections were digitized with a Zeiss Axioskop2 and customized macros.
Anatomical analysis
All cell number quantification was done on a series of coronal sections spaced 200–210 μm apart through the hypothalamus in a minimum of three Lhx9 knockout and three wild-type mice, typically littermates. Counts from two independent investigators, blinded to condition, were averaged for each mouse. For Hcrt, Mch, and Th staining, all neuron labeling with any antibody was counted in any section showing any Hcrt neuronal labeling. Statistical comparisons were with unpaired two-tailed t-tests, as calculated in Microsoft Excel. For Dlk1, Celf6, Nnat, Ngb, Gpx3, Psck1, Stat5b, and Lhx9, cells were counted from confocal images of all available Hcrt-expressing regions from three wild-type mice. Antibodies used are detailed in Supplemental Table S4.
For Allen Brain Atlas analysis, the 188 TRAP transcripts were mixed with the top 188 transcripts from the Pabp study “Supplemental Excel File 1” from Cvetkovic-Lopes et al. (2010) and 188 transcripts selected at random from the genes present on the microarray and evaluated by two blinded reviewers using the following criteria to identify a HLP. First, transcripts without coronal sections were excluded, as were any ISH with no detectable signal anywhere in tissue. Then, if there was broad labeling of cells in the hypothalamus (or indeed, the entire brain), then they were scored as a 4: broadly expressed, including HLP (e.g., Snap25). If there was strong labeling in some population somewhere in the brain and absolutely no labeling in the region of an HLP, then they were scored as a 5: not expressed in HLP (e.g., Agrp). Looking at the entire brain, if the only signal was in a HLP, then they were scored as a 1: unique to Hcrt neurons (e.g., Hcrt). Looking just within the hypothalamus and ignoring the rest of the brain, if the only signal was in a HLP, then they were scored as a 2: unique within hypothalamus (e.g., Igfbp3, which showed an HLP plus labeling of choroid). Finally, looking just within the hypothalamus, if the labeling overlapped with HLP but also showed labeling in just a few other populations in hypothalamus, then it was scored as a 3: enriched in Hcrt neurons (e.g., Pdyn and Nptx2). Generally, markers from the literature for Hcrt neurons received scores between 1 and 3. For the purpose of statistical analysis, scores of 1–3 were pooled, and χ2 tests were calculated in Microsoft Excel, comparing observed distributions (TRAP or Pabp) with those expected by chance (random).
ISH
ISH was performed as described previously (VanDunk et al. 2011) on coronal sections of P28 wild-type mouse brains that were immersion-fixed in 4% paraformaldehyde for 20 min and cryoprotected in sucrose before sectioning. Digoxigenin-labeled riboprobe was transcribed with T7 RNA polymerase using the templates described (Supplemental Table S3). Hybridization was performed with 1–2 μg/mL probe for 12–16 h at 65°C. Slides were then washed with 2× SSC for 15 min at 62°C followed by two washes in 0.2× SSC for 30 min each at 62°C. After blocking in PBS, Triton (PBT), and 10% normal horse serum, slides were incubated in alkaline phosphatase-conjugated anti-DIG antibody (1:2000) overnight at 4°C. The following day, slides were washed twice in PBT for 30 min each, and color development proceeded in the dark using NBT and BCIP as a substrate. Color was allowed to develop for 2–20 h, depending on the abundance of the message. After three washes in PBS, slides were fixed in 4% paraformaldehyde for 15 min and stained for Hcrt following standard immunofluorescence protocol, and data were acquired as above. NBT and BCIP signal was inverted, pseudocolored green, and overlayed on Hcrt staining in ImageJ.
TRAP assays and analysis
Four replicate pools of four to six mixed-sex 6- to 12-wk-old mice from the brightest transgenic line were killed, and brains were removed and transferred to ice-cold dissection buffer containing cycloheximide; diencephalon was harvested with the aid of a dissecting microscope. TRAP was conducted as described (Heiman et al. 2008). Briefly, each pool was homogenized for 12 strokes in a glass Teflon homogenizer on ice in buffer (10 mM HEPES at pH 7.4, 150 mM KCl, 5 mM MgCl2, 0.5 mM dithiothreitol, 100 μg/mL cycloheximide, protease inhibitors, recombinant RNase inhibitors). Nuclei and debris were removed with centrifugation at 2000g for 10 min at 4°C. DHPC (Avanti) and NP-40 (Ipgal-ca630, Sigma) were added to the supernatant to final concentrations of 30 mM and 1%, respectively. After 5 min of incubation on ice, the supernatant was centrifuged at 20,000g for 15 min, and the pellet was discarded. The supernatant was mixed with protein G-coated magnetic beads (Invitrogen/Life Technologies), previously conjugated with a mix of two monoclonal anti-GFP antibodies (Doyle et al. 2008), and incubated with rotation for 30 min at 4°C. Beads were washed three times with high-salt wash buffer (10 mM HEPES at pH 7.4, 350 mM KCl, 5 mM MgCl2, 1% NP-40, 0.5 mM dithiothreitol, 100 μg/mL cycloheximide), and RNA was purified from ribosomes using Trizol (Invitrogen) following the manufacturer's protocols, followed by DNase treatment, further purification, and concentration with Rneasy min-elute columns following the manufacturer's protocol (Qiagen). RNA was also harvested in parallel from each unbound fraction of affinity purification as a measure of total tissue RNA. RNA concentration of all samples was measured with a Nanodrop spectrophotometer, and integrity was confirmed with PicoChips on the Agilent BioAnalyzer (RIN>8).
Fifteen nanograms of each RNA sample was amplified with the Affymetrix Two-Cycle amplification kit following the manufacturer's instructions, and the quality of labeled aRNA was assessed with Bioanalyzer. Labeled RNA from immunoprecipitated ribosomes and total tissue RNA were hybridized to separate Affymetrix Mouse Genome 430 2.0 arrays and scanned following the manufacturer's protocols. Raw data are available from the Gene Expression Omnibus (GSE38668).
Data were analyzed using the Bioconductor module within the statistical package R. Data quality was assessed by examining box plots, histograms, correlation coefficients, false positive rates, and scatter plots comparing replicate experiments. Data were normalized as described but using Affymetrix chip definition files. Briefly, GC robust multiarray average (GCRMA) was used to normalize within replicates and to biotinylated spike in probes between conditions. In Supplemental Table S2, fold change (FC), specificity index (SI), and pSI are reported for all genes with expression >50 arbitrary fluorescent units and a fold change greater than the background threshold of 0.5 (demarked at the mean plus two standard deviations of the glial gene fold changes) (Dougherty et al. 2010).
Gene list analysis was conducted using two tools in parallel. Cytoscape 2.8.2 with the BiNGO plugin (2.4) (Maere et al. 2005) was run to identify those GO biological processes enriched (P < 0.005) in the 188 Hcrt neuron transcripts using a hypergeometic test with B-H correction. The same list was analyzed using the functional annotation clustering tool of the DAVID interface (Dennis et al. 2003).
List-wise comparison with previous studies was conducted in Excel using the data from Table 3 of Honda et al. (2009) and “Supplemental Excel File 1” from Cvetkovic-Lopes et al. (2010). Markers considered for χ2 were Gal, Hcrt, Pdyn, Igfbp3, and Cartpt.
Sleep physiology
Polysomnographic sleep–wake cycle analysis of mice was performed as described previously (Bero et al. 2011; Roh et al. 2012). Briefly, EEG and electromyogram (EMG) electrodes were implanted simultaneously 10 d prior to recording. For EEG recording, two stainless steel screws attached to wire electrodes were placed over the right frontal and parietal bone. EMG was recorded by two wire electrodes directly inserted into the neck muscles. The ground electrode was placed on the skull over the cerebellum. Insulated leads from the EEG and EMG electrodes were soldered to a miniconnector. After 10 d of habituation after surgery, mice were transferred to recording cages maintained in 12-h light/12-h dark conditions (light on at 6:00 a.m.), and the miniconnector was connected to flexible recording cables. Mice were habituated to the recording cages for three more days. At the end of the habituation period, EEG and EMG recordings began. EEG and EMG recordings were assessed using a P511K A.C. Preamplifier (Grass-Telefactor Instruments), digitized with a DigiData 1440A data acquisition system (Molecular Devices), and recorded digitally using pClamp 10.2 (Molecular Devices). Using sleep scoring software (SleepSign, Kissei Comtec Co. Ltd.), EEG and EMG signals were binned into 10-sec epochs as wakefulness, REM sleep, and NREM sleep based on the standard criteria of rodent sleep. Semiautomatic sleep scoring was visually inspected and corrected when appropriate. The automatic analysis and visual inspection were performed in a blinded state to the genotypes of mice. Episodes of cataplexy, defined as described (Scammell et al. 2009), were not seen in Lhx9 knockout mice.
Generation of plasmids
Lhx9 (Open Biosystems, clone 40117467, BC131622.1) was cloned with Gateway technology into a custom Gateway-compatible lentiviral vector derived from the previously published FCIV (Li et al. 2010) by replacing the IRES-Venus sequence with a Gateway acceptor cassette. This plasmid was validated with antibody staining for Lhx9 expression following both transient transfection and lentiviral transduction. For the luciferase assays, a fragment of the mouse promoter corresponding to the 3 kb upstream of the translation start site was cloned by PCR into the pGL3 basic vector (Invitrogen).
Overexpression of Lhx9 in the unilateral lateral hypothalamus
To investigate whether the overexpression of Lhx9 augments Hcrt signaling in the brain, Lhx9 lentiviral vector under the control of a ubiquitin promoter (1.5 μL, 1.9 × 109 IU/mL) was unilaterally injected right above the left side lateral hypothalamus (anterioposterior [AP] −1.82, mediolateral [ML] −0.3, dorsoventral [DV] 5.1 mm, directed vertically) in C57/Bl6 female mice. For a control group, GFP lentiviral vector (1.5 μL, 5 × 107 IU/mL) was infused in the same way in C57/Bl6 female mice. After 8 d of viral vector infusion, mice were perfused, and brain tissue was obtained and stained as described. Immunofluorescence for either GFP or Lhx9 was used to confirm accurate targeting of viral injections. Two independent researchers blinded to the treatment manually counted the total number of cells stained with Hcrt.
Forty-eight-hour activity monitoring
The activity monitoring procedure was adapted from previously published methods (Dougherty et al. 2013). Briefly, Lhx9 knockout and wild-type mice were placed in transparent polystyrene chambers (47.6 × 25.4 × 20.6 cm) containing food and clean home cage bedding spread on the floor and a water bottle attached to one wall. A metal grid containing a 4 × 8 matrix of photobeam pairs surrounded each chamber and was connected to a computer equipped with software (MotorMonitor, Kinder Scientific) that used an algorithm to quantify photobeam breaks as horizontal ambulations (an estimate of wakefulness) and the lack of photobeam breaks as time at rest (an estimate of sleep). A rest threshold of 40 sec was used based on previous findings that this threshold is optimal for photobeam assessment of inactivity as an estimate of sleep (Pack et al. 2007). Activity monitoring began halfway through the light cycle and continued for 48 h. The testing room lighting was a 12-light:12-h dark cycle (light on at 6:00 a.m.). Activity monitoring was conducted in two sessions separated by 1 mo, during which the Lhx9 knockout mice received Lhx9 or GFP lentiviral vector injections. SPSS statistics software was used for data analyses. To determine differences between genotypes during session 1, one-way ANOVAs were conducted on average time at rest and ambulations per hour, a repeated measures ANOVA was conducted on 6-h blocks of total ambulations across the light/dark cycle, and paired t-tests were conducted on time at rest within genotypes to assess circadian rhythms. One wild-type mouse was excluded from ambulation analyses as an outlier (z-score > 2.0). For analysis of the influence of Lhx9 lentiviral vector injections on time at rest and ambulations of Lhx9 knockout mice, repeated measure ANOVAs were conducted on pre- and post-injection performance. One Lhx9-injected mouse was excluded because the injection was off-target.
Viral rescue of Lhx9 in Lhx9 knockout mice
To investigate whether the rescue of Lhx9 affects behavioral phenotype as well as Hcrt signaling in Lhx9 knockout mice, Lhx9 or GFP lentiviral vector was bilaterally injected as described above in Lhx9 knockout mice. On day 13 of viral vector infusion, mice underwent behavioral tests as described above followed by perfusion at the end of tests. After confirmation of target engagement and viral vector expression in each mouse, total numbers of Hcrt neurons were counted manually by two researchers blinded to the treatment.
Acknowledgments
We acknowledge the Rockefeller University Genomics Resource Center and Rockefeller University Bioimaging Core; the Hope Center Viral Vector and Animal Surgery Cores (supported by NIH P30 NS057105); the Animal Behavior Core (NIH P30 HO062171); Sujata Bupp, Sukrit Singh, Allyson Mallya, Matt Corley, Ashish Heda, Sarah Pyfrom, Ronaldo Perez, and Alexandra Fen for technical assistance; and Jane Dodd, Paul Shaw, and Andres Burkhalter for reagents, advice and assistance. We thank Emmanuel Mignot (Stanford University) for providing detailed array data. This work was funded by a Clinical Research Training Fellowship from the American Academy of Neurology and American Brain Foundation (R.J.H.), the National Institute of Neurological Disorders and Stroke (4R00NS067239-03) and Mallinckrodt Foundation (J.D.D.), the National Human Genome Research Institute (R25 HG006687) (W.B.J.), National Institute of Arthritis and Musculoskeletal and Skin Diseases (R00AR055948, to C.d.G.S.), National Institutes of Health (HL 089742 to P.A.G.), and the Adelson Medical Research Foundation and the Howard Hughes Medical Institute (N.H.).
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.207654.112.
References
- Adamantidis AR, Zhang F, Aravanis AM, Deisseroth K, de Lecea L 2007. Neural substrates of awakening probed with optogenetic control of hypocretin neurons. Nature 450: 420–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer L, Eggermann E, Serafin M, Grivel J, Machard D, Muhlethaler M, Jones BE 2005. Opposite effects of noradrenaline and acetylcholine upon hypocretin/orexin versus melanin concentrating hormone neurons in rat hypothalamic slices. Neuroscience 130: 807–811 [DOI] [PubMed] [Google Scholar]
- Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, Lee JM, Holtzman DM 2011. Neuronal activity regulates the regional vulnerability to amyloid-β deposition. Nat Neurosci 14: 750–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertuzzi S, Porter FD, Pitts A, Kumar M, Agulnick A, Wassif C, Westphal H 1999. Characterization of Lhx9, a novel LIM/homeobox gene expressed by the pioneer neurons in the mouse cerebral cortex. Mech Dev 81: 193–198 [DOI] [PubMed] [Google Scholar]
- Birk OS, Casiano DE, Wassif CA, Cogliati T, Zhao L, Zhao Y, Grinberg A, Huang S, Kreidberg JA, Parker KL, et al. 2000. The LIM homeobox gene Lhx9 is essential for mouse gonad formation. Nature 403: 909–913 [DOI] [PubMed] [Google Scholar]
- Borgland SL, Labouebe G 2010. Orexin/hypocretin in psychiatric disorders: Present state of knowledge and future potential. Neuropsychopharmacology 35: 353–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgin P, Zeitzer JM, Mignot E 2008. CSF hypocretin-1 assessment in sleep and neurological disorders. Lancet Neurol 7: 649–662 [DOI] [PubMed] [Google Scholar]
- Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, Richardson JA, Williams SC, Xiong Y, Kisanuki Y, et al. 1999. Narcolepsy in orexin knockout mice: Molecular genetics of sleep regulation. Cell 98: 437–451 [DOI] [PubMed] [Google Scholar]
- Crocker A, Espana RA, Papadopoulou M, Saper CB, Faraco J, Sakurai T, Honda M, Mignot E, Scammell TE 2005. Concomitant loss of dynorphin, NARP, and orexin in narcolepsy. Neurology 65: 1184–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvetkovic-Lopes V, Bayer L, Dorsaz S, Maret S, Pradervand S, Dauvilliers Y, Lecendreux M, Lammers GJ, Donjacour CE, Du Pasquier RA, et al. 2010. Elevated Tribbles homolog 2-specific antibody levels in narcolepsy patients. J Clin Invest 120: 713–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauvilliers Y, Montplaisir J, Cochen V, Desautels A, Einen M, Lin L, Kawashima M, Bayard S, Monaca C, Tiberge M, et al. 2010. Post-H1N1 narcolepsy-cataplexy. Sleep 33: 1428–1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Herran-Arita AK, Zomosa-Signoret VC, Millan-Aldaco DA, Palomero-Rivero M, Guerra-Crespo M, Drucker-Colin R, Vidaltamayo R 2011. Aspects of the narcolepsy-cataplexy syndrome in O/E3-null mutant mice. Neuroscience 183: 134–143 [DOI] [PubMed] [Google Scholar]
- de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett FS 2nd, et al. 1998. The hypocretins: Hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci 95: 322–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA 2003. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 4: 3. [PubMed] [Google Scholar]
- Dougherty JD, Schmidt EF, Nakajima M, Heintz N 2010. Analytical approaches to RNA profiling data for the identification of genes enriched in specific cells. Nucleic Acids Res 38: 4218–4230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty JD, Maloney SE, Wozniak DF, Rieger MA, Sonnenblick L, Coppola G, Mahieu NG, Zhang J, Cai J, Patti GJ, et al. 2013. The disruption of Celf6, a gene identified by translational profiling of serotonergic neurons, results in autism-related behaviors. J Neurosci (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle JP, Dougherty JD, Heiman M, Schmidt EF, Stevens TR, Ma G, Bupp S, Shrestha P, Shah RD, Doughty ML, et al. 2008. Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell 135: 749–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerashchenko D, Kohls MD, Greco M, Waleh NS, Salin-Pascual R, Kilduff TS, Lappi DA, Shiromani PJ 2001. Hypocretin-2-saporin lesions of the lateral hypothalamus produce narcoleptic-like sleep behavior in the rat. J Neurosci 21: 7273–7283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray PA, Fu H, Luo P, Zhao Q, Yu J, Ferrari A, Tenzen T, Yuk DI, Tsung EF, Cai ZH, et al. 2004. Mouse brain organization revealed through direct genome-scale TF expression analysis. Science 306: 2255–2257 [DOI] [PubMed] [Google Scholar]
- Hallmayer J, Faraco J, Lin L, Hesselson S, Winkelmann J, Kawashima M, Mayer G, Plazzi G, Nevsimalova S, Bourgin P, et al. 2009. Narcolepsy is strongly associated with the T-cell receptor α locus. Nat Genet 41: 708–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara J, Beuckmann CT, Nambu T, Willie JT, Chemelli RM, Sinton CM, Sugiyama F, Yagami K-i, Goto K, Yanagisawa M, et al. 2001. Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron 30: 345–354 [DOI] [PubMed] [Google Scholar]
- Hara J, Gerashchenko D, Wisor JP, Sakurai T, Xie X, Kilduff TS 2009. Thyrotropin-releasing hormone increases behavioral arousal through modulation of hypocretin/orexin neurons. J Neurosci 29: 3705–3714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suarez-Farinas M, Schwarz C, Stephan DA, Surmeier DJ, et al. 2008. A translational profiling approach for the molecular characterization of CNS cell types. Cell 135: 738–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda M, Eriksson KS, Zhang S, Tanaka S, Lin L, Salehi A, Hesla PE, Maehlen J, Gaus SE, Yanagisawa M, et al. 2009. IGFBP3 colocalizes with and regulates hypocretin (orexin). PLoS ONE 4: e4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima M, Lin L, Tanaka S, Jennum P, Knudsen S, Nevsimalova S, Plazzi G, Mignot E 2010. Anti-Tribbles homolog 2 (TRIB2) autoantibodies in narcolepsy are associated with recent onset of cataplexy. Sleep 33: 869–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller M, Ruegg A, Werner S, Beer HD 2008. Active caspase-1 is a regulator of unconventional protein secretion. Cell 132: 818–831 [DOI] [PubMed] [Google Scholar]
- Kornum BR, Kawashima M, Faraco J, Lin L, Rico TJ, Hesselson S, Axtell RC, Kuipers H, Weiner K, Hamacher A, et al. 2011. Common variants in P2RY11 are associated with narcolepsy. Nat Genet 43: 66–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Szymusiak R, Bashir T, Rai S, McGinty D, Alam MN 2007. Effects of serotonin on perifornical-lateral hypothalamic area neurons in rat. Eur J Neurosci 25: 201–212 [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. 2007. Genome-wide atlas of gene expression in the adult mouse brain. Nature 445: 168–176 [DOI] [PubMed] [Google Scholar]
- Li M, Husic N, Lin Y, Christensen H, Malik I, McIver S, LaPash Daniels CM, Harris DA, Kotzbauer PT, Goldberg MP, et al. 2010. Optimal promoter usage for lentiviral vector-mediated transduction of cultured central nervous system cells. J Neurosci Methods 189: 56–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim AS, Scammell TE 2010. The trouble with Tribbles: Do antibodies against TRIB2 cause narcolepsy? Sleep 33: 857–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Faraco J, Li R, Kadotani H, Rogers W, Lin X, Qiu X, de Jong PJ, Nishino S, Mignot E 1999. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell 98: 365–376 [DOI] [PubMed] [Google Scholar]
- Maere S, Heymans K, Kuiper M 2005. BiNGO: A Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21: 3448–3449 [DOI] [PubMed] [Google Scholar]
- Miao EA, Rajan JV, Aderem A 2011. Caspase-1-induced pyroptotic cell death. Immunol Rev 243: 206–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morairty SR, Revel FG, Malherbe P, Moreau JL, Valladao D, Wettstein JG, Kilduff TS, Borroni E 2012. Dual hypocretin receptor antagonism is more effective for sleep promotion than antagonism of either receptor alone. PLoS ONE 7: e39131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriguchi T, Sakurai T, Takahashi S, Goto K, Yamamoto M 2002. The human prepro-orexin gene regulatory region that activates gene expression in the lateral region and represses it in the medial regions of the hypothalamus. J Biol Chem 277: 16985–16992 [DOI] [PubMed] [Google Scholar]
- Nakagawa Y, O'Leary DD 2001. Combinatorial expression patterns of LIM-homeodomain and other regulatory genes parcellate developing thalamus. J Neurosci 21: 2711–2725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino S, Ripley B, Overeem S, Lammers GJ, Mignot E 2000. Hypocretin (orexin) deficiency in human narcolepsy. Lancet 355: 39–40 [DOI] [PubMed] [Google Scholar]
- Pack AI, Galante RJ, Maislin G, Cater J, Metaxas D, Lu S, Zhang L, Von Smith R, Kay T, Lian J, et al. 2007. Novel method for high-throughput phenotyping of sleep in mice. Physiol Genomics 28: 232–238 [DOI] [PubMed] [Google Scholar]
- Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, Kilduff TS 1998. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci 18: 9996–10015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retaux S, Rogard M, Bach I, Failli V, Besson MJ 1999. Lhx9: A novel LIM-homeodomain gene expressed in the developing forebrain. J Neurosci 19: 783–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reti IM, Reddy R, Worley PF, Baraban JM 2002. Selective expression of Narp, a secreted neuronal pentraxin, in orexin neurons. J Neurochem 82: 1561–1565 [DOI] [PubMed] [Google Scholar]
- Roh JH, Huang Y, Bero AW, Kasten T, Stewart FR, Bateman RJ, Holtzman DM 2012. Disruption of the sleep–wake cycle and diurnal fluctuation of β-amyloid in mice with Alzheimer's disease pathology. Sci Transl Med 4: 150ra122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai T, Mieda M 2011. Connectomics of orexin-producing neurons: Interface of systems of emotion, energy homeostasis and arousal. Trends Pharmacol Sci 32: 451–462 [DOI] [PubMed] [Google Scholar]
- Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, et al. 1998. Orexins and orexin receptors: A family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell 92: 573–585 [DOI] [PubMed] [Google Scholar]
- Scammell TE, Willie JT, Guilleminault C, Siegel JM 2009. A consensus definition of cataplexy in mouse models of narcolepsy. Sleep 32: 111–116 [PMC free article] [PubMed] [Google Scholar]
- Silva JP, von Meyenn F, Howell J, Thorens B, Wolfrum C, Stoffel M 2009. Regulation of adaptive behaviour during fasting by hypothalamic Foxa2. Nature 462: 646–650 [DOI] [PubMed] [Google Scholar]
- Thannickal TC, Moore RY, Nienhuis R, Ramanathan L, Gulyani S, Aldrich M, Cornford M, Siegel JM 2000. Reduced number of hypocretin neurons in human narcolepsy. Neuron 27: 469–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thannickal TC, Nienhuis R, Siegel JM 2009. Localized loss of hypocretin (orexin) cells in narcolepsy without cataplexy. Sleep 32: 993–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda H, Tanaka S, Miyagawa T, Honda Y, Tokunaga K, Honda M 2010. Anti-Tribbles homolog 2 autoantibodies in Japanese patients with narcolepsy. Sleep 33: 875–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujino N, Yamanaka A, Ichiki K, Muraki Y, Kilduff TS, Yagami K, Takahashi S, Goto K, Sakurai T 2005. Cholecystokinin activates orexin/hypocretin neurons through the cholecystokinin A receptor. J Neurosci 25: 7459–7469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanDunk C, Hunter LA, Gray PA 2011. Development, maturation, and necessity of transcription factors in the mouse suprachiasmatic nucleus. J Neurosci 31: 6457–6467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SI, Shafer B, Lee KJ, Dodd J 2008. A molecular program for contralateral trajectory: Rig-1 control by LIM homeodomain transcription factors. Neuron 59: 413–424 [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Ueta Y, Hara Y, Serino R, Nomura M, Shibuya I, Shirahata A, Yamashita H 2000. Postnatal development of orexin/hypocretin in rats. Brain Res Mol Brain Res 78: 108–119 [DOI] [PubMed] [Google Scholar]
- Zhang S, Lin L, Kaur S, Thankachan S, Blanco-Centurion C, Yanagisawa M, Mignot E, Shiromani PJ 2007. The development of hypocretin (orexin) deficiency in hypocretin/ataxin-3 transgenic rats. Neuroscience 148: 34–43 [DOI] [PMC free article] [PubMed] [Google Scholar]