

Background: PRC, a coactivator of respiratory chain expression, also acts as a sensor of metabolic stress.
Results: The PRC stress program is induced by oxidant and replication stress associated with apoptosis and premature senescence.
Conclusion: PRC plays an adaptive role in the response to cellular dysfunction.
Significance: Elucidating the PRC stress response enhances our understanding of cellular dysfunction in the etiology of age-related diseases.
Keywords: Coactivator Transcription, Gene Regulation, Metabolic Regulation, Signaling, Stress, SN-38, c-MYC, Meclizine
Abstract
PGC-1-related coactivator (PRC), a growth-regulated member of the PGC-1 coactivator family, contributes to the expression of the mitochondrial respiratory apparatus. PRC also orchestrates a robust response to metabolic stress by promoting the expression of multiple genes specifying inflammation, proliferation, and metabolic reprogramming. Here, we demonstrate that this PRC-dependent stress program is activated during apoptosis and senescence, two major protective mechanisms against cellular dysfunction. Both PRC and its targets (IL1α, SPRR2D, and SPRR2F) were rapidly induced by menadione, an agent that promotes apoptosis through the generation of intracellular oxidants. Menadione-induced apoptosis and the PRC stress program were blocked by the antioxidant N-acetylcysteine. The PRC stress response was also activated by the topoisomerase I inhibitor 7-ethyl-10-hydroxycamptothecin (SN-38), an inducer of premature senescence in tumor cells. Cells treated with SN-38 displayed morphological characteristics of senescence and express senescence-associated β-galactosidase activity. In contrast to menadione, the SN-38 induction of the PRC program occurred over an extended time course and was antioxidant-insensitive. The potential adaptive function of the PRC stress response was investigated by treating cells with meclizine, a drug that promotes glycolytic energy metabolism and has been linked to cardio- and neuroprotection against ischemia-reperfusion injury. Meclizine increased lactate production and was a potent inducer of the PRC stress program, suggesting that PRC may contribute to the protective effects of meclizine. Finally, c-MYC and PRC were coordinately induced under all conditions tested, implicating c-MYC in the biological response to metabolic stress. The results suggest a general role for PRC in the adaptive response to cellular dysfunction.
Introduction
Members of the PGC-12 family of regulated coactivators (PGC-1α, PGC-1β, and PGC-1-related coactivator (PRC)) play important roles in metabolic regulation by serving as intermediaries between physiological signals and the transcriptional apparatus governing metabolic gene expression (1–4). These coactivators enhance gene expression by targeting transcription factors that regulate mitochondrial biogenesis, fatty acid oxidation, thermogenesis, gluconeogenesis, and cell proliferation. The proteins themselves can be induced transcriptionally by growth or metabolic regulatory signals, and in some cases, their function is known to be modulated post-transcriptionally by phosphorylation or acetylation (5, 6). PGC-1α, the founding member of the family, coactivates mitochondrial biogenesis by binding transcription factors directly associated with mitochondrial respiratory function, including the nuclear respiratory factors (NRF-1 and NRF-2), estrogen-related receptor α, YY1, peroxisome proliferator-activated receptor α, and MEF2C (1).
We originally identified and characterized PRC as a member of the PGC-1 coactivator family (7). Although divergent from PGC-1α and -β, it shares significant sequence conservations within discrete domains, including a potent amino-terminal activation domain and the carboxyl-terminal RNA recognition motif (7). The structural conservation and spatial arrangement of these domains are consistent with related function (8). However, despite similarities between PRC and PGC-1α in the activation of respiratory gene expression (9), PRC is not an equivalent isoform of PGC-1α but rather a functionally distinct molecule. Unlike PGC-1α, PRC is not induced significantly in brown fat during adaptive thermogenesis but is expressed at high levels upon the initiation of cell proliferation (7, 10). The rapid induction of PRC by serum growth factors in the absence of de novo protein synthesis combined with its relatively short mRNA half-life places the PRC gene (PPRC1) in the class of immediate early genes (or primary response genes) (10, 11).
PRC silencing in cultured human cells leads to respiratory chain dysfunction accompanied by abundant atypical mitochondria (12). This phenotype is commonly observed upon tissue-specific disruption of nuclear genes whose products are localized to mitochondria and are required by the mitochondrial genetic system (13–15). A gene array revealed numerous mitochondrion-related genes whose expression was altered significantly upon efficient PRC silencing (8, 12). Although germ line homozygous knock-outs of PGC-1α or -β are viable and fertile with no global changes in mitochondrial number or morphology (16–19), a germ line knock-out of the PPRC1 gene in mice results in peri-implantation lethality (20). Embryonic lethality is observed in mouse knock-outs of NRF-1, GABP(NRF-2), YY1, and other nuclear transcription factors associated with mitochondrial biogenesis (21, 22). Thus, the in vivo phenotype is consistent with the mitochondrial functional and morphological defects associated with PRC silencing.
We used carbonyl cyanide m-chlorophenylhydrazone (CCCP), a respiratory chain uncoupler, to determine the effects of mitochondrial stress on PRC expression in human U2OS cells (23). PRC is growth-regulated in these cells, and efficient PRC silencing results in phenotypic changes typical of mitochondrial dysfunction in vivo (12, 23). This cell line is also known to undergo premature senescence (24). Thus, U2OS cells represent a good model for investigating early regulatory events mediated by PRC.
In untreated cells, PRC protein levels are high upon the initiation of cell growth and drop precipitously upon achieving growth equilibrium. CCCP treatment elicits a rapid, robust, and persistent induction of PRC, a striking departure from its normal transient expression pattern (10). The PRC response was not restricted to CCCP but rather was a generalized response to multiple forms of metabolic stress, including glucose deprivation, dinitrophenol treatment (another uncoupler), and overexpression of dominant-negative NRF-1 (an inhibitor of respiratory gene expression and mitochondrial biogenesis) (23).
Differential expression screening revealed that the induction of PRC by uncoupler was accompanied by a PRC-dependent program of gene expression. This program was markedly diminished in independent lentiviral transductants in which PRC is silenced (23). The genes in the PRC stress program are involved predominantly in inflammation, cell growth, and metabolic reprogramming. Many of these PRC stress genes are common to the chronic inflammation associated with multiple age-related diseases (25). Some are postulated to promote cell survival under adverse conditions by enhancing cell growth and migration, by conferring resistance to apoptosis, and by stimulating angiogenesis (26). Several have been associated with the inflammatory microenvironment in human cancers (26–28), which is consistent with the up-regulation of PRC in human tumors (22, 29).
Notable among the PRC stress genes are those encoding IL1α and members of the small proline-rich proteins SPRR2D and SPRR2F (23). IL1α is a cytokine that mediates innate immune responses but also has an intranuclear function in controlling cell migration, proliferation, and apoptosis (30, 31). IL1-responsive genes include those encoding IL8 and cyclooxygenase 2 (31), which were also originally identified as PRC stress genes (23). SPRR2D and SPRR2F are associated with the response to DNA damage elicited by ultraviolet light exposure and exit from the cell cycle (32, 33). They provide a protective antioxidant barrier to cellular damage and thereby promote tissue remodeling in response to tissue damage in multiple systems (34, 35).
Here, we investigated the induction of the PRC stress program by agents that induce either apoptosis or senescence, two major protective mechanisms against cellular dysfunction (36). PRC and several of its target genes were rapidly induced in response to intracellular oxidants generated by menadione, a potent inducer of apoptosis (37). The PRC stress program was also activated in an oxidant-independent fashion by the topoisomerase I inhibitor 7-ethyl-10-hydroxycamptothecin (SN-38), an inducer of premature senescence in tumor cells (38). The results are consistent with a role for PRC in cellular adaptive responses triggered by oxidant and replication stress associated with apoptosis and senescence.
EXPERIMENTAL PROCEDURES
Cell Culture
U2OS cells were obtained from ATCC and maintained in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) with 10% fetal bovine serum (HyClone) and 1% penicillin-streptomycin (Invitrogen). Lentiviral shRNA U2OS cell transductants designated as control shRNA and PRC shRNA1 were described previously (12) and grown in the same medium with the inclusion of blasticidin to maintain selection. Cells were plated at a density of 1 × 106 cells/10-cm dish, grown for 24–48 h, and then subjected to treatment with various agents as follows: 40 μm CCCP (Sigma) in DMSO, 20 μm menadione (Sigma) in DMSO, 400 ng/ml SN-38 (Sigma) in DMSO, 5 mm N-acetyl-l-cysteine (NAC; Sigma) in H2O, and 50 μm meclizine dihydrochloride (MP Biomedicals, LLC) in DMSO for various times. Vehicle controls were treated with either DMSO or H2O as appropriate. Cell viability was assayed by the trypan blue dye exclusion method using a Beckman Coulter Vi-Cell. Senescence-associated β-galactosidase staining was performed at pH 6.0 using a senescence β-galactosidase staining kit (Cell Signaling Technology).
Immunoblotting
Whole cell lysates were prepared in Nonidet P-40 lysis buffer as described previously (7). Extracts were subjected to denaturing gel electrophoresis, and the proteins were transferred to nitrocellulose membranes (Schleicher & Schuell) as described (12, 23). The primary antibodies used were rabbit anti-PRC (1047-1379) (10), mouse anti-c-MYC (9E10) (Thermo Scientific), rabbit anti-NRF2α (39), rabbit anti-poly(ADP-ribose) polymerase (PARP) (cleaved; Cell Signaling Technology), mouse anti-lamin B (40) (a gift from Robert D. Goldman, Northwestern University), and mouse anti-HSP70 (a gift from Richard Morimoto, Northwestern University).
TUNEL Assay
U2OS cells were plated at a density of 106 cells/10-cm dish. After 48 h, cells were either untreated, treated with menadione, or treated with menadione plus NAC for 8 h. Cells were subjected to TUNEL assay using the APO-BrdU TUNEL assay kit (Invitrogen) according to the manufacturer's protocol. The percentage of TUNEL-positive cells was determined by flow cytometry.
ROS Assay
ROS levels were measured using 2′,7′-dichlorofluorescein diacetate (Sigma) (41). U2OS cells were plated at a density of 15,000 cells/well in a 96-well microtiter plate and allowed to grow for 24 h. Cells were washed and pretreated with 100 μm 2′,7′-dichlorofluorescein diacetate for 30 min at 37 °C. 2′,7′-Dichlorofluorescein diacetate was removed, and cells were either left untreated or treated with 20 μm menadione for various times. 2′,7′-Dichlorofluorescein fluorescence at 530 nm was measured in a Spectramax fluorescence microplate reader using an excitation wavelength of 485 nm.
Quantitative Real Time PCR
Total RNA was purified using TRIzol reagent (Invitrogen), and quantitative real time PCR was carried out as described (12, 23). The sequences of new primers designed for this study are as follows: human c-MYC: sense, 5′-CGTCTCCACACATCAGCACAA; antisense, 5′-CACTGTCCAACTTGACCCTCTTG; human HK2: sense, 5′-GAGCCACCACTCACCCTACT; antisense, 5′-CCAGGCATTCGGCAATGTG; and human NAMPT: sense, ATCCTGTTCCAGGCTATTCTGT; antisense, 5′-CCCCATATTTTCTCACACGCAT.
Lactate Assay
U2OS WT and the lentiviral transductants expressing the control shRNA or PRC shRNA1 (12) were grown in DMEM for 24 h and subjected to either SN-38 or meclizine treatment for 72 h. The concentration of l-lactate released to the culture medium was measured using a Lactate Assay kit (Biomedical Research Service Center, University at Buffalo).
RESULTS
Kinetics of PRC Stress Program Induction by CCCP
The PRC-dependent inflammatory/stress program was originally defined in U2OS cells as a response to treatment with the respiratory chain uncoupler CCCP (23). Loss of PRC function through efficient PRC silencing blocks the induction of the PRC-dependent genes by CCCP (23). The data in Fig. 1 show that the kinetics of PRC protein induction by CCCP relative to the NRF-2α negative control (Fig. 1A) coincide with the kinetics of induction of representative PRC-dependent stress genes, IL1α, SPRR2D, and SPRR2F (Fig. 1B). The PRC and TFAM negative control RNAs showed little or no change, confirming that the induction of these genes is not the result of a generalized transcriptional response. The robust induction of PRC protein in the absence of a significant change in PRC mRNA expression is consistent with post-transcriptional regulation. Interestingly, c-MYC protein was coordinately induced along with PRC, suggesting that c-MYC participates in the response to CCCP. Both PRC and c-MYC are early response genes, and MYC has been implicated in other inflammatory/stress pathways (27, 42). However, in contrast to PRC, c-MYC mRNA was also induced, suggesting that transcriptional mechanisms contribute to the up-regulation of c-MYC. The tight temporal link between the induction of PRC protein and the PRC stress genes provides corroborating proof that the program is PRC-dependent.
FIGURE 1.
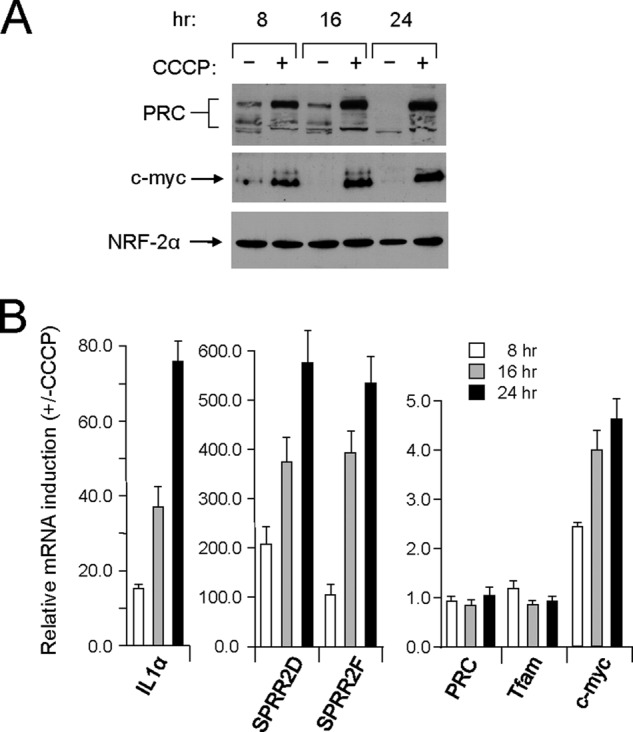
Kinetics of PRC-dependent stress program induction by CCCP. A, human log phase U2OS cells were plated and 24–48 h later (T = 0 h) were treated with either vehicle (−) or CCCP (+) for the indicated times. Total cell extracts from subconfluent cells were subjected to immunoblotting using rabbit anti-PRC, mouse anti-c-MYC, or rabbit anti-NRF-2α antibodies. B, the time course of mRNA induction for representative PRC-dependent stress genes (IL1α, SPRR2D, and SPRR2F) by CCCP was compared with that of PRC, TFAM, and c-MYC by quantitative real time PCR. RNA induction for each gene is expressed relative to the untreated control. Values are the averages ± S.E. (error bars) for at least three separate determinations.
Menadione, an Inducer of Apoptosis and the PRC Stress Program
The PRC stress response to CCCP was completely inhibited by the antioxidant NAC, suggesting that the program is redox-sensitive (23). Thus, it was of interest to determine whether the direct generation of intracellular oxidants could induce the program. Menadione can serve as a vitamin K precursor and is known to generate intracellular oxidants at multiple sites through futile redox cycling (43, 44). Menadione treatment of U2OS cells led to cell death as evidenced by cell shrinkage and the loss of viable, adherent cells (data not shown). There was an ∼25% loss of cell viability after 16 h of menadione treatment (Fig. 2A). This menadione-induced cell death did not occur in the presence of NAC (Fig. 2A), indicating that death occurs through menadione-induced oxidant toxicity.
FIGURE 2.
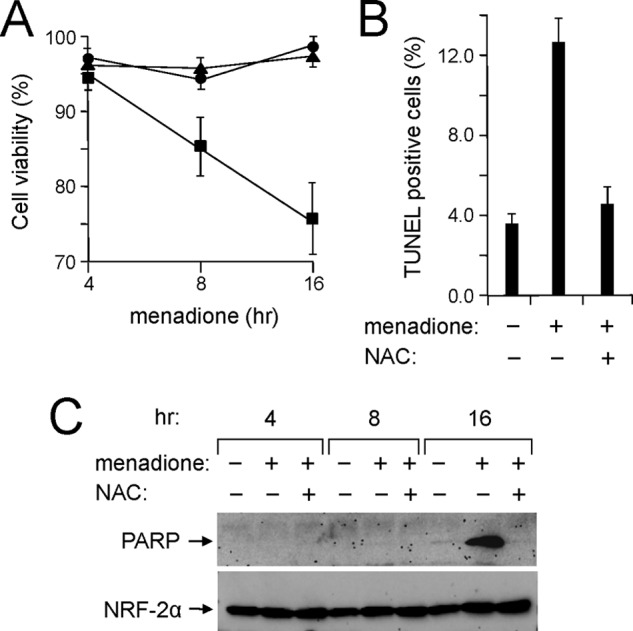
Oxidant-dependent induction of apoptosis by menadione. A, human log phase U2OS cells were plated as in Fig. 1A and treated with vehicle, menadione, or menadione plus NAC for the indicated times. Cell viability was determined by trypan blue exclusion assay. Results are presented as the percentage of viable cells relative to total cells (adherent and non-adherent) for cells treated with vehicle (filled circles), menadione (filled squares), or menadione plus NAC (filled triangles). Values are the averages ± S.E. (error bars) for at least three separate determinations. B, cells treated with menadione as in A were subjected to the TUNEL apoptosis assay. Values are the averages ± S.E. (error bars) for at least three separate determinations. C, total cell extracts from cells treated as in A were subjected to immunoblotting using rabbit anti-PARP (cleaved) or rabbit anti-NRF-2α antibodies.
Apoptotic cell death was confirmed by a severalfold increase in TUNEL-positive cells (Fig. 2B) and by the appearance of the cleaved, inactivated form of PARP (Fig. 2C), a well known marker of apoptosis (45). Cleaved PARP appeared at ∼16 h of menadione treatment when the morphological changes characteristic of apoptosis were most evident (Fig. 2C). As with the loss of cell viability, inclusion of NAC completely blocked PARP cleavage and markedly diminished the fraction of TUNEL-positive cells to control levels (Fig. 2, B and C). Thus, NAC inhibits the menadione-induced oxidant toxicity that leads to apoptotic cell death.
The effect of menadione on the PRC-dependent stress response was determined by assaying PRC protein levels by immunoblotting and the expression of representative PRC stress genes by quantitative real time PCR. As shown in Fig. 3A, menadione treatment resulted in a rapid and robust induction of PRC protein expression. As observed with CCCP, c-MYC was coordinately induced with PRC, but in contrast to PRC, c-MYC mRNA was also induced. This suggests mechanistic differences between the control of PRC and c-MYC.
FIGURE 3.

Kinetics of PRC-dependent stress program induction by menadione. A, human log phase U2OS cells were plated as in Fig. 1A and treated with vehicle, menadione, or menadione plus NAC for the indicated times. Total cell extracts from subconfluent cells were subjected to immunoblotting using rabbit anti-PRC, mouse anti-c-MYC, or rabbit anti-NRF-2α antibodies. B, ROS generation in cells treated with menadione as in A for the indicated times was measured using 2′,7′-dichlorofluorescein (DCF) diacetate. C, cells were plated and treated with menadione as in A for 16 h. Total cell extracts were subjected to immunoblotting using mouse anti-HSP70 or rabbit anti-NRF-2α antibodies. D, cells were treated as in A for the indicated times. The time course of mRNA induction for representative PRC-dependent stress genes (IL1α, SPRR2D, and SPRR2F) by menadione was compared with that of PRC, TFAM, and c-MYC by quantitative real time PCR. RNA induction for each gene is expressed relative to the untreated control. A significant difference between NAC-treated and untreated samples is indicated by an asterisk denoting a p value of <0.03. Values are the averages ± S.E. (error bars) for at least three separate determinations.
Menadione treatment was accompanied by a marked increase in menadione-dependent ROS production as measured by 2′,7′-dichlorofluorescein fluorescence (Fig. 3B). Elevated ROS levels are consistent with the inhibition of both PRC and c-MYC expression by NAC (Fig. 3A). The induction of the NRF-2α negative control was relatively unaffected by these treatments, demonstrating a high degree of specificity. The oxidant dependence was further confirmed by the finding that HSP70, a protein known to respond to oxidative stress (46), was also induced by menadione, and its induction was sensitive to NAC (Fig. 3C).
The rapid induction of PRC coincided with the marked up-regulation of the PRC-dependent stress genes, and this increase in gene expression was also completely abrogated by NAC (Fig. 3D). Although the magnitude of IL1α induction by CCCP and menadione is similar, the ∼70-fold increase in SPRR2 gene expression is about an order of magnitude lower with menadione. As with CCCP, the TFAM and PRC negative control mRNAs showed comparatively little induction by menadione. Thus, there is a strong temporal correlation between the onset of apoptosis and the specific activation of the PRC stress program by menadione. The sensitivity of both of these events to NAC supports the conclusion that they are both mediated by the menadione-dependent production of intracellular oxidants.
Premature Senescence and the PRC Stress Response
Apoptosis is a major protective mechanism against cellular dysfunction through the elimination of cells damaged by severe environmental, metabolic, or genetic stress. The current results along with previous findings (23) suggest that the PRC stress program is activated as an adaptive response to multiple forms of cellular stress. Premature senescence is a second major mechanism that protects against cellular dysfunction by driving damaged cells into a non-replicative state (36). It was of interest to determine whether the PRC stress response is activated upon growth inhibition associated with premature senescence.
SN-38 is a topoisomerase I inhibitor that induces premature senescence in tumor cells (38). SN-38 treatment of U2OS cells resulted in immediate growth arrest and a very pronounced senescent phenotype characterized by an enlarged, flattened, and granular cellular morphology (data not shown), a classic indicator of premature senescence (47). These morphological changes were accompanied by the expression of senescence-associated β-galactosidase activity (Fig. 4A), a widely used senescence marker (47). To varying degrees, nearly all of the SN-38-treated cells displayed senescence-associated β-galactosidase as punctate blue cytoplasmic staining typical of senescent cells. Senescence was further confirmed by the down-regulation of lamin B1 (Fig. 4B), a recently identified marker of senescence (40).
FIGURE 4.

Induction of premature senescence by SN-38. A, human log phase U2OS cells were plated as in Fig. 1A and treated with either vehicle or SN-38 for 96 h. Cells were stained for senescence-associated (SA) β-galactosidase activity and visualized by phase-contrast microscopy at 200×. B, total cell extracts were prepared from cells treated as in A and subjected to immunoblotting using rabbit anti-lamin B (LB) or rabbit anti-NRF-2α antibodies.
The emergence of this senescent phenotype coincided with a robust induction of PRC protein (Fig. 5A). As observed with the response to CCCP and menadione, c-MYC was coordinately induced along with PRC, and the NRF-2α negative control was relatively unaffected (Fig. 5A). Although maximal CCCP or menadione induction of PRC occurred within 8–24 h, maximal SN-38 induction of PRC occurred more gradually, peaking within 72–96 h (Fig. 5A). Also, in contrast to CCCP or menadione, neither the induction of PRC (Fig. 5B) nor the senescent phenotype (data not shown) was affected by NAC. This indicates that premature senescence mediated by SN-38 occurs through a ROS-independent mechanism. Notably, the up-regulation of the representative PRC stress genes by SN-38 followed the same extended time course as PRC protein induction (Fig. 5C), again providing a kinetic link between protein and RNA induction.
FIGURE 5.
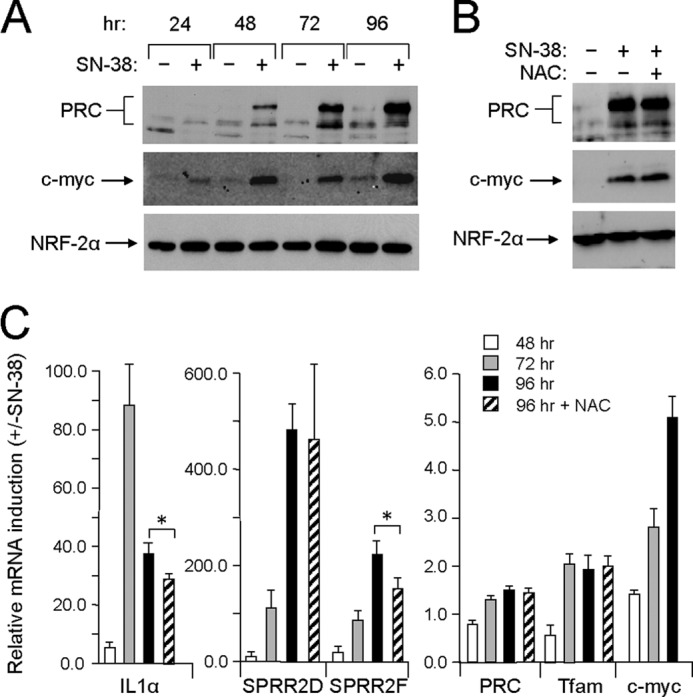
Kinetics of PRC-dependent stress program induction by SN-38. A, human log phase U2OS cells were plated as in Fig. 1A and treated with either vehicle (−) or SN-38 (+) for the indicated times. Total cell extracts from subconfluent cells were subjected to immunoblotting using rabbit anti-PRC, mouse anti-c-MYC, or rabbit anti-NRF-2α antibodies. B, cells plated as in A were treated with vehicle, SN-38, or SN-38 plus NAC for 96 h. Total cell extracts from subconfluent cells were subjected to immunoblotting using rabbit anti-PRC, mouse anti-c-MYC, or rabbit anti-NRF-2α antibodies. C, cells were treated as in A for the indicated times. The time course of mRNA induction for representative PRC-dependent stress genes (IL1α, SPRR2D, and SPRR2F) by SN-38 or SN-38 plus NAC was compared with that of PRC, TFAM, and c-MYC by quantitative real time PCR. RNA induction for each gene is expressed relative to the untreated control. A significant difference between NAC-treated and untreated samples is indicated by an asterisk denoting a p value of <0.07. Values are the averages ± S.E. (error bars) for at least three separate determinations.
The SPRR mRNAs exhibited a gradual increase between 48 and 96 h, coinciding with the appearance of PRC protein. IL1α increased between 48 and 72 h, but its expression diminished by 96 h, possibly reflecting its regulatory role as a primary inducer of inflammatory responses. In addition, the expression of the PRC stress genes showed either no or modest inhibition by NAC at 96 h (Fig. 5C), which is consistent with the observation that the senescent phenotype and PRC expression (Fig. 5B) were also unaffected by NAC. Again, in contrast to PRC, c-MYC induction was accompanied by a temporal increase in c-MYC mRNA (Fig. 5C). Thus, the induction kinetics of PRC by three different agents (CCCP, menadione, and SN-38) coincides with the induction kinetics of the PRC-dependent stress genes. This temporal relationship among kinetically different responses (rapid for CCCP and menadione and extended for SN-38) lends further credence to the causal link between PRC and the stress response program. The results establish the association of the PRC stress program with premature senescence mediated by the replication stress induced by topoisomerase I inhibition.
The connection between the senescent phenotype and the PRC stress program was examined further by assaying SN-38 induction of PRC and the PRC-dependent stress genes in a lentiviral transductant in which PRC is efficiently silenced (12). As observed in wild-type cells, PRC was markedly induced by SN-38 in a lentiviral transductant expressing a negative control shRNA (Fig. 6A). The time course of PRC induction was similar to that observed in wild-type cells (Fig. 5A). PRC expression and its induction by SN-38 were greatly reduced in the lentiviral transductant expressing PRC shRNA1 (Fig. 6A), a transductant in which PRC expression is efficiently silenced (12). The NRF-2α negative control was expressed at similar levels in both transductants and was relatively unaffected by SN-38 treatment (Fig. 6A).
FIGURE 6.

Effect of PRC silencing on the senescence-associated PRC stress program. A, lentiviral transductants of U2OS cells stably expressing either the control shRNA or PRC shRNA1 were subjected to SN-38 treatment and immunoblotting as described in the legend to Fig. 5A. B, lentiviral transductants were treated as in A for 48 or 96 h. The induction of mRNAs by SN-38 of representative PRC-dependent stress genes (IL1α, SPRR2D, and SPRR2F) was compared with that of PRC, TFAM, and c-MYC by quantitative real time PCR. RNA induction for each gene at 48 and 96 h is expressed relative to the untreated control. A significant difference between control shRNA and PRC shRNA1 transductants is indicated by an asterisk denoting a p value of <0.04. Values are the averages ± S.E. (error bars) for at least three separate determinations. C, lentiviral transductants expressing either the control shRNA or PRC shRNA1 were treated with vehicle or SN-38 for 96 h and stained for senescence-associated β-galactosidase activity. D, lentiviral transductants were treated with SN-38 for 72 h, and cell extracts were subjected to immunoblotting as described in A. Densitometric intensities of PRC or c-MYC chemiluminescence signals were normalized to that of NRF-2α using identical blotting and exposure conditions for three independent trails. A significant difference between control shRNA and PRC shRNA1 transductants is indicated by an asterisk denoting a p value of <0.02. Values are the averages ± S.E. (error bars) for three separate determinations.
The diminished SN-38 induction of PRC in the PRC shRNA1 transductant correlated with diminished induction of the PRC stress genes (Fig. 6B). This is consistent with a PRC requirement for the maximal expression of these genes. However, the effect of PRC silencing on the stress program was less pronounced than that observed with CCCP (23). This may result from the more extended time course of PRC induction by SN-38, allowing time for modest levels of PRC to accumulate in the PRC shRNA1 transductant. TFAM mRNA was expressed at similar levels in the two transductants. Although PRC mRNA was modestly induced in both transductants, its induction was far below that of PRC protein. Moreover, SN-38 treatment of both transductants resulted in the cessation of cell growth associated with a flattened, granular, senescent morphology and no difference in senescence-associated β-galactosidase staining (Fig. 6C). Therefore, PRC is required for maximal induction of the stress program but is not required for premature senescence mediated by SN-38.
PRC and the Induction of c-MYC
Interestingly, c-MYC was induced in both the control and PRC shRNA1 transductants, suggesting that PRC is not absolutely required for the up-regulation of c-MYC by SN-38. In addition, the c-MYC gene was not among the PRC-dependent genes identified in the initial screening (23), suggesting that PRC does not regulate c-MYC expression.
To estimate the potential contribution of PRC to c-MYC expression, protein extracts were prepared in triplicate from cells either left untreated or treated with SN-38 for 72 h and subjected to immunoblotting. The densitometric intensity of the PRC, c-MYC, and NRF-2α bands was determined under identical blotting and exposure conditions. As shown in Fig. 6D, the expression of PRC relative to NRF-2α in the PRC shRNA1 transductant was ∼17-fold lower than in the control shRNA transductant as expected from the efficient silencing of PRC in this cell line (12). By contrast, c-MYC expression relative to the NRF-2α control was reduced by ∼2-fold, arguing that a large reduction in PRC expression has a relatively modest effect on c-MYC induction. Thus, PRC and c-MYC appear to be regulated independently in response to SN-38. The results establish that PRC is required for maximal induction of the PRC-dependent stress program in cells experiencing premature senescence as a result of topoisomerase I inhibition.
Induction of Additional PRC Stress Genes by Menadione and SN-38
The degree of similarity between the PRC stress response elicited by menadione and that elicited by SN-38 was examined by assaying the expression of additional genes from the three main functional subcategories of the program (23). These included several more inflammatory genes (ESM1, CHRNA9, and CCL20), two genes associated with cell signaling (GNG11 and BMP6), and two genes involved in metabolism (HK2 and NAMPT). All but GNG11 were induced by menadione (Fig. 7A), whereas all seven were induced by SN-38 (Fig. 7B). As observed for the SPRR genes, the response to menadione was more blunted compared with that obtained with SN-38. For example, the inflammatory genes (ESM1, CHRNA9, and CCL20) were induced tens of fold by menadione (Fig. 7A) but hundreds of fold by SN-38 (Fig. 7B). This may reflect the overall cellular dysfunction associated with the toxicity of menadione as an apoptotic agent. The exception was HK2, which was induced to similar levels by both agents.
FIGURE 7.
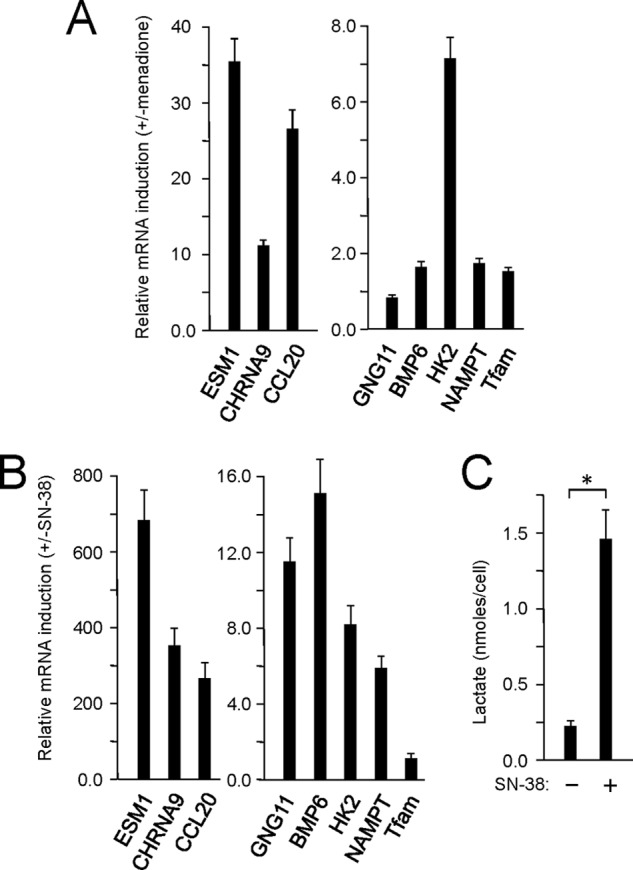
Activation of additional PRC stress response genes by menadione and SN-38. A, U2OS cells were treated with menadione as in Fig. 3 for 16 h, and the induction of additional PRC stress genes involved in inflammation (ESM1, CHRNA9, and CCL20), cell proliferation (GNG11 and BMP6), and metabolism (HK2 and NAMPT) was measured by quantitative real time PCR. B, U2OS cells were treated with SN-38 for 72 h as in Fig. 5, and the induction of the same collection of PRC stress genes as in A was measured by quantitative real time PCR. C, lactate production on a per cell basis was measured after 72 h of SN-38 treatment and compared with the vehicle-treated control. A significant difference between SN-38-treated and untreated samples is indicated by an asterisk denoting a p value of <0.03. Values in each panel are the averages ± S.E. (error bars) for at least three separate determinations.
Interestingly, c-MYC, which is an activator of both HK2 and NAMPT as well as other glycolytic genes, was induced at the RNA level by both agents (Figs. 3B and 5C), suggesting that the PRC program may be associated with a shift to glycolytic metabolism. This is consistent with the observation that lactate production on a per cell basis was markedly increased in the medium of SN-38-treated cells (Fig. 7C). This suggests that in addition to up-regulating genes involved in inflammation and cell signaling part of the adaptive function of the PRC stress program may be to facilitate metabolic reprogramming.
Induction of the PRC Stress Program by Meclizine
A recent screen of over 3500 compounds identified meclizine as an agent that inhibits oxidative phosphorylation and facilitates a shift to predominantly glycolytic metabolism (48). Meclizine accomplishes this metabolic shift without inhibiting the mitochondrial respiratory chain directly. As shown in Fig. 8A, meclizine treatment of U2OS cells resulted in a severalfold increase in lactate production, indicating that it promotes glycolysis in these cells. Because several PRC stress genes are linked to metabolic reprogramming (23), it was of interest to determine whether meclizine can induce the PRC stress response. As shown in Fig. 8B, meclizine was a potent inducer of PRC protein expression. PRC was induced at 24 h of meclizine treatment, and this induction was enhanced at 72h. As observed with the other agents, c-MYC protein expression was elevated coordinately along with that of PRC under conditions where the NRF-2α negative control was relatively unchanged.
FIGURE 8.

Activation of the PRC stress program by meclizine. A, lactate production on a per cell basis was measured in wild-type U2OS cells after 72 h of meclizine treatment and compared with that of the vehicle-treated control. A significant difference between meclizine-treated and untreated samples is indicated by an asterisk denoting a p value of <0.0001. Values are the averages ± S.E. (error bars) for three separate determinations. B, human log phase U2OS cells were plated as in Fig. 1A and treated with either vehicle (−) or meclizine (+) for the indicated times. Total cell extracts from subconfluent cells were subjected to immunoblotting using rabbit anti-PRC, mouse anti-c-MYC, or rabbit anti-NRF-2α antibodies. C, human log phase U2OS cells were plated as in Fig. 1A and treated with either vehicle (−) or meclizine (+) for 24 h to induce PRC. Cells were then washed and grown in medium containing meclizine (+ meclizine, −cycloheximide), in medium in which meclizine was removed (−meclizine, −cycloheximide), or in medium containing cycloheximide (−meclizine, + cycloheximide). Total cell extracts were prepared at various times and subjected to immunoblotting using rabbit anti-PRC or rabbit anti-NRF-2α antibodies. D, U2OS cells were treated with either vehicle or meclizine for 72 h. The induction of representative PRC-dependent stress genes (IL1α, SPRR2D, SPRR2F, HK2, and NAMPT) was compared with that of PRC, TFAM, and c-MYC by quantitative real time PCR. Values are the averages ± S.E. (error bars) for at least three separate determinations. E, lentiviral transductants expressing either the control shRNA or PRC shRNA1 were treated as in D for 72 h. The meclizine induction of the same genes in D was determined by quantitative real time PCR for each transductant. Values represent the percent decrease of meclizine induction in the PRC shRNA1 transductant relative to the control. A significant difference in meclizine induction between the control shRNA and PRC shRNA1 transductants is indicated by an asterisk denoting a p value of <0.0002. Values are the averages ± S.E. (error bars) for at least three separate determinations. F, lactate production on a per cell basis was measured after 72 h of meclizine treatment as in A for lentiviral transductants expressing either the control shRNA or PRC shRNA1. A significant difference between meclizine-treated and untreated samples is indicated by an asterisk denoting a p value of <0.04. Values are the averages ± S.E. (error bars) for at least three separate determinations.
PRC protein exhibited a time-dependent increase in expression when meclizine persisted in the growth medium (Fig. 8C). However, when PRC was induced for 24 h followed by removal of meclizine, PRC protein declined to uninduced levels within 16 h. Thus, meclizine induction of PRC is reversible, and the relatively rapid turnover of PRC is similar to that observed following the initiation of cell growth (23). When meclizine was replaced by cycloheximide, PRC protein levels initially declined but then increased in a time-dependent fashion most likely because of the stress imposed by the inhibition of protein synthesis (Fig. 8C). Thus, de novo protein synthesis is not required for PRC induction, which is consistent with a post-translational mode of regulation.
The induction of PRC by meclizine was accompanied by the induction of representative PRC stress genes (Fig. 8D). The inflammatory genes IL1α, SPRR2D, and -F were dramatically up-regulated along with more modest increases in HK2 and NAMPT. PRC and TFAM displayed less than a 2-fold induction. The effect of PRC silencing on the regulation of the PRC stress genes was determined by assaying the induction of these genes in lentiviral transductants in which PRC is normally expressed (control shRNA) or silenced (PRC shRNA1). The results are presented as the percentage of inhibition of meclizine inducibility by PRC silencing. The data in Fig. 8E show that meclizine induction of the PRC stress genes (IL1α, SPRR2D, and -F) was nearly completely blocked by PRC silencing. Meclizine induction of HK2 and NAMPT was also inhibited by PRC silencing although to a lesser degree than the inflammatory genes under conditions where PRC showed no inhibition. Although TFAM was only modestly induced by meclizine, its induction was diminished by PRC silencing. These results demonstrate that the induction of the PRC stress response by meclizine is PRC-dependent and that it is associated with a shift to increased lactate production.
The potential direct contribution of PRC to glycolytic function was tested by comparing the level of lactate production on a per cell basis in lentiviral transductants in which PRC is either normally expressed (control shRNA) or efficiently silenced (PRC shRNA1). The data in Fig. 8F show that in the absence of meclizine lactate production was about 2-fold higher in the transductant in which PRC is silenced by PRC shRNA1. This difference likely reflects an adaptation to the severe mitochondrial dysfunction in this cell line (12, 23). However, meclizine treatment increased the level of lactate production by 2-fold in both transductants, arguing that PRC silencing alone does not block the shift to increased glycolysis. This may be explained by a compensatory effect mediated by the induction of c-MYC, which is a transcriptional activator of several glycolytic genes (49). Thus, meclizine is a potent inducer of the PRC stress program, but PRC alone does not appear to mediate the meclizine-dependent transition to glycolytic metabolism.
DISCUSSION
This work demonstrates that a PRC-dependent stress response is temporally linked to apoptosis or premature senescence brought about by oxidative or replication stress, respectively. We initially defined this PRC response as a rapid and robust increase in PRC protein expression by the respiratory chain uncoupler CCCP. The up-regulation of PRC was accompanied by the induction of PRC-dependent genes identified by exploiting the differential response to uncoupler-induced metabolic stress in lentiviral transductants in which PRC is either normally expressed (control shRNA) or efficiently silenced (PRC shRNA1) (23). A subgroup of inflammation/stress genes displayed the highest degree of PRC dependence. The results presented here suggest that this PRC stress program may be part of an adaptive response linked to apoptosis and premature senescence, two physiologically significant protective mechanisms against cellular dysfunction (36).
The PRC stress response to CCCP was completely abolished when cells were treated in the presence of the antioxidant NAC, suggesting that signaling to PRC may occur through ROS (23). Menadione, a precursor in the synthesis of vitamin K, generates intracellular ROS through redox cycling and is used to generate intracellular oxidants at multiple sites (43, 44). It is a potent inducer of apoptosis and has been used as a chemotherapeutic agent (37, 44). Cancer cells display a high sensitivity to oxidative stress, and there has been interest in using menadione as a proapoptotic agent that selectively targets cancer cells (50).
The rapid induction of PRC and several representative PRC stress genes in response to menadione was similar to that observed with CCCP. The induction of the PRC stress response and the apoptotic effects of menadione were associated with increased ROS levels and were completely blocked by NAC, arguing that intracellular oxidants are potent mediators of the PRC stress program. The induction of the program by uncoupler likely operates through intracellular oxidant production as well. The implication is that other agents and conditions that generate oxidative stress are likely to trigger the PRC stress response. It is possible that one function of PRC is to confer resistance to oxidant stress possibly through the massive up-regulation of the SPRR genes. Thus, inhibitors of PRC may increase the potency of therapeutic agents that work through metabolic or replication stress. This program may partly account for the chronic inflammation that is a hallmark of aged tissues (51).
A second major protective mechanism for neutralizing the effects of severe stress is premature senescence (36). One strategy for blocking oncogenic proliferation is to utilize agents that drive cells into a nonreplicative, senescent state (52), although it is not clear that this is universally beneficial (53). Nevertheless, human U2OS cells are known to undergo premature senescence, demonstrating that this program can be reactivated in these tumor cells (24). SN-38 is the active metabolite of irinotecan, a topoisomerase I inhibitor that is used in the treatment of colorectal cancer (54).
Here, we show that SN-38 is a potent inducer of both premature senescence and the PRC stress program in U2OS cells. Cells treated with SN-38 exited the cell cycle and displayed the flattened, granular, and enlarged morphology that is typical of senescent cells. Although there is a strong kinetic relationship between the induction of PRC protein and several representative PRC-dependent stress genes by SN-38, the more extended time course of the response compared with that with CCCP or menadione is suggestive of mechanistic differences in signaling.
The fact that the response to SN-38 was antioxidant-insensitive may indicate that the direct topoisomerase I inhibition by SN-38 is downstream from ROS signaling. Although it is generally believed that intracellular ROS plays an important role in inducing cellular senescence (55), hyperoxia-induced premature senescence that is independent of mitochondrial ROS has been observed (55, 56). The different kinetic responses may also reflect differences in the site of action (nuclear or cytoplasmic) of the inducer. Despite these differences, there is a striking correlation between the appearance of PRC protein and the induction of the stress response genes by each agent. This along with the fact that the PRC program was inhibited by PRC silencing argues that PRC mediates the effects on gene expression.
A hallmark of senescent cells is the senescence-associated secretory phenotype defined by a marked increase in the secretion of proinflammatory cytokines (51). This senescence-associated cytokine network is in part regulated by IL1α, a cytokine that functions as an upstream regulator of the secretion of the proinflammatory cytokines, IL6, and IL8. The latter are thought to contribute to senescence growth arrest (57). It is of interest that both IL1α and IL8 were identified as PRC-dependent inflammatory stress genes (23). Induction of IL1α expression in senescent U2OS cells was particularly sensitive to PRC silencing. In addition, the PRC stress gene CCL20 was abundantly up-regulated in senescent U2OS cells (Fig. 7B). CCL20 is one of the CCL chemokine family members that are frequently up-regulated in senescent cells (51). These findings suggest that PRC may play a role in promoting a senescence-associated secretory phenotype-like response.
It has been suggested that the senescence-associated secretory phenotype may signal cellular damage to surrounding tissue for the purpose of stimulating repair (58). Potential PRC involvement in this function is consistent with the finding that the SPRR2D and -F genes are among the most highly induced PRC stress genes (23). The SPRR family is associated with the response to DNA damage and exit from the cell cycle (32, 33). They also provide a protective antioxidant barrier to cellular damage, thereby promoting tissue remodeling (34, 35). This appears to be a generalized response to stress or injury in many tissues (34). Thus, the PRC stress program may be part of an integrated response to cellular damage.
A potential protective function for PRC is supported by the observation that meclizine was a very effective inducer of PRC and the PRC stress genes. As with the other agents, meclizine induction of the program was highly sensitive to PRC silencing. The induction of PRC by meclizine was reversible, indicating that PRC turns over rapidly in the absence of inducer. Moreover, PRC induction occurred in the presence of cycloheximide, indicating that de novo protein synthesis is not required for PRC up-regulation. These observations along with the fact that PRC expression is massively increased by proteasome inhibitors (23) are consistent with a post-translational regulatory mechanism.
Meclizine was identified as a Food and Drug Administration-approved drug that has low toxicity and redirects energy metabolism toward glycolysis. This occurs through an unknown mechanism that does not involve inactivation of the hypoxia-inducible factor pathway (48). Shifting cellular metabolism from mitochondrial respiration to glycolysis may have therapeutic potential for protecting tissues from ischemia-reperfusion injury (48). Although HK2 was identified as a PRC stress gene in U2OS cells (23), PRC silencing did not inhibit the meclizine-induced increase in lactate production. Thus, although HK2 gene expression was increased upon PRC induction by all of the agents described here, PRC may not be limiting for a shift to glycolysis in this system. It is possible that c-MYC plays a role because it was also strongly induced by meclizine and acts on the promoters of a number of glycolytic genes, including HK2 (49). PRC may contribute to the protective effects of meclizine by inducing IL1α, the SPRR2 genes, or other genes associated with increased PRC expression.
A generalized response to oxidative stress by the SPRR proteins may be protective through their recently characterized antioxidant properties (35). The SPRR proteins are expressed in many tissues subjected to stress or injury (34), and SPRR1A has been identified as a stress-inducible cardioprotective protein in hearts subjected to ischemic injury (59). Interestingly, cytokines have been implicated in the myocardial stress response, and SPRR1A is a downstream target of IL6 signaling through the gp130 receptor. Thus, potential PRC control of SPRR gene expression may be an adaptive mechanism of protection from oxidative damage. However, this pathway may also be maladaptive in fostering the survival of damaged cells that are destined for neutralization by apoptosis or senescence. This may explain the increased PRC expression found in human cancers (22, 29) and the expression of several PRC stress genes as part of the inflammatory microenvironment in human tumors (26–28).
Several transcription factors associated with the regulation of mitochondrial biogenesis (PRC, NRF-1, NRF-2α, NRF-2β, Sp1, c-MYC, and TFAM) were tested for their inducibility by uncoupler (23). Of these, PRC and c-MYC were the only factors induced by CCCP. Here, we establish a tight kinetic link between the induction of PRC and the induction of c-MYC in response to CCCP, menadione, SN-38, and meclizine in spite of the fact that these agents act on different cellular subcompartments and have different mechanisms of action. PRC and c-MYC were coordinately up-regulated rapidly during menadione-induced apoptosis, and both were equally sensitive to antioxidant. Moreover, they both followed the same extended time course of induction in response to SN-38-mediated premature senescence. However, the two proteins differ in that c-MYC protein and mRNA were induced by each treatment, whereas PRC protein induction appears to be entirely post-transcriptional with little or no change in PRC mRNA expression. This suggests that there are differences in the signaling mechanisms that control expression. There may also be a post-transcriptional component to c-MYC induction because both proteins appeared concomitantly. MYC stabilization by the proteasome system has been linked to tumor cell proliferation (60).
These results are suggestive of a potential functional relationship between PRC and c-MYC in mediating the response to cellular stress. It seems unlikely that PRC is absolutely required for c-MYC induction because a large decrease in PRC protein levels through stable PRC silencing had a comparatively modest effect on the induction of c-MYC protein levels by SN-38. Also, because c-MYC is a transcription factor, it seems unlikely to act as a direct regulator of the rapid post-transcriptional induction of PRC. It is possible that PRC acts as a c-MYC coactivator to enhance its transcriptional activity or to modulate promoter specificity. Recent studies have implicated MYC in promoting DNA replication through checkpoint activation under conditions of replication stress (61, 62). Such a function may account for the robust induction of c-MYC by a wide range of stress inducers. It is notable in this context that PRC exists in a complex with host cell factor, an abundant chromatin-associated protein that is required for cell cycle progression (39, 63). It is tempting to speculate that PRC is part of the mechanism controlling the cell cycle under conditions of metabolic stress.
Acknowledgments
We acknowledge the assistance of the Center for Genetic Medicine and the Flow Cytometry Facility at Northwestern Medical School. We also thank Northwestern University colleagues Dr. Paul Schumacker for helpful discussions and Drs. Robert Goldman and Richard Morimoto for antibodies.
This work was supported, in whole or in part, by National Institutes of Health Grant GM 32525-29 from the NIGMS.
- PGC
- peroxisome proliferator-activated receptor γ coactivator
- PRC
- PGC-1-related coactivator
- CCCP
- carbonyl cyanide m-chlorophenylhydrazone
- NAC
- N-acetyl-l-cysteine
- PARP
- poly(ADP-ribose) polymerase
- NRF
- nuclear respiratory factor
- TFAM
- mitochondrial transcription factor A
- ROS
- reactive oxygen species
- SN-38
- 7-ethyl-10-hydroxycamptothecin
- SPRR
- small proline-rich.
REFERENCES
- 1. Scarpulla R. C., Vega R. B., Kelly D. P. (2012) Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab. 23, 459–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Patten I. S., Arany Z. (2012) PGC-1 coactivators in the cardiovascular system. Trends Endocrinol. Metab. 23, 90–97 [DOI] [PubMed] [Google Scholar]
- 3. Giguère V. (2008) Transcriptional control of energy homeostasis by the estrogen-related receptors. Endocr. Rev. 29, 677–696 [DOI] [PubMed] [Google Scholar]
- 4. Rodgers J. T., Lerin C., Gerhart-Hines Z., Puigserver P. (2008) Metabolic adaptations through the PGC-1α and SIRT1 pathways. FEBS Lett. 582, 46–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cantó C., Gerhart-Hines Z., Feige J. N., Lagouge M., Noriega L., Milne J. C., Elliott P. J., Puigserver P., Auwerx J. (2009) AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458, 1056–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dominy J. E., Gerhart-Hines Z., Puigserver P. (2011) Nutrient-dependent acetylation controls basic regulatory metabolic switches and cellular reprogramming. Cold Spring Harb. Symp. Quant. Biol. 76, 203–209 [DOI] [PubMed] [Google Scholar]
- 7. Andersson U., Scarpulla R. C. (2001) Pgc-1-related coactivator, a novel, serum-inducible coactivator of nuclear respiratory factor 1-dependent transcription in mammalian cells. Mol. Cell. Biol. 21, 3738–3749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Scarpulla R. C. (2011) Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 1813, 1269–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gleyzer N., Vercauteren K., Scarpulla R. C. (2005) Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol. Cell. Biol. 25, 1354–1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vercauteren K., Pasko R. A., Gleyzer N., Marino V. M., Scarpulla R. C. (2006) PGC-1-related coactivator: immediate early expression and characterization of a CREB/NRF-1 binding domain associated with cytochrome c promoter occupancy and respiratory growth. Mol. Cell. Biol. 26, 7409–7419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fowler T., Sen R., Roy A. L. (2011) Regulation of primary response genes. Mol. Cell 44, 348–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vercauteren K., Gleyzer N., Scarpulla R. C. (2009) Short hairpin RNA-mediated silencing of PRC (PGC-1-related coactivator) results in a severe respiratory chain deficiency associated with the proliferation of aberrant mitochondria. J. Biol. Chem. 284, 2307–2319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Larsson N. G., Wang J., Wilhelmsson H., Oldfors A., Rustin P., Lewandoski M., Barsh G. S., Clayton D. A. (1998) Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 18, 231–236 [DOI] [PubMed] [Google Scholar]
- 14. Park C. B., Asin-Cayuela J., Cámara Y., Shi Y., Pellegrini M., Gaspari M., Wibom R., Hultenby K., Erdjument-Bromage H., Tempst P., Falkenberg M., Gustafsson C. M., Larsson N. G. (2007) MTERF3 is a negative regulator of mammalian mtDNA transcription. Cell 130, 273–285 [DOI] [PubMed] [Google Scholar]
- 15. Metodiev M. D., Lesko N., Park C. B., Cámara Y., Shi Y., Wibom R., Hultenby K., Gustafsson C. M., Larsson N. G. (2009) Methylation of 12S rRNA is necessary for in vivo stability of the small subunit of the mammalian mitochondrial ribosome. Cell Metab. 9, 386–397 [DOI] [PubMed] [Google Scholar]
- 16. Lin J., Wu P. H., Tarr P. T., Lindenberg K. S., St-Pierre J., Zhang C. Y., Mootha V. K., Jäger S., Vianna C. R., Reznick R. M., Cui L., Manieri M., Donovan M. X., Wu Z., Cooper M. P., Fan M. C., Rohas L. M., Zavacki A. M., Cinti S., Shulman G. I., Lowell B. B., Krainc D., Spiegelman B. M. (2004) Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1α null mice. Cell 119, 121–135 [DOI] [PubMed] [Google Scholar]
- 17. Leone T. C., Lehman J. J., Finck B. N., Schaeffer P. J., Wende A. R., Boudina S., Courtois M., Wozniak D. F., Sambandam N., Bernal-Mizrachi C., Chen Z., Holloszy J. O., Medeiros D. M., Schmidt R. E., Saffitz J. E., Abel E. D., Semenkovich C. F., Kelly D. P. (2005) PGC-1α deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 3, e101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lelliott C. J., Medina-Gomez G., Petrovic N., Kis A., Feldmann H. M., Bjursell M., Parker N., Curtis K., Campbell M., Hu P., Zhang D., Litwin S. E., Zaha V. G., Fountain K. T., Boudina S., Jimenez-Linan M., Blount M., Lopez M., Meirhaeghe A., Bohlooly-Y M., Storlien L., Strömstedt M., Snaith M., Oresic M., Abel E. D., Cannon B., Vidal-Puig A. (2006) Ablation of PGC-1β results in defective mitochondrial activity, thermogenesis, hepatic function, and cardiac performance. PLoS Biol. 4, e369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sonoda J., Mehl I. R., Chong L. W., Nofsinger R. R., Evans R. M. (2007) PGC-1β controls mitochondrial metabolism to modulate circadian activity, adaptive thermogenesis, and hepatic steatosis. Proc. Natl. Acad. Sci. U.S.A. 104, 5223–5228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. He X., Sun C., Wang F., Shan A., Guo T., Gu W., Cui B., Ning G. (2012) Peri-implantation lethality in mice lacking the PGC-1-related coactivator protein. Dev. Dyn. 241, 975–983 [DOI] [PubMed] [Google Scholar]
- 21. Scarpulla R. C. (2008) Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 88, 611–638 [DOI] [PubMed] [Google Scholar]
- 22. Scarpulla R. C. (2012) Nucleus-encoded regulators of mitochondrial function: integration of respiratory chain expression, nutrient sensing and metabolic stress. Biochim. Biophys. Acta 1819, 1088–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gleyzer N., Scarpulla R. C. (2011) PGC-1-related coactivator (PRC), a sensor of metabolic stress, orchestrates a redox-sensitive program of inflammatory gene expression. J. Biol. Chem. 286, 39715–39725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bihani T., Mason D. X., Jackson T. J., Chen S. C., Boettner B., Lin A. W. (2004) Differential oncogenic Ras signaling and senescence in tumor cells. Cell Cycle 3, 1201–1207 [PubMed] [Google Scholar]
- 25. Chung H. Y., Cesari M., Anton S., Marzetti E., Giovannini S., Seo A. Y., Carter C., Yu B. P., Leeuwenburgh C. (2009) Molecular inflammation: underpinnings of aging and age-related diseases. Ageing Res. Rev. 8, 18–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mantovani A., Garlanda C., Allavena P. (2010) Molecular pathways and targets in cancer-related inflammation. Ann. Med. 42, 161–170 [DOI] [PubMed] [Google Scholar]
- 27. Borrello M. G., Degl'Innocenti D., Pierotti M. A. (2008) Inflammation and cancer: the oncogene-driven connection. Cancer Lett. 267, 262–270 [DOI] [PubMed] [Google Scholar]
- 28. Grivennikov S. I., Greten F. R., Karin M. (2010) Immunity, inflammation, and cancer. Cell 140, 883–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Savagner F., Mirebeau D., Jacques C., Guyetant S., Morgan C., Franc B., Reynier P., Malthièry Y. (2003) PGC-1-related coactivator and targets are upregulated in thyroid oncocytoma. Biochem. Biophys. Res. Commun. 310, 779–784 [DOI] [PubMed] [Google Scholar]
- 30. Luheshi N. M., Rothwell N. J., Brough D. (2009) Dual functionality of interleukin-1 family cytokines: implications for anti-interleukin-1 therapy. Br. J. Pharmacol. 157, 1318–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Weber A., Wasiliew P., Kracht M. (2010) Interleukin-1 (IL-1) pathway. Sci. Signal. 3, cm1. [DOI] [PubMed] [Google Scholar]
- 32. Lohman F. P., Medema J. K., Gibbs S., Ponec M., van de Putte P., Backendorf C. (1997) Expression of the SPRR cornification genes is differentially affected by carcinogenic transformation. Exp. Cell Res. 231, 141–148 [DOI] [PubMed] [Google Scholar]
- 33. Tesfaigzi J., Carlson D. M. (1999) Expression, regulation, and function of the SPR family of proteins. A review. Cell Biochem. Biophys. 30, 243–265 [DOI] [PubMed] [Google Scholar]
- 34. Vermeij W. P., Backendorf C. (2010) Skin cornification proteins provide global link between ROS detoxification and cell migration during wound healing. PLoS One 5, e11957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vermeij W. P., Alia A., Backendorf C. (2011) ROS quenching potential of the epidermal cornified cell envelope. J. Invest. Dermatol. 131, 1435–1441 [DOI] [PubMed] [Google Scholar]
- 36. Kuilman T., Michaloglou C., Mooi W. J., Peeper D. S. (2010) The essence of senescence. Genes Dev. 24, 2463–2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Criddle D. N., Gillies S., Baumgartner-Wilson H. K., Jaffar M., Chinje E. C., Passmore S., Chvanov M., Barrow S., Gerasimenko O. V., Tepikin A. V., Sutton R., Petersen O. H. (2006) Menadione-induced reactive oxygen species generation via redox cycling promotes apoptosis of murine pancreatic acinar cells. J. Biol. Chem. 281, 40485–40492 [DOI] [PubMed] [Google Scholar]
- 38. te Poele R. H., Okorokov A. L., Jardine L., Cummings J., Joel S. P. (2002) DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res. 62, 1876–1883 [PubMed] [Google Scholar]
- 39. Vercauteren K., Gleyzer N., Scarpulla R. C. (2008) PGC-1-related coactivator complexes with HCF-1 and NRF-2β in mediating NRF-2(GABP)-dependent respiratory gene expression. J. Biol. Chem. 283, 12102–12111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shimi T., Butin-Israeli V., Adam S. A., Hamanaka R. B., Goldman A. E., Lucas C. A., Shumaker D. K., Kosak S. T., Chandel N. S., Goldman R. D. (2011) The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev. 25, 2579–2593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang H., Joseph J. A. (1999) Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radic. Biol. Med. 27, 612–616 [DOI] [PubMed] [Google Scholar]
- 42. Whitfield J. R., Soucek L. (2012) Tumor microenvironment: becoming sick of Myc. Cell. Mol. Life Sci. 69, 931–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Verrax J., Cadrobbi J., Marques C., Taper H., Habraken Y., Piette J., Calderon P. B. (2004) Ascorbate potentiates the cytotoxicity of menadione leading to an oxidative stress that kills cancer cells by a non-apoptotic caspase-3 independent form of cell death. Apoptosis 9, 223–233 [DOI] [PubMed] [Google Scholar]
- 44. Loor G., Kondapalli J., Schriewer J. M., Chandel N. S., Vanden Hoek T. L., Schumacker P. T. (2010) Menadione triggers cell death through ROS-dependent mechanisms involving PARP activation without requiring apoptosis. Free Radic. Biol. Med. 49, 1925–1936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Oliver F. J., de la Rubia G., Rolli V., Ruiz-Ruiz M. C., de Murcia G., Murcia J. M. (1998) Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson from an uncleavable mutant. J. Biol. Chem. 273, 33533–33539 [DOI] [PubMed] [Google Scholar]
- 46. Calabrese V., Cornelius C., Cuzzocrea S., Iavicoli I., Rizzarelli E., Calabrese E. J. (2011) Hormesis, cellular stress response and vitagenes as critical determinants in aging and longevity. Mol. Aspects Med. 32, 279–304 [DOI] [PubMed] [Google Scholar]
- 47. Muller M. (2009) Cellular senescence: molecular mechanisms, in vivo significance, and redox considerations. Antioxid. Redox. Signal. 11, 59–98 [DOI] [PubMed] [Google Scholar]
- 48. Gohil V. M., Sheth S. A., Nilsson R., Wojtovich A. P., Lee J. H., Perocchi F., Chen W., Clish C. B., Ayata C., Brookes P. S., Mootha V. K. (2010) Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nat. Biotechnol. 28, 249–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim J. W., Zeller K. I., Wang Y., Jegga A. G., Aronow B. J., O'Donnell K. A., Dang C. V. (2004) Evaluation of myc E-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol. Cell. Biol. 24, 5923–5936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Verrax J., Pedrosa R. C., Beck R., Dejeans N., Taper H., Calderon P. B. (2009) In situ modulation of oxidative stress: a novel and efficient strategy to kill cancer cells. Curr. Med. Chem. 16, 1821–1830 [DOI] [PubMed] [Google Scholar]
- 51. Davalos A. R., Coppe J. P., Campisi J., Desprez P. Y. (2010) Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. 29, 273–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Roninson I. B. (2003) Tumor cell senescence in cancer treatment. Cancer Res. 63, 2705–2715 [PubMed] [Google Scholar]
- 53. Kahlem P., Dörken B., Schmitt C. A. (2004) Cellular senescence in cancer treatment: friend or foe? J. Clin. Investig. 113, 169–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gilbert D. C., Chalmers A. J., El-Khamisy S. F. (2012) Topoisomerase I inhibition in colorectal cancer: biomarkers and therapeutic targets. Br. J. Cancer 106, 18–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Stöckl P., Zankl C., Hütter E., Unterluggauer H., Laun P., Heeren G., Bogengruber E., Herndler-Brandstetter D., Breitenbach M., Jansen-Dürr P. (2007) Partial uncoupling of oxidative phosphorylation induces premature senescence in human fibroblasts and yeast mother cells. Free Radic. Biol. Med. 43, 947–958 [DOI] [PubMed] [Google Scholar]
- 56. Klimova T. A., Bell E. L., Shroff E. H., Weinberg F. D., Snyder C. M., Dimri G. P., Schumacker P. T., Budinger G. R., Chandel N. S. (2009) Hyperoxia-induced premature senescence requires p53 and pRb, but not mitochondrial matrix ROS. FASEB J. 23, 783–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Orjalo A. V., Bhaumik D., Gengler B. K., Scott G. K., Campisi J. (2009) Cell surface-bound IL-1α is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc. Natl. Acad. Sci. U.S.A. 106, 17031–17036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Campisi J. (2011) Cellular senescence: putting the paradoxes in perspective. Curr. Opin. Genet. Dev. 21, 107–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pradervand S., Yasukawa H., Muller O. G., Kjekshus H., Nakamura T., St Amand T. R., Yajima T., Matsumura K., Duplain H., Iwatate M., Woodard S., Pedrazzini T., Ross J., Firsov D., Rossier B. C., Hoshijima M., Chien K. R. (2004) Small proline-rich protein 1A is a gp130 pathway- and stress-inducible cardioprotective protein. EMBO J. 23, 4517–4525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Popov N., Wanzel M., Madiredjo M., Zhang D., Beijersbergen R., Bernards R., Moll R., Elledge S. J., Eilers M. (2007) The ubiquitin-specific protease USP28 is required for MYC stability. Nat. Cell Biol. 9, 765–774 [DOI] [PubMed] [Google Scholar]
- 61. Eilers M., Eisenman R. N. (2008) Myc's broad reach. Genes Dev. 22, 2755–2766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Herold S., Herkert B., Eilers M. (2009) Facilitating replication under stress: an oncogenic function of MYC? Nat. Rev. Cancer 9, 441–444 [DOI] [PubMed] [Google Scholar]
- 63. Wysocka J., Herr W. (2003) The herpes simplex virus VP16-induced complex: the makings of a regulatory switch. Trends Biochem. Sci. 28, 294–304 [DOI] [PubMed] [Google Scholar]